
Fig. 18.1 Haematopoiesis. Reproduced from Qasim et al. Normal haematopoiesis and the concept of stem cell transplantation. Expert Reviews in Molecular Medicine. Vol. 6; Issue 13; 2 July 2004.
Haematology is a unique speciality that combines both medicine and pathology. Haematologists manage patients with diseases affecting the bone marrow, blood, and lymphatic system. As well as treating patients with primary haematological disease, haematologists are essential to ensure the safe and effective running of hospitals by managing anticoagulation and transfusion services.
With most haematological malignancies, the diagnosis and initial assessment can be made either as an inpatient or outpatient. In the presence of certain complications, these patients will need to be treated as inpatients, as well as with some more intensive regimens of chemotherapy.
Patients often present to hospital with signs and symptoms of bleeding although investigations and treatment can often occur as an outpatient.
Is a rare but fatal disorder of haemostasis that presents acutely to hospitals and requires urgent treatment.
Patients attend clinic regularly and are admitted for management of complications such as a sickle cell chest crisis or strokes.
Patients with a variety of medical problems require anticoagulation. Understanding the rationale and duration of anticoagulation, as well as the recommended agent and reversal of anticoagulation, is important.
Blood transfusions are commonly administered on surgical wards, ICUs, and medical units. Understanding the theory behind transfusions, the indications for transfusions, and the recognition and management of complications is vital.
Is an essential diagnostic tool in medicine and haematology and provides important qualitative and quantitative information.
Is a liquid sample of bone marrow taken and smeared on a slide and stained to primarily observe the morphology of the bone marrow cells.
A bone marrow biopsy (or trephine) is a core of bone tissue which allows the observation of the architecture of the tissue as well as morphology.
Are an important place to learn about haematology. Most hospitals will have a laboratory and blood bank and the scientists there will be happy to teach you about the laboratory and techniques used. Understanding how a test works and why it is used is an excellent way to gain a deeper knowledge of medicine and haematology.
Most haematology departments have a day unit where patients attend for chemotherapy, blood products, and other treatments. Attending the day unit will give you an opportunity to speak to and examine more physiologically stable patients.
As with all specialities, attending a MDT meeting will allow you to see a breadth of cases and to observe the diverse expertise required to care for patients with haematological disease. Get a list of cases beforehand to familiarize yourself with the cases using the patient’s notes.
Blood films examination of a peripheral blood film is an essential part of a haematologist’s work. Take time to read the following terms, see examples and to think about why the abnormalities occur, what the abnormalities mean, and in which diseases the abnormalities are detected. Morphological abnormalities occur in primary haematological disorders as well as disorders of other systems. The following are examples of morphological terms that are of importance and terms you need to be familiar with.
• Anisocytosis: variation in RBC size—iron deficiency, thalassaemia, vitamin B12/folate deficiency.
• Blasts: refers to immature blood precursor cells not normally present in the blood: leukaemia, myelofibrosis.
• Fragmented RBC: irregular, broken RBC—microangiopathic haemolysis.
• Haematopoiesis: the process of haematopoiesis is an important concept in haematology.
• Howell–Jolly bodies: DNA inclusions in the RBCs—hyposplenism.
• Hypochromia: pale RBCs due to reduced haemoglobinization—iron deficiency, thalassaemia.
• Left shifted: immature neutrophils present in the peripheral blood—sepsis.
• Leucoerythroblastic: early immature white and red blood cells present in the peripheral blood—marrow infiltration, e.g. cancer.
• Macrocytic RBCs: large RBCs—vitamin B12/folate deficiency, myelodysplastic syndromes (MDS), alcohol/liver disease, haemolysis (increase in reticulocytes).
• Microcytic RBCs: small RBCs—iron deficiency, thalassaemia, anaemia of chronic disease.
• Pencil cells: elongated RBCs—iron-deficiency anaemia, thalassaemia.
• Poikilocytosis: variation in RBC shape, e.g. sickle cell disease, iron deficiency anaemia.
• Polychromatic RBC: immature RBCs (blue tinge)—haemorrhage, haemolysis, bone marrow infiltration.
• Rouleaux: stacking of RBCs—chronic inflammation, paraproteinaemias (e.g. multiple myeloma).
• Sickle cells: sickle-shaped RBCs—sickle cell disease.
• Spherocytes: round RBCs—hereditary spherocytosis, haemolysis, post-splenectomy.
• Target cells: RBCs with area of central staining—liver disease, hyposplenism.
• Teardrop RBC: teardrop-shaped RBCs—myelofibrosis, marrow infiltration by malignancy.
Each cell lineage has a complex maturation process which is assisted by growth factors as seen in Fig. 18.1. In haematological malignancy, disruption of this process results in excess proliferation without maturation. Identifying the lineage from which this occurs is essential in understanding haematological malignancy and its treatment.
Fig. 18.1 Haematopoiesis. Reproduced from Qasim et al. Normal haematopoiesis and the concept of stem cell transplantation. Expert Reviews in Molecular Medicine. Vol. 6; Issue 13; 2 July 2004.
A patient can present with symptoms due to bone marrow dysfunction. Think about the three main types of cells the bone marrow produces (Fig 18.1) and you can extrapolate these symptoms: (1) anaemia—fatigue/shortness of breath or polycythaemia—headaches/blurred vision, (2) thrombocytopenia—bruising/bleeding or thrombocytosis—thrombosis, and (3) leucopaenia—infection; or leucocytosis—headaches) and infiltration of any organs (e.g. hepatomegaly, lymphadenopathy), ± adjacent tissue compression (e.g. spinal cord compression 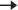 leg weakness).
leg weakness).
There are several important diagnostic methods utilized in malignant haematology. No one technique is used in isolation: multiple techniques are used to accurately identify the malignancy and provide information which influences treatment decisions.
• Morphology: characterizing the appearance of cells in the blood or bone marrow.
• Immunohistocytochemistry: detecting a protein in a tissue section.
• Cytogenetic analysis and fluorescent in situ hybridization (FISH): detecting the karyotype and chromosomal abnormalities (e.g. translocations).
• Flow cytometry: detecting antigen expression on or in cells (‘the immunophenotype’).
• Molecular studies: detecting DNA sequences or expression of genes in malignant cells.
Honours
This was developed to ascertain a consensus on the diagnosis of malignancy in the haemopoietic and lymphoid tissues. The classification stratifies neoplasms based on their cell lineage of origin (e.g. myeloid, lymphoid, or histiocytic). Within each category, diseases are further classified based on their morphology, immunophenotype and genetic abnormalities.
The leukaemias are a group of highly heterogeneous diseases characterized by the accumulation of a clonal population of white blood cells in the bone marrow and/or lymph nodes and can spill into the blood. They are classified according to the cell of origin (myeloid vs lymphoid (i.e. T or B cells)), and as usual the diagnosis is established on bone marrow aspiration and biopsy.
Commonest acute leukaemia in adults with increasing incidence with age; presents with acute signs/symptoms of bone marrow failure (although WCC can be high due to the malignany myeloid cells spilling into the blood). Chemotherapy is used to produce remission, followed by further of chemotherapy cycles as consolidation. You may see patients receiving an allogenic stem cell transplant (SCT) which is reserved for patients who have aggressive disease (see Honours: Haemopoietic stem cell transplantation (HSCT) p. 376).
Commonest cancer in children. These malignant cells are lymphoid (unlike AML) and can be either B or T cells. Similar presentation with bone marrow failure, but high WCC or lymphadenopathy may be seen. Treatment is as for AML, with allogenic SCT or maintenance chemotherapy offered for high-risk disease.
The malignant cells are mature B cells, and this is the most common adult leukaemia in the West. While they may present with bone marrow failure symptoms, these patients are often asymptomatic and diagnosis is made after a blood test which shows lymphocytosis. These patients are treated with chemotherapy although newer ‘targeted’ treatments are now being used.
The lymphomas are a heterogeneous group of disorders characterized by accumulation of malignant lymphocytes in the lymph nodes and/or extranodal sites including the CNS. They are broadly classified into Hodgkin lymphoma (HL; characterized by the presence of Reed–Sternberg cells) and non-Hodgkin lymphoma (NHL).
HL occurs in a bimodal age distribution and is associated with EBV infection. The malignant cell is derived from the B-cell lineage. Classical HL is characterized by the multinucleated Reed–Sternberg cell. Nodular lymphocyte-predominant HL is characterized by the lymphocytic and histiocytic cells.
Patients typically present with painless lymphadenopathy and may have ‘B’ symptoms such as weight loss and high temperatures, and are diagnosed by core biopsy or lymph node excision. Try to see a CT or positron emission tomography (PET)-CT which is used to stage the disease and ask the radiologist to talk you through his/her assessment of the size and location of lymphadenopathy (they may discuss the Ann Arbor Classification with Cotswold modification).
Chemotherapy ± radiotherapy. When this fails or in relapse, haemopoietic SCT is offered. Find out about the short- and long-term complications associated with these treatments such as second cancers and heart failure.
Are a very diverse group of clonal disorders of B- and T-cell lymphoid cells (if you think it includes every lymphoma other than the relatively rarer Hodgkin). You should be able to name a few common types: diffuse large B-cell lymphoma, follicular lymphoma, and Burkitt’s lymphoma.
Irrespective of the type, the presentation is largely dependent on the grade of disease. ‘Low-grade’ lymphoma typically presents with widespread disease with an indolent course, whereas ‘high-grade’ lymphoma typically presents with a rapidly enlarging lymph node, organ infiltration, and ‘B’ symptoms.
Treatment is dependent on the type and distribution of NHL. Broadly speaking, chemotherapy and radiotherapy are used to induce disease control or remission. However, monoclonal antibodies and ‘targeted therapies’ are being employed in isolation or in combination with traditional treatment modalities.
Honours
Monoclonal antibodies are monospecific antibodies. Monoclonal antibodies are designed to target a specific protein and are used therapeutically. For example, rituximab specifically targets CD20, a protein found on B cells. This contrasts with chemotherapeutic agents which have the potential to destroy all dividing cells as defined by their mechanism of action. Monoclonal antibodies are in widespread use in haematology and in other medical specialities.
In healthy individuals, plasma cells are responsible for producing antibodies and are derived from the B cells. MM is a haematological malignancy characterized by clonal proliferation of plasma cells, usually in the bone marrow, the presence of a monoclonal protein in the serum/urine, and tissue damage (CRAB: Calcium (elevation), Renal Impairment, Anaemia, Bone disease—often resulting in lyric lesions and fractures). Multiple cycles of combination chemotherapy and an autologous stem cell transplant are used to prolong remission but there is no cure. Find radiographs of patients who have these typically appearing lytic lesions and fractures. Bisphosphonates are used to prevent the progression of bone disease.
The MPNs are a group of disorders characterized by clonal proliferation of haemopoietic cells with preserved maturation (lots of ‘normal looking’ cells) resulting in an increase in the number of cells of a particular lineage. This can affect the erythroid, granulocytic, or megakaryocytic lineage. Gene mutations in the JAK2 and CALR genes are common in MPNs. Patients with an MPN are at increased of developing an acute myeloid leukaemia (‘transformed disease’).
CML is a clonal disorder of the myeloid progenitor cell. It is characterized by the Philadelphia chromosome t(9; 22) (q34;q11) abnormality resulting in an abnormal fusion protein, BCR-ABL. Clinical features are high WCC and splenomegaly. Tyrosine kinase inhibitors (e.g. imatinib), are used to inhibit the the activity of the BCR-ABL fusion protein.
This is the commonest MPN and is characterized by an erythrocytosis. Do not forget to think about differential diagnoses of a secondary polycythaemia (e.g. secondary to chronic hypoxia in COPD) as well as a relative erythrocytosis secondary to reduced plasma volume (e.g. dehydration). Thrombosis or transformation to myelofibrosis or AML are significant complications. Venesection is the main treatment along with cytoreductive therapy, aspirin, and a JAK2 inhibitor.
PMF is a clonal proliferation of megakaryocytes and monocytes, which results in activation of fibroblasts initiating a fibrotic response within the bone marrow. Clinical features include splenomegaly and bone marrow failure, and is treated with cytoreductive therapy, or an allogenic SCT.
ET is characterized by an elevation in platelet count in the absence of a secondary cause. Patients are often asymptomatic or present with vasomotor symptoms such as headaches, thrombosis, or paradoxical bleeding; treated with aspirin and cytoreductive medication.
The myelodysplastic syndromes are a group of clonal disorders of the bone marrow characterized by ineffective haematopoiesis and cytopenias, classified by clinical features, morphology (from blood film/bone marrow aspirate), karyotype, and molecular tests. There may also be a relative excess of myeloblasts (immature myeloid cells, although still <20% of all nucleated cells on a bone marrow aspirate), and MDS can progress to AML. Previous chemotherapy family history and age are risk factors, and presentations vary between bone marrow failure and being asymptomatic.
Options are tailored to each individual, but can be categorized as follows:
• Supportive: blood product support, iron chelation therapy.
• Low intensity: demethylating agents such as azacytidine (epigenetic modification).
• High intensity: chemotherapy and/or an allogenic SCT.
Honours
The aim of haemopoietic stem cell transplantation is to achieve reconstitution of haemopoiesis by the transfer of haemopoietic stem cells. Broadly speaking, this has two sources: autologous—from oneself; allogeneic—from another (sibling, unrelated). Stem cells can be harvested from the umbilical cord, bone marrow, or peripheral blood (after the administration of G-CSF) and can be stored frozen or given fresh.
HSCT can be used in the following circumstances:
• Following high (myeloablative) doses of chemotherapy in the treatment of malignant disease.
• Less commonly to replace abnormal bone marrow/immune system/inherited errors in metabolism (e.g. thalassaemia, lysosomal storage disorders).
Allogenic SCT has an additional therapeutic benefit not restricted to high-dose chemotherapy: i.e. graft vs disease effect. However, this is associated with a major short- and long-term toxicity: i.e. graft vs host effect. This effect can be modulated post transplant by adjusting immunosuppression.
Cases are discussed at regional MDT meetings and this is an opportunity for doctors to review clinical details and investigations and agree a management plan.
Patients with haematological malignancy are at risk of infection due to the disease and the treatment administered. These infections are often not seen in immunocompetent people. Haematologists work closely with microbiologists to treat infection.
Central venous access (‘line’) is often required to ensure safe administration of chemotherapeutic agents, blood products, and antibiotics.
Patients often require transfusion support as part of their management due to the myelosuppressive nature of their disease and the treatment administered.
The initiation of treatment can result in rapid destruction of tumour cells which release metabolites into the bloodstream, which can be fatal. Fluid hydration as well as specific agents such as allopurinol and rasburicase can prevent the sequelae of tumour destruction.
Psychological and social support is required to minimise the impact of a diagnosis of haematological disease, as well as treatment, on a patient’s life.
Cytotoxic treatment is associated with infertility. Patients should be counselled about this and be offered reproductive conservation.
Malignancy can affect the CNS and specific treatment is required to prevent and treat this (e.g. intrathecal methotrexate).
The haemostatic system is tightly regulated to ensure an appropriate cessation of bleeding in haemorrhage with appropriate clot dissolution. Clotting should not occur in the absence of haemorrhage. When a blood vessel is damaged, there are three distinct phases:
1. Vascular phase: vascular spasm reduces blood loss.
2. Platelet phase: platelet aggregation acts as a plug as well as releasing platelet factor 3, which acts to promote the coagulation cascade.
3. Coagulation cascade: biological amplification process acts to create fibrin which stabilizes the platelet plug. Diseases can be thought of as thrombophilic (increased propensity to clot) or haemophilic (increased propensity to bleed). These can be inherited or acquired.
Coagulation screening tests are designed to identify which part of the clotting cascade is dysfunctional (Fig 46.2, although it is important to remember, these tests are performed on blood that has been removed from the body and are therefore surrogate tests for the haemostatic performance in a human body):
• Activated partial thromboplastin time (APTT): the time taken in seconds for a citrated blood sample to clot after calcium and contact factor are added. The APTT is a functional determination of the intrinsic and common pathways. Prolonged by unfractionated heparin, DIC, dilutional coagulopathy, factor VIII, IX, XI deficiencies, antiphospholipid antibodies (although in vivo is procoagulant).
• Prothrombin time (PT): time taken in seconds for blood to clot after calcium and thromboplastin (a preparation of tissue factor) is added. The PT is a functional determination of the ‘extrinsic pathway’. Prolonged by warfarin, vitamin K deficiency, and liver disease.
• INR: the thromboplastin used in the PT is not the same in different laboratories and this alters the result of the PT, making results incomparable. The INR is calculated as a ratio of the patient’s PT and a standard and thus permits comparison.
• Thrombin time: time in seconds for a citrated blood sample to clot after thrombin is added. Prolonged in unfractionated heparin and hypofibrinogenaemia.
• Mixing studies: a patient’s plasma (with APTT prolongation) is mixed with normal plasma (usually a 1:1 ratio). If the mixture fails to correct, this suggests a coagulation factor inhibitor is present in the patient’s serum (as a factor deficiency would correct with replacement via the normal plasma).
• Fibrinogen: levels are low in DIC (consumption) and dilutional coagulopathy.
Deep vein thrombosis (DVT) of the lower limb and PE are the most common sites of venous thrombosis encountered in clinical practice. Referring to Virchow’s triad is a sensible way to classify the risk factors associated with thrombosis:
• Hypercoagulability: malignancy, oral contraceptive pill, hyperviscosity.
• Stasis of blood: prolonged immobility, AF, varicose veins.
• Vessel wall injury: trauma, infection/inflammation, catheters.
VTE is a preventable cause of hospital death. Patients are assessed based on their risk of developing thrombosis and bleeding. Interventions include thromboembolic deterrent stockings, heparin injections, and mechanical compression devices. It is important to note that the approach in medical, surgical, and obstetric patients is different.
• Symptoms: pain, swelling, and discolouration of the leg.
• Investigations: the Wells score is used to provide a ‘pretest probability’ of DVT. The D-dimer test is a measure of fibrin degradation products and is positive in thrombosis as well as malignancy, infection, and pregnancy—it is therefore not specific for thrombosis.
• In patients with a low pretest probability and a negative D-dimer—a DVT may be excluded. Compression ultrasonography should be carried out on patients with a low pretest probability with a positive D-dimer or those with a moderate/high pretest probability.
• If a negative initial ultrasound result is obtained in selected patients, a repeat ultrasound in 1 week may be required to exclude a DVT.
• Differential diagnosis: muscle strain, cellulitis, ruptured Baker’s cyst.
• Symptoms: shortness of breath, chest pain, haemoptysis.
• Investigations: Wells score is used to provide a ‘pretest probability’ of PE.
• In patients with a low pretest probability and a negative D-dimer—a PE may be excluded. CT pulmonary angiography should be carried out on patients with a low pretest probability with a positive D-dimer or those with a high pretest probability. If a negative CT pulmonary angiogram is obtained, PE is excluded.
• Differential diagnosis: acute coronary syndrome, costochondritis, pneumonia, aortic aneurysm.
Treatment of DVT and PE: anticoagulation initially with LMWH (or unfractionated heparin if significant renal dysfunction) and then with orally administered anticoagulation for 3–6 months (duration variable and dependent on bleeding risk) for first thrombosis and for longer if a subsequent thrombosis. Consideration must be given for any patient with VTE as to whether a secondary reversible cause exists, e.g. malignancy or inherited thrombophilia.
Honours
Selected patients should be investigated for inherited thrombophilia, e.g.:
• First thrombosis <40 years without a major provoking factor.
• Individuals from apparent thrombosis-prone families (more than two other symptomatic family members).
• Multiple unexplained miscarriages before 12 weeks of gestation.
• Consists of antithrombin, protein C and protein S, factor V Leiden and activated protein C ratio, prothrombin gene mutation, lupus anticoagulant, and anticardiolipin antibodies.
• Heritable thrombophilias include factor V Leiden, prothrombin gene mutation, protein C or protein S deficiency, antithrombin deficiency.
• Thrombophilia testing should not be carried out in the acute episode and should ideally be deferred till 8 weeks after pregnancy or 4 weeks after cessation of anticoagulation.
Mechanism: inhibits the COX enzyme, irreversibly reducing platelet thromboxane A2. Indications: ischaemic stroke, acute coronary syndrome.
Mechanism: ADP receptor antagonist. Indications: ischaemic stroke, acute coronary syndrome.
Indications: therapeutic anticoagulation in patients with significant renal impairment, anticoagulation perioperatively. Mechanism: potentiates the activity of antithrombin III. Pharmacokinetics: quick onset of action and has a short half-life. Renally eliminated. Given as a bolus and infusion. Patients commenced on a heparin infusion are closely monitored and the infusion is titrated to APTT results. Reversal agent: protamine
E.g. enoxaparin. Indications: therapeutic anticoagulation in VTE, thromboprophylaxis. Mechanism: potentiates the effect of antithrombin III. Renally eliminated. Given as a SC injection once a day. Reversal agent: protamine, recombinant factor VIIa.
E.g. warfarin. Indications: therapeutic anticoagulation in VTE, AF, anticoagulation for metallic heart valves. Pharmacokinetics: inter-patient variable dose determined (dose adjusted as per INR). Patients are required to attend an anticoagulant clinic where the INR is measured and appropriate dose of warfarin prescribed.
E.g. rivaroxaban. Indications: stroke prevention in non-valvular AF. Mechanism of action: dependent on specific agent. Pharmacokinetics: dependent on specific agent. Oral tablet once/twice per day. Reversal agent: are currently under development and are specific to each agent.
Reversal of anticoagulation is considered in patients who present with haemorrhage or require an urgent procedure. This requires a balance of risks and benefits and is a frequent topic of discussion for the on-call haematologist.
This is a rare but often fatal disease characterized by: (1) thrombocytopenia, (2) microangiopathic haemolytic anaemia (physical destruction of RBCs by fibrin mesh on small blood vessels), (3) fluctuating neurological signs, (4) renal impairment, and (5) fever. Antibodies are found to ADAMTS13 (which breaks down large von Willebrand factor (vWF) multimers) in adults with idiopathic TTP. Untreated TTP has a high mortality rate and the treatment is with urgent plasma exchange.
This has features in common with TTP although with predominant renal dysfunction and diarrhoea. Many cases are associated with Escherichia coli and other organisms such as Shigella. Treatment is with BP control and supportive therapy.
A specialist haemophilia centre should manage patients with inherited bleeding disorders.
This is one of the most common heritable bleeding disorder characterized by reduced vWF levels and/or dysfunction in vWF due to a genetic mutation (inheritance often autosomal dominant). vWF is produced by endothelial cells and megakaryocytes and is responsible for promoting platelet adhesion and for preventing factor VIII destruction. vWF is classified as follows:
• Type I: quantitative deficiency in vWF (accounts for 75% of cases).
• Type II: qualitative dysfunction of VWF (further subclassified).
• Type III: absence of vWF (rare).
Clinical features: variable depending on the type and the causative mutation. Mucocutaneous bleeding and excessive bleeding following cuts and surgery. Haemarthrosis occurs in type III vWD. Treatment: mild bleeding—antifibrinolytics (e.g. tranexamic acid). Moderate disease and minor surgery—desmopressin (DDAVP®, releases vWF from endothelial cells). Major surgery—significant bleeding, severe disease (vWF concentrate).
X-linked bleeding disorders that are distinguishable on specific clotting factor assays. The genetic defect results in a low level of plasma factor VIII and factor IX respectively. Clinical features: soft tissue bleeding is noted which may lead to compartment syndrome. Recurrent joint bleeding causes chronic arthropathy secondary to haemarthroses and large encapsulated haematomas. Investigations: see Table 18.1 for a comparison of the inherited bleeding disorders.
Table 18.1 Comparison of haemophilia types
| Laboratory test | Haemophilia A | Haemophilia B | von Willebrand disease |
| Platelet count |
|
|
|
| PFA-100 (platelet function test) |
|
|
Prolonged |
| PT |
|
|
|
| APTT | Prolonged | Prolonged | Prolonged/
|
| Factor VIII | Low |
|
 |
| Factor IX |
|
Low |
|
| vWF |
|
|
Low/abnormal function |
Desmopressin and tranexamic acid for minor surgery. Recombinant factor concentrates are recommended (donor-derived products have a risk of virus transmission) but are not accessible worldwide.
Honours
A complication of haemophilia treatment is the development of antibodies (inhibitors) to infused factor concentrates, rendering the patient refractory to additional replacement therapy. Factor VIII bypassing agents can be used in those with inhibitors who are bleeding.
DIC is a medical emergency and is characterized by systemic activation of coagulation resulting in consumption of platelets and coagulation factors which leads to excessive bleeding. Causes include sepsis, trauma, organ necrosis, malignancy. Investigations:  PT, APTT, fibrin degradation products, fibrinogen, thrombocytopenia. Treatment: treatment of the underlying cause with blood product support.
PT, APTT, fibrin degradation products, fibrinogen, thrombocytopenia. Treatment: treatment of the underlying cause with blood product support.
ITP is a disorder characterized by peripheral platelet destruction with a reduction in platelet production by megakaryocytes (immune mediated). Clinical features: patients typically present with non-palpable purpura (in contrast to vasculitis) and is located in dependent areas of the body. Investigations: exclude other causes of thrombocytopenia—causative drugs, autoimmune screen, viral screen, thyroid screen, infection screen. A blood film should demonstrate true thrombocytopenia (with no platelet clumping). There must be an absence of a secondary condition associated with destruction of platelets (e.g. SLE) and no other unexplained cytopenias must be present. Bone marrow tests are performed in those >60 years of age or to exclude another cause. Differential diagnosis: DIC, TTP/HUS, myelodysplasia, splenic sequestration. Treatment: adults with a platelet count of <30 are treated due to a risk of bleeding whereas children often require no treatment as the majority undergo spontaneous remission. IV immunoglobulin, steroids, and anti-rhesus (Rh)-D have all been used.
• Haemoglobin (Hb): measures blood concentration of Hb (g/L).
• Haematocrit (Hct): proportion of blood occupied by RBC (%).
• Mean corpuscular volume (MCV): average volume of RBC (fL).
• Mean cell haemoglobin: average mass of Hb in the average RBC (pg).
• Mean cell haemoglobin concentration: average concentration of Hb in the average RBC (g/L).
• Red cell distribution width: variation in the size of RBCs (%).
• Reticulocyte count: measure of immature red cells (% or ×109/L).
In adults is defined by WHO as Hb <12 in women or Hb <13 in men. It can be classified as per the MCV as microcytic (iron deficiency anaemia, anaemia of chronic disease, thalassaemia), normocytic (recent bleeding, anaemia of chronic disease, combined iron and folate/vitamin B12 deficiency, leukaemia, aplastic anaemia), and macrocytic (vitamin B12/folate deficiency, alcohol abuse, hypothyroidism, liver disease, myelodysplasia).
The recent discovery of the protein hepcidin and its elevation in inflammatory disorders is thought to be crucial in the pathogenesis of ACD. However, the pathogenesis of ACD is complex and involves the dysregulation of iron absorption, transport, and storage. Diagnosis: microcytic anaemia with an elevated ferritin.
The most common cause of a microcytic anaemia is IDA—although this is not a diagnosis and a cause must be established. Causes include hookworm infestation, chronic blood loss (e.g. bleeding from the GI tract secondary to cancer, menorrhagia), malabsorption, and dietary deficiency. Diagnosis: microcytic anaemia with a low serum ferritin level. Serum ferritin is also elevated in inflammatory states and in patients with coexisting IDA and systemic inflammation (common), iron indices are useful. Treatment: iron can be replaced through oral iron salts although unwanted side effects are common. Reducing the dose or liquid iron salts can be helpful. IV iron is an alternative. Iron indices are useful in establishing ACD vs IDA. (See Table 18.2.)
Table 18.2 Comparing anaemia subtypes
| IDA | ACD | |
| Iron | |
|
| Transferrin | /  |
|
| Transferrin saturation | |
|
| Hepcidin | |
|
Vitamin B12 is required for DNA synthesis and is important in haematopoiesis and neurological function. Vitamin B12 deficiency presents with symptoms of anaemia (which is macrocytic) and neurological symptoms (peripheral neuropathy affecting proprioception and vibration sense). Vitamin B12 is absorbed at the terminal ileum following binding to intrinsic factor (produced by gastric parietal cells). Causes include malabsorption (pernicious anaemia, gastrectomy, ileal disease, coeliac disease) and dietary deficiency. Treatment: vitamin B12 replacement can be given IM and orally (although less well absorbed).
Causes a derangement in haematological parameters indistinguishable from vitamin B12 deficiency. Folate absorption occurs in the proximal jejunum and is required for DNA synthesis. Causes include malabsorption (coeliac disease, Crohn’s disease), dietary deficiency, requirements (pregnancy, haemolysis), and drugs (methotrexate, barbiturates).
Honours
In patients with folate deficiency, vitamin B12 deficiency should be excluded prior to administering folate replacement therapy. This is because isolated folate replacement in patients with a combined folate and vitamin B12 deficiency can result in subacute combined degeneration of the spinal cord.
The haemoglobinopathies are a group of conditions characterized by mutations in the genes which are responsible for the synthesis of Hb. This can result in either a qualitative change in Hb function (e.g. sickle cell disease) or a quantitative change (e.g. thalassaemia). There are areas of prevalence of haemoglobinopathies due to selection pressures (e.g. malaria infection). Hb electrophoresis is used in the diagnosis of haemoglobinopathies. The Hb is separated across a gel based on electrical charge and size.
Is prevalent in West Africa, Middle East, and parts of India. The mutation is a single base change in the beta globin gene resulting in the amino acid at position 6, glutamine, being substituted for valine. The resulting haemoglobin polymerizes in hypoxic environments resulting in sickling of the RBCs. Heterozygotes tend to be asymptomatic. Despite a specific genetic mutation, there is a broad phenotype associated with homozygosity for the sickle cell mutation.
Sickle cells cause blockage of blood vessels resulting in tissue ischaemia. This can affect any tissue (e.g. skin—ulcers, brain—stoke, chest—embolism, bone—avascular necrosis of the hip, renal—papillary necrosis, spleen—hyposplenism).
Sudden drop in Hb production by the bone marrow due to nutritional deficiency or infection with parvovirus.
Sudden drop in Hb due to breakdown of RBCs. Long-term haemolysis predisposes to gallstone formation.
Mainly occurs in children and can be precipitated by a viral infection. Pooling of blood occurs in the liver and spleen resulting in hypotension and profound anaemia.
Sickledex®, haemoglobin electrophoresis, sickle cells demonstrated on blood film.
Of an acute sickle cell crisis is a medical emergency. Oxygen, fluids, analgesia, and antibiotics if infection is suspected. Transfusions are recommended for red cell aplasia secondary to parvovirus infection, sequestration crisis, and chest crisis. Repeated transfusions are associated with antibody formation making subsequent transfusions complex. Exchange transfusion is reserved for patients with a chest crisis or for patients with a CVA. Hydroxycarbamide can be used long term and works via multiple mechanisms including increasing fetal Hb levels. Phenoxymethylpenicillin is given (long term) as patients are hyposplenic.
The thalassaemias are prevalent in the Mediterranean, Middle East, and Indian subcontinent. Adult haemoglobin is formed from two α-globin chains and two β-globin chains. Diminished or absent production of these chains result in thalassaemia. The globin chains in excess form tetramers and precipitate within RBS chronic haemolysis.
Two α-globin genes are present on each chromosome 16 resulting in a total of four α-globin genes per cell (−α/αα).
• Silent α-thalassaemia (−α/αα): one gene is deleted. Asymptomatic.
• α-Thalassaemia trait (−−/αα) or (−α/−α): two genes deleted. Hb and MCH. Requires no treatment.
• Haemoglobin H (HbH) disease (−−/−α): three genes deleted. Moderate anaemia with Hb, mean corpuscular Hb (MCH), and MCV. Blood film shows reticulocytes, hypochromia, target cells. Brilliant cresyl blue stain on peripheral blood shows HbH inclusions (tetramers of β-globin). Clinical features: hepatosplenomegaly (extramedullary haematopoiesis) and jaundice (haemolysis). Treatment: folic acid supplementation and prompt treatment of infection.
• Haemoglobin Bart’s (−−/−−): four genes deleted. γ-globin chains form tetrameters which have a high affinity for oxygen resulting in tissue hypoxia. The result is a stillborn fetus or one that dies soon after birth. Intrauterine transfusions have been used.
One β-globin gene is present on each chromosome 11 resulting in a total of two β-globin genes per cell.
• β-Thalassaemia trait: heterozygous for a β-globin gene mutation. Mild anaemia (microcytic). Blood film: microcytic, hypochromic RBCs with target cells. Clinical features: asymptomatic. No treatment required.
• β-Thalassaemia intermedia: arises through a variety of different genetic mutations: by definition these patients do not require regular transfusions. Clinical features: moderate anaemia with hepatosplenomegaly, iron overload. Some patients demonstrate skeletal abnormalities, chronic leg ulceration, and impaired growth. Treatment: iron chelation, folic acid supplementation, and prompt management of infection
• β-Thalassaemia major: abnormality of both β-globin genes. Clinical features: severe anaemia with MCV, MCH, reticulocytosis. Extramedullary haematopoiesis, hepatosplenomegaly, and skeletal abnormalities. Blood film: anisopoikilocytosis, target cells, nucleated RBCs. Methyl blue stain demonstrates α-tetrameter inclusions within the RBC. Treatment: regular lifelong blood transfusions every 2–4 weeks. Iron chelation. Splenectomy may reduce transfusion requirements. HSCT has been used.
Haemolysis is the premature destruction of RBCs (normal life span ~120 days). This can be classified as hereditary vs acquired, extravascular vs intravascular, or immune vs non-immune.
• Red cell membrane disorders (e.g. hereditary spherocytosis).
• Red cell enzyme disorders (e.g. glucose-6-phosphotase deficiency).
• Abnormal haemoglobin e.g. (sickle cell disease, thalassaemia).
• Alloimmune: e.g. haemolytic disease of the newborn—anti Rh antibodies from a Rh-negative mother crossing the placenta and causing haemolysis of the fetus
• Autoimmune: e.g. warm autoimmune haemolytic anaemia in SLE, cold haemagglutinin disease, Mycoplasma infection.
E.g. microangiopathic haemolytic anaemia (MAHA), TTP/HUS, prosthetic heart valves, malaria, paroxysmal nocturnal haemoglobinuria.
Confirm whether haemolysis is occurring: Hb, reticulocyte count, serum bilirubin, lactate dehydrogenase (released from RBC), haptoglobin (binds free Hb). Blood film: polychromasia, spherocytes, fragmentation, helmet cells, echinocytes. Is the haemolysis immune mediated? Direct antiglobulin test (DAT): a positive test indicates the red cells are coated with antibodies and the haemolysis is immune mediated.
Treat underlying cause. Give folic acid and iron supplementation.
A transfusion involves the safe transfer of blood products from a donor to a recipient. Blood product transfusion should only be administered when the benefit outweighs the risk and there are no other options (such as cell salvage, haematinics replacement). The benefits and risks of transfusion should be discussed with the patient and the indication for transfusion documented in the notes. The decision to transfuse is based on clinical assessment of the patient and the application of evidence, not the results of a laboratory test in isolation. Errors related to blood product transfusions are associated with significant morbidity and mortality.
Honours
This is the ‘systematic surveillance of adverse reactions and adverse events related to transfusion’ with the overall aim of reducing the risk associated with transfusion. Transfusion reactions and adverse events should be investigated by the hospital as well as reported to the Serious Hazards of Transfusion (SHOT) scheme.
These are proteins present on the surface of RBCs and some are present on platelets as well as other tissues in the body. There are >300 human blood groups described. In clinical practice, the ABO and Rh systems are of most importance.
These are produced when an individual is exposed to blood of a different group or during pregnancy. Antibodies to AB antigens are naturally occurring and are found in all adults.
There are four main blood groups: A, B, AB, O. An individual will have antibodies to the A or B antigens that are not present on their own red cells (see Table 18.3).
Table 18.3 Comparing ABO subtypes
| Blood group | Antigens on red cells | Antibodies in plasma | Compatible red cell product |
| O | None | Anti-A and anti-B | O |
| A | A | Anti-B | A, O |
| B | B | Anti-A | B, O |
| AB | A and B | None | AB, A, B, O |
Consists of five main Rh antigens, the most clinically important is RhD. Unlike the ABO system, anti-RhD antibodies are only present in RhD-negative individuals who have been exposed to RhD-positive blood (transfusions or fetomaternal haemorrhage).
Transfusion of an incompatible blood group to a patient can be fatal by causing activation of the immune system and intravascular haemolysis. It is therefore important to accurately determine the recipient’s blood group. Hospitals have a strict policy as to how this is done, how samples are taken and processed, and when the samples are taken with respect to when the blood is transfused.
The recipient’s blood group is established and their plasma tested for antibodies.
The recipients plasma is mixed with a panel of red cells to ensure no significant antibodies are present. In certain circumstances, this process is now being replaced with a computer cross-matching system, which is quicker.
Processing whole blood into different components allows the transfusion of specific components to patients. Whole blood is filtered to remove white blood cells and was introduced in 1998 to reduce the risk of variant CJD, febrile transfusion reactions, and alloimmunization. Here are the most commonly used products:
Are used to restore the oxygen carrying capacity of the blood in patients with blood loss or anaemia where alternative interventions are not appropriate. They are stored at 4°C for 35 days.
Are used to prevent or stop bleeding in patients who are thrombocytopenic (one pool is often sufficient). They should be matched for ABO groups as they have reduced survival if incompatible. A pool of platelets consists of platelets from four donations. If a patient’s platelet count fails to increment with a transfusion of a pool of platelets, a single donor (apheresis) unit or HLA-matched units can be used. Platelets are stored at room temperature on an agitator. The introduction of bacterial screening can increase the shelf life from 5 days to 7 days.
Is used to replace clotting factors in patients who are actively bleeding with a derangement in their clotting factors (e.g. in DIC). The recommended dose is 12–15 mL/kg. FFP can be stored up to 36 months at <−25°C but once thawed to 4°C, it must be used within 24 hours.
Is a fibrinogen-rich product derived from FFP. It is used as a concentrated method to replace fibrinogen.
If a transfusion reaction is suspected, the patient should be reviewed and action taken quickly with frequent clinical assessment. Ensure the laboratory is aware as additional investigations and specific action will need to be taken. Seek help early.
Honours
There are a number of blood product special requirements and anyone that can prescribe products should be aware of these. Here are some examples:
Are used for patients at risk of transfusion-associated graft vs host diseases. This includes patients who are undergoing haemopoietic stem cell transplantation, patients with HL, and patients treated with purine analogues.
Negative products should be provided for intrauterine transfusions and neonates as well as pregnant women (unless in an emergency).
Symptoms: an isolated urticarial skin rash. Management: chlorphenamine and reducing the rate of transfusion.
Symptoms: a temperature rise of <1.5°C. Management: paracetamol and reducing the rate of transfusion.
Symptoms: angio-oedema, pain, hypotension, bronchospasm. Management: stop the transfusion and return the bag and set to the blood bank. Administer oxygen, salbutamol nebulizer, adrenaline (epinephrine), chlorphenamine.
Symptoms: pyrexia, shortness of breath, pain. Management: stop the transfusion and return the bag and set to the blood bank. Give oxygen, antibiotics, fluid support, and maintain satisfactory urine output.
Symptoms: shortness of breath, hypoxia, elevated JVP, fluid overload. Management: administer oxygen and diuretics.
Symptoms: shortness of breath, hypoxia, pyrexia, JVP not elevated. Management: stop the transfusion and return the bag and set to the laboratory. Administer oxygen and contact ITU.
Each hospital has a major haemorrhage protocol, which consists of quick and safe access to compatible (O RhD-negative) blood products in the absence of a cross-match.
This is defined as the presence of signs and symptoms of infection in a patient with an absolute neutrophil count of <0.5 × 109/L. This is one of the most common emergencies in haematology and in 2012 accounted for two deaths per day in England and Wales. Patients who are neutropenic are susceptible to invasive infection and can deteriorate rapidly. Many patients do not exhibit classic features of sepsis (e.g. pyrexia), often due to concomitant chemotherapy and steroid therapy.
Investigations: FBC, U&E, LFTs, CRP, bone profile, coagulation studies, blood cultures (peripherally and from any lumens of the line), ABG (beware patients with low platelets—ensure pressure applied after), urine dipstick and culture, CXR.
If possible, take a brief history from the patient ascertaining the chronicity of symptoms, whether the patient has had previous infections and source, history of malignancy (if so, which type, treatment stage, last dose of chemotherapy, physician they are under the care of), and allergies.
Cultures should ideally be taken prior to the administration of antibiotics but cultures should not delay the administration of antibiotics. Antibiotics administered should provide an anti-microbial effect against a large number of bacteria (so called ‘broad spectrum’). Individual hospitals have local policies but a suggestion for empirical antibiotic therapy is piperacillin/tazobactam and amikacin (beware aminoglycosides and renal impairment). Previous cultures if available are informative (e.g. previous MRSA) and may alter antibiotic therapy and for patients who have a prolongued history of neutropaenia, other opportunisitic infections should be considered (e.g. infection with pneumocystis jiroveci). Consider removing the line if this is the source of sepsis and discussing the patient with the intensive care unit if the patient is significantly unwell or at risk of further clinical deterioration.
Honours
G-CSF is a growth factor which stimulates the bone marrow to produce and release neutrophils into the blood stream. A significant side effect ties bone pain/discomfort. Different preparations are available but are given subcutaneously. Consider giving G-CSF in patients with neutropenic sepsis providing the patient does not have active leukaemia. G-CSF is however not recommended for prophylaxis.
As haematological disease can affect the whole body, it is important that you can demonstrate the ability to perform an accurate and systematic history and clinical examination of a patient. One must be able to identify the signs associated with other diseases and be able to perform an examination of the reticuloendothelial system.
• Symptoms related to anaemia and chronicity: tiredness, shortness of breath, reduced functional ability, peripheral oedema, angina.
• Symptoms related to immunocompromise: pyrexia, specific sites of infection, sore throat.
• Symptoms related to haemostatic dysfunction: e.g. bruising, bleeding, joint swelling, leg swelling. Assess for haemostatic function when the patient has been ‘challenged’, e.g. surgery, trauma.
• Symptoms related to organ infiltration: CNS (headache, leg weakness), lymphadenopathy (abdominal pain/swelling), chronicity.
• Past medical history: other diseases are associated with haematological conditions (e.g. autoimmune disorders) or may have a relation to their presenting complaint (e.g. anaemia secondary to chronic blood loss).
• Past surgical history: surgical procedures may be related to haematological disease (e.g. gastrectomy and vitamin B12 deficiency) and represent an event where the body is challenged. Identifying complications such as bleeding and thrombosis during these periods is useful.
• Past obstetric history: number of pregnancies, complications, and relationship to symptoms.
• Past transfusion history: have they received blood products? If so, when, where, which products, and for what reason?
• Drug history and allergies: prescribed and non-prescribed medications can cause haematological abnormalities.
• Social history: haematological diseases can interfere with daily functioning. This interference can provide considerable insight into the severity of disease. Management of haematological disease is often long term and the impact on patients’ lives can be profound. Haematological disease can be associated with particular jobs. Lifestyle factors can predispose patients to certain infections.
• Tobacco and alcohol: consumption can affect the haematological system.
• Travel history: important when considering infections.
• Family history: many haematological diseases are inherited or demonstrate familial susceptibility.
• General examination: vital signs, pallor, jaundice, weight, skin rashes, pigmentation.
• Mouth: dentition, gingival hypertrophy, ulcers, bleeding.
• Abdomen: abdominal scars (splenectomy), assess for hepatomegaly and splenomegaly.
• Lymphadenopathy: examine the cervical, axillary, and inguinal nodes. Quantify the location, the number, and size of the lymph nodes. Note the consistency and whether tender.
• Bones and joints: joint swelling and range of movement (important in haemophilia) and bony tenderness (malignancy).
• Optic fundi: examine for signs of hyperviscosity (e.g. hyperleucocytosis in leukaemia with a high WCC, paraproteinaemias).
• Neurological exam: peripheral neuropathy in vitamin B12 deficiency.
There are clinical scenarios which frequently present to haematology clinics, one of which is anaemia. The second is lymphadenopathy. Understanding the differential diagnosis will assist in planning the necessary investigations.
Lymphadenopathy can be caused by a wide range of different pathological processes.
• Bacterial, e.g. tonsillitis, cellulitis, TB, syphilis.
• Viral, e.g. CMV, EBV, HIV, hepatitis B and C, rubella.
• Other, e.g. toxoplasmosis, histoplasmosis.
— Leukaemia (CLL and ALL, rarely AML).
— Systemic disorders, e.g. RA.
— FBC and peripheral blood film examination.
— U&E, LFTs, CRP, bone profile.
• Imaging: consider CT of the chest abdomen and pelvis to assess extent of lymphadenopathy (lymphadenopathy may be deep, e.g. para-aortic and therefore not palpable).
• Lymph node biopsy (core or excision): to achieve a definitive diagnosis.
• Microbiology: blood and urine cultures, serology, consider sending tissue samples, TB testing.
• Bone marrow examination: not necessary unless staging as part of haematological malignancy.
The key to being successful when interpreting laboratory results is to develop a systematic method. Clinical correlation is important as well as thinking about further informative tests to help achieve a diagnosis.
• Microcytic : low Fe, thalassaemia, chronic disease, sideroblastic.
• Normocytic: leukaemia, aplastic anaemia, haemolysis, acute blood loss, pregnancy.
• Macrocytic: vitamin B12/folate deficiency, myelodysplasia, reticulocytosis, cytotoxic, hypothyroidism, liver disease.
• Primary: myeloproliferative neoplasms.
• Secondary: bacterial infection, inflammation, e.g. MI, trauma, surgery, burns.
• Reduced production: viral infection, sepsis, bone marrow failure (e.g. leukaemia), cytotoxic drugs.
• Destruction: viral infection, sepsis, antineutrophil antibodies, splenic sequestration.
• Primary: leukaemia and lymphomas, e.g. CLL.
• Secondary : acute viral infections, chronic infections, e.g. TB.
• Reduced production : HIV, uraemia, bone marrow failure (e.g. leukaemia), cytotoxic drugs.
• Destruction: HIV, steroid therapy, SLE.
• Production: vitamin B12/folate deficiency, leukaemia or myelodysplastic syndromes, reduced production of thrombopoietin in liver failure, sepsis, hereditary syndromes, e.g. congenital amegakaryocytic thrombocytopenia.
• Destruction: ITP, TTP/HUS, DIC, SLE, post-transfusion purpura, neonatal alloimmune thrombocytopenia, splenic sequestration, dengue fever, HIV.
• Primary: leukaemia and lymphomas, hypereosinophilic syndrome.