2. Reduction–oxidation reactions
Freshwater lakes provide an ideal example for considering the carbon cycle and other biogeochemical cycles. A range of redox conditions exists in lakes that allows observation of numerous chemical and biochemical processes. The processes are not limited to freshwater lakes, and similar examples can be found in marine and terrestrial systems. The cycles of carbon and other elements are closely linked. Production of organic carbon depends on cycling of nutrients such as nitrogen and phosphorus. Respiration of organic carbon alters the redox condition, which in turn influences the cycling of nutrients. Many other elements can be influenced by redox conditions (e.g., sulfur, iron, and mercury). The elements can have indirect effects on carbon cycling or can be deleterious to organisms present in the ecosystem.
airshed. A region sharing a common flow of air
biogeochemistry. The scientific study of the physical, chemical, geological, and biological processes and reactions that govern the cycles of matter and energy in the natural environment
ecosystem. A natural unit consisting of all plants, animals and microorganisms (biotic factors) in an area functioning together with all of the nonliving physical and chemical (abiotic) factors of the environment.
lake. A body of water of considerable size surrounded entirely by land
micronutrient. A chemical element necessary in relatively small quantities for organism growth
nutrient. A chemical element necessary for organism growth
watershed. The area of land where all of the water that is under it or drains off of it goes into the same place
Streams, rivers, lakes, and wetlands constitute some of the most obvious natural freshwater ecosystems. Additionally, groundwater and intermittent pools can be considered in this context. Artificial ecosystems, such as small impoundments, large reservoirs, and engineered wetlands, also are labeled as freshwater ecosystems.
There are many physical, chemical, and biological differences among these varied ecosystems. Hydrology offers one brief indication of the differences. Water residence time, the average time a molecule of water spends in an ecosystem, varies from minutes to years to millennia in streams, lakes, and ancient groundwater. Related to the water residence time is the movement of water. At one end of the spectrum are streams, which have strong unidirectional flow governed by gravity. At the other end of the spectrum, lakes typically show little directional flow.
There is one key commonality that links all the freshwater ecosystems. Indeed, this commonality also links marine and terrestrial counterparts. The cycling of elements in all ecosystems is mainly controlled by aqueous chemical reactions, including those reactions mediated by organisms. In that vein, the cycles examined in this chapter are considered mainly in the context of freshwater lakes.
Figure 1. Typical zones describes in lakes.
Lakes provide an ideal example to consider element cycling for several reasons. Because lakes often have easily defined ecosystem boundaries and fairly long water residence times, studying the internal cycling of elements becomes more tractable. Also, lakes contain many habitats that are similar to habitats in other freshwater ecosystems. A brief description of these habitats is warranted (figure 1).
Two important zones, the photic and aphotic zones, are present in many lakes with sufficient depth. The photic zone represents the surface water of a lake where light is sufficient for photosynthesis to occur. The aphotic zone is defined as the volume of water where photosynthetically available radiation is less than 1% of that present at the surface. Because light limits photosynthesis in the aphotic zone, respiratory processes usually dominate. Processes dominant in the aphotic zone of a lake are also likely to occur in groundwater. In streams, the hyporheic zone, an area below the streambed that contains the interaction between surface and groundwater, has similar processes as the aphotic zone. Although relatively unstudied, lakes, like streams, also contain an area where lake and groundwater interact.
Lake habitats can be further divided into littoral and pelagic zones. The littoral zone is the region of the lake, near shore, where there is significant interaction between bottom substrates and the photic zone. Surface water in streams and wetlands is often subject to similar light and temperature regimes as the littoral zone of a lake. Because rooted plants can occupy the littoral zone of a lake, the littoral zone provides many similarities to wetlands, and many times there is little distinction between the two. The pelagic zone is the region of the lake beyond the littoral zone.
One unique feature of lakes is the potential for layers of water to stratify because of differences in density. In most freshwater lakes, temperature causes the difference in density. For brevity, only considering lakes warmer than about 4°C, the warmer the water, the less dense it is. Just as oil floats on vinegar in a salad dressing, less-dense water floats above more dense water. Shaking the bottle of salad dressing to mix it is akin to the energy that must be applied in order to mix a stratified water column. This energy usually comes from wind, which causes waves and currents to be set in motion. Density differences and stratification caused by temperature depend on seasonal cycles. A common seasonal cycle for lakes is as follows. (1) In spring, the lake is isothermal, there is little difference in density throughout the water column, and the lake is easily mixed by wind. (2) In summer, increased solar radiation warms the surface waters, decreasing their density to the point where wind energy is insufficient to completely mix the water column. (3) In the fall, the combination of surface water cooling and bottom water warming causes the density difference in lakes to decrease to the point where wind energy can mix the lake again.
Three zones can be identified with respect to summer thermal stratification. The epilimnion is the surface water that has relatively higher, and fairly uniform, temperatures. The metalimnion is the layer of transition between the warm surface water and the colder deep water. It is characterized by a strong thermal and density gradient. The hypolimnion consists of colder water, and temperature tends to decrease with depth. Stratification is important because the hypolimnion can effectively be sealed off from interactions with the atmosphere, because diffusion of gases (e.g., O2) or solutes through the metalimnion is extremely slow. Additionally, the depth of the top of the hypolimnion may fall below the photic zone, resulting in an absence of photosynthesis. With limited supply of O2 to the hypolimnion but continued respiration, O2 can become depleted in the hypolimnia of some lakes.
Oxygen is of key importance to biogeochemical cycling in all freshwater environments. Oxygen is only sparingly soluble in water and diffuses much more slowly in water than in air. Not only can O2 be extremely low in wet environments, but strong gradients of O2 can exist. Gradients from O2 saturation to depletion can exist within centimeters in sediments of lake, streams, and wetlands. A similar gradient might exist over the distance of several meters in the metalimnion of a lake or reservoir. These oxic/anoxic interfaces are important sites of chemical transformations because of the unique reduction/oxidation potential (see below) that exists in some of these environments.
Over the course of this chapter we explore the cycles of iron and mercury (Fe, Hg), phosphorus (P), nitrogen (N), sulfur (S), and finally carbon (C). Oxygen and reduction/oxidation potentials will be important in many of these sections.
The broad concepts captured by this chapter are that freshwater ecosystems receive fluxes of solutes and nutrients from the surrounding watershed and airshed. These solutes and nutrients can be significantly cycled and transformed in freshwater ecosystems. In addition, these nutrients provide substrate for primary production and respiration, ultimately impacting the carbon cycle of the recipient ecosystem. In a landscape, lakes and wetlands can store significant amounts of C, P, and N. However, because of terrestrial subsidies, a large amount of carbon can also be lost from freshwaters in the form of carbon dioxide (CO2) and methane (CH4). Nitrogen can also be lost from aquatic ecosystem in gaseous form (N2). Cycling of metals plays a significant part in the functioning of freshwaters as micronutrients, as potential toxins, and because of their interactions with other cycles. Finally, humans have significantly altered the cycling of many of these elements, not only by altering their input to freshwater ecosystems but also by creation or destruction of freshwater ecosystems.
Just as the flow of electrons from a battery provides the energy to drive an electric motor, a flow of electrons is needed to drive the metabolic machinery of organisms. For this flow to occur, there must be an electrochemical gradient. In aqueous environments, this gradient derives from the mixture of reduced and oxidized compounds that exist in a particular location. For instance, the most basic equation of heterotrophic respiration,
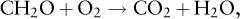
is the coupled oxidation of organic carbon (CH2O) to CO2 and reduction of O2 to H2O. Biological enzymes are needed to catalyze this reaction, but there is an overall release of energy for metabolism. Just as the electrons flow in the battery, in the process of respiration, electrons (coupled with hydrogen) flow from the organic carbon to the O2. In all redox reactions there must be a source of electrons and an electron acceptor. Alternatively, the creation of organic matter through photosynthesis,

reduces CO2 and oxidizes water. Photosynthesis requires the input of energy as photons from the sun and is catalyzed by enzymes in the cell. In another analogy, this input of energy to create organic carbon is similar to the input of energy that occurs when a battery is charged. The organic matter then stores this potential energy for later release in reactions such as respiration, above. When oxygen is not present, other elements or compounds can act as electron acceptors and become reduced during anaerobic respiration. For example, iron in its oxidized form [Fe(III)] can be used as an electron acceptor and reduced to Fe(II). Iron is extremely electroactive (easily exchanges electrons), and the reduced form of iron [Fe(II)] is oxidized in the presence of dissolved O2 fairly rapidly, even without the presence of biological enzymes. During the oxidation of Fe(II), energy is released, and although the amount of energy is modest, and the reaction does not need to be catalyzed by enzymes, there are bacteria that take advantage of this flow of electrons for energy. Generally, if significant energy can be gained from a redox reaction, microorganisms have evolved to enzymatically exploit it. These reduction and oxidation reactions are central to the cycling of several elements including C, N, S, Fe, and Hg.
The ability of an environment to donate or accept electrons is the redox potential (Eh; expressed in millivolts, mV). This potential is derived from the oxidation state of the constituents in the mixture. Recall from general chemistry the oxidation state of some familiar nitrogen molecules (e.g., NH4+ : N = –III, H = +I; N2 : N = 0; NO3– : N = +III, O = –II). In practice, redox potential is measured with a platinum electrode attached to a voltmeter. Because the environment contains a mixture of many different reducing and oxidizing species, not always in equilibrium, measurements from electrodes only give a relative indication of the true redox potential. These measurements can still shed light on reactions that are likely to occur under certain situations. For instance, the surface waters of lakes or streams generally have a large positive redox potential (e.g., above +400 mV), indicative of an oxidizing environment (oxygen is the most energetically favorable electron acceptor), whereas water in organic sediments can have very large negative redox potential (e.g., more negative than –400 mV), indicative of a strongly reducing environment (oxygen is not present, and many of the other electron acceptors have also been reduced).
In general, sunlight provides the energy to create large amounts of reduced organic carbon and a reservoir of oxygen. The presence of these two products is a prime example of the nonequilibrium conditions that often exist with respect to redox conditions in natural waters. Respiratory, fermentative, and other non-photosynthetic organisms take advantage of the energy stored in the reduced carbon and enzymatically catalyze a myriad of reactions that tend to restore equilibrium. Table 1 lists several of these potential reactions, many of which are further explored in this chapter. Therefore, the carbon cycle, which depends not only on the cycles of other nutrients, can alter the cycle of these nutrients through their reduction–oxidation reactions.
Table 1. Sequence of electron acceptors used for respiratory oxidation of organic carbon

The cycles of two metals, iron and mercury, are of special interest in freshwater ecosystems. Iron is required as an important micronutrient in many cellular functions of both autotrophs and heterotrophs. Iron cycling has additional ramifications because of its interaction with other cycles, namely phosphorus and sulfur. Mercury concentrations have been increasing in freshwater ecosystems as a result of human activities. Because of its toxicity, the cycling of Hg warrants special attention. All of these metals are highly electroactive, so their cycling is closely linked to redox conditions.
Iron exists in freshwater environments in both the oxidized [Fe(III)] and reduced [Fe(II)] forms. The primary factor controlling the cycling of Fe is the difference in solubility between the oxidized and reduced moieties. The reduced forms tend to be much more soluble than the oxidized forms. In addition to the cycling within an aquatic ecosystem, the difference in solubility also determines how these metals are transported to the ecosystem. Dissolved organic matter (DOM), because of its ability to bind with metals, can also influence the transport and cycling of Fe. Although it is not considered in this chapter, manganese has very similar properties to Fe, and the cycles are nearly similar.
Rust is a familiar form of oxidized iron. Oxidized iron, also called ferric iron, has relatively low solubility in most surface freshwaters and is generally in the solid form of Fe(OH)3. Therefore, it is mainly transported to lakes via streams or atmospheric deposition in particulate or colloidal form or bound or chelated to DOM. On the other hand, reduced iron (ferrous iron) is much more soluble and can be transported in its ionic form (Fe2+), as long as the water is anoxic or strongly acidic. Therefore, groundwater with low concentrations of oxygen can carry reduced iron to lakes. The solubility of Fe2+ is mainly controlled by FeCO3 and FeS.
In well-oxygenated waters such as the epilimnion, ferrous iron is oxidized quickly to form the precipitate, Fe(OH)3. Other ions can be precipitated with the ferric ion or become adsorbed onto the ferric hydroxide, most notably orthophosphate (see section 4). Surface waters of lakes thus tend to have low concentrations of iron because of its low solubility and the tendency for particulate iron to leave the surface water via sedimentation. In some instances, the low concentration of iron in the surface water can directly limit algal primary production, as is the case in some parts of the ocean (chapter III.13).
Oxygen consumption by heterotrophic bacteria usually creates substantial redox gradients in the sediments or hypolimnia of lakes. This redox gradient affects the internal cycling of Fe significantly. When oxygen becomes depleted, iron can act as an electron acceptor for respiration, and iron is reduced. Because reduced iron tends to be more soluble, Fe2+ can diffuse upward through the sediments toward the overlying water column. When it reaches an oxygenated strata, the solid Fe(OH)3 is again formed. When this reaction occurs in the water column, the precipitate again sediments out of the water column. Whether the location of the zone of iron oxidations is within the sediments or in the water column has large ramifications for the cycling of P. The repeated cycling of iron oxidation states is sometimes referred to as the “ferrous wheel” (figure 2). Some bacterial assemblages take advantage of the ferrous wheel to continually supply a source of energy to fix carbon from the oxidation of reduced iron and, conversely, to supply an electron acceptor for respiration during the reduction of oxidized iron. In the presence of reduced forms of sulfur, ferrous iron can also form amorphous FeS, which is fairly insoluble and tends to remove iron from further cycling. The production of FeS can be a significant factor in the cycling of both Fe and S.
Figure 2. The iron cycle. External inputs and outputs are not considered. The impact of the iron cycle on both orthophosphate and sulfide is noted in this figure.
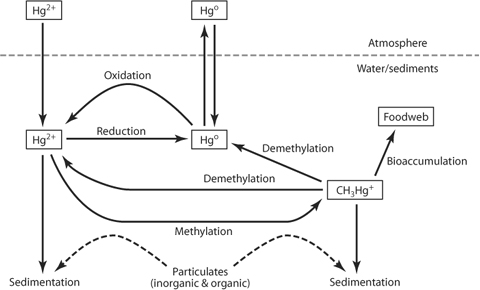
Mercury mainly exists in two different oxidation states in the environment: elemental mercury (Hg0) and the more oxidized mercuric mercury (Hg2+) (figure 3). Mercury occurs naturally in the environment, most often as inorganic salts [e.g., HgCl2, Hg(OH)2, HgS]. Elemental mercury, the familiar silver-colored liquid in thermometers, is rare in the environment. However, the presence of total mercury in the environment has been increasing mainly because of the combustion of fossil fuels and other industrial processes. In the atmosphere mercury can exist in both oxidation states; however, Hg0 has a longer residence time in the atmosphere and can be transported large distances. Mercuric Hg is more quickly removed from the atmosphere in precipitation, and both forms can be deposited on land and water as dry deposition. The solubility and movement of Hg from land to water are strongly controlled by its complexation with DOM. Once in a lake or wetland, mercury can undergo changes in oxidation state, similar to Fe. Elemental mercury is not very soluble in water and has a tendency to volatilize back into the atmosphere. This can be one significant pathway of loss in aquatic ecosystems. Another significant loss of mercury from a lake is via sedimentation of organic or inorganic materials that have complexed with Hg. Neither of the inorganic forms of Hg is extremely toxic to biota.
One peculiar facet of mercuric mercury, however, is its ability to form covalent bonds as opposed to ionic bonds. One covalently bonded molecule in particular, methyl mercury (CH3Hg+), is extremely toxic. In addition, methyl mercury tends to bioaccumulate in organisms. This has led to advisories suggesting limited consumption of fish from certain areas. The balance between inflows and losses, and the processes that methylate and demethylate mercury, control the concentrations of toxic methyl mercury in fish. Two correlates with high fish methyl mercury concentrations are high dissolved organic carbon concentrations and low pH (although these two parameters are often correlated).
Methylation of mercury is thought to be carried out predominately by sulfate-reducing bacteria (see section 6, below). Once it has been methylated, methyl mercury still has a large affinity for complexing with particles, such as bacteria and algae. This may be an important mechanism for methyl mercury to enter the food web. Once in an organism, methyl mercury is lipophilic and is not easily excreted. This leads to the bioaccumulation that poses increasing threats for large predators, including avian predators that rely on a large portion of fish for their diet. Several bacteria are also able to demethylate mercury, returning it to either Hg0 or Hg2+. Additionally, reactions with UV light can lead to photodegradation of methyl mercury. There are still large uncertainties in the specific details of many aspects of mercury cycling. The uncertainty in these processes and the need for sound policy regarding mercury emissions have created contention among state, federal, and international authorities.
Phosphorus is often found to be limiting to primary production in many freshwater ecosystems, especially lakes. Nitrogen, another key limiting nutrient is discussed below. The phosphorus cycle has been greatly augmented by man. The use of phosphorus as a fertilizer and detergent has led to increased loading into freshwater ecosystems. The consequence of this excess phosphorus is cultural eutrophication (see chapter VII.5).
Although the P cycle is closely linked to redox conditions, phosphorus in water is mainly found at one oxidation state in the form of orthophosphate (PO43–). At this oxidation state, P exists in several different inorganic and organic forms, either dissolved or particulate. The primary P-containing geologic mineral is apatite. Sedimentary rocks such as CaCO3 also can contain significant amounts of P. Inorganic forms include dissolved orthophosphate or particulate orthophosphate adsorbed to clay, carbonates, ferric hydroxides, or organic particles. Phosphorus is found in cellular organic material such as nucleic acid, phosphoproteins, phospholipids, phosphate esters, or nucleotide phosphates (e.g., ATP).
Figure 3. The mercury cycle. Additional stream and groundwater sources are not displayed.
In the absence of human influence, most terrestrial landscapes substantially retain P. Phosphorus is largely held in the vegetation, in soil organic matter, or adsorbed to inorganic particles. In these situations, the primary flux of P to aquatic ecosystems comes from atmospheric deposition or fluvial inputs of dissolved organic P. In the presence of human activity, the forms and quantities of P loading change. Use of P fertilizer can increase the amount of dissolved orthophosphate in streams and groundwater. Land use practices that cause erosion increase the proportion of P that enters aquatic ecosystems adsorbed or contained in particulates.
The simplest example of the aquatic P cycle occurs in lakes with high levels of oxygen throughout the water column. These conditions are usually found in oligotrophic lakes, lakes with low levels of nutrients that limit primary productivity. In oligotrophic lakes, very little phosphorus is found in the dissolved form, and most is contained in particulate forms. Phosphorus is rapidly and efficiently cycled in the biota. There is strong competition between algae and bacteria for limited P resources. Algae and bacteria also produce enzymes to release bound phosphorus (phosphatases). Losses of particulate P via sedimentation constitute the dominant loss term in this oligotrophic P cycle.
Several conditions such as increased productivity, large external loading of organic carbon, or a high ratio of sediment surface area to hypolimnetic volume can lead to oxygen depletion in the hypolimnion. The change in redox conditions and zone of Fe oxidation in the sediments can significantly alter the P cycle (figure 2). Under oxic conditions, orthophosphate can be adsorbed with iron hydroxide or precipitated as FePO4. When Fe is reduced in anoxic sediments, soluble Fe2+ is produced, and PO43– is also released. If the zone of Fe oxidation occurs within the sediments, PO43– is effectively “trapped” in the sediments as it readsorbs with iron precipitates. Under extremely anoxic conditions, there is no zone of oxidation in the sediments, and the PO43– can be returned to the water column. If reduced iron is buried as FeS, PO43– can escape, but there will be little iron available to be reoxidized, allowing the PO43– to remain in solution. Therefore, P cycling can be linked to both Fe and S cycling.
The recycling of orthophosphate from the sediments represents an internal feedback mechanism that has significant impact on the mitigation of human-caused eutrophication. The recycling of P from the sediments sustains high levels productivity and reinforces the conditions for P recycling. In these cases, mitigation of the original P pollution will have only a limited effect on reducing the problems of eutrophication (see chapter VII.5).
Nitrogen exists in several different oxidation states, and the variation in redox conditions in freshwater ecosystems creates an environment where transformations between oxidation states are significant. Most nitrogen exists in its molecular form, N2, as a gas. Biological fixation of N2 to organic forms is therefore a significant pathway in the N cycle. Humans have invented methods for N2 fixation, and these methods have contributed more fixed nitrogen to the environment than forms of biological fixation. The effect of this added N has had large ramifications for many environments including freshwater ecosystems (see chapter III.10).
The forms of nitrogen found in water include dissolved gases such as N2, N2O (nitrous oxide), and (ammonia), dissolved ions of NH4+ (ammonium), NO2– (nitrite), and NO3– (nitrate), and dissolved and particulate organic forms. The reactive inorganic forms of N (species of N, excluding N2) most prominent in the environment are NH4+ and NO3–. The organic forms range from amino acids, amines, and proteins to complex organic compounds with low nitrogen content.
Nitrogen is often considered a key limiting nutrient for terrestrial ecosystems. Therefore, N tends to be highly retained in vegetated landscapes. As with P, this terrestrial retention can cause N to be limiting (or co-limiting with P) to aquatic primary production. For lakes in pristine environments, terrestrial landscapes do contribute some dissolved and particulate organic nitrogen. Atmospheric deposition is a significant source of reactive inorganic nitrogen (NH4+ or NO3–). Biological fixation of N2 by cyanobacteria and other heterotrophic bacteria can also be important.
Several sources of N pollution significantly alter the flux of N to recipient freshwater ecosystems. Fertilizer use can increase the flux of both NH4+ and NO3– from streams and groundwater. Agricultural practices also tend to increase the amount of gaseous NH3, which can be deposited on the lake surface. Of particular interest is the emission of nitrogen oxides (NOx) emanating from high-temperature combustion of fossil fuels, mainly attributed to automobiles. Nitrogen oxides are transformed in the atmosphere or in the lake and become nitric acid, an important component of acid rain. All these inputs can increase the productivity of the lake, with the side effect of increased respiratory loss of O2 in the aphotic zone.
Fixation of N2 by cyanobacteria is a key process in supplying nitrogen to freshwater ecosystems when external sources are small. Nitrogen fixation by cyanobacteria requires nitrogenase enzyme, organic carbon, and light energy. In most cyanobacteria, nitrogen fixation takes place in specialized cells called heterocysts. Because nitrogenase enzyme is inactivated by O2, the heterocysts provide a microenvironment of low oxygen to allow for nitrogen fixation. Heterocysts are only found in filamentous cyanobacteria; however, not all species of filamentous cyanobacteria have heterocysts. Also, some unicellular cyanobacteria have the ability to fix N2 without the presence of heterocysts. Nitrogen fixation generally increases when inorganic nitrogen concentration is low or when the N:P ratio is decreased, sometimes because of P pollution.
Within a lake, the major transformations of N are largely maintained by microbial processes (figure 4). Algae can use both NH4+ and NO3– to form N-containing organic compounds, most notably amino acids. However, NO3– must first be reduced to NH4+ before it can be incorporated as an amine group. This process is known as assimilatory (because the nitrogen is incorporated in the biomass) nitrate reduction. Nitrate assimilation is therefore more energetically costly than ammonium assimilation, and ammonium is preferentially used, if available. Because of the preferential uptake of ammonium by plants and algae, it generally has very low concentrations in surface waters. Another process that keeps ammonium at relatively low concentrations is nitrification.
Figure 4. The nitrogen cycle. External inputs of inorganic and organic nitrogen from streams, groundwater, and precipitation are not displayed; however, they are often much more significant than N2 fixation.
Nitrification is the process of NH4+ oxidation. The oxidation is biologically mediated by a relatively small number of bacterial species and has two distinct steps. These reactions are a form of chemoautotrophic production. The first step,

is carried out mainly buy Nitrosomonas bacteria. The second step,

is carried out by Nitrobacter. The overall reaction,

points to some interesting aspects of nitrification. First, nitrification is an aerobic process, and although it is autotrophic, it actually consumes 2 moles of O2 per mole of N oxidized. In the absence of O2, nitrification can not proceed, and it is possible for relatively higher concentrations of NH4+ to accumulate (e.g., in sediments or hypolimnia that experience anoxia). Second, nitrification is an acidifying process; 2 moles of H+are produced per mole of N oxidized. Therefore, NH4+ pollution can indirectly lead to acidification after nitrification.
Particulate organic nitrogen has two significant fates. First, the particulate organic nitrogen (also dissolved organic nitrogen) can be decomposed by hetero-trophic bacteria. If the N in the detrital organic matter is in excess of the needs of the bacteria, it will be released as NH4+. The release of NH4+ from organic material decomposition is called mineralization or ammonification. At this point the NH4+ can again be assimilated by microorganisms or be nitrified. The second major fate of particulate organic nitrogen is sedimentation. As bacteria preferentially decompose simpler compounds or molecules where N is more accessible, there is a decrease in the quality of the remaining organic matter for further degradation. The N bound in this organic matter is then permanently buried in the sediments.
One additional process is key to understanding N cycling in freshwater ecosystems. Denitrification is the reduction of NO3– with the concomitant oxidation of organic matter. Nitrate is used as a terminal electron acceptor when oxygen becomes depleted (table 1) and yields nearly as much free energy as aerobic respiration. Denitrification is carried out by many facultative anaerobic bacteria. During denitrification, NO3– is sequentially reduced to N2 following these intermediate steps:

The process can be interrupted at any of the intermediate steps, but generally there is little accumulation of either NO2– or N2O. Denitrification tends to be limited to certain areas because it requires a source of NO3–, organic carbon, and low oxygen. Sediments of lakes and wetlands often have high rates of denitrification, as do the hyporheic zones of streams and rivers. The loss of N2 to the atmosphere after denitrification is a permanent loss of nitrogen from the system. Because denitrification decreases concentrations of inorganic nitrogen, wetlands are often constructed with the specific purpose of denitrifying large amounts of nitrate in order to reduce the load of nitrate to downstream ecosystems. Denitrification is also important in regard to the impacts of acid deposition because the process consumes hydrogen ions.
A large interest in the sulfur cycle of lakes developed after the recognition of the impacts of acid precipitation, which is often dominated by sulfuric acid. Sulfur dioxide (SO2), mainly emitted by coal combustion and ore smelting, is oxidized to form sulfuric acid (H2SO4) in the atmosphere or in the lake. Acidic precipitation has increased the flux of sulfate ions (SO42–) from both direct inputs to a lake. Some regions can have naturally high concentrations of SO42–. Sedimentary deposits often contain significant amounts of calcium sulfate. Weathering and oxidation of sulfur (S0) or sulfide (S2–)-containing minerals will also produce sulfate ions and acidity.
Lakes have some natural ability to mitigate the inputs of SO42–. Just as the reduction of nitrate consumes H+, sulfate reduction also consumes acidity (2 moles of H + per mole of SO42– reduced). The reduction of SO42– also has important connections to Fe, Hg, and P cycling. Sulfate can be used as a terminal electron acceptor for anaerobic respiration by sulfate-reducing bacteria (table 1). Sulfate-reducing bacteria produce hydrogen sulfide (H2S). Hydrogen sulfide is quickly oxidized if it diffuses into oxygenated waters. Or, the reduced sulfide can react with reduced Fe to form fairly insoluble precipitates such as FeS. The formation of FeS, if it occurs beyond the zone of oxidation in the sediments, can represent a permanent burial of S. Sulfur-containing organic matter can be formed by reduction of sulfate and by abiotic reactions of sulfide and organic matter. This organic bound sulfur tends to be less prone to reoxidation in the sediments than FeS.
Regions with high loading of SO42– ions and significant sulfate reduction can precipitate a large quantity of iron as FeS. The relationship between Fe and S cycling also impacts P cycling (figure 2). With large amounts of sulfide and FeS formation, very little Fe2 diffuses to the water column, but PO43– still escapes. It has been speculated that increased flux of SO42– from acid rain could exhaust the supply of iron in some lakes. The ramifications of this are twofold. First, if FeS can no longer be formed because of limited Fe, sulfide will remain as H2S. Because H2S can be easily reoxidized, there will be little permanent loss of sulfate, and the consumption of acidity by sulfide burial will be reduced. Second, the loss of Fe2 that can be oxidized could possibly increase the release of PO43– from sediments, affecting eutrophication. Additionally, sulfate-reducing bacteria play a significant role in the methylation of Hg, and increased SO42– concentrations could alter the cycling of Hg.
Two groups of sulfur-oxidizing bacteria can use H2S. Chemosynthetic sulfur-oxidizing bacteria derive energy from the H2S and oxidize it to elemental sulfur or sulfate. Photosynthetic sulfur bacteria also oxidize H2S. Both the green and purple photosynthetic sulfur bacteria are anaerobes and require light as a source of energy. Hydrogen sulfide is used as an electron donor, with the concomitant production of elemental sulfur, just as water is used as an electron donor in oxygen-producing photosynthesis.
Central to inorganic C cycling is the role of carbonate chemistry. Carbonate chemistry describes which species of inorganic carbon are present in water. To start the explanation of carbonate chemistry, it is probably easiest to consider a very simplistic example of water in a beaker. This removes some of the complexities of biogeochemistry that are considered later.
Carbon dioxide (CO2) from the atmosphere is soluble in water. Once dissolved in water CO2 is hydrated by water to yield carbonic acid:

The equilibrium concentration of carbonic acid is considerably less than that of dissolved CO2, such that the amount of carbonic acid is negligible compared to the other constituents. Carbonic acid can dissociate to form bicarbonate ions (HCO3–) and carbonate ions (CO32–).
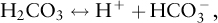
and

The sum of these three carbonate species—dissolved CO2, bicarbonate, and carbonate—are often described as the dissolved inorganic carbon (DIC).
The speciation of the different carbonate compounds is pH dependent, and if the pH of the beaker is altered, the amount of DIC and the relative proportion of each of the species will change. At an approximate pH of 6.4, the concentrations of bicarbonate and dissolved CO2 are equal, and the relative contribution of carbonate ions to the amount of DIC is negligible. At an approximate pH of 10.4, the concentrations of bicarbonate and carbonate are equal, and the relative contribution from dissolved CO2 is negligible.
The range of pH observed in freshwater systems is generally between 4 and 10. For a simple approximation, one can assume that if the pH of the system is below 5, dissolved CO2 dominates the DIC. As pH increases, the proportion of CO2 declines, and bicarbonate dominates between pH 7 and 9. As pH further increases, carbonate begins to become appreciable above pH 9.5.
Adding to the complexity of the beaker example, the addition of some other ions can influence the carbonate chemistry. Calcium has a large interaction with carbonate chemistry. In the presence of high concentrations of calcium and high pH, calcium carbonate (CaCO3) can precipitate from the system:

The reverse of this reaction can also take place, with CaCO3 becoming soluble with increased acidity or the presence of increasing CO2 (which increases the acidity through carbonic acid).
The final layer of complexity includes the biological processes that make a lake infinitely more interesting than the beaker example (figure 5). The main biological processes of interest are those that consume or generate CO2, namely, photosynthesis and respiration. Photosynthesis consumes CO2, and this can alter the carbonate chemistry in several ways. As CO2 is consumed, pH increases, and at very high rates of photosynthesis pH can increase to greater than 9. In the presence of enough calcium, during these events of high productivity, CaCO3 can precipitate. This tends to restore the system to lower pH, as CO2 is released during the precipitation of calcium carbonate. The precipitation of calcium carbonate can be an important sedimentary process in a lake. The sedimentation of CaCO3 can increase the burial of organic carbon as well as orthophosphate, which can coprecipitate with the CaCO3. Respiration, on the other hand, releases CO2 and will cause the pH to decline.
Figure 5. The carbon cycle. This very generalized diagram does not consider other forms of autotrophic production besides photosynthesis, but in general, these processes are much less significant.
We must keep in mind that the patterns of dissolved gases, such as O2 and CO2 are also modulated by the physics of gas exchange. If the concentration is higher than equilibrium with the atmosphere, gas will be lost to the atmosphere, and vice versa. The rate of this flux is dependent on the concentration gradient between air and water and the amount of turbulence, most often caused by wind, at the air–water interface.
The source of inorganic carbon in lakes is often much greater than just the amount coming from atmospheric exchange, however. Inflowing streams and groundwater can import large quantities of inorganic carbon. Groundwater is unique in that it can often be much more supersaturated in CO2 than surface waters because of a predominance of respiration and very slow atmospheric gas exchange.
There are two sources of organic carbon for lakes: primary production of carbon within the lake (autochthonous carbon) and inputs of external sources of carbon, primarily from the terrestrial component of the watershed (allochthonous carbon). Organic carbon can either be dissolved or particulate (DOC and POC). Particulate carbon can be further categorized as living organisms or detritus (nonliving organic carbon). Once present in the lake, organic carbon has several fates (figure 5). The organic carbon can be exported downstream or buried in the sediments of the lake. Also, the organic carbon can be respired to inorganic compounds (CO2 or CH4) and lost via gaseous exchange with the atmosphere. Therefore, freshwater ecosystems can have an interesting role in the landscape, capable of both burying organic carbon and releasing CO2 back to the atmosphere.
The balance of organic carbon burial and loss of CO2 to the atmosphere is represented by net ecosystem production (NEP). NEP is the difference of total respiration (RT) from gross primary production (GPP):

Positive NEP means that GPP exceeds RT and that CO2 is taken from the atmosphere and buried as organic carbon. Alternatively, negative NEP means that RT exceeds GPP. In order for RT to exceed GPP, there must be a subsidy of allochthonous carbon. With sufficient external inputs of organic carbon, there can be burial of organic carbon and release of inorganic carbon from aquatic ecosystems.
Respiration of organic carbon in aerobic conditions produces CO2 and is the most energetically favorable. Under anaerobic conditions, other electron acceptors besides O2 must be used with declining energetic benefit (table 1). In many freshwater ecosystems that experience oxygen depletion, fermentation and methanogenesis represent the final steps in organic matter decomposition. During fermentation, organic carbon acts as both an electron donor and acceptor. A common example of fermentation is the breakdown of glucose, which produces acetic acid, CO2, and hydrogen (H2). Fermentation produces very little energy compared to other forms of respiration, but it is a significant process because fermentative organisms can degrade many organic compounds that are not accessible to other nonfermentative organisms.
The final step in organic matter decomposition is methanogenesis, which is closely tied to fermentation. Two pathways of methanogenesis are prevalent. The first pathway, CO2 reduction, is a chemoautotrophic process. In this pathway,

hydrogen, supplied from fermentation, is a source of energy, and CO2 is both an electron acceptor and the source for cellular carbon production. In the second pathway, acetate is decomposed to CO2 and methane:

Methane can be further cycled in the water column. Methane can be oxidized by chemoautotrophic organisms. Also, methane is not very soluble in water, and in sediments where production of methane is high, methane comes out of solution to form bubbles. When the bubbles escape the sediments, they bypass the opportunity for oxidation and escape to the atmosphere (ebullition). The contribution of freshwater ecosystems, especially wetlands, to the global flux of methane is significant. Because methane is about 20 times more effective than CO2 as a greenhouse gas, the cycling of methane in freshwaters is of special interest.
The cycling of organic carbon in freshwater ecosystems is closely tied to the cycling of other elements through changing redox conditions in different environments. Conversely, the supply of many elements is critical for the production of new organic carbon. An important question is whether the carbon and biogeochemical cycling of freshwater ecosystems is significant on a large, even global, scale. This question is especially interesting in the light of understanding the global carbon budget. In the case of lakes, many are supersaturated in CO2, implying that RT exceeds GPP, and NEP is negative. A significant amount of allochthonous carbon must be respired for this to occur. This appears to be a common occurrence in many freshwater ecosystems. As organic carbon moves from land to the ocean through a series of aquatic ecosystems, a significant portion of the carbon is returned to the atmosphere as CO2.
But freshwater ecosystems can also bury significant amounts of carbon produced either internally or externally. As redox conditions decrease in the anoxic zones of freshwater ecosystems, decomposition of organic carbon becomes less and less energetically favorable, and carbon can be effectively buried. As part of a working group at the National Center for Ecological Analysis and Synthesis, Cole and colleagues (2007), estimated globally that aquatic ecosystems received about 1.9 Pg C year–1 from adjacent terrestrial ecosystems. Of this amount, about 0.8 Pg C year–1 was lost to the atmosphere as CO2, and about 0.2Pg C year–1 was buried as sediments. The remainding 0.9 Pg C year–1 was supplied to the oceans from the worlds rivers as organic or inorganic carbon. Evidence is mounting that carbon and biogeochemical cycling of elements in freshwater ecosystems matters, even at the large, global scale.
Allan, J. D., and M. M. Castillo. 2007. Stream Ecology: Structure and Function of Running Waters. Dordrecht: Springer.
Cole, J. J., Y. T. Prairie, N. F. Caraco, W. H. McDowell, L. J. Tranvik, R. G. Striegl, C. M. Duarte, P. Kortelainen, J. A. Downing, J. J. Middelburg, and J. Melack. 2007. Plumbing the global carbon cycle: Integrating inland waters into the terrestrial carbon budget. Ecosystems 10: 171–184.
Mitsch, W. J., and J. G. Gosselink. 2000. Wetlands. New York: John Wiley & Sons.
Schlesinger, W. H. 1997. Biogeochemistry: An Analysis of Global Change. San Diego: Academic Press.
Wetzel, R. G. 2001. Limnology: Lake and River Ecosystems. San Diego: Academic Press.