Induced Innate Immunity Effector Mechanisms
Thus far in this chapter we have discussed the anatomical barriers that provide the first line of defense against infection and have introduced the six families of pattern recognition receptors and the signaling pathways they activate that induce the cellular innate immune responses that constitute the second line of defense. We will now present the induced effector mechanisms by which innate immune responses protect us. Some are molecules that are directly antimicrobial, while others are cellular responses that eliminate pathogens or infected cells. In general, these effector mechanisms are effective against the invading pathogen that induced them, such as antiviral interferons for viruses, phagocytosis for extracellular bacteria, or cell death for infected cells, but of course they are not specific for individual pathogens, as are adaptive immune responses. Overview Figure 4-15 summarizes these innate immunity effector mechanisms.
Expression of Innate Immunity Proteins Is Induced by PRR Signaling
The PRR-activated signaling pathways described above activate transcription factors that turn on genes encoding an arsenal of proteins that help us to mount protective responses. Some of the induced proteins are antimicrobial and directly combat pathogens, while others serve key roles in activating and enhancing innate and adaptive immune responses. There is tremendous variation in what proteins are made in response to different pathogens, reflecting their PAMPs as well as the responding cell types and their arrays of PRRs. Some of the most common proteins and peptides that are secreted by cells following PAMP activation of PRRs and that contribute to innate and inflammatory responses are listed in Table 4-4.
| Peptides/proteins | Produced by | Act on | Immune/inflammatory effects | |
|---|---|---|---|---|
| Antimicrobials | Defensins and cathelicidin* |
Epithelia (e.g., oro/nasal, respiratory, intestinal, reproductive tracts; skin keratinocytes, kidney); neutrophils, NK cells |
Pathogens Monocytes, immature dendritic cells, T cells Mast cells |
Inhibit, kill Chemoattractant; activate cytokine production Activate degranulation |
Interferons α and β |
Virus-infected cells, macrophages, dendritic cells, NK cells |
Virus-infected cells NK cells Macrophages, T cells |
Inhibit virus replication Activate Regulate activity |
|
| Cytokines | IL-1 |
Monocytes, macrophages, dendritic cells, keratinocytes, epithelial cells, vascular endothelial cells |
Lymphocytes Bone marrow Vascular endothelium Liver Hypothalamus |
Enhances activity Promotes neutrophil production Activates; increases vascular permeability Induces acute-phase response Fever |
IL-6 |
Monocytes, macrophages, dendritic cells, NK cells, epithelial cells, vascular endothelial cells |
Lymphocytes Bone marrow Vascular endothelium Liver Hypothalamus |
Regulates activity Promotes hematopoiesis → neutrophils Activates; increases vascular permeability Induces acute-phase response Fever |
|
TNF-α |
Monocytes, macrophages, dendritic cells, mast cells, NK cells, epithelial cells |
Macrophages Vascular endothelium Liver Hypothalamus Tumors |
Activates Activates, increases vascular permeability, fluid loss, local blood clotting Induces acute-phase response Fever Cytotoxic for many tumor cells |
|
GM-CSF |
Macrophages, vascular endothelial cells |
Bone marrow |
Stimulates hematopoiesis → myeloid cells |
|
IL-12, IL-18 |
Monocytes, macrophages, dendritic cells |
Naïve CD4 T cells Naïve CD8 T cells, NK cells |
Induce TH1 phenotype, IFN-γ production Activate |
|
IL-10 |
Macrophages, dendritic cells, and mast cells; NK, T, and B cells |
Macrophages, dendritic cells |
Antagonizes inflammatory response, including production of IL-12 and TH1 cells |
|
| Chemokines | Example: IL-8 (CXCL8)† |
Macrophages, dendritic cells, vascular endothelial cells |
Neutrophils, basophils, immature dendritic cells, T cells |
Chemoattract cells to infection site |
Antimicrobial Peptides
Defensins and cathelicidins were mentioned earlier as being important in barrier protection, such as on the skin and the mucosal epithelial layers connected to the body’s openings (see Table 4-2). Some cells and tissues constitutively express these peptides. For example, human intestinal epithelial Paneth cells constitutively express α-defensins and some β-defensins. In addition, some defensins and the cathelicidin LL-37 are constitutively synthesized and packaged in the granules of neutrophils, ready to kill phagocytosed bacteria, fungi, viruses, and protozoan parasites.
However, in some other cell types, such as mucosal and glandular epithelial cells, skin keratinocytes, and NK cells, the expression of these antimicrobial peptides is induced or enhanced by signaling through PRRs, in particular TLRs and NLRs. Macrophages do not produce these antimicrobial peptides following PRR activation; however, there is an indirect pathway by which microbes induce LL-37 in macrophages. Binding of microbial ligands to macrophage TLRs induces increased expression of receptors for vitamin D; binding of vitamin D to these receptors activates the macrophages to produce LL-37, which then can help the macrophages kill the pathogens.
Type I Interferons
Another major class of antimicrobial proteins transcriptionally induced directly by PRRs is the type I interferons, of which the major representatives are IFN-α and IFN-β. As summarized in Table 4-4, type I interferons are produced in two situations. When infected with a virus, many cell types are induced to make IFN-α and/or IFN-β following binding of cytosolic viral PAMPs, usually nucleic acids, to intracellular PRRs such as ALRs, RLRs, and cGAS. These PRRs activate the IRF transcription factors that induce expression of IFN genes. In addition, many uninfected cells express cell surface TLRs that recognize extracellular viral PAMPs, and/or they internalize virus without necessarily being infected, allowing endosomal TLRs to recognize viral components. Signaling from these TLRs activates the IRFs and IFN-α and IFN-β production.
One type of dendritic cell, called the plasmacytoid dendritic cell (pDC) because of its shape, is a particularly effective producer of type I IFNs (and also type III IFNs such as IFN-l, which have similar functions). The pDCs endocytose virus that has bound to various cell surface proteins (including CLRs such as DC-SIGN, which, e.g., binds HIV). TLR7 and TLR9 in the endosomes then are activated by viral PAMPs (ssRNA and viral DNA, respectively), leading to IRF activation and type I IFN production.
IFN-α and IFN-β exert their antiviral and other effects by binding to a specific receptor called IFNAR (IFN-alpha receptor) that is expressed by most cell types. Like many cytokines, IFNs are dimers. Binding of the IFN dimer to IFNAR induces receptor dimerization and activation of the JAK/STAT signaling pathway, used by many cytokines to activate specific responses, as was introduced in Chapter 3 (see Figure 3-25). The IFNAR dimer activates the Janus kinases JAK1 and TYK2, which recruit and phosphorylate inactive STAT transcription factors (Figure 4-16). Phosphorylated STAT1 and STAT2 dimerize and change conformation, revealing a nuclear localization signal that allows the dimer to enter the nucleus, where it initiates transcription of specific genes.

FIGURE 4-16 Induction of antiviral activities by type I interferons. Interferons α and β bind to and dimerize IFNAR (the IFN-alpha receptor), which then recruits and activates the JAK1 and TYK2 protein kinases. They bind and phosphorylate STAT1 and STAT2, which dimerize, enter the nucleus, and stimulate expression of proteins that activate antiviral effects. Four are shown in this figure. Protein kinase R (PKR) binds viral dsRNA and inhibits the activity of the eIF2α translation initiation factor. 2’,5’-Oligoadenylate synthetase synthesizes 2’,5’-oligoadenylate, which activates a ribonuclease, RNase L, that degrades viral and cellular mRNAs. Mx proteins self-assemble into ringlike structures that inhibit viral replication and the formation of new viral particles. IFIT proteins inhibit translation of viral proteins by binding to viral RNA and to eIF3, a translation initiation factor.
Genes turned on by IFN are known as interferon-stimulated genes (ISGs). Four ISGs important for inhibiting viral replication are shown in Figure 4-16:
- Protein kinase R (PKR) binds and is activated by dsRNA; it then blocks viral (and cellular) protein synthesis by inhibiting the translation initiation factor eIF2α.
- 2’,5’-Oligoadenylate A synthetase (OAS) is a nucleotidyltransferase structurally related to cGAS. After its expression is induced by IFN, OAS binds cytosolic dsRNA, which activates it to generate 2’,5’-oligoadenylate from ATP. Oligoadenylate then binds RNase L and induces it to degrade viral RNA.
- Mx group proteins inhibit both the transcription of viral genes into mRNAs and the assembly of virus particles.
- The IFIT (IFN-induced proteins with tetratricopeptide repeats) proteins bind dsRNA, blocking viral RNA translation. Several IFITs also bind and inactivate the eIF3 translation initiation factor.
Reflecting the potent antiviral activities of type I interferons, they are used to treat some viral infections, such as hepatitis B and C. In addition to their key roles in controlling viral infections, type I interferons have several other beneficial immune-related activities. They increase expression of MHC class I proteins, making the cells better targets for T cell–mediated killing; activate NK cells; and regulate the activities of macrophages and T cells. Treatment with IFN-β has been shown to have beneficial effects in some forms of multiple sclerosis, a T cell–mediated autoimmune disease with inflammatory involvement, probably by inhibiting production of proinflammatory cytokines, including IL-1 and others produced by T cells (see Chapter 16).
Cytokines
Among the proteins transcriptionally induced by PRR activation are several key cytokines, which—while not directly antimicrobial—activate and regulate a wide variety of cells and tissues involved in innate, inflammatory, and adaptive responses. Cytokines function as the protein hormones of the immune system, produced in response to stimuli and acting on a variety of cellular targets. Several key examples of cytokines induced by PRR activation during innate immune responses are listed in Table 4-4, along with their effects on target cells and tissues.
Three of the most important cytokines are IL-1, TNF-α, and IL-6, the major proinflammatory cytokines. They act locally on blood vessels to increase vascular permeability and also on other cells, including lymphocytes, to recruit and activate them at sites of infection. They also have systemic effects (see below), including inducing fever and feeding back on bone marrow hematopoiesis to enhance production of neutrophils and other myeloid cells that will contribute to pathogen clearance.
IL-1 exists in two forms, IL-1α and IL-1β; the latter is more common and is what we refer to as IL-1. Its gene is activated by transcription factors downstream of many PRRs; as was discussed in Advances Box 4-2, the initial large pro-IL-1 precursor must be processed into the smaller form by caspases, usually part of activated inflammasomes. The IL-1 receptor (see Figure 3-19) has two chains that have immunoglobulin (Ig)-like domains and a cytoplasmic TIR domain (remember that TIR stands for TLR and IL-1R). Because of its TIR domain, after binding IL-1 the IL-1R recruits MyD88 to the receptor and activates the same signaling pathways, transcription factors, and genes for antimicrobial and proinflammatory proteins as do plasma membrane TLRs (see Figure 4-8). IL-1 induces its own synthesis, an example of a proinflammatory positive feedback loop.
TNF-α (often called just TNF) activates cells via the trimeric TNF receptor (Figure 4-17). Although it can induce apoptosis of some cells, such as some tumors, it activates macrophages to be more active in innate immunity, becoming more efficient at phagocytosis and generating antimicrobial molecules and cytokines. As shown in Figure 4-17a, binding of TNF-α to its receptor triggers unique receptor-proximal steps that activate TAK1 and downstream signaling pathways and transcription factors common to TLR and IL-1R signaling pathways.

FIGURE 4-17 Signaling through TNF receptors. Signaling through TNF receptors (TNFRs) can lead to different outcomes, depending on the cells and their environment. (a) Binding of TNF, which is a trimer, to the receptor activates the receptor’s cytoplasmic DD (death domain) region and recruits the TRADD (TNF receptor–associated death domain) adaptor. TRADD then binds the kinase RIP1, TRAF2 (TNF receptor–associated factor 2), and several other proteins. The complex recruits and activates TAK1 as well as the IKK complex of the NF-κB pathway and MAPK pathways. NF-κB and AP-1 can activate the same genes as in TLR and IL-1 signaling pathways. Among other prosurvival effects, NF-κB activates the transcription of the cFLIP protein, which inhibits TNF-induced apoptosis. (b) In certain cells, the RIP1/TRADD/TRAF2 complex dissociates from the receptor and migrates to the cytoplasm, where it binds the adapter protein FADD, which in turn binds procaspase-8. Its clustering activates formation of the active caspase-8 protease, which cleaves other enzymes and initiates apoptosis.
In contrast, IL-6, a class 1 cytokine, stimulates different responses. As is true for other class 1 cytokines and interferons, IL-6 binding to its receptor activates a JAK/STAT pathway and several other pathways, including MAPK pathways. IL-6 contributes to local and systemic inflammatory responses, as will be discussed below.
Activation of monocytes, macrophages, and dendritic cells by the binding of some PAMPs to certain TLRs also induces production of IL-12 and IL-18, cytokines that play key roles in driving the subsequent adaptive response by influencing the differentiation of activated T cells toward proinflammatory adaptive responses. IL-10 is another important cytokine specifically induced by some TLRs in macrophages, dendritic cells, other myeloid cells, and in subsets of T, B, and NK cells. IL-10 is anti-inflammatory, in that it inhibits macrophage activation and the production of proinflammatory cytokines by other myeloid cells. IL-10 levels increase over time and contribute to controlling the extent of inflammation-caused tissue damage. Roles of pathogen-induced cytokines in regulating T-cell adaptive responses will be discussed later in the chapter.
Chemokines
These small protein chemoattractants (agents that induce cells to move toward higher concentrations of the agent) recruit cells into, within, and out of tissues (see Chapter 14 and Appendix III). Some chemokines are responsible for constitutive (homeostatic) migration of white blood cells throughout the body. Other chemokines, produced in response to PRR activation, have key roles in the early stages of immune and inflammatory responses in that they attract cells that contribute both to clearing the infection or damage and to amplifying the response.
The first chemokine to be cloned, IL-8 (also called CXCL8), is produced in response to activation—by PAMPs, DAMPs, or some cytokines—of a variety of cells at sites of infection or tissue damage, including macrophages, dendritic cells, epithelial cells, and vascular endothelial cells. One of IL-8’s key roles occurs in the initial stages of infection or tissue damage; it serves as a chemoattractant for neutrophils, recruiting them from the blood to sites of infection. Other chemokines are specifically induced by PRR activation of epithelial cells in certain mucosal tissues and serve to recruit cells specifically to those sites, where they generate immune responses appropriate for clearing the invading pathogen.
Enzymes: iNOS and COX2
Two enzymes produced in response to PRR-activated signaling pathways—inducible nitric oxide synthase (iNOS) and cyclooxygenase-2 (COX2)—have key roles in the generation of antimicrobial and proinflammatory mediators. The iNOS enzyme catalyzes an important step in the formation of nitric oxide, which kills phagocytosed microbes as will be discussed below. COX2, whose synthesis is induced by PRR activation in monocytes, macrophages, neutrophils, and mast cells, is key to converting the lipid intermediate arachidonic acid to prostaglandins, potent proinflammatory mediators.
Phagocytosis Is an Important Mechanism for Eliminating Pathogens
Phagocytic cells make up an important line of defense against pathogens that have penetrated the epithelial cell barriers. Monocytes in the blood and macrophages, neutrophils, and dendritic cells in tissues are the main cell types that carry out phagocytosis, the cellular uptake (eating) and destruction of particulate materials greater than 0.5 microns (µm) in size, such as bacteria. This major role of phagocytic cells at the site of invading organisms is evolutionarily ancient, present in invertebrates as well as vertebrates. Elie Metchnikoff initially described the process of phagocytosis in the 1880s, using cells from starfish (echinoderm invertebrates), which are similar to vertebrate white blood cells (see Figure 1-3a). From these observations he concluded that phagocytosis has a major role in immunity. He was correct in this conclusion; we now know that defects in phagocytosis lead to severe immunodeficiency.
As described in Chapter 2, most tissues contain resident populations of macrophages that function as sentinels for the innate immune system. These macrophages are derived from embryonic precursors that seeded the tissues during early development. During an infection, monocytes (which circulate in the blood and can phagocytose blood-borne pathogens) are recruited into the infection site, where they differentiate into mature macrophages for fighting the infection. Through various cell surface receptors, macrophages recognize microbes, such as bacteria. This recognition activates signaling pathways that induce the polymerization of actin microfilaments, extending the phagocyte’s plasma membrane to engulf and internalize the microbes into phagosomes (endosomes resulting from phagocytosis; see Figure 4-18). Lysosomes then fuse with the phagosomes, delivering agents that kill and degrade the microbes. These lethal agents include hydrolytic enzymes that are activated by the increasing acidity of lysosomes as protons are pumped inside.

FIGURE 4-18 Phagocytosis. (a) Scanning electron micrograph of macrophage phagocytosis of bacteria. (b) Steps in the phagocytosis of a bacterium. PRRs on the macrophage directly recognize PAMPs on a bacterial cell, or opsonin receptors recognize opsonins attached to the bacterial cell. The receptors then trigger actin filament–mediated internalization and formation of a phagosome. The phagosome fuses with a lysosome; the acidic pH activates the lysosomal hydrolytic enzymes, which along with other mechanisms contribute to killing the bacterium (see text). Digestion products may be released from the macrophage.
Neutrophils are a second major type of phagocyte, usually recruited early from the blood to sites of infection. Their granules contain pre-packaged antimicrobial proteins and peptides, which fuse with phagosomes. Finally, dendritic cells also can bind and phagocytose microbes.
As will be described later in this chapter and more extensively in Chapters 7 and 10, uptake and degradation of microbes by dendritic cells play key roles in the initiation of adaptive immune responses. In addition to triggering phagocytosis, various receptors on phagocytes recognize microbes and activate the production of a variety of molecules that contribute in other ways to eliminating infection.
Phagocytic Receptors
How does a phagocytic cell recognize microbes, triggering their phagocytosis? Phagocytes express on their surfaces a variety of receptors, many of which are PRRs that directly recognize PAMPs on the surfaces of microbes, such as cell wall components of bacteria and fungi. Some but not all PRRs induce phagocytosis; TLRs are a major class of PRRs that do not induce phagocytosis. PRRs that bind microbes and trigger phagocytosis of the bound microbes are listed at the top of Table 4-5, along with the PAMPs they recognize. As we will see later, there are other PRRs that, after PAMP binding, do not activate phagocytosis but trigger other types of responses. Most PAMPs that induce phagocytosis are cell wall components, including complex carbohydrates such as mannans and b-glucans, lipopolysaccharides (LPS), other lipid-containing molecules, peptidoglycans, and surface proteins.
| Receptor type on phagocytes | Examples | Ligands |
|---|---|---|
| Pattern recognition receptors | Microbial ligands (found on microbes) | |
C-type lectin receptors (CLRs) |
Mannose receptor Dectin-1 DC-SIGN |
Mannans (bacteria, fungi, parasites) β-Glucans (fungi, some bacteria) Mannans (bacteria, fungi, parasites) |
Scavenger receptors |
SR-A |
Lipopolysaccharide (LPS), lipoteichoic acid(LTA) (bacteria) |
SR-B |
LTA, lipopeptides, diacylglycerides (bacteria),β-glucans (fungi) |
|
Opsonin receptors |
Microbe-binding opsonins (soluble; bind to microbes) |
|
Collagen-domain receptor |
CD91/calreticulin |
Collectins SP-A, SP-D, MBL; L-ficolin; C1q |
Complement receptors |
CR1, CR3, CR4, CRIg, C1qRp |
Complement components and fragments* |
Immunoglobulin Fc receptors |
FcαRs FcγRs |
Specific IgA antibodies bound to antigen† Specific IgG antibodies bound to antigen†; C-reactive protein |
Activation of phagocytosis can also occur indirectly, by phagocyte recognition of soluble proteins that have bound to microbial surfaces, thus enhancing phagocytosis; this process is called opsonization (from the Greek word for “to make tasty”) (see Overview Figure 4-15). Many of these soluble phagocytosis-enhancing proteins (called opsonins) also bind to conserved, repeating components on the surfaces of microbes such as carbohydrate structures, lipopolysaccharides, and viral proteins; hence they are sometimes referred to as soluble pattern-recognition proteins. Once bound to microbe surfaces, opsonins are recognized by membrane opsonin receptors on phagocytes, activating phagocytosis (see Table 4-5, bottom).
A variety of soluble proteins function as opsonins; many also play other roles in innate immunity. Mannose-binding lectin (MBL), a collectin with opsonizing activity, is found in the blood (where it can also activate the complement pathway) and respiratory fluids (Figure 4-19a). The complement component C1q also functions as an opsonin, binding bacterial cell wall components such as lipopolysaccharides and some viral proteins (see Figure 4-19b); C1q is recognized by the CR1 opsonin receptor, triggering phagocytosis. Another interesting example is the two surfactant collectin proteins, SP-A and SP-D, mentioned earlier in the context of their protective roles in mucosal secretions in the lungs and elsewhere. They are found in the blood, where they function as opsonins. After binding to microbes they are recognized by the CD91 opsonin receptor (see Table 4-5) and promote phagocytosis by alveolar and other macrophage populations. This function of SP-A and SP-D contributes to clearance of the fungal respiratory pathogen Pneumocystis jirovecii, a major cause of pneumonia in individuals with acquired immunodeficiency syndrome (AIDS). MBL (and other collectins), ficolins, and C1q share structural features, including similar polymeric structures with collagen-like shafts, but have recognition regions with different binding specificities (see Figure 4-19). As a result of their structural similarities, all are bound by the CD91 opsonin receptor (see Table 4-5) and activate pathogen phagocytosis.
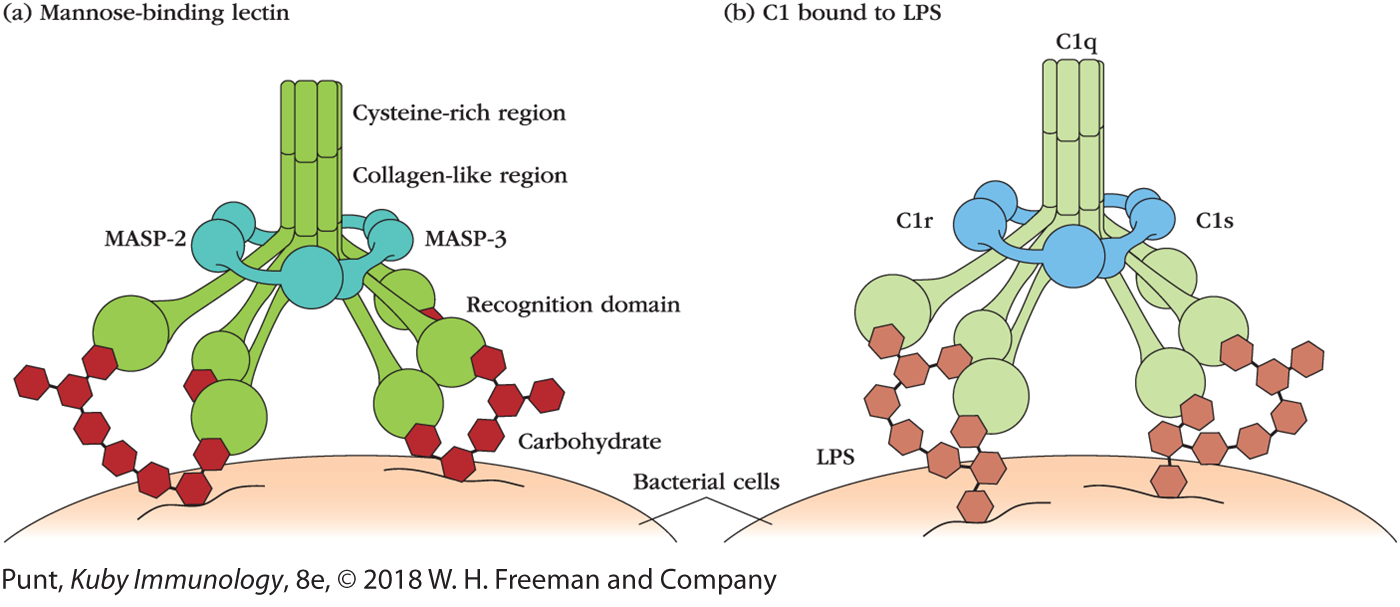
FIGURE 4-19 Structures of opsonins. (a) Mannose-binding lectin (MBL), a collectin, is a complex of multiple polypeptide chains, each containing an N-terminal cysteine-rich region followed by a collagen-like region, an α-helical neck region (not visible), and a recognition domain. Bound to the MBL are MBL-associated serine proteases (MASPs), which become active after the recognition domains bind to specific carbohydrate residues on pathogen surfaces. The MASPs then can activate the complement pathway. (b) C1, the first component of complement, has a multimeric structure similar to that of MBL. The C1q portion binds LPS on bacterial cell walls. MBL and C1q bound to microbes are then recognized by opsonin receptors, triggering phagocytosis.
Another opsonin, C-reactive protein (CRP), recognizes phosphocholine and carbohydrates on bacteria, fungi, and parasites (and some dead body cells) and is then bound by Fc receptors (FcRs), receptors that also bind the constant (Fc) region of antibodies and that are found on most phagocytes. Fc receptors are important for the opsonizing activity of several classes of antibodies, an important mechanism of adaptive immunity. As mentioned above, among the most effective opsonins are several components of the complement system, which is described in detail in Chapter 5. Present in both invertebrates and vertebrates, complement straddles both the innate and adaptive immune systems, indicating that it is ancient and important. As we will see in Chapter 5, phagocytosis is one of many important antimicrobial effects resulting from complement activation.
The importance of MBL as both an opsonin and a complement activator has been indicated by the effects of MBL deficiencies, which affect about 25% of the population. Individuals with MBL deficiencies are predisposed to severe respiratory tract infections, especially pneumococcal pneumonia. Interestingly, MBL deficiencies may be protective against tuberculosis, probably reflecting MBL’s opsonizing role in enhancing the phagocytosis of Mycobacterium tuberculosis, the route by which it infects macrophages, potentially leading to tuberculosis.
Processes That Kill Phagocytosed Microbes
The binding of microbes—bacteria, fungi, protozoan parasites, and viruses—to phagocytes via pattern recognition receptors, or via opsonins and opsonin receptors, activates signaling pathways that initiate phagocytosis. The phagosomes then fuse with lysosomes and, in neutrophils, with preformed primary and secondary granules (see Figure 2-4a). The resulting phagolysosomes contain an arsenal of antimicrobial agents that then kill and degrade the internalized microbes. These agents include antimicrobial proteins and peptides (including defensins and cathelicidins), low pH (due to the activity of a vacuolar ATPase proton pump), hydrolytic enzymes including lysozyme and proteases activated by the increasing acidity of the phagolysosomes, and specialized molecules that mediate oxidative attack.
Oxidative attack on the phagocytosed microbes, which occurs in neutrophils, macrophages, and dendritic cells, employs highly toxic reactive oxygen species (ROS) and reactive nitrogen species (RNS), which damage microbial membranes and intracellular components (Figure 4-20). The reactive oxygen species are generated by the phagocytes’ unique NADPH oxidase enzyme complex (also called phagosome NADPH oxidase), which is activated when microbes bind to the phagocytic receptors. The oxygen consumed by phagocytes to support ROS production by NADPH oxidase is provided by a metabolic process known as the respiratory burst, during which oxygen uptake by the cell increases severalfold. NADPH oxidase converts oxygen to superoxide ion (•O2-); other ROS generated by the action of additional enzymes are hydrogen peroxide (H2O2), and hypochlorous acid (HClO), the active component of household bleach.
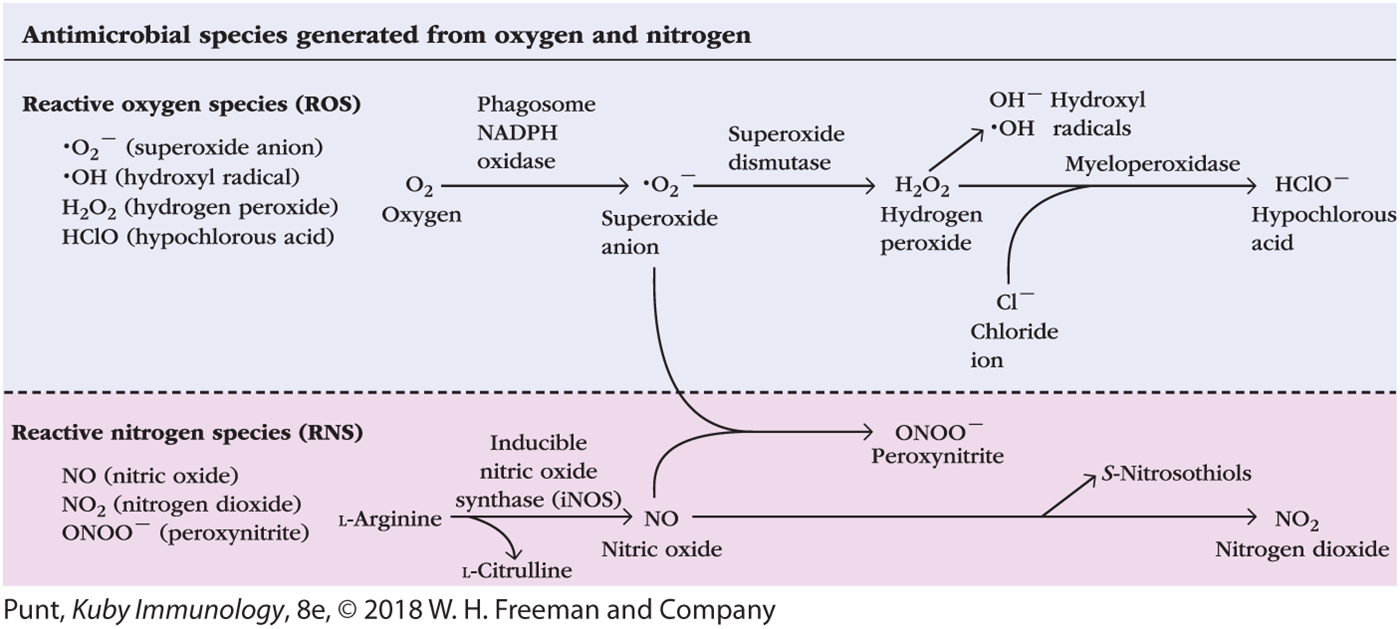
FIGURE 4-20 Generation of antimicrobial reactive oxygen and nitrogen species. In the cytoplasm of neutrophils, macrophages, and dendritic cells, several enzymes, including phagosome NADPH oxidase, transform molecular oxygen into highly reactive oxygen species (ROS) that have antimicrobial activity. One of the products of this pathway, superoxide anion, can interact with a reactive nitrogen species (RNS; in this case, nitric oxide [NO]) generated by inducible nitric oxide synthase (iNOS); the result is peroxynitrite (ONOO–), another RNS. NO can also undergo oxidation to generate the RNS nitrogen dioxide (NO2).
The generation of RNS requires the transcriptional activation of the gene for the enzyme inducible nitric oxide synthase (iNOS, or NOS2)—called that to distinguish it from related nitric oxide synthases in other tissues. Expression of iNOS is activated by microbial PAMPs binding to various PRRs. iNOS oxidizes L-arginine to yield L-citrulline and nitric oxide (NO), a potent antimicrobial agent (see Figure 4-20). In combination with superoxide ion (•O2‒) generated by NADPH oxidase, NO produces an additional reactive nitrogen species, peroxynitrite (ONOO–), and toxic S-nitrosothiols. Collectively, ROS and RNS are highly toxic to phagocytosed microbes because of their alteration of microbial molecules through oxidation, hydroxylation, chlorination, nitration, and S-nitrosylation, along with the formation of sulfonic acids and destruction of iron-sulfur clusters in proteins. One example of how these oxidative species may be toxic to pathogens is the oxidation by ROS and RNS of cysteine sulfhydryl groups that are present in the active sites of many enzymes, inactivating the enzymes. ROS and RNS also can be released from activated neutrophils and macrophages and kill extracellular pathogens.
Evidence from genetic defects in humans and mice highlights the critical roles of these reactive chemical species in microbial elimination by phagocytic cells. The importance to antimicrobial defense of phagosomal NADPH oxidase and its products, ROS and RNS, is illustrated by chronic granulomatous disease (CGD). Patients afflicted with this disease have dramatically increased susceptibility to some fungal and bacterial infections, caused by defects in subunits of NADPH oxidase that destroy its ability to generate oxidizing species. In addition, studies with mice in which the genes encoding iNOS were “knocked out” have shown that nitric oxide and substances derived from it account for much of the antimicrobial activity of macrophages against bacteria, fungi, and parasites. These mice lost much of their usual ability to control infections caused by such intracellular pathogens as M. tuberculosis and Leishmania major, the intracellular protozoan parasite that causes leishmaniasis.
Elimination of Intracellular Pathogens by Autophagy
Some bacterial pathogens, such as Listeria, escape immune effector mechanisms such as phagocytosis by replicating in the cytosol. However, they can’t escape an intracellular form of phagocytosis called autophagy, in which membrane derived from the endoplasmic reticulum envelopes the bacteria, forming an autophagosome. This vesicle then fuses with lysosomes, leading to the destruction of the pathogens as discussed above. As mentioned earlier, autophagy is an innate effector function activated by the NLRs NOD1 and NOD2.
Cell Turnover and the Clearance of Dead Cells
Our discussion of phagocytosis thus far has focused on its essential roles in killing pathogens. As the body’s main scavenger cells, macrophages also use their phagocytic receptors to take up and clear cellular debris, cells that have died from damage or toxic stimuli (necrotic cell death) or from apoptosis (programmed cell death), and aging red blood cells.
Considerable progress has been made in recent years in understanding the specific markers and receptors that trigger macrophage phagocytosis of dead, dying, and aging cells. Collectively, the components of dead/dying cells and damaged tissues that are recognized by PRRs, leading to their clearance, are sometimes referred to as damage-associated molecular patterns (DAMPs). As the presence of these components may also be an indicator of conditions harmful to the body or may contribute to harmful consequences (such as autoimmune diseases), the “D” in DAMP also can refer to a “Danger” signal. Phagocytosis is the major mode of clearance of cells that have undergone apoptosis as part of developmental remodeling of tissues, normal cell turnover, or killing of pathogen-infected or tumor cells by innate or adaptive immune responses.
Apoptotic cells attract phagocytes by releasing the lipid mediator lysophosphatidic acid, which functions as a chemoattractant. These dying cells facilitate their own phagocytosis by expressing on their surfaces an array of molecules not expressed on healthy cells, including phospholipids (such as phosphatidylserine and lysophosphatidylcholine), proteins (annexin I), and altered carbohydrates. These DAMPs are recognized directly by phagocytic receptors such as the phosphatidylserine receptor and scavenger receptor SR-A1. Other DAMPs are recognized by soluble pattern recognition molecules that function as opsonins, including the collectins MBL, SP-A, and SP-D mentioned earlier; various complement components; and the pentraxins C-reactive protein and serum amyloid protein. These opsonins are then recognized by opsonin receptors, activating phagocytosis and degradation of the apoptotic cells.
An important additional activity of macrophages in the spleen and those in the liver (known as Kupffer cells) is to recognize, phagocytose, and degrade aging and damaged red blood cells. As these cells age, novel molecules that are recognized by phagocytes accumulate in their plasma membrane. Phosphatidylserine flips from the inner to the outer leaflet of the lipid bilayer and is recognized by phosphatidylserine receptors on phagocytes. Modifications of erythrocyte membrane proteins have also been detected that may promote phagocytosis.
Obviously it is important that normal cells not be phagocytosed, and accumulating evidence indicates that whether or not a cell is phagocytosed is controlled by sets of “eat me” signals—the altered membrane components (DAMPs) described above—and “don’t eat me” signals expressed by normal cells. Young, healthy erythrocytes avoid being phagocytosed by not expressing “eat me” signals, such as phosphatidylserine, and also by expressing a “don’t eat me” signal, the protein CD47. CD47, expressed on many cell types throughout the body, is recognized by the SIRPα (signal regulatory protein α) receptor on macrophages, which transmits signals that inhibit phagocytosis. Recent studies have shown that tumors use elevated CD47 expression to evade tumor surveillance and phagocytic elimination by the immune system. Increased expression of CD47 on all or most human cancers is correlated with tumor progression, probably because the CD47 activates SIRPα–mediated inhibition of the phagocytosis of tumor cells by macrophages. This understanding of the role of CD47 in preventing phagocytosis is being used to develop novel therapies for certain cancers, such as using antibodies to block CD47 on tumor cells, which should then allow them to be phagocytosed and eliminated.
Regulated Cell Death Contributes to Pathogen Elimination
In addition to the induced expression of antimicrobial and proinflammatory proteins and peptides, PAMPs also induce unusual responses, resulting in cell death, that can be beneficial in the control of infections. Cell death induced by receptor-activated signaling pathways is called regulated cell death. One form, apoptosis, is programmed cell death; its induction by TNF binding to the TNF receptor and by NK cells and cytotoxic T cells is an essential mechanism of cell-mediated immunity against altered self-cells (see Chapter 12). Several additional forms of regulated cell death exist; two of them—NETosis and pyroptosis—are forms of innate immune response.
NETs and NETosis
Several years ago neutrophils activated through various PRRs were shown to expel filaments of chromatin (the compacted DNA with bound proteins, includinghistones, that make up chromosomes) and other cellular debris that entrap and kill pathogens, with the neutrophil dying in the process. These filaments, which can extend to 10 to 15 times the size of the cell they originate from, are called neutrophil extracellular traps (NETs), and the accompanying cell death is called NETosis (Figure 4-21a and b). NET formation requires activation of NADPH oxidase and the generation of ROS such as superoxides, which initiate damage of intracellular components, as mentioned earlier (and see Figure 4-21c). Enzymes, including neutrophil elastase and myeloperoxidase, enter the nucleus, modify histones, and trigger chromosome decondensation. Cell membranes disintegrate, and cytoplasmic and nuclear contents are expelled to form NETs. The NETs, containing DNA and other chromatin components, trap bacterial, fungal, and parasite cells, preventing their spread. In addition, associated with the fibrils of NETs are a variety of antimicrobial neutrophil proteins, including lysozyme, proteases, antimicrobial peptides, ion chelators, complement components, and histones (whose cationic properties give them antimicrobial activity). NETs also contribute to innate immunity indirectly, as DNA and other released chromatin components can serve as DAMPs, further activating local innate and inflammatory responses. Recent studies have shown that other granulocytes—eosinophils, mast cells, and basophils—can also form extracellular traps.

FIGURE 4-21 Neutrophil extracellular traps (NETs) and NETosis. (a) Fibers of NETs with trapped Salmonella bacteria. (b) A NET produced by an activated human neutrophil (dying due to NETosis), and four nonactivated neutrophils. Red stain: histones in decondensed chromatin of the NET. Green stain: Neutrophil elastase (visible in granules of nonactivated neutrophils and faintly in the NET). Blue stain: DNA. (c) Formation of NETs and NETosis.
Pyroptosis
A second type of regulated cell death functioning in innate immunity, pyroptosis (typically of macrophages), is induced by inflammasome activation, described earlier in Advances Box 4-2. Pyroptosis contributes to pathogen elimination in several ways. First, the death of infected macrophages prevents further spread of intracellular bacteria, such as Salmonella and Listeria, that replicate in these cells. Second, as mentioned earlier, pyroptosis appears to be an important mechanism for the release of mature IL-1β and IL-18, generated by inflammasome-associated caspase-1– or caspase-11–mediated cleavage from large precursors. These are important cytokines for promoting beneficial inflammatory responses.
Local Inflammation Is Triggered by Innate Immune Responses
When the outer barriers of the innate immune system—skin and other epithelial layers—are damaged, the resulting innate responses to infection or tissue injury can induce a complex cascade of events known as the inflammatory response. Inflammation may be acute (short-term effects contributing to combating infection, followed by healing)—for example, in response to local tissue damage—or it may be chronic (long term, not resolved), contributing to conditions such as arthritis, inflammatory bowel disease, cardiovascular disease, and type 2 diabetes.
The hallmarks of a localized inflammatory response were first described by the Roman physician Celsus in the first century A.D. as rubor et tumor cum calore et dolore (redness and swelling with heat and pain). An additional mark of inflammation added in the second century A.D. by the physician Galen is loss of function (functio laesa). Today we know that these symptoms reflect an increase in vascular diameter (vasodilation), resulting in a rise of blood volume in the area. Higher blood volume heats the tissue and causes it to redden. Vascular permeability also increases, leading to leakage of fluid from the blood vessels, resulting in an accumulation of fluid (edema) that swells the tissue. Within a few hours, leukocytes also enter the tissue from the local blood vessels. These hallmark features of inflammatory responses result from the activation of innate immune responses in the vicinity of the infection or wound.
When there is local infection, tissue damage, or exposure to some harmful substances (such as asbestos or silica crystals in the lungs), sentinel cells residing in the epithelial layer—macrophages, mast cells, and dendritic cells—are activated by PAMPs, DAMPs, crystals, and so on to start phagocytosing the offending invaders (Figure 4-22). The cells are also activated by the PRR signaling pathways discussed earlier in this chapter to release innate immunity mediators, including cytokines and chemokines that trigger a series of processes that collectively constitute the inflammatory response.

FIGURE 4-22 Initiation of a local inflammatory response. Bacterial entry through wounds activates initial innate immune mechanisms, including activation of phagocytosis by resident cells, such as macrophages and dendritic cells. Recognition of bacteria by cellular pattern receptors initiates production of cytokines, chemokines and other mediators, triggering changes in vascular endothelial cells that lead to an influx from the blood of fluid (containing antimicrobial substances) and phagocytes (first neutrophils and then monocytes) to the site of infection. These and subsequent events cause the redness, swelling, heat, and pain that are characteristic of local inflammatory responses.
The recruitment of various leukocyte populations from the blood into the site of infection or damage is a critical early component of inflammatory responses. PRR signaling activates resident macrophages, dendritic cells, and mast cells to release the initial components of cellular innate immune responses, including the proinflammatory cytokines TNF-α, IL-1β, and IL-6; chemokines; prostaglandins (following the induced expression of the COX2 enzyme); and histamine and other mediators released by mast cells. These factors act on the vascular endothelial cells of local blood vessels, increasing vascular permeability and the expression of cell adhesion molecules (CAMs) and chemokines such as IL-8. The affected epithelium is said to be inflamed or activated. Fluid enters the tissue, delivering antimicrobial molecules such as complement components and causing swelling. Cells flowing through the local capillaries are induced by chemokines and cell adhesion molecule interactions to adhere to vascular endothelial cells in the inflamed region and pass through the walls of capillaries and into the tissue spaces, a process called extravasation that will be described in Chapter 14. Neutrophils are the first to be recruited to a site of infection, where they enhance local innate responses, followed by monocytes that differentiate into macrophages; the macrophages participate in clearance of pathogens and cellular debris and help initiate wound healing.
In addition to these key events at the site of infection or damage, the key cytokines made early in response to innate and inflammatory stimuli—TNF-α, IL-1β, and IL-6—also have systemic effects, which will be described in more detail in Chapter 15. They induce fever (a protective response, as elevated body temperature inhibits replication of some pathogens) by inducing COX2 expression, which activates prostaglandin synthesis, as mentioned above. Prostaglandin E2 (PGE2) acts on the hypothalamus (the brain center controlling body temperature), causing fever. The three proinflammatory cytokines (TNF-α, IL-1β, and IL-6) also act on the liver, inducing the acute-phase response, which involves production by the liver of proteins that contribute to the elimination of pathogens (including opsonins and proteins that activate the complement system, such as MBL) and the resolution of the inflammatory response.