Helper CD4+ T-Cell Differentiation
One to two days after successful engagement with a dendritic cell in the T-cell zone of a secondary lymphoid organ, a naïve T cell enlarges into a blast cell and undergoes repeated rounds of cell division. Signals 1 plus 2 induce up-regulation of expression and activity of prosurvival genes (e.g., Bcl-2), as well as the transcription of genes for both IL-2 and the α chain (CD25) of the high-affinity IL-2 receptor (Figure 10-8). The combined effect on a naïve T cell results in robust proliferation. Activated T cells divide two or three times per day for 4 to 5 days, generating a clone of progeny cells.

FIGURE 10-8 Activation and differentiation of naïve T cells into effector and memory T cells. Signal 1 and Signal 2 cooperate to enhance the transcription and stability of mRNA for IL-2 and the high-affinity IL-2R chain (also referred to as CD25). Secreted IL-2 binds the IL-2R, which generates signals that enhance the entry of the T cell into the cell cycle. T cells undergo many rounds of proliferation, during which they differentiate into memory cell subsets and effector helper or cytotoxic cells.
Activated T cells and their progeny gain unique functional abilities, becoming memory and effector helper or cytotoxic T cells (see Figure 10-8). These coordinate efforts with other immune cells to both indirectly and directly clear infections. CD8+ cytotoxic T cells leave the secondary lymphoid tissues and circulate to sites of infection, where they bind and kill infected cells. CD4+ helper T cells secrete cytokines that orchestrate the activity of several other cell types, including B cells, macrophages, and other T cells. Some CD4+ T cells, particularly those that help B cells and those that become central memory T cells, stay within the secondary lymphoid tissue. Others return to the sites of infection and enhance the activity of macrophages and cytotoxic cells. Still others circulate to other tissues to join the first lines of attack against re-infection.
Whereas naïve T cells can live for months, effector cells tend to be short-lived and have life spans that range from a few days to a few weeks. Memory cells typically live longer, and some extend their lives by dividing over the many months or even years that they are present in an organism.
Effector T cells come in many more varieties than originally anticipated, and each subset plays a specific and important role in the immune response. The first effector cell distinction to be recognized, of course, was between CD8+ T cells and CD4+ T cells. Activated CD8+ T cells acquire the ability to induce the death of target cells, becoming “killer” or “cytotoxic” T lymphocytes (CTLs; also called TC cells). Because cytotoxic CD8+ T cells recognize peptide bound to MHC class I, which is expressed by almost all cells in the body, they are perfectly poised to clear the body of cells that have been internally infected with the pathogen that resulted in their activation. On the other hand, activated CD4+ T cells, or T helper (TH)cells, acquire the ability to secrete factors that enhance the activation and proliferation of other cells. Specifically, they regulate the activation and antibody production of B cells; enhance the phagocytic, antimicrobial, cytolytic, and antigen-presenting capacity of macrophages; and are required for the development of B-cell and CD8+ T-cell memory.
As immunologists developed and adopted more tools to distinguish proteins expressed and secreted by T cells, it became clear that CD4+ helper T cells are particularly diverse, differentiating into several different subtypes, each of which secretes a signature set of cytokines. The cytokines secreted by CD4+ TH cells act either directly on the same cell that produced them (i.e., in an autocrine fashion) or indirectly by binding to receptors on cells in the vicinity (i.e., in a paracrine fashion). Below we describe the major features and functions of the best-characterized CD4+ helper T-cell subsets. Although CD8+ cytotoxic T cells also secrete cytokines, and there are indications that CD8+ T cells may also differentiate into more than one killer subtype, the diversity of CD8+ T-cell effector functions is clearly more restricted than that of CD4+ TH cells. The generation and activity of CTLs are described in more detail in Chapters 12 and 13.
Helper T Cells Can Be Divided into Distinct Subsets and Coordinate Type 1 and Type 2 Responses
Tim Mosmann, Robert Coffman, and colleagues can be credited with one of the earliest and most elegant experiments that demonstrated the diversity of helper CD4+ T cells. Earlier investigations revealed that helper T cells produced a diverse array of cytokines. Although this could mean that each cell made many different cytokines, Mosmann and Coffman tested a different possibility: that the CD4+ cell population was, itself, heterogeneous and made of distinct helper subtypes that made distinct sets of cytokines.
Specifically, they isolated individual T-cell clones from the polyclonal mixture of CD4+ T cells generated in the spleen of an immunized mouse. At the time, the immunology community did not have the tools to identify most cytokines directly. However, Mosmann and Coffman developed clever (and elaborate) strategies to define which cytokines each clone generated. To the surprise of many, they showed that the individual clones did not make all of the cytokines; instead, they fell into two distinct categories, which they named TH1 and TH2.
Because TH1 and TH2 subsets were originally identified from in vitro studies of cloned T-cell lines, some researchers doubted that they represented true in vivo subpopulations. They suggested instead that these subsets might represent different maturational stages of a single lineage. In addition, the initial failure to locate either subset in humans led some to believe that TH1, TH2, and other subsets of CD4+ T helper cells were not present in our species. Further research corrected these views. In many in vivo systems, the full commitment of populations of T cells to either a TH1 or TH2 phenotype occurs late in an immune response. Hence, it was difficult to find clear TH1 and TH2 subsets in studies on healthy human subjects, where these cells would not have developed.
These experiments set the stage for the discovery that naïve CD4+ T cells can adopt not just two but six or more distinct helper fates, depending on the signals they receive during activation. TH1 and TH2 subpopulations were quickly joined by the TH17, TFH, and TREG subpopulations, each of which produces a distinct cytokine profile and regulates distinct activities within the body (Overview Figure 10-9). The genotype, phenotype, and function of these five populations are well described and will be the focus of this section. However, other helper subpopulations continue to reveal themselves. Whether these merit their own category or are variants within the major subgroups is not always clear. Nonetheless, two additional CD4+ T helper subpopulations, TH9 and TH22, have recently gained prominence and will be discussed briefly.
In retrospect, we probably should have anticipated the heterogeneity of helper responses, which allows an organism to “tailor” a response to a particular type of pathogen. The type of effector TH cell that a naïve T cell (also called a TH0 cell) becomes depends largely on the kind of infection that occurs. For example, worm (helminth) infections stimulate the differentiation of activated CD4+ T cells into TH2 cells, which help B cells to produce IgE antibodies that bind the parasite and trigger the release of damaging agents from granulocytes.
On the other hand, infection with an intracellular virus or bacterium induces differentiation of CD4+ T cells into TH1 helpers that enhance the cytolytic activity of macrophages and CD8+ T cells, which then kill infected cells. Responses to fungi stimulate the differentiation of other helper responses than the responses to extracellular bacteria, and so on.
The reality is, of course, even more complex. Infections evoke the differentiation of more than one helper subtype and recruit other T-cell subsets and ILCs (see Chapters 4 and 13) that both overlap in function and add unique features to the immune response. Investigators are now finding it useful to divide the complex responses into two major types (type 1 and type 2). Type 1 responses are triggered by viral and many bacterial infections and polarize CD4+ T cells to the TH1 and TH17 helper subsets. These work with other immune cells (including ILC1s and ILC3s) to generate protective cytotoxic responses. Type 2 responses are triggered by larger parasites, including worms, protozoa, and allergens. These polarize CD4+ T cells to TH2 and TH9 helper subsets, which work with other populations of immune cells (including ILC2s) to generate an IgE response.
What regulates the differentiation of helper T cells to each effector subset and their participation in type 1 or type 2 responses is still being actively investigated. We describe the fundamentals of our current understanding in this section.
The Differentiation of Helper T-Cell Subsets Is Regulated by Polarizing Cytokines
As we have seen, T-cell activation requires TCR and costimulatory receptor engagement, both of which are supplied by an activated APC. It is now clear that the functional fate of activated T cells is determined by signals they receive from additional cytokines generated during the response. These cytokines (Signal 3) are referred to as polarizing cytokines because they are responsible for guiding a helper T cell toward one of several different effector fates (Figure 10-10).

FIGURE 10-10 General events and factors that drive TH subset polarization. Interaction of pathogen with pattern recognition receptors (PRRs) on dendritic cells and other neighboring immune cells determines which polarizing cytokines are produced and, hence, into which T helper subset a naïve T cell will differentiate. In general, polarizing cytokines that arise from dendritic cells or other neighboring cells interact with cytokine receptors and generate signals that induce transcription of unique master gene regulators. These master regulators, in turn, regulate expression of various genes, including effector cytokines, which define the function of each subset.
For example, T cells that are activated in the presence of IL-12 and IFN-γ tend to differentiate, or polarize, to the TH1 lineage, whereas T cells that are activated in the presence of IL-4 and IL-6 polarize to the TH2 lineage (Figure 10-11). Overview Figure 10-9 and Table 10-3 show the polarizing cytokines responsible for the development of each CD4+ helper T-cell subset. These will be discussed in greater detail shortly.

FIGURE 10-11 Initiation of TH1 and TH2 responses by pathogens. (a) Intracellular pathogens activate a cascade of signals that polarize cells to the TH1 lineage and initiate type 1 responses (which can also activate TH17 cells). For example, viruses interact with PRRs (e.g., TLR3) that induce dendritic cells to generate IL-12. This binds to receptors on naïve T cells (which have also engaged MHC-peptide and costimulatory ligands on the DC), activating a cytokine signal transduction pathway mediated by STAT4 that induces expression of the master regulator T-Bet. T-Bet, in turn, activates expression of effector cytokines, including IFN-γ, which define the TH1 subset’s functional capacities as a type 1 responder. (b) Extracellular pathogens activate signal cascades that polarize naïve T cells to the TH2 lineage and initiate a type 2 response. Parasitic worms interact with PRRs on neighboring immune cells (such as mast cells, basophils, or germinal center B cells), triggering the release of the signature TH2 polarizing cytokine IL-4. This interacts with receptors on T cells that activate STAT6, up-regulating expression of the transcriptional regulator GATA-3. GATA-3, in turn, induces expression of the TH2 and type 2 effector cytokines, including IL-4, IL-5, and IL-13.
Polarizing cytokines |
Master gene regulators |
Effector cytokines |
Functions |
|
TH1 |
IL-12 IFN-γ IL-18 |
T-Bet |
IFN-γ TNF |
Enhances APC activity Enhances TC activation Protects against intracellular pathogens Involved in delayed type hypersensitivity, autoimmunity |
TH2 |
IL-4 |
GATA-3 |
IL-4IL-5 IL-13 |
Protects against extracellular pathogens(particularly IgE responses) Involved in allergy |
TH9 |
IL-4TGF-β |
PU.1 |
IL-9 |
Protects against extracellular pathogensInvolved in mucosal autoimmunity |
TH17 |
TGF-β IL-6 (IL-23) |
RORγt |
IL-17A IL-17F IL-22 |
Protects against fungal and extracellular bacterial infections Contributes to inflammation, autoimmunity |
TH22 |
IL-6 TNF-α |
AHR |
IL-22 |
Protects against extracellular pathogens Involved in inflammatory skin disease |
TREG |
TGF-β IL-2 |
FoxP3 |
IL-10 TGF-β |
Inhibits inflammation Inhibit antitumor responses |
TFH |
IL-6 IL-21 |
Bcl-6 |
IL-4 IL-21 |
B-cell help in follicles and germinal centers |
Polarizing cytokines can be generated by the stimulating APC itself, or by neighboring innate and adaptive immune cells that have also been activated by antigen. Which polarizing cytokines are produced during an immune response depends on which cell has been alerted (DC, macrophage, B cell, NK cell, etc.), what pathogen has alerted it, and what pattern recognition receptors have been stimulated, and its tissue context. Innate interactions therefore play a critical role in determining the outcome of adaptive responses, by shaping the information that an antigen-presenting cell conveys to naïve T cells (see Figure 4-23).
Recall from Chapter 4 that APCs and other innate immune cells are activated by interaction with pathogens bearing pathogen-associated molecular patterns (PAMPs). These PAMPs bind PRRs, including (but not limited to) Toll-like receptors (TLRs). PRR interactions activate dendritic cells by stimulating the up-regulation of MHC and costimulatory proteins. They also determine the type of cytokine(s) that dendritic cells and other immune cells secrete. These polarizing cytokines, in turn, dictate the fate a T cell will adopt following activation (see Overview Figure 10-9).
For example, double-stranded RNA, a product of many viruses, binds TLR3 receptors on dendritic cells, initiating a signaling cascade that results in the production of IL-12, which directly promotes TH1 differentiation and a type 1 response. On the other hand, worms stimulate PRRs on innate immune cells, including mast cells, which generate IL-4. IL-4 directly promotes polarization of activated T cells to the TH2 subset, which coordinates a type 2 response and IgE-mediated killing of worms (see Figure 10-11). In this case, the main polarizing cytokine is not made by the activating dendritic cell, but is generated by a neighboring immune cell. The pathogen interactions that give rise to the polarizing cytokines that drive T helper cell differentiation are often complex and continue to be actively investigated.
Adjuvants, which have been used for decades to enhance the efficacy of vaccines, are now understood to exert their influence on the innate immune system by regulating the expression of costimulatory ligands and polarizing cytokines by APCs, events that ultimately shape the consequences of T-cell activation. PAMPs and cytokines such as IL-12, produced by APCs themselves, are considered natural adjuvants. Dead mycobacteria, which clearly activate many PRRs, have long been used as a very potent adjuvant for immune responses in mice. Very few adjuvants are approved for human vaccination, but given our new and evolving understanding of the molecules that stimulate PRRs and the consequences of that stimulation, investigators expect that we will one day be able to shape the effector response to vaccine antigens by varying the adjuvants—natural and/or synthetic—included in vaccine preparations (see Chapter 17).
Each Effector Helper T-Cell Subset Has Unique Properties
Each helper T-cell subset is defined by an array of features, the details of which can rapidly overwhelm a new (or old) student of immunology. Understanding these specifics, however, helps clarify the role each subset plays in resolving infection and causing disease. And having a ready reference to them makes it easier to decipher the primary literature describing advances. However, some generalizations provide a useful conceptual framework for organizing some of these details. Consider the following:
- Each of the major T helper cell subsets is characterized by (1) a distinct set of polarizing cytokines that induce the expression of (2) a master gene regulator that regulates expression of (3) a signature set of effector cytokines the T-cell population produces once it is fully differentiated.
- Which effector subset an activated helper cell becomes a member of depends on the quality and quantity of signals its naïve cell precursor receives from APCs in a secondary lymphoid organ; that activity, in turn, depends on the nature of the pathogen the APC encountered at the site of infection.
- Broadly speaking, TH1 cells regulate immunity to intracellular bacteria and viruses, TH17 cells regulate immunity to extracellular bacteria and fungi, TH2 (and perhaps TH9) cells regulate the response to worms, and TFH cells regulate humoral immunity (B cells). It is important to recognize that multiple CD4+ effector T-cell subsets may have the potential to provide help to B cells. TH1 and TH17 subsets generally encourage B cells to produce antibodies that contribute to cell-mediated immunity (e.g., isotypes like IgG2a that can “arm” NK cells for cytotoxicity; see Chapter 12). TH2 cells encourage B cells to produce antibodies that mediate the clearance of extracellular pathogens (e.g., isotypes like IgE that induce the release of molecules that harm extracellular parasites). TREG cells are inhibitory and play a role in terminating healthy immune responses and inhibiting autoimmunity.
- Helper T-cell subsets often “cross-regulate” each other. The cytokines they secrete typically enhance their own differentiation and expansion, while inhibiting commitment to other helper T-cell lineages. This is particularly true of the TH1 and TH2 subset pair, as well as the TH17 and TREG subset pair.
- Helper T cell lineages may not be fixed; some subsets can assume the cytokine secretion profile of other subsets if exposed to a different set of cytokines, particularly early in the differentiation process.
- The precise biological function and sites of differentiation and activity of each subset continue to be actively investigated. Much remains unknown.
We start our deeper discussion of helper cell subset characteristics by focusing on the first two subsets to be identified: TH1 and TH2 cells. They provide an illustrative example of the features that distinguish T helper cells as well as the relationship between subsets. We follow this section with summaries of what is currently understood about the more recently characterized helper subsets: TH17, TREG, TFH, TH9, and TH22. Overview Figure 10-9 and Table 10-3 provide useful summaries of the information presented here, which probes some of the complexities of origin and function that are being continually updated in this rapidly moving field.
The Differentiation and Function of TH1 and TH2 Cells
The key polarizing cytokines that induce differentiation of naïve T cells into T helper type 1 (TH1) cells are IL-12, IL-18, and IFN-γ (see Overview Figure 10-9 and Figure 10-11). IL-12 is produced by dendritic cells after an encounter with pathogens via PRRs, including TLR4 and TLR3. IL-12 expression is also up-regulated in response to IFN-γ, which is generated by activated T cells and activated NK cells. IL-18, which is also produced by dendritic cells, promotes proliferation of developing TH1 cells and enhances their own production of IFN-γ. These polarizing cytokines trigger signaling pathways in naïve T cells that up-regulate expression of the master gene regulator T-Bet. This master transcription factor induces expression of signature type 1 effector cytokines, including IFN-γ and TNF, and differentiation of CD4+ T cells to the TH1 lineage.
IFN-γ is a particularly potent type 1 effector cytokine. It activates macrophages, stimulating these cells to increase microbicidal activity, up-regulate the level of MHC class II molecules, and, as mentioned above, secrete cytokines such as IL-12, which further enhance TH1 differentiation. IFN-γ secretion also induces antibody class switching in B cells to IgG classes (such as IgG2a in mice) that support phagocytosis and fixation of complement. Finally, IFN-γ secretion promotes the differentiation of fully cytotoxic TC cells from CD8+ precursors by activating the dendritic cells that engage naïve TC cells. These combined effects make the TH1 subset particularly well suited to respond to viral infections and other intracellular pathogens. They also contribute to the pathological effects of TH1 cells, which are also involved in the delayed-type hypersensitivity response to poison ivy (see Chapter 15).
Just as differentiation to the TH1 subset is promoted by IL-12 and IFN-γ, differentiation of naïve T cells into T helper type 2 (TH2) cells is promoted by a defining and potent polarizing cytokine, IL-4 (see Overview Figure 10-9 and Figure 10-11). Exposing naïve helper T cells to IL-4 at the beginning of an immune response causes them to differentiate into TH2 cells and, interestingly, TH2 development is greatly favored over TH1 development. Even in the presence of IFN-γ and IL-12, naïve T cells will differentiate into TH2 effectors if IL-4 is present. IL-4 triggers a signaling pathway within the T cell that up-regulates the master gene regulator GATA-3, which, in turn, regulates expression of type 2–specific cytokines, including IL-4, IL-5, and IL-13.
Investigators are still working to understand which cells produce the polarizing cytokine IL-4. Several may be responsible, depending on the origin and type of infection. Recent data suggest that type 2 innate lymphoid cells (ILC2) are a major source of IL-4 in lymphoid tissue responding to an intestinal worm infection. However, other studies suggest that ILC2s may not be required. Many innate cells, such as mast cells, basophils, eosinophils, γδ T cells, and natural killer T (NKT) cells, can be induced to make IL-4 after exposure to pathogen and could influence helper T-cell development if they make their way to secondary lymphoid tissue. Cells that are more routinely found in secondary lymphoid tissue, including germinal-center B cells, TFH cells, and a specialized DC subset, can also produce IL-4 and may also play a role in polarizing naïve T cells. Finally, TH2 cells themselves are an excellent source of additional IL-4 and may enhance polarization events of neighboring naïve T cells. Clarity will require more research.
The type 2 effector cytokines produced by TH2 cells help clear extracellular parasitic infections, including those caused by worms. IL-4, the defining TH2 effector cytokine, acts on both B cells and eosinophils. It induces eosinophil differentiation, activation, and migration and promotes B-cell activation and class switching to IgE. These effects act synergistically because eosinophils express IgE receptors (FcεR) that, when cross-linked, release inflammatory mediators that are particularly good at attacking roundworms. Thus, IgE antibody can form a bridge between the worm and the eosinophil, binding to worm antigens via its variable regions and binding to FcεR via its constant region. IgE antibodies also mediate allergic reactions, and the role of TH2 activity in these pathological responses is described in Chapter 15.
IL-5 can also induce B-cell class switching to IgG subclasses that do not activate the complement pathway (e.g., IgG1 in mice), and IL-13 functions largely overlap with IL-4. Finally, IL-4 itself can suppress the expansion of TH1-cell populations.
TH1 and TH2 Cross-Regulation
The major effector cytokines produced by TH1 and TH2 subsets (IFN-γ and IL-4, respectively) not only influence the overall immune response, but also influence the fate and function of helper T cells, themselves. First, they promote the growth and enhance the polarization of the subset that produces them; second, they inhibit the development and activity of the opposite subset, an effect known as cross-regulation (Figure 10-12). For instance, IFN-γ (secreted by the TH1 subset) inhibits proliferation of the TH2 subset, and IL-4 (secreted by the TH2 subset) down-regulates the secretion of IL-12 by APCs, thereby inhibiting TH1 differentiation. Furthermore, IL-4 enhances TH2-cell development by making TH cells less susceptible to the TH1-promoting cytokine signals (and vice versa).
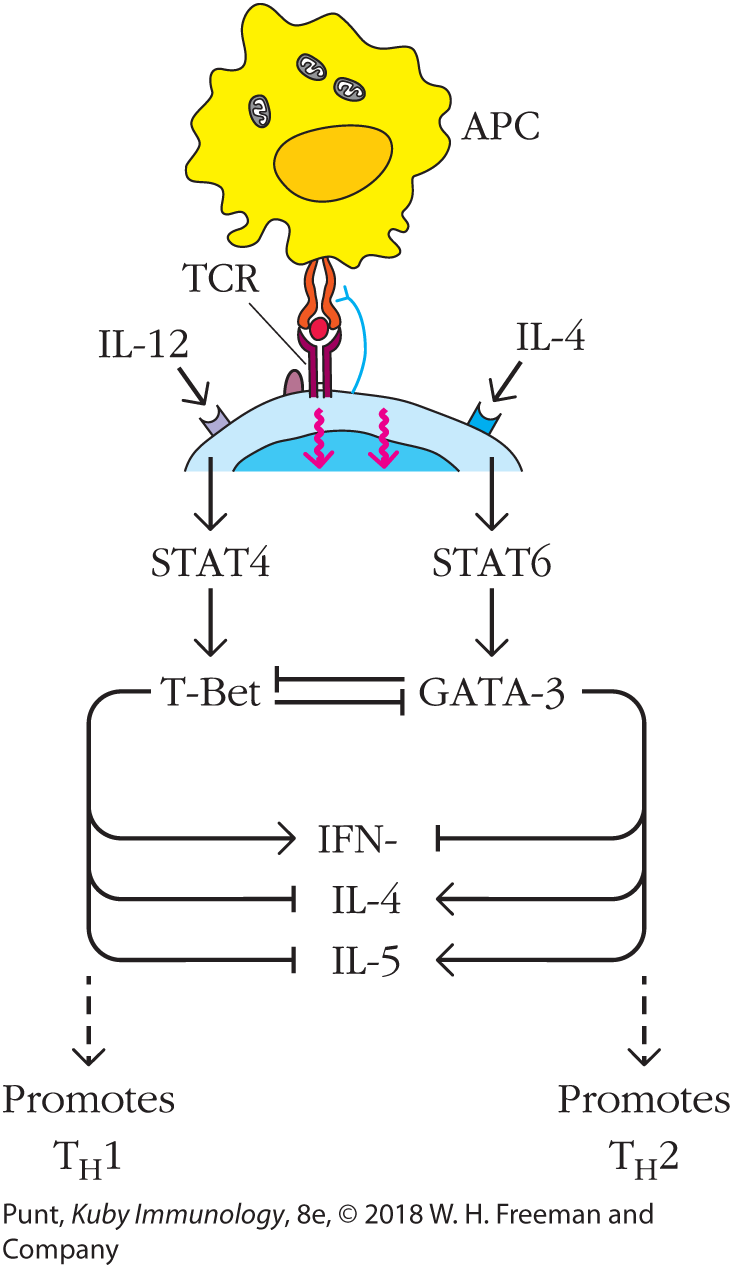
FIGURE 10-12 Cross-regulation of T helper cell subsets by transcriptional regulators. GATA-3 and T-Bet reciprocally regulate differentiation of TH1 and TH2 lineages. IL-12 promotes the expression of the TH1-defining transcription factor, T-Bet, which induces expression of TH1 effector cytokines, including IFN-γ. At the same time, T-Bet represses expression of the TH2-defining master transcriptional regulator, GATA-3, as well as expression of the effector cytokines IL-4 and IL-5. Reciprocally, IL-4 promotes expression of GATA-3, which up-regulates the synthesis of IL-4 and IL-5, and at the same time represses expression of T-Bet and the TH1 effector cytokine IFN-γ.
Similarly, these cytokines have opposing effects on target cells other than TH subsets. In mice, where the TH1 and TH2 subsets have been studied most extensively, the cytokines have distinct effects on the type of antibody made by B cells. For example, antibody isotype IgG2a enhances cell-mediated immunity by arming NKT cells, whereas the isotypes IgG1 and IgE enhance humoral immunity by their activities on extracellular pathogens. IFN-γ secreted by the TH1 subset promotes IgG2a production by B cells but inhibits IgG1 and IgE production. On the other hand, IL-4 secreted by the TH2 subset promotes production of IgG1 and IgE and suppresses production of IgG2a. Thus, these effects on antibody production are consistent with the TH1 and TH2 subsets’ overall tendencies to promote cell-mediated versus humoral immunity, respectively.
IL-10 was once considered a signature effector cytokine for TH2 cells. However, we now know that many other cell types and multiple helper subsets also produce IL-10. IL-10 inhibits TH1-cell differentiation, although not directly. Instead, IL-10 acts on monocytes and macrophages, interfering with their ability to activate the TH1 subset by abrogating their activation, specifically by (1) inhibiting expression of MHC class II molecules, (2) suppressing production of bactericidal metabolites (e.g., nitric oxide) and various inflammatory mediators (e.g., IL-1, IL-6, IL-8, GM-CSF, G-CSF, and TNF-β), and even by (3) inducing apoptosis.
The master regulators T-Bet and GATA-3 also play an important role in cross-regulation (see Figure 10-12). Specifically, the expression of T-Bet drives expression of genes required for T-cell differentiation into the TH1 lineage and represses genes involved in differentiation along the TH2 pathway. Expression of GATA-3 does the opposite, promoting TH2-specific gene expression and repressing genes associated with TH1 differentiation. Consequently, cytokine signals that induce one of these transcription factors set in motion a chain of events that represses the other.
TH17 Cells
The discovery of the T helper type 17 (TH17) cell subset was inspired in part by the recognition that IL-12, one of the polarizing cytokines that induces TH1 development, was a member of a larger family of cytokines that shared the same subunit (p40), including IL-23. At first investigators assumed that the defects in helper T-cell activity in p40 knockout mice were due entirely to a deficiency of TH1 cells. However, once it was understood that p40 was required not only for IL-12, but also for IL-23 production, investigators quickly realized that p40 knockout mice also failed to produce a T-cell subset that required IL-23. Some of the functions originally attributed to IL-12–induced TH1 cells were actually carried out by an IL-23–induced T helper-cell subpopulation now referred to as the TH17 lineage because of their ability to secrete IL-17.
Several cytokines are involved in polarizing naïve T cells to the TH17 lineage, including TGF-β, IL-6, and IL-23 (see Overview Figure 10-9). TH17 cells have dual roles and can adopt both a protective and pathogenic function; the cytokine mixtures that lead to both types are still being clarified. Activation of naïve T cells in the presence of TGF-β and IL-6 appears to induce development to the more protective form of TH17, which can produce IL-10 and patrol our barrier tissues (Clinical Focus Box 10-3; and see Chapter 13). Activation in the presence of IL-23, however, drives development of a more inflammatory version of TH17 cells. IL-23 is produced by dendritic cells in response to antigens produced by some bacteria and fungi. Alone, IL-23 cannot polarize naïve T cells to the TH17 lineage; rather, it enhances their ability to produce inflammatory cytokines. Inflammatory TH17 is an important participant in the protective response against fungi and some bacteria at mucosal barriers (see Chapter 13). Not only do TH17 cells produce IL-17 (also called IL-17A), but they also produce IL-17F and IL-22. Like all of our effector T-cell subsets, they can also go off-script and are one of several culprits behind chronic inflammatory and autoimmune disorders, including inflammatory bowel disease, arthritis, and multiple sclerosis.
TH17-cell differentiation is controlled by the master transcriptional regulator, RORγt, an orphan steroid receptor. RORγt knockout mice are less susceptible to autoimmune disease, including experimental autoimmune encephalitis (EAE, a mouse model of multiple sclerosis), largely because of the reduction in pathogenic TH17 cells.
Because of the pathogenic potential of inflammatory TH17 cells, many researchers are interested in identifying approaches to inactivate them. IL-23 inhibitors are already in clinical trials. Dectin-1, a PRR that binds fungal antigens and triggers IL-23 production by TH17 cells, may also be a promising new drug target.
Peripheral TREG Cells
Another major CD4+ T-cell subset negatively regulates T-cell responses and plays a critically important role in peripheral tolerance by limiting autoimmune T-cell activity. This subset of T cells, called peripheral TREG (pTREG) cells (previously known as induced TREG [iTREG] cells), is similar in function to the thymic TREG cells (tTREGs) (previously known as natural TREG [nTREG] cells) that differentiate to this lineage in the thymus (see Chapter 8). Peripheral TREG cells, however, arise from naïve T cells that are activated in secondary lymphoid tissue in the presence of TGF-β (see Overview Figure 10-9). The vitamin A metabolite retinoic acid (RA) also contributes to TREG polarization.
TGF-β triggers a signaling cascade that up-regulates expression of FoxP3, the master transcriptional regulator responsible for pTREG commitment. pTREG cells secrete the effector cytokines IL-10 and TGF-β, which down-regulate inflammation via their inhibitory effects on APCs, and can also exert their suppressive function by interacting directly with T cells. The depletion of pTREG cells in otherwise healthy animals leads to multiple autoimmune outbreaks, revealing that even healthy organisms are continually warding off autoimmune responses. A reduction in pTREG cells also increases women’s susceptibility to pre-eclampsia, a pregnancy complication thought to be due to an autoimmune response that impairs blood flow at the placenta. Advances Box 10-4 describes discoveries that revealed the importance of this subset of TREG cells in maintaining a mother’s tolerance to her developing fetus.
TH17 and TREG Cross-Regulation
Just as TH1 and TH2 cells reciprocally regulate each other, TREG and TH17 cells also cross-regulate each other. TGF-β plays a role in the differentiation of both TREG and TH17 cells. Alone, it induces a program that leads cells to the TREG fate. When accompanied by IL-6, however, TGF-β induces TH17 differentiation. How is this possible? TGF-β appears to up-regulate expression of both master regulators FoxP3 and RORγt, which control TREG and TH17 differentiation, respectively. In combination with signals generated by IL-6, TGF-β actually inhibits FoxP3 expression, letting RORγt dominate and induce TH17 development. Other variables play a role, too, including the presence of IL-21 and IL-23, which favor the differentiation of TH17 cells. High levels of TGF-β and the availability of retinoic acid favor the differentiation of TREG cells.
The cross-regulation of anti-inflammatory and inflammatory cell populations is elegantly adaptive, particularly at barrier tissues. Anti-inflammatory TH17 and pTREG populations dominate the barrier tissues of healthy animals, where innate cells generate immunosuppressive cytokines and metabolites (like TGF-β and RA). Activation of antigen-presenting cells by invading pathogens induces the release of a variety of inflammatory cytokines, which shift the development of cells away from pTREGs and anti-inflammatory TH17s toward the pro-inflammatory TH17 cells, so a proper defense can be mounted.
TFH Cells
T follicular helper (TFH) cells are now firmly established as a distinct helper T-cell subset. Whereas TH2 cells focus their help on the response to worms and enhance IgE production, TFH cells provide cognate help to a wide range of B cells and enhance switching to a variety of antibody classes. TFH cells are required for the formation of germinal centers and provide the signals that drive affinity maturation of B cells.
Cytokines that polarize activated T cells toward the TFH lineage include IL-6 and IL-21, both of which are produced by a variety of activated antigen-presenting cells. Together with signals produced by the TCR and costimulatory molecules, these cytokines induce the expression of Bcl-6, a repressor that is the master transcriptional regulator of TFH cells (see Overview Figure 10-9). Cross-regulation is also a feature of TFH function: Bcl-6 expression inhibits T-Bet, GATA-3, and RORγt expression, thus inhibiting TH1, TH2, and TH17 differentiation, respectively, while inducing TFH polarization. On the other hand, IL-2 production inhibits differentiation to the TFH lineage.
Although both TFH and TH2 cells secrete IL-4, TFH cells are best characterized by their secretion of IL-21, which contributes to B-cell differentiation. They also express high levels of CD40L, which is required for cognate B-cell help (Figure 10-13). TFH cells are also uniquely characterized by the expression of CXCR5, the chemokine receptor that attracts them to the B-cell follicle, where they can help establish germinal centers.

FIGURE 10-13 Examples of how TFH and TH1 T cells provide help in the immune response. (a) TFH cells interact directly with B cells and generate effector cytokines, such as IL-21 and IL-4, which induce B-cell proliferation and differentiation into antibody-producing plasma cells. (b) TH1 cells provide indirect help to CD8+ T cells by interacting with antigen-presenting cells and producing effector cytokines, such as IFN-γ, that license the antigen-presenting cell to finalize CD8+ T-cell differentiation into cytotoxic T cells. TH1 cells also produce IL-2, which enhances CD8+ T-cell proliferation.
Interestingly, CD28 isn’t the only costimulatory molecule involved in the activation of naïve T cells destined to differentiate into the TFH lineage. ICOS (discussed earlier) plays an additional and unique role. CD28 appears to play the key role in early activation events that result in up-regulation of Bcl-6. ICOS cooperates with the TCR later in the developmental program and stabilizes the cell’s commitment to the TFH lineage. Specifically, ICOS appears to contribute signals that relieve repression of TFH-specific genes, including CXCR5. In its absence, TFH cells leave the germinal center and lose their signature TFH functions.
TH9 Cells
Although TH2 cells were once thought to be the source of both IL-4 and IL-9, investigators have discovered that these cytokines originate from distinct helper T-cell subsets. The helper cells that produce IL-9 are now known as T helper type 9 (TH9) cells. Cytokines that polarize naïve T cells to the TH9 lineage include IL-2, and the combination of TGF-β and IL-4. (IL-1 also plays a role in enhancing polarization.) These cytokines induce expression of the transcriptional regulators IRF4 and PU.1, which are both responsible for driving TH9 differentiation. IL-9 plays a role in expelling worms and contributes to some antitumor responses. It is also involved in a variety of allergic, asthmatic, and autoimmune responses and may be a promising target for therapy.
Other Helper T-Cell Subsets
Another helper T-cell subset that may become a definitive member of the helper T-cell family is the T helper type 22 (TH22) cell subset. As its name suggests, this subset’s signature effector cytokine is IL-22. IL-22 can also be made by other T-cell subsets including TH17. However, TH22 cells appear quite distinct from TH17 cells and do not express RORγt or make IL-17. In addition to IL-22, they secrete IL-13 and express homing receptors for the skin, where they join the cells that protect the epithelium. What polarizes naïve T cells to the TH22 subset? TNF, IL-6, and IL-23 appear to act in concert to induce up-regulation of the aryl hydrocarbon receptor (AHR), which may be the master transcriptional regulator for TH22 cells. The function of TH22 cells in healthy animals is still not clear; however, they contribute to the inflammation associated with multiple autoimmune disorders.
Ongoing investigations may identify even more helper T-cell subsets. Distinguishing bona fide new lineages from functional or developmental variants of currently defined subsets remains a challenge. As you have seen above, some effector cytokines are secreted by more than one subset and some cytokines contribute to the polarization of more than one lineage. Adding to the complication is the growing awareness that the relationship among subsets is plastic and the identity of a cell may not be stable (as we’ll see shortly). Our understanding of the developmental relationship among effector subtypes will continue to evolve as new technologies allow us access to information about complex cell populations in vivo.
Helper T Cells May Not Be Irrevocably Committed to a Lineage
As indicated earlier, investigations now suggest that the relationship among TH-cell subpopulations may be more plastic than previously suspected. At early stages in differentiation, at least, helper cells may be able to shift their commitment and produce another subset’s signature cytokine(s). For example, when exposed to IL-12, young TH2 cells can be induced to express the signature TH1-cell cytokine, IFN-γ. Young TH1 cells can also be induced to express the signature TH2 cytokine, IL-4, under TH2 polarizing conditions.
Interestingly, once they commit to their lineage TH1 and TH2 cells seem rather inflexible and are unable to adopt TH17 or pTREG characteristics. On the other hand, TH17 and pTREG cells are more versatile and appear to be able to adopt the cytokine expression profiles of other subsets, including TH1 and TH2. The TH17 subset may be the most unstable or “plastic” lineage and can be induced to express type 1 (IFN-γ) and type 2 (IL-4) cytokines, depending on environmental input. Some pTREG cells can also be induced to express IFN-γ, and some can be redirected toward a TH17 phenotype if exposed to IL-6 and TGF-β. TFH cells appear to lose their subset identity after a B-cell response has concluded. These are just some examples of the plasticity that has been observed, which adds to the complexity of determining the independence of specific helper cell lineages.
Helper T-Cell Subsets Play Critical Roles in Immune Health and Disease
Helper T-cell differentiation is typically initiated in secondary lymphoid tissue, but can also be completed at the site of infection, where these effector cells are most needed. How do helper T cells actually provide help? T cells that provide B-cell help interact directly with the B cell, providing what is known as cognate help (see Figure 10-13 and Chapter 11). This interaction is similar to the interaction between a naïve T cell and its activating APC, and also involves the formation of an immune synapse.
A helper T cell, often a TFH cell, first recognizes peptide-MHC complexes that have been processed by the B cell and at the same time engages costimulatory ligands on the B-cell surface. TCR/costimulatory receptor co-engagement induces the helper T cell to release its effector cytokines. Helper T cells also express CD40L that binds CD40 on the B-cell surface. Without this critical engagement, B cells do not differentiate into high-affinity, long-lived plasma cells. Figure 10-13a shows the interaction between a virus-specific B cell and helper TFH cell, which produces several effector cytokines, including IL-21 and IL-4.
TH1 cells deliver a different and more indirect kind of help to CD8+ cytotoxic T cells, as shown in Figure 10-13b. They interact with and license macrophages, which in turn provide cytokines and signals that finalize CD8+ T-cell activation. TH1 cells can also provide CD40L directly to a neighboring CD8+ T cell.
Studies in both mice and humans also show that the balance of activity among T-cell subsets can significantly influence the outcome of the immune response. A now classic illustration of the influence of T-cell subset balance on disease outcome is provided by leprosy, which is caused by Mycobacterium leprae, an intracellular pathogen that can survive within the phagosomes of macrophages. Leprosy is not a single clinical entity; rather, the disease presents as a spectrum of clinical responses, with two major forms of disease, tuberculoid and lepromatous, at each end of the spectrum. In tuberculoid leprosy, cell-mediated immune responses destroy most of the mycobacteria. Although skin and peripheral nerves are damaged in tuberculoid leprosy, it progresses slowly and patients usually survive. In contrast, lepromatous leprosy is characterized by a humoral response; cell-mediated immunity is depressed. The humoral response sometimes results in markedly high levels of immunoglobulin (hypergammaglobulinemia). This response is not as effective in inhibiting disease, and mycobacteria are widely disseminated in macrophages, often reaching numbers as high as 1010 per gram of tissue. Lepromatous leprosy progresses into disseminated infection of the bone and cartilage with extensive nerve and tissue damage.
The development of lepromatous or tuberculoid leprosy depends, in part, on the balance between TH1 and TH2 responses (Figure 10-14). In tuberculoid leprosy, the immune response is characterized by a TH1-type response and high circulating levels of IL-2, IFN-γ, and lymphotoxin-α (LT-α). In lepromatous leprosy, there is a TH2-type immune response, with high circulating levels of IL-4 and IL-5. IL-10 is also produced and may come from other helper subsets, including TREG cells. This cytokine profile explains the diminished cell-mediated immunity and increased production of serum antibody in lepromatous leprosy.
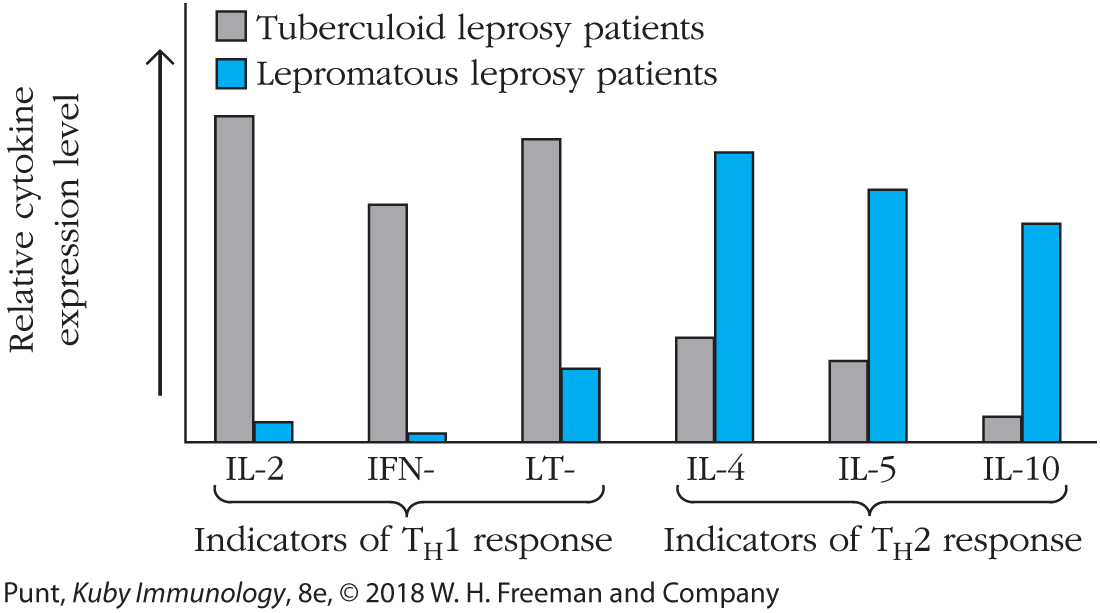
FIGURE 10-14 Correlation between type of leprosy and relative TH1 or TH2 activity. Messenger RNA isolated from lesions from patients with tuberculoid and lepromatous leprosy was analyzed by Southern blotting, using the cytokine probes indicated. The results are represented in this graph. Cytokines characteristic of TH1 cells and type 1 responses (e.g., IFN-γ and LT-α [also known as TNF-β]) predominate in the patients with tuberculoid leprosy, whereas cytokines characteristic of TH2 cells and type 2 responses (e.g., IL-4) predominate in the patients with lepromatous leprosy. [Data from Sieling, P.A., and Modlin, R.L., “Cytokine patterns at the site of mycobacterial infection,” Immunobiology, 1994, October, 191(4–5):378–87.]
Presumably each of these patients was exposed to the identical bacterial pathogen. Why do some patients develop an effective TH1 response while others do not? Studies suggest that genetic differences among human hosts play a role. For example, differences in susceptibility may correlate with individual differences in the expression of PRRs (specifically, TLR1, TLR2, and NOD2) by innate cells. This makes sense given that interactions between pathogen and innate immune cells determine the cytokine environment that influences the outcome of T-cell polarization. Changes in PRR expression or activity could alter either the quality or quantity of cytokines produced.
Progression of HIV infection to AIDS is also characterized by upsets in the balance among T-cell subsets. All CD4+ T cells are susceptible to HIV infection. In the early stages of infection, our bodies are still able to generate new CD4+ T cells to replace those that have died. However, inflammation induced by HIV infection generates cytokines and other factors that favor polarization of these new T cells to the TREG lineage, reducing the body’s ability to fight the infection. Even as total T-cell numbers decline, the proportion of TREG cells remains high in infected patients.
Some pathogens have evolved to directly influence the activity of the TH subsets. The Epstein-Barr virus, for example, produces a homolog (mimic) of human IL-10 called viral IL-10 (vIL-10). Like cellular IL-10, vIL-10 suppresses helper T-cell and macrophage activity in a variety of ways, reducing the cell-mediated response to the Epstein-Barr virus and giving it a survival advantage.
TH17 cells first received attention because of their association with chronic autoimmune disease. Mice that were unable to make IL-23, a cytokine that contributes to TH17 polarization, were found to be resistant to autoimmunity. TH17 cells and their defining effector cytokine, IL-17, are often found in inflamed tissue associated with rheumatoid arthritis, inflammatory bowel disease, multiple sclerosis, psoriasis, and asthma. The role TH17 cells played in protecting organisms from infection was not immediately obvious until studies of individuals with an autosomal dominant form of a disease known as hyper-IgE syndrome or Job syndrome confirmed that TH17 cells were important in controlling infections by extracellular bacteria and fungi (see Clinical Focus Box 10-3).
These are just some examples of the profound influence helper T-cell subsets have on disease progression. It is important to recognize that our current perspectives on the roles of helper subsets in disease and health remain simplistic. Our appreciation of the complex interplay among subsets will continue to improve and add more subtlety to our explanations in the future.