Cell-Mediated Effector Responses
Cytotoxic effector cells include the CD8+ cytotoxic T lymphocytes (CTLs) of the adaptive immune system; NKT lymphocytes, which bridge the innate and adaptive immune systems; and NK cells, which were once associated strictly with the innate immune system but are now known to share intriguing functional features with their adaptive immune lymphocyte relatives. CTL, NKT, and NK effectors all induce cell death by triggering apoptosis in their target cells. Not only do these cytotoxic cells eliminate targets infected with intracellular pathogens (viruses and bacteria), but they also play a critical role in eliminating tumor cells and cells that have been stressed by extreme temperatures or trauma (Table 12-3). CTL and NK cells also play a less desirable role in rejecting cells from allogeneic organ transplants. Essentially, the cell-mediated immune response is prepared to recognize and attack any cell that exhibits “nonself” or “altered-self” characteristics.
| Cell type | Effector molecules produced | Mechanism of killing |
|---|---|---|
| CTL (typically CD8+ T cell) | Cytotoxins (perforins and granzymes), IFN-γ, TNF, Fas ligand (FasL) | Cytotoxic granule release; and FasL-Fas interactions |
| NKT cell | IFN-γ, IL-4, GM-CSF, IL-2, TNF, FasL | FasL interactions predominantly; can activate NK cells indirectly via cytokines |
| NK cell | Cytotoxins (perforins and granzymes), IFN-γ, TNF, FasL | Cytotoxic granule release; and FasL-Fas interactions |
Cytotoxic T Lymphocytes Recognize and Kill Infected or Tumor Cells Via T-Cell Receptor Activation
The importance of cytotoxic T lymphocytes in the cell-mediated immune response to pathogens is underscored by the pathologies that afflict those defective in T-cell generation. Children with DiGeorge syndrome are born without a thymus and therefore lack the T-cell component of the cell-mediated immune system. They are generally able to cope with extracellular bacterial infections because their innate and T cell—independent antibody responses are intact, but they cannot effectively eliminate intracellular pathogens such as viruses, intracellular bacteria, and fungi. The severity of the cell-mediated immunodeficiency in these children is such that even the attenuated virus present in a vaccine, which is capable of only limited growth in normal individuals, can produce life-threatening infections.
T cell—mediated immune responses can be divided into two major categories according to the different effector populations that are mobilized: cytotoxic T cells and helper T cells. The differentiation and activity of helper T-cell subsets was discussed in detail in Chapter 10, and their roles in macrophage activation and delayed-type hypersensitivity will be described in Chapter 15. Here, we focus on the immune response mediated by cytotoxic T lymphocytes (TC cells or CTLs), which can be divided into two phases. In the first phase, naïve TC cells undergo activation and differentiation into functional effector CTLs within secondary lymphoid tissue. As you learned from Chapter 10, differentiation of a naïve CD8+ T cell into an effector CTL involves recognition of MHC class I—peptide complexes on an activated dendritic cell as well as help from TH1 effector CD4+ T cells. This help is provided both indirectly, via the ability of CD4+ T cells to enhance dendritic cell function, and directly, via the release of cytokines by helper CD4+ T cells. These events usually occur in secondary lymphoid tissues, such as lymph nodes and the spleen.
In the second phase, effector CTLs recognize MHC class I—peptide antigen complexes on specific target cells in the periphery, such as virus-infected or tumor cells, an event that ultimately induces the apoptosis of the target cells. Since virtually all nucleated cells in the body express MHC class I molecules, a CTL can recognize and eliminate almost any cell in the body that displays the specific foreign antigen—derived peptide recognized by that CTL in association with an MHC class I molecule.
Effector CTL Generation from CTL Precursors
Naïve TC cells are incapable of killing target cells and are therefore also referred to as CTL precursors (CTL-Ps) to denote their functionally immature state. Only after a CTL-P has been activated does the cell differentiate into a functional CTL with cytotoxic activity. As is true for naïve CD4+ T cells (see Chapter 10), three signals are required for CTL-P activation (Figure 12-6):

FIGURE 12-6 Generation of effector CTLs. The differentiation of a naïve CD8+ T cell (a CTL precursor, or CTL-P) into a functional CTL requires several events. The CTL precursor, in this case specific for a viral antigen, must engage MHC class I—peptide complexes and costimulatory ligands on a “licensed” antigen-presenting cell (dendritic cell). Licensing of dendritic cells occurs either through engagement with an activated, CD40L+ helper T cell (e.g., a TH1 cell) or through signals from pattern recognition receptors (e.g., via Toll-like receptors). (a) Sequential activation. Left: CD+ T-cell activation of an unlicensed dendritic cell can occur prior to naïve CD8+ T-cell engagement by the dendritic cell. Right: The licensed dendritic cell then activates a naïve CD8+ T cell. (b) Simultaneous activation. Alternatively, activation can occur at the same time as the CTL-P engages the dendritic cell. Dendritic cells play a key role in the activation of naïve CTL-Ps because licensing enables dendritic cells to cross-present peptides from internalized viral particles on MHC class I proteins that then are recognized by the CTL-P’s T-cell receptors, providing signal 1 for T-cell activation. In addition, licensing induces dendritic cell expression both of membrane CD80/86, which provides signal 2 to the CTL-P, and of secreted cytokines (IL-12), which contribute to T-cell activation. Simultaneous engagement of a dendritic cell by both helper CD4+ and precursor CD8+ T cells would allow delivery to the pre-CTL of IL-2 generated by the helper CD4+ T cells. In response to TCR stimulation, precursor CTLs begin to up-regulate expression of the IL-2 receptor and begin to make more of their own IL-2. IL-2, which provides signal 3 to the T cells, is critical for the induction of proliferation and successful differentiation into functional CTL and memory cells.
- Signal 1: An antigen-specific signal transmitted by the TCR complex on recognition of an MHC class I—peptide complex on a “licensed” APC, usually a dendritic cell (licensing is explained below)
- Signal 2: A costimulatory signal transmitted when CD28 on the CTL-P surface binds CD80/86 (B7) on the dendritic cell
- Signal 3: A signal provided by the CTL-P’s high-affinity IL-2 receptor binding to IL-2, generated by helper T cells as well as by the CD8+ T cell itself.
These three signals induce the proliferation and differentiation of the antigen-activated CTL-P into effector CTLs and memory cells.
Differentiation of a CTL-P is initiated by its recognition of antigen on an APC presented in the context of an MHC class I molecule, but other factors are also required. For CTL-Ps to mature into cytotoxic cells they need to recognize peptide—MHC class I complexes presented by a licensed APC, usually a dendritic cell. A dendritic cell can be licensed in several ways—by a CD4+ helper T cell (TH) usually of the TH1 subset, or by engagement with microbial products, which activate Toll-like receptors. What does a CD4+ T cell provide that is so important for optimal activation of a CD8+ T cell? Recall from Chapter 10 that helper T cells generate cytokines (e.g., IFN-γ) that activate antigen-presenting cells. However, activated helper T cells also express CD40 ligand (CD40L, also known as CD154), a member of the TNF family of proteins, which provides an all-important costimulatory signal to the APC. CD40L is recognized by CD40, a TNF receptor family member expressed by activated professional APCs. When it binds CD40L, CD40 initiates a signaling cascade within the APC that increases the expression of costimulatory ligands (CD80 and CD86), chemokines, and cytokines, significantly enhancing the APC’s ability to activate the CD8+ T cell.
In addition, licensing enables the dendritic cell to carry out cross-presentation. As described in Chapter 7, in order for naïve CTL-Ps to be activated by viral or tumor antigens, peptide fragments must be presented on MHC class I proteins. If the dendritic cell is not itself infected or malignant, which will be the case most of the time, it will be taking up virus or antigens released from tumor cells from the environment. But exogenous antigens are typically processed and presented in the context of MHC class II molecules. How, then, can those antigens be processed in a way that allows their peptides to be presented by MHC class I proteins? Recall from Chapter 7 that cross-presentation is the process by which endocytosed exogenous proteins are degraded and their peptides loaded onto MHC class I proteins, rather than on MHC class II. Only licensed dendritic cells can carry out the cross-presentation necessary for the activation of naïve CTL-Ps.
Investigators envision a simultaneous interaction among three cells that results in CD8+ T-cell activation: the TH cell, which interacts with and licenses the APC, which, in turn, interacts with and activates the CTL-P. In fact, fluorescence imaging studies have provided direct evidence of the formation of three-cell complexes of a dendritic cell, a CD4+ T cell, and a CD8+ T cell during the T-cell response to viral antigen (see Figure 12-6b and Figure 14-17). It is important to realize, however, that the interactions between the dendritic cell and the two different T cells may not have to be simultaneous; rather, a dendritic cell “licensed” by a TH cell could retain its capacity to activate CTL-Ps for some period after the TH cell disengages (see Figure 12-6a). In this case, specific TH cells could move on to sequentially activate other dendritic cells, amplifying the activation response.
Consequences of CTL Activation
CTL differentiation is accompanied by several changes. CTL precursors do not express the high-affinity IL-2 receptor α chain (CD25), nor do they produce much IL-2. They do not proliferate, and do not display cytotoxic activity. Signals 1 and 2 induce expression of both the IL-2Rα chain (generating the high-affinity IL-2 receptor) and IL-2 itself (the principal cytokine required for full proliferation and differentiation of effector CTLs). The importance of IL-2 in CTL function is underscored in IL-2 knockout mice, which are markedly deficient in CTL-mediated cytotoxicity. Fully activated CTLs turn on expression of proteins, including granzyme B and perforin, that are packaged into lytic granules and, when released, will induce apoptosis of the target cell.
CTLs are potentially very dangerous to an organism, and the stringent requirements for activation help prevent unwanted cellular destruction. Thus, the requirement that both TH and TC cells recognize antigen before the TC cell is optimally activated provides a safeguard against inappropriate self-reactivity by cytotoxic cells. The fact that the IL-2 receptor is not expressed until after a naïve CD8+ T cell has been activated by T-cell receptor engagement also ensures that only the antigen-specific CTL-Ps proliferate and become cytotoxic.
Expansion of Antigen-Specific CD8+ T Cells
For many years the study of the CTL response to viruses or tumors was hampered by the inability to identify CD8+ T cells specific for viral or tumor antigens. As these T cells recognize antigen only as peptide fragments bound to MHC proteins, and as TCRs have much lower affinity for their MHC-peptide ligands compared with the much higher affinity of BCRs for intact antigens, one could not simply use fluorescent antigens to detect antigen-specific T cells. Fortunately, a technique was developed that uses tetramers of MHC-peptide complexes to identify T cells specific for an MHC-peptide complex. This technique allowed identification of antigen-specific T cells (see Advances Box 12-2).
With this powerful tool, one can directly measure the increase in antigen-specific CD8+ T cells in response to exposure to pathogens such as viruses or cancer-associated antigens, and trace their tissue distribution. For instance, researchers infected mice with vesicular stomatitis virus (VSV) and, using tetramer technology, examined all organs for the presence of CD8+ cells specific for a VSV-derived peptide—MHC complex. This study demonstrated that during acute infection with VSV, VSV-specific CD8+ cells migrate away from the lymphoid system and are distributed widely, with large numbers found in the liver and kidneys, but antigen-specific cells present virtually everywhere (Figure 12-7a). Follow-up studies showed that, regardless of the original site of infection, effector CD8+ T cells distribute themselves throughout the body. Similar analyses over the course of an infection have shown that the dramatically increased numbers of virus-specific CD8+ T cells that arise soon after infection, many of which are CTL effector cells, decline as the virus is eliminated. Most effector CTLs have short half-lives, with only 5% to 10% remaining after the virus is cleared; the bulk of the surviving virus-specific cells are an expanded population of memory cells (Figure 12-7b).
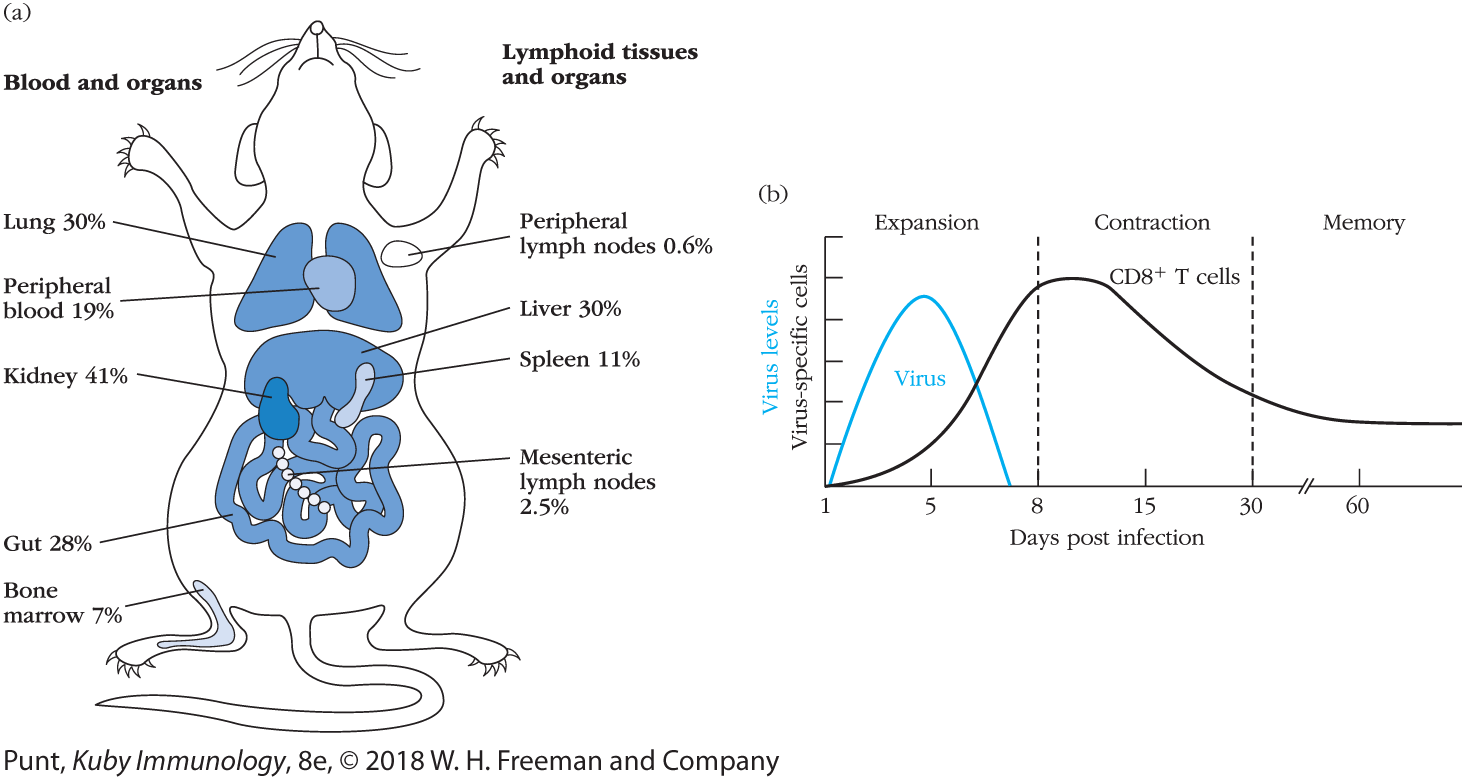
FIGURE 12-7 Localizing antigen-specific CD8+ T-cell populations in vivo (a) Mice were infected with vesicular stomatitis virus (VSV), and during the course of the acute stage of the infection cell populations were isolated from the tissues indicated. These cells were then incubated with fluorescent tetramers containing VSV peptide—MHC complexes. Flow cytometric analysis allowed determination of the percentages of CD8+ T cells that were VSV specific in each of the populations examined. (b) General pattern of changes in levels of virus and numbers of virus peptide—MHC tetramer—positive CD8+ T cells in mice following acute viral infections. [(a) Data from Klenerman, P., V. Cerundolo, and P. R. Dunbar. 2002. Tracking T cells with tetramers: new tales from new tools. Nature Reviews Immunology 2:263. (b) Data from Kaech, S. M., and W. Cui. 2012. Transcriptional control of effector and memory CD8+ T cell differentiation. Nature Reviews Immunology 12:749.]
How CTLs Kill Cells
A CTL can kill a target in two major ways: either via the directional release of granule contents, or by a Fas-FasL membrane signaling interaction. Rather than inducing cell lysis, both of these processes induce the target cell to undergo apoptosis, typically within an hour of contact with the cytotoxic cell.
Regardless of which method is employed, CTL killing involves a carefully orchestrated sequence of events that begins when the attacking cell binds to the target cell (Figure 12-8) and forms a cell-cell conjugate, an event so intimate that it is sometimes referred to as the “kiss of death.” Formation of a CTL—target cell conjugate is followed within several minutes by a Ca2+-dependent, energy-requiring step in which, ultimately, the CTL induces death of the target cell. The CTL then dissociates from the target cell and may go on to bind another target cell.

FIGURE 12-8 Stages in CTL-mediated killing of target cells. (a) T-cell receptors on a CTL interact with processed antigen—MHC class I complexes on an appropriate target cell, leading to formation of a CTL—target cell conjugate. The centrosome (also called the microtubule organizing center, or MTOC) polarizes to the site of interaction, repositioning the Golgi stacks and granules toward the point of contact with the target cell, where the granules’ contents are released by exocytosis onto the surface of the target cell, which is induced to apoptose. After dissociation of the conjugate, the CTL is recycled and the target cell dies by apoptosis. (b) Fluorescence image of a CTL—target cell conjugate, showing the enrichment of the protein kinase Lck (green) under the CTL membrane at the synapse between the two cells, where it is contributing to activation of the CTL; the centrosome (yellow), which is located under the synapse; and secretory granules (red), which are migrating on microtubule tracks up to the junction between the cells. (Note: the red granules in the target cell are probably lysosomes. Nuclei are blue.)
The specific signaling events involved in establishing the CTL—target cell interaction are very similar to those associated with an activating T cell—APC interaction. The TCR-CD3 membrane complexes on a CTL recognize peptide antigen in association with MHC class I molecules on the target cell. This recognition event triggers the development of a highly organized immunological synapse (see Chapter 10) characterized by a central ring of TCR molecules, surrounded by a peripheral ring of adhesion molecules, formed primarily by interactions between the integrin receptor LFA-1 on the CTL membrane and the intracellular adhesion molecules (ICAMs) on the target cell membrane (see Figure 10-3).
TCR signals directly enhance the adhesion between killer and target by converting LFA-1 from a folded, low-affinity state to an extended, high-affinity state (Figure 12-9a). This change is a consequence of “inside-out signaling,” in which an intracellular signal cascade (generated by the TCR in this example, but can also be activated by some cytokine and chemokine receptors) acts on the intracellular portion of LFA-1 and induces a conformational change that straightens out the extracellular region of LFA so that its high-affinity face is accessible to ICAM. LFA-1 persists in its high-affinity state for only 5 to 10 minutes after antigen-mediated activation, and then it returns to the low-affinity state. This downshift in LFA-1 affinity may facilitate dissociation of the CTL from the target cell.

FIGURE 12-9 Effect of antigen activation on the ability of CTLs to bind to the intercellular cell adhesion molecule ICAM-1. (a) TCR signals induce a conformational change in LFA-1 molecules from a folded state to an extended state that allows them to bind to ICAM with high affinity. (b) The importance of TCR signaling in inducing LFA-1—mediated adhesion is illustrated by an experiment in which resting mouse CTLs were first incubated with anti-CD3 antibodies. Cross-linkage of CD3 molecules on the CTL membrane by anti-CD3 has the same activating effect as interaction with MHC class I—peptide complexes on a target cell. Adhesion was assayed by binding radiolabeled CTLs to microwells coated with ICAM-1. Antigen activation increased CTL binding to ICAM-1 more than 10-fold. The presence of excess monoclonal antibody to LFA-1 or ICAM-1 in the microwell abolished binding, demonstrating that both molecules are necessary for adhesion.
The importance of TCR signals in promoting CTL adhesion was demonstrated by an experiment in which purified ICAM protein was coated onto plastic and the ability of resting CTLs versus TCR-stimulated CTLs to adhere was measured. As you can see from Figure 12-9b, more than 10 times the number of TCR-stimulated CTLs (versus unstimulated CTLs) bound to the ICAM-coated plastic. Antibodies that could block the interaction between LFA-1 and ICAM abrogated the effect of TCR stimulation, showing that the adhesion was, in fact, LFA-1/ICAM specific.
Granzyme- and perforin-mediated cytolysis
Many CTLs initiate killing of their targets via the delivery of proapoptotic molecules. These molecules are packaged within granules that can be visualized by microscopy (Figure 12-10). Investigators originally isolated CTL granules by subcellular fractionation and showed that they could induce target cell damage directly. Analysis of their contents revealed 65-kDa monomers of a pore-forming protein called perforin and several serine proteases called granzymes. CTL-Ps lack cytoplasmic granules; however, once activated, CTLs begin to form cytoplasmic granules that include perforin monomers and granzyme molecules.

FIGURE 12-10 Formation of a conjugate between a CTL and a target cell and reorientation of CTL cytoplasmic granules as recorded by time-lapse photography. (a) A motile mouse CTL (thin arrow) approaches an appropriate target cell (TC). The thick arrow indicates the direction of movement. (b) Initial contact of the CTL and target cell has occurred. (c) Within 2 minutes of initial contact, the membrane-contact region has broadened and the rearrangement of dark cytoplasmic granules within the CTL (thin arrow) is underway. (d) Further movement of dark granules toward the target cell is evident 10 minutes after initial contact.
Almost immediately after conjugate formation, CTL granules containing granzyme and perforin are brought to the site of interaction between a killer and target (see Figure 12-8a) via the activity of the centrosome (also called the microtubule organizing center), which polarizes to this synapse in response to TCR stimulation. The secretory granules migrate on the microtubules up to the site of contact with the target cell and the vesicles fuse with the outer membrane, releasing perforin monomers and granzyme proteases into the space at the junction between the two cells.
As the perforin monomers contact the target cell membrane, they undergo a conformational change, exposing an amphipathic domain that inserts into the target cell membrane; the monomers then polymerize (in the presence of Ca2+) to form cylindrical pores with an internal diameter of 5 to 20 nm (Figure 12-11). Perhaps not surprisingly, perforin exhibits some sequence homology with the terminal C9 component of the complement system, which forms the membrane attack complex that causes complement-mediated lysis. The importance of perforin to CTL-mediated killing is demonstrated in perforin-deficient knockout mice, which are unable to eliminate lymphocytic choriomeningitis virus (LCMV) from the body even though they mount a significant CD8+ cell immune response to virally infected cells.

FIGURE 12-11 CTL-mediated pore formation in target cell membrane. (a) In this model, a rise in intracellular Ca2+ triggered by CTL—target cell interaction (1) induces exocytosis, in which the granules fuse with the CTL cell membrane (2) and release monomeric perforin into the small space between the two cells (3). The released perforin monomers undergo a Ca2+-induced conformational change that allows them to insert into the target cell membrane (4). In the presence of Ca2+, the monomers polymerize within the membrane (5), forming cylindrical pores (6). (b) Electron micrograph of perforin pores on the surface of a rabbit erythrocyte target cell. The arrow indicates a single pore.
Although granzyme B was once thought to gain entry into the target cell via surface perforin pores, it is now thought that it enters mostly via endocytic processes. Many target cells express the mannose 6-phosphate receptor on their surface, which binds granzyme B. Complexes of granzyme B bound to mannose 6-phosphate receptor are internalized and appear inside endosomal vesicles. Perforin internalized at the same time then forms pores that release granzyme B from the endosomal vesicle into the cytoplasm of the target cell.
Regardless of the mechanism of entry, once in the cytoplasm, granzyme B and other granzymes initiate a cascade of reactions that result in the fragmentation of target cell DNA into oligomers of 200 base pairs (bp); this type of DNA fragmentation is typical of apoptosis. Granzyme proteases do not directly mediate DNA fragmentation. Rather, they activate an apoptotic pathway within the target cell. This apoptotic process does not require mRNA or protein synthesis in either the CTL or the target cell. Within several minutes of CTL contact, target cells begin to exhibit DNA fragmentation. Interestingly, viral DNA within infected target cells has also been shown to be fragmented during this process. This observation shows that CTL-mediated killing not only kills virus-infected cells but also can destroy the viral DNA in those cells directly. The rapid onset of DNA fragmentation after CTL contact may prevent continued viral replication and assembly in the period before the target cell is destroyed.
How do CTLs protect themselves from their own perforin and granzyme activity? Studies show that CTLs are more resistant to the activities of granzyme and perforin than their targets. The strategies they use are not fully understood, but investigators have found that CTLs express high levels of serine protease inhibitors (serpins), which protect them from granzyme B activity. In support of a role for serpins in protecting CTLs from their own proapoptotic functions, CTLs do not survive in mice deficient for one of the most common serpins expressed by CD8+ T cells, Spi-1.
Fas/FasL-mediated cytolysis
Some potent CTL lines have been shown to lack perforin and granzymes. In these cases, cytotoxicity is mediated by Fas (CD95). This transmembrane protein, expressed by many cell types, is a member of the TNF receptor family and can deliver a death signal when cross-linked by its natural ligand, a member of the TNF family called Fas ligand (FasL). FasL is synthesized by activated CTLs and localizes in the granule membrane, which becomes part of the CTL membrane after granule fusion. The interaction of FasL with Fas on a target cell triggers apoptosis.
Fas mutations lead to multiple disorders in both mice and humans. Mice that are homozygous for the lpr (lymphoproliferation) mutation express little or no Fas on their cell membranes, and have remarkably large lymph nodes. In more rigorous terms, they are afflicted with a lymphoproliferative disease characterized by the accumulation of mature, activated T and B lymphocytes in their lymph nodes. CTLs in lpr mutant mice can be induced to express FasL; however, these CTLs cannot kill targets, as they express no Fas. These mice also develop autoimmune diseases because peripheral autoreactive lymphocytes are not eliminated. A very similar disorder, now known as autoimmune lymphoproliferative syndrome (ALPS or Canale-Smith syndrome), has been identified in human patients who have genetic defects in Fas, Fas ligand, or Fas signaling pathways.
The critical importance of both the perforin and the Fas-FasL systems in CTL-mediated cytolysis was revealed by experiments with two types of mutant mice: perforin knockout mice and the Fas-deficient lpr strain (Figure 12-12). Investigators wanted to determine the relative importance of each of these molecules and developed a system in which they generated CTLs by coculturing T cells from H2b mice with killed cells from H2k mice. CTLs generated in such mixed-lymphocyte reactions reacted strongly against allogeneic MHC and could kill H2k-expressing cells. With this system, investigators first asked whether CTLs could kill target cells generated from lpr mice (which expressed no Fas). In fact, they could, demonstrating that Fas-FasL interactions were not absolutely required for CTL activity. Next they asked whether perforin was required and generated CTLs from perforin knockout mice. They found that these cells could also kill targets, demonstrating that perforin wasn’t absolutely required. Finally, they asked whether killing occurred in the absence of both perforin and Fas-FasL interactions. They generated CTLs from perforin knockout mice and determined their ability to kill target cells from the Fas-deficient mice. No killing was observed. These and other observations indicate that CTLs employ these and only these two methods—perforin-mediated and Fas-mediated apoptosis—to kill their targets.

FIGURE 12-12 Experimental demonstration that CTLs use Fas and perforin pathways. (a) Lymphocytes were harvested from mice of H2b and H2k MHC haplotypes. H2k haplotype cells were killed by treatment with mitomycin C and cocultured with H2b haplotype cells to stimulate the generation of anti-H2k CTLs. If the H2b lymphocytes were derived from normal mice, they gave rise to CTLs that had both perforin and Fas ligand. If the CTLs were raised by stimulation of lymphocytes from perforin knockout (KO) mice, they expressed Fas ligand but not perforin. (b) Interaction of CTLs with Fas+ and Fas— targets. Normal H2b anti-H2k CTLs that express both Fas ligand and perforin kill normal H2k target cells and H2k lpr mutant cells, which do not express Fas. In contrast, H2b anti-H2k CTLs from perforin KO mice kill Fas+ normal cells by engagement of Fas with Fas ligand but are unable to kill the lpr cells, which lack Fas.
Induction of apoptosis
Both the perforin and Fas-FasL killing strategies activate signaling pathways in the target cell that induce apoptosis (Figure 12-13). A central feature of cell death by apoptosis is the involvement of the caspase family of cysteine proteases, which cleave after an aspartic acid residue. The term caspase incorporates each of these elements (cysteine, aspartate, protease). Normally, caspases are present in the cell as inactive proenzymes—procaspases—that require proteolytic cleavage for conversion to the active forms. Cleavage of a procaspase produces an active caspase, which cleaves other procaspases, thereby activating their proteolytic activity, which results in a cascade of events that systematically disassemble the cell—the hallmark of apoptosis. More than a dozen different caspases have been found, each with its own specificity. They are typically divided into two categories: those that initiate the caspase cascade (initiator caspases) and those that directly initiate apoptosis (effector caspases).

FIGURE 12-13 Two pathways of CTL-activated target cell apoptosis. (a) The Fas pathway. Ligation of the trimeric Fas protein by the CTL’s Fas ligand (FasL) delivered by the granule membrane leads to the association of Fas with the adapter molecule FADD, which in turn results in a series of reactions that activate a caspase cascade, leading to apoptosis of the target cell. (b) The perforin-granzyme pathway. Granule exocytosis releases granzymes and perforin from the CTL into the space between the CTL and the target cell. Granzyme B enters the target cell by endocytosis and then passes into the cytoplasm through perforin pores. Granzyme B can cleave and activate the proapoptotic Bcl-2 family member Bid, which stimulates mitochondria to release cytochrome c. Cytochrome c, a molecule called Apaf, and procaspase-9 assemble into an apoptosome, leading to caspase-9 activation and cleavage of procaspase-3, which activates death pathways. Granzyme B can also cleave and partially activate caspase-3. Release of cytochrome c and activation of caspase-3 are both required to initiate the caspase cascade that leads to target cell apoptosis.
What strategies do CTLs use to initiate caspase activation in target cells? The engagement of Fas on a target cell by Fas ligand on a CTL first induces the activation of an initiator caspase in the target cell. Fas is associated with a protein known as Fas-associated protein with death domain (FADD), which in turn associates with procaspase-8 (see Figure 12-13a). On Fas cross-linking, procaspase-8 is converted to caspase-8 and initiates an apoptotic caspase cascade.
The CTL-derived granzymes, which enter target cells through perforin pores in endosomal vesicles as mentioned earlier, are proteolytic and have several targets (see Figure 12-13b). They can directly cleave procaspase-3, an event that appears to only partially activate this effector caspase. They can also cleave the protein Bid, which induces mitochondrial release of cytochrome c. The latter assembles with Apaf-1 (apoptosis protease activating factor-1), ATP, and procaspase-9 to form an apoptosome, activating caspase-9. Caspase-9 cleaves procaspase 3, activating its apoptosis-inducing functions. Both granzyme activities—activating caspase-3 directly and indirectly via the apoptosome—appear to be required for optimal proapoptotic activity.
The end result of both perforin-granzyme and Fas-mediated pathways is, therefore, the activation of apoptotic programmed cell death pathways that are present in the target cell. As one immunologist has so aptly put it, CTLs don’t so much kill target cells as persuade them to commit suicide.
TC1 and TC2: Two Subsets of Effector CTLs
As you know from Chapter 10, CD4+ effector T helper cells can differentiate into several subsets, each of which secretes a distinct panel of cytokines. Effector CD8+ cytotoxic cells are not as diverse, but can develop into two distinct subsets: TC1 cells and TC2 cells. These subtypes loosely resemble TH1 and TH2 cells in terms of the cytokines they generate as well as the cytokines that promote their development (see Table 10-3). CTLs are biased toward becoming TC1 cells, which secrete IFN-γ but not IL-4. In the presence of IL-4, CTLs develop into TC2 cells, which secrete much more IL-4 and IL-5 than IFN-γ. Both subsets are potent killers, although TC1 cells can use both perforin/granzyme- and FasL-mediated strategies, whereas TC2 cells appear to use only perforin and granzymes. Studies suggest that these two subsets play different roles in regulating disease, but results are not yet sufficient to generate confidence in one particular model.
Natural Killer Cell Activity Depends on the Balance of Activating and Inhibitory Signals
Another cell type involved in cell-mediated immunity, the natural killer (NK) cell, initiates apoptotic pathways in target cells using very similar mechanisms as CTLs, but via very different receptors. NK cells were discovered essentially by accident when immunologists were measuring the cytolytic ability of lymphocytes isolated from mice with tumors. They originally predicted that these lymphocytes would exhibit a specific ability to kill the tumor cells to which they had been exposed. To do their experiments they included multiple controls, comparing the activity of these lymphocytes of interest with those taken from mice that had no tumors, as well as mice that had unrelated tumors. Much to their surprise, the investigators discovered that even the control lymphocytes, which either had not been exposed to any tumor or had been exposed to a very different type of tumor, were able to kill the tumor cells. In fact, the killing they were seeing did not seem to be following the rules of specificity that are the hallmark of a lymphocyte response to conventional antigens and pathogens. Further study of this nonspecific tumor-cell killing revealed that, in fact, neither T nor B lymphocytes were involved at all. Instead, a population of larger, more granular lymphocytes was responsible. Similar nonspecific and rapid tumor-cell killing was observed with human peripheral blood lymphocytes, even from people without cancer.
The cells, called “natural killer” (NK) cells for their ability to kill tumor cells without prior exposure, make up 5% to 10% of the circulating lymphocyte population. Despite the absence of antigen-specific receptors (i.e., membrane antibodies, T-cell receptors), they play a major role in immune defenses against infected cells, stressed cells, and tumor cells. They can also contribute to autoimmunity when dysregulated. More versatile than originally anticipated, NK cells also play a regulatory role in both innate and adaptive immune responses to conventional antigens by secreting cytokines that regulate immune responses. They have also recently been shown to be critically important for the development of a normal placenta, not via their cytotoxic ability, but via their ability to recognize the presence of a different (paternal) MHC and initiate the remodeling of blood vessels. As introduced in Chapter 4, NK cells are innate lymphoid cells (ILCs) that play a variety of roles in early protection against infection, in stimulating and regulating adaptive immune responses, and in tissue development and remodeling (see Table 4-6).
The importance of NK cells in our defense against infections is compellingly illustrated by the case of a young woman with a disorder that resulted in a complete absence of these cells. Even though this patient had normal T- and B-cell counts, she suffered severe Varicella virus (chickenpox) infections and a life-threatening cytomegalovirus infection. We now know that NK cells are a critical first line of defense against infection with intracellular pathogens (viruses and some bacteria) by killing infected cells early and thereby controlling pathogen replication during the 7 days it takes CTL-Ps to develop into functional CTLs. NK cell cytotoxic activity is stimulated by the innate immune cytokines IFN-α, IFN-β, and IL-12, which all rise rapidly during the early stages of a viral infection. The wave of NK cell activity peaks subsequent to this rise, about 3 days after infection (Figure 12-14).

FIGURE 12-14 Time course of responses to viral infection. IFN-α and IFN-β (dashed curve) are released from virus-infected cells soon after infection. These cytokines stimulate the NK cells, quickly leading to a rise in the NK-cell population (blue curve) from the basal level. NK cells help contain the infection during the period required for the generation of CTLs (black curve). Once the CTL population reaches a peak, the virus titer (blue area) rapidly decreases.
As we mentioned, NK cells also produce an abundance of immunologically important cytokines that can indirectly but potently influence both innate and adaptive immune responses. IFN-γ production by NK cells enhances the phagocytic, microbicidal, and antigen presentation activities of macrophages. IFN-γ derived from NK cells also influences the differentiation of CD4+ T helper subsets, stimulating TH1 development via induction of IL-12 production by macrophages and dendritic cells and inhibiting TH2 proliferation (see Chapter 10). NK cells also secrete TNF-α, GM-CSF, and chemokines that attract and activate macrophages, contributing to the local innate immune response.
NK cells are potent enough that, in conjunction with other protective mechanisms provided by the innate immune system, they can protect animals totally lacking in adaptive immunity. This is nicely illustrated by RAG1 knockout mice, which have no antigen-specific B or T lymphocytes yet are healthy, active, and able to fend off many infections. These animals do not fare nearly as well when NK-cell development is also impaired. Interestingly, humans appear to be more dependent on their adaptive immune systems and suffer more without B and T lymphocytes than do their murine relatives.
Phenotype of NK Cells
Where do NK cells come from, and what do they look like? Like B cells, T cells, and other ILCs, NK cells are derived from the common lymphoid progenitor (CLP) in the bone marrow. Although some NK cells develop in the thymus, this organ is not required for NK cell maturation. Nude mice, which lack a thymus and have few or no T cells, have functional NK-cell populations. Unlike T cells and B cells, NK cells do not undergo rearrangement of receptor genes; NK cells still develop in mice in which the recombinase genes RAG1 and RAG2 have been knocked out, preventing the rearrangement and expression of antibody and TCR genes.
NK cells are quite heterogeneous. Various subpopulations can be distinguished on the basis of differences in expression and secretion of specific immunologically relevant molecules. Whether this heterogeneity reflects different stages in their activation or maturation or truly distinct subpopulations remains unclear.
Most murine NK cells express CD122 (the 75-kDa β subunit of the IL-2 receptor), NK1.1 (a member of the NKR-P1 family), and CD49b (an integrin). NK cells also typically express CD2 and FcγRIII (CD16). In fact, cell depletion with monoclonal anti-FcγRIII antibody removes almost all NK cells from the circulation. Human NK cells also express IL-2 receptors and FcγRIII, but do not express NK1.1. Rather, they are distinguished from other lymphocytes by expression of the adhesion molecule CD56, which varies in expression depending on the maturation and activity state of an NK cell (CD56 high expressers tend to produce cytokines and may differentiate into CD56 low expressers, which exhibit more cytolytic activity).
Perhaps the most distinctive phenotypic characteristic of NK cells is their expression of a set of unique activating and inhibiting NK receptors (NKRs). These receptors are responsible for determining which targets NK cells will kill. Interestingly, NK cells from mice and humans use mostly distinct sets of receptors to accomplish the same thing; however, the principles driving NK activation remain the same. In addition, the number and type of activating and inhibitory receptors expressed by NK cells vary widely even within an individual, and we know now that it is the balance of signals received through these receptors that determines whether or not an NK cell will kill a target cell.
How NK Cells Recognize Targets: The Missing Self and Balanced Signals Models
As NK cells do not express antigen-specific receptors, the mechanism by which these cells recognize tumor or infected cells and distinguish them from normal body cells baffled immunologists for years. Klas Kärre advanced an interesting hypothesis that formed the foundations for our understanding of how NK cells distinguish self from nonself (or altered self). He proposed that NK cells kill when they do not perceive the presence of normal self-MHC proteins on a cell; this was known as the missing self model. The implied corollary to the proposal is that recognition of self inhibits the ability to kill.
Clues to the origins of such inhibitory signals came from early studies looking at which tumor targets NK cells killed best. Investigators examined multiple variables associated with NK-cell preferences and discovered that killing was inversely correlated with levels of MHC class I molecules expressed on tumor cells. In one study, they examined the ability of CTLs and NK cells to kill B cells that were transformed into tumor cells by infection with Epstein-Barr virus (EBV). CTLs were unable to recognize and lyse these B cells. However, NK cells were very effective killers of these tumor cells. Ultimately, investigators realized that EBV infection down-regulated MHC class I, allowing the cells to evade CD8+ T-cell recognition. However, this absence of class I made them perfect targets for NK cells, which responded to the “missing self.” The investigators “clinched” a role for MHC class I by transfecting the B-cell tumors with human MHC class I genes. NK cells were no longer able to kill the cells. The subsequent discovery of receptors on NK cells that produce inhibitory signals when they recognize MHC molecules on potential target cells provided direct support for this model. These inhibitory receptors were shown to prevent NK-cell killing, proliferation, and cytokine release.
As many virus-infected and tumor cells have decreased MHC expression, the missing self model (i.e., basing the decision to kill on whether a target expresses sufficient levels of MHC class I, a ubiquitous self protein) makes good physiological sense. Although the fundamental paradigm has stood the test of time, not surprisingly the real situation is more complicated (Figure 12-15). It turns out that NK cells express two different categories of receptors: one that delivers signals that inhibit the cytotoxic activity of NK cells (receptors that recognize MHC class I proteins) and another that delivers signals that stimulate cytotoxic activity (receptors that recognize ligands up-regulated on infected, stressed, and tumor cells). NK cells distinguish healthy cells from infected or cancerous ones by monitoring and integrating both sets of signals; whether or not a target is killed depends on the balance between activating and inhibitory ligands that it expresses. Inhibitory signals, in general, trump activating signals; thus, NK cells are tolerant of cells that express normal levels of unaltered self MHC class I molecules (see Figure 12-15a). However, consistent with the original missing self model, if a cell has lost MHC class I expression, such as through infection with a virus that down-regulates MHC class I as part of its immune evasion strategies, then expression on the target cell of a ligand for an activating receptor can trigger activation and killing or cytokine production (see Figure 12-15b). More recent findings that most NK cells express multiple inhibitory and activating receptors have led to a modified model for the control of NK cell activation, called the balanced signals model. In this model it is the balance of inhibitory-versus-activating signals coming in from receptors recognizing a variety of ligands on the target cell that determines whether the NK cell is activated (see Figure 12-15c).

FIGURE 12-15 How NK cytotoxicity is restricted to altered self cells: missing self model and balanced signals model. The balance of signals from activating and inhibitory receptors determines whether an NK cell will kill a target cell. (a) Tolerance: no killing. An activating receptor on NK cells interacts with its ligand on normal cells, inducing an activation signal. However, engagement of inhibitory NK-cell receptor(s) by self MHC class I molecules delivers inhibitory signals that counteract the activation signal, and the cell is not killed. (b) Missing self model. Because MHC class I expression is often decreased on altered self cells, such as virus-infected and tumor cells, the activating signal predominates, leading to target cell destruction. (c) As some cells that are killed by NK cells are not always MHC class I-negative and express multiple activating and inhibitory receptors, the balanced signals model was developed. Infected, stressed, or tumor cells up-regulate expression of certain proteins that are recognized by activating receptors on the NK cell. If the activating signals are more extensive than inhibitory receptors, the NK cell becomes activated.
NK Cell Receptor Families
NK receptors (NKRs) fall into two functional categories: inhibitory receptors that bind MHC class I and create signals that block the NK cell from killing, and activating receptors that induce signals that trigger NK-cell cytotoxicity if sufficient inhibitory signals are not received. Each of these functional NKR groups includes two types of receptors: members with lectin-like extracellular regions and members with immunoglobulin-like extracellular domains (Table 12-4). Note that although lectins typically bind carbohydrates, most of the lectin-like NK-cell receptors actually recognize proteins.
| Family | Species | Structure | Ligands | Activating or inhibitory | Examples |
|---|---|---|---|---|---|
| NKG2A-D | Mouse and human | Lectin-like | MHC class I—like | Most (but not all) activating | NKG2D (activating) binds MHC class I—like MICA, MICB, and ULPB family members (human) or H60, Mult1, Rae-1 family members (mouse)
CD94-NKG2A (inhibiting, human) binds HLA-E with bound signal peptide |
| Natural cytotoxicity receptors (NCRs) | Human | Immunoglobulin family | Viral Ags and proteins up-regulated on infected and tumor cells | Activating | NKp30 recognition of BAT3/BAG6 on tumor cells
NKp44 and NKp46 recognition of influenza virus HA and Sendai virus HN |
| KIR (many) | Human | Immunoglobulin family | MHC class I (HLA-B and HLA-C) | Both activating and inhibitory | KIR2DL1 (inhibitory) interacts with HLA-C
KIR2DS4 (activating) interacts with HLA-Cw4 and other non-HLA proteins in tumors |
| Ly49A-P | Mouse | Lectin-like | MHC class I and homologs | Both activating and inhibitory | Ly49A,C (inhibitory) recognize various MHC class I
Ly49H (activating) recognizes MCMV protein m157, a viral homolog of MHC class I |
The extracellular structure of the NK-cell receptor does not immediately identify it as inhibitory or activating. NK receptors with similar extracellular structures can have different intracellular domains and therefore different signaling properties. As with FcRs, the intracellular sequences of the activating NK receptors or their associated signaling chains generally contain ITAMs, and the intracellular sequences of inhibitory NK receptors contain ITIMs.
Inhibitory NK receptors and ligands
Inhibitory receptors, which bind to MHC class I molecules, are the most important determinant of an NK cell’s decision whether or not to kill a target cell. In humans, inhibitory receptors are members of a diverse family of proteins with immunoglobulin-like domains, known as the killer-cell immunoglobulin-like receptors, or KIRs. Surprisingly, the KIR family appears to have evolved extremely rapidly. Functional KIRs, found in primates, do not exist in rodents. Mice use a distinct family of receptors, the lectin-like Ly49 family, to achieve the same function as KIRs, namely inhibition of NK-cell cytolytic activity through binding to MHC class I molecules on healthy cells. Functional Ly49 receptors do not exist in humans.
Both KIR and Ly49 family members are highly diverse and polymorphic. These inhibitory receptors are generally specific for polymorphic regions of MHC class I molecules (H2-K or H2-D in mice; particular alleles of HLA-A, HLA-B, or HLA-C in humans). Figure 12-16a shows the human KIR3DL1 inhibitory receptor bound to the HLA-B*5701 class I allelic protein (allotype). Note that while the HLA-B*5701 class I protein has a bound peptide, as do all MHC proteins expressed on the cell surface, recognition by the KIR3DL1 receptor is only partially specific for the peptide. Only a fraction of MHC class I and class I—like ligands for these diverse receptors have been identified. Given the genetic polymorphism of both KIRs and their MHC class I ligands, it is not surprising that individuals can inherit KIR—MHC class I combinations that are not ideal and could increase their susceptibilities to disease.

FIGURE 12-16 Structures of NK inhibitory and activating receptors bound to their ligands. (a) Human KIR3DL1 inhibitory receptor bound to a normal MHC class I protein, HLA-B*5701 with a bound peptide. The three Ig-like domains (D0, D1, D2) bind to various portions of the MHC class I molecule; recognition is not peptide-specific. (b) Human NKG2D activating receptor bound to MICA, a nonclassical MHC class I protein that does not have bound peptides.
An exception to the rule of thumb that human NK inhibitory receptors are Ig-like molecules is the lectin-like inhibitory receptor CD94-NKG2A, a disulfide-bonded heterodimer made up of two glycoproteins: CD94 and NKG2A. The inhibitory receptor CD94-NKG2A is a member of the NKG2 family; the other members of this family are all activating receptors (see Table 12-4). Whereas KIRs typically recognize polymorphisms of HLA-B or HLA-C, CD94-NKG2A receptor recognizes the MHC class I—related protein HLA-E on potential target cells. HLA-E is not transported to the surface of a cell unless it has bound a peptide derived from the HLA-A, HLA-B, or HLA-C proteins: the leader (or signal) peptide cleaved from the nascent MHC leader peptide as it enters the rough endoplasmic reticulum. Thus the amount of HLA-E on the surface serves as an indicator of the overall level of MHC class I biosynthesis in the cells. The inhibitory CD94-NKG2A receptor recognizes surface HLA-E and sends inhibitory signals to the NK cell, with the net result that killing of potential target cells is inhibited if they are expressing adequate levels of MHC class I.
Unlike the antigen receptors expressed by B cells and T cells, NK receptors are not subject to allelic exclusion, and NK cells can express several different KIRs or Ly49 receptors, each specific for a different MHC molecule or for a set of closely related MHC molecules. Individual human NK cells expressing the CD94-NKG2A receptor and as many as six different KIRs have been found. The ability of each NK cell to express multiple KIRs or NKG2A receptors improves its chances of recognizing the polymorphic MHC class I variants expressed by an individual’s cells, therefore preventing NK cells from killing healthy cells.
Activating NK receptors and ligands
Most activating receptors expressed by murine and human NK cells are structurally similar and many are C-lectin-like—so named because they have domains related to calcium-dependent carbohydrate recognition domains, although the NK receptors recognize protein determinants. One major family in both humans and mice is the NKG2 family. NKG2D has emerged as one of the most important activating receptors in humans and mice; it acts through a signaling cascade similar to that initiated by CD28 in T cells. The ligands for NKG2D are nonpolymorphic MHC class I—like molecules that do not associate with β2-microglobulin. These ligands are often induced on cells undergoing stress, such as that caused by DNA damage or infection. Their binding to NKG2D activates NK-cell functions—cytotoxicity and cytokine production at sites of infection, malignancy, and tissue damage. The structure of the NKG2D activating receptor bound to MICA, an MHC class I—like protein encoded in the class I region of the HLA gene complex, is shown in Figure 12-16b. One particularly interesting example of an activating lectin-like NK receptor is Ly49H, a member of a second family of activating receptors, the Ly49 receptors, that are found in mice but not in humans. Ly49H binds an MHC class I—like protein, m157, encoded by the mouse virus murine cytomegalovirus (MCMV). Thus this activating receptor directly recognizes a pathogen-derived activating ligand. In the absence of this receptor, mice are poorly protected from this common virus.
Some receptors that are not limited to NK cells also can serve as activating receptors to enhance NK activity. Perhaps the most important is FcγRIII (CD16), which binds IgG antibodies associated with cell-surface antigens (including viral and tumor antigens). This binding provides key activating signals that trigger NK cytotoxicity, an important example of antibody-dependent cell-mediated cytotoxicity (ADCC) by the NK cell. As described earlier in this chapter, ADCC is a potent immune effector response to infection and malignancy. Cells infected with virus, for instance, often express viral envelope proteins on their surface. Antibodies produced by B cells that responded to these proteins will bind the cell surface and recruit NK cells, which will induce apoptosis of the infected cell.
Other activating receptors on NK cells include CD2 (the receptor for the adhesion molecule LFA-3) and receptors for inflammatory cytokines. The involvement of these proteins as activating receptors again makes biological sense, enabling NK cells to participate in clearance of infected or tumor cells.
How NK Cells Induce Apoptosis of Their Targets
Regardless of which receptors are involved in regulating NK-cell lytic activity, NK cells kill targets by processes similar to those employed by CTLs (see Table 12-3). Like CTLs, the cytoplasm of NK cells also has numerous granules containing perforin and granzymes. NK cells develop an organized immunological synapse at the site of contact with a target cell, after which degranulation occurs, with release of perforin and granzymes at the junction between the interacting cells. Perforin and granzymes are thought to play the same roles in NK cell—mediated induction of target cell apoptosis as they do in the CTL-mediated killing process. In addition, also similar to CTLs, NK cells express FasL and readily induce death of Fas-bearing target cells.
NK Cell Licensing and Regulation
Even newly formed NK cells have large granules in their cytoplasm, and it was traditionally thought that NK cells were capable of killing from the moment they matured. It is now thought that most NK cells need to be licensed before they can use their cytotoxic machinery on any target. NK cell licensing is thought to occur via a first engagement of their inhibitory, MHC class I—binding receptors. This event can be considered a way for the immune system to test an NK cell’s ability to restrain its killing activity when interacting with normal cells and an important strategy for maintaining NK-cell tolerance to self. Specifically, licensing allows only those cells that have the capacity to be disarmed via inhibitory interactions, usually from the recognition of self MHC class I proteins, to become armed and ready to kill.
Once licensed, NK cells are thought to continuously browse tissues and potential target cells via their multiple inhibitory and activating receptors. Engagement of activating ligands on the surface of a tumor cell, virus-infected cell, or otherwise stressed cell signals NK cells to kill the target cell. If the NK cells’ inhibitory receptors detect normal levels of MHC class I on potential target cells, these inhibitory signals override the activation signals. This would not only prevent the death of the target cell but would also abrogate NK-cell proliferation and the production of cytokines such as IFN-γ and TNF-α. The overall consequence of this strategy is to spare cells that express critical indicators of normal self, the MHC class I molecules, and to kill cells that lack indicators of self and/or also express high levels of activating ligands that indicate that they are infected, malignant, or dangerous in other ways.
NK-Cell Memory
The distinctions between NK cells and B and T lymphocytes continue to blur. Clearly, B and T lymphocytes are unique in their generation of clonally restricted antigen-specific receptors through V(D)J gene rearrangements. But B and T cells were also once considered the only cells in the immune system that could generate memory responses. However, recent data indicate that NK cells can also generate a memory response to antigen. The evidence for this important and unexpected property came from experiments showing that NK cells expressing a receptor that binds a viral protein could transfer memory of this antigen exposure to naïve animals that had not been previously exposed or infected (see Classic Experiment Box 12-3).
These observations show that at least some NK cells possess the developmental machinery to become memory cells, in other words to increase their number and longevity (although perhaps not as long as B and T lymphocytes) and thereby improve their responses over time. Recent studies have also documented the induction of memory NK cells in humans. For example, acute infection with human cytomegalovirus induces expansion and persistence of NKG2C+ NK cells that are activated not only by that virus but also by hantavirus. Evidence for NK memory has also been gathered for influenza, herpes simplex, and vaccinia viruses. This raises the exciting idea that it may be possible to immunize people to enhance their NK-cell memory against viruses and possibly even against certain tumors, potentially providing an expanded army of NK cells for early innate protection against infection or malignancy.
NKT Cells Bridge the Innate and Adaptive Immune Systems
Thus far, this chapter’s discussions of cell-mediated immunity covered the CTL, an important component of adaptive immunity that expresses an antigen-specific TCR; and the NK cell, a cell with both innate and adaptive properties that bears receptors that recognize inhibitory self ligands and activating ligands on altered cells. A third type of cytolytic lymphocyte has been identified with characteristics shared by both the CTL and the NK cell. This cell type, designated the NKT cell to reflect its hybrid quality, develops in the thymus, and, strictly speaking, is a member of the adaptive immune system. It undergoes antigen-receptor gene rearrangements and expresses an αβ TCR complex on its surface. However, it also exhibits characteristics of cells in the innate immune system:
- The T-cell receptor on many human NKT cells is invariant, with the TCR α and TCR β chains encoded by specific gene segments (Vα24-Jα18 and Vβ 11, respectively) within the germ-line DNA; the cells expressing this αβ TCR combination are therefore sometimes referred to as invariant NKT (iNKT) cells. Similar cells occur in mice.
- The TCR on NKT cells does not recognize MHC-bound peptides but rather glycolipid antigens presented by the nonpolymorphic MHC class I—related CD1 molecule, as described in Chapter 7 (Figure 7-19).
- NKT cells can act both as helper cells (secreting cytokines that shape responses) and as cytotoxic cells (killing target cells).
- NKT cells include both CD4+ and CD4− subpopulations, which may also differ in their cytokine production.
- NKT-cell killing appears to depend predominantly on FasL-Fas interactions.
- NKT cells do not form memory cells.
- NKT cells do not express a number of markers characteristic of T lymphocytes but do express multiple proteins characteristic of NK cells. For example, mouse NKT cells express the NK1.1 protein.
The exact role of NKT cells in immunity remains to be defined. However, experiments show that mice lacking NKT cells are deficient in their response to certain low-dose infections of bacteria that express glycolipids that can be recognized by NKT-cell receptors (e.g., Sphingomonas and Ehrlichia). Interestingly, high-dose Sphingomonas infection leads to sepsis and death in wild-type mice, but those lacking NKT cells survive this challenge, suggesting that the NKT cells may also contribute to pathology if they secrete excessive levels of inflammatory cytokines. (See Chapter 15 for a description of the role of proinflammatory cytokines in the onset of sepsis and the exacerbation of disease.)
Other data implicate NKT cells in immunity to tumors and suggest that NKT cells recognize lipid antigens specific to tumor cells. Finally, NKT cells also appear to contribute to viral immunity, despite the fact that viruses do not typically express glycolipids. NKT cells may play an indirect role in shaping the viral immune responses via the production of cytokines, including IFN-γ, IL-2, TNF-α, and IL-4.