Vaccines
Preventive vaccines have led to the control or elimination of many infectious diseases that once claimed millions of lives. Since October 1977, not a single naturally acquired smallpox case has been reported anywhere. On the heels of the global victory over smallpox, the program to eradicate polio went into overdrive. That campaign started in 1998 and, led largely by the WHO and several large philanthropic donors, has reduced the number of polio cases worldwide by over 99%. Worldwide vaccination campaigns can also be credited with the control of at least 10 other major infectious diseases (measles, mumps, rubella, typhoid, tetanus, diphtheria, pertussis, influenza, yellow fever, and rabies), many of which previously affected and killed many, mostly babies and young children.
Still, a need remains for vaccines against many other diseases, including malaria, tuberculosis, and AIDS, among others. More work is needed for existing vaccines as well: to improve the safety and efficacy of some, or to lower the cost and ensure delivery of existing vaccines to those most in need, especially in developing countries. Even today, based on WHO data, millions of infants still die of diseases that could be prevented by existing vaccines. The good news is that decades spent on basic research aimed at characterizing the mammalian immune system and the recent “omics” boom (genomics, transcriptomics, proteomics, and even immunomics) are bearing clinical fruit. We have entered a new age of “rational design” for drugs and vaccines, aimed at maximizing the impact on immune function.
In this section, we describe the most common vaccine strategies, some vaccines presently in use, and new avenues of research under development. Keep in mind: no one strategy, additive, or administration route is likely to work for all infectious agents, or even for all members of one type of pathogen, and many of these approaches can be applied in an à la carte fashion, depending on the situation.
Basic Research and Rational Design Advance Vaccine Development
Development of effective new vaccines is a long, complicated, and costly process, rarely even reaching the final stage of years-long clinical trials. Many vaccine candidates that were successful in laboratory and animal studies fail to prevent disease in humans, have unacceptable side effects, or worsen the disease they were meant to prevent. Stringent testing is an absolute necessity, because approved vaccines will be given to large numbers of healthy people. Clear information for consumers about adverse side effects (even those that occur at very low frequency), contraindications (in what situations the vaccine is ill-advised), and potential interactions with other drugs must be made available and carefully balanced with the potential benefit of protection by the vaccine.
Vaccine development begins with years of basic research. For example, characterization of the SARS virus would never have moved with such speed if not for decades of previous work understanding other, less pathogenic, coronaviruses. Along with rhinovirus, coronavirus is one of several causes for the coldlike symptoms we all experience from time to time. Intensive research such as this has led to an appreciation of the features of immunogens, epitopes on a pathogen that can be recognized by T and B cells. This has enabled immunologists to design vaccine candidates that maximize activation of key cellular and humoral elements that recognize these immunogens. In animal models, new adjuvants, or additives, are being tested as a means to maximize antigen presentation, overcome original antigenic sin, or to activate the most productive immune pathways. Newer targeting strategies to elicit protection at mucosal surfaces, the most common site of infection, are also underway.
No matter the approach, the first crucial step in the path to a new vaccine is to define specific immunologic targets. These targets are called correlates of immune protection, and they represent the specific immunologic goals or markers that scientists believe will result in protection (immunity) from infection or disease upon natural encounter with a pathogen. For instance, sometimes high circulating levels of IgG against a specific surface protein may be required to protect the host from infection, while other times mucosal IgA is more protective. To be immune, we may need activated macrophages to assist in the destruction of a vesicular infection, and not cytotoxic T cells looking to kill infected cells. In other words, we must first ask the following question: what specific memory response do we need to have on hand before we encounter the real pathogen in order to be protected? The aim of the latter phases of clinical trials is to determine this empirically; have we in fact immunized individuals against this infectious agent? Of course, we are more likely to hit our targets (often, decades later) if we actually aim at them in the first place!
Protective Immunity Can Be Achieved by Active or Passive Immunization
Immunization is the process of eliciting a state of protective immunity against a disease-causing pathogen. Exposure to the live pathogen followed by recovery is one route to immunization. However, while highly effective, this can also be dangerous. Vaccination, or intentional exposure to modified forms or parts of a pathogen that do not cause disease (a vaccine), is another. In an ideal world, both will engage antigen-specific lymphocytes and result in the generation of memory cells, providing long-lived protection. However, vaccination does not always ensure immunity. Thus, vaccination is an event, whereas immunization (the development of a protective memory response) is a potential outcome of that event.
A state of at least temporary immune protection can also be achieved by means other than infection or vaccination: for example, the transfer of antibodies from mother to fetus or the injection of antiserum against a pathogen or a toxin to provide immune protection (passive immunization). Without the development of memory B or T cells specific to the organism, however, this state of immunity is only temporary. In this section, we describe current use of immunization techniques, both passive and active.
Passive Immunization by Delivery of Preformed Antibody
Edward Jenner and Louis Pasteur are recognized as the pioneers of vaccination for their documented attempts to induce active immunity, although prior civilizations had employed similar protective strategies (see the chapter introduction). Recognition also is due to Emil von Behring and Kitasato Shibasaburō for their contributions to passive immunity. These latter two investigators were the first to show that immunity elicited in one animal can be transferred to another by injecting serum taken from the first.
Passive immunization, in which preformed antibodies are transferred to a recipient, occurs naturally when maternal IgG crosses the placenta to the developing fetus. Maternal antibodies to diphtheria, tetanus, streptococci, measles, mumps, and poliovirus all afford passively acquired protection to the developing fetus and for months in the newborn. Maternal antibodies present in breast milk can also provide passive immunity to the infant in the form of maternally produced IgA. The latter, however, enters the baby’s digestive tract and therefore has a different and complementary effect to maternal IgG circulating in the blood.
Passive immunization can also be achieved by injecting a recipient with preformed antibodies, called antiserum, from other immune individuals. Before vaccines and antibiotics became available, passive immunization was the only effective therapy for some otherwise fatal diseases, such as diphtheria, providing much needed humoral defense (see Chapter 1, Clinical Focus Box 1-2). Currently, several conditions still warrant the use of passive immunization, including the following:
- Immune deficiency, especially congenital or acquired B-cell defects
- Toxin or venom exposure with immediate threat to life
- Exposure to pathogens that can cause death faster than an effective immune response can develop
Babies born with congenital immune deficiencies are frequently treated by passive immunization, as are children experiencing acute respiratory failure caused by respiratory syncytial virus (RSV). Passive immunity is used in unvaccinated individuals exposed to the organisms that cause botulism, tetanus, diphtheria, hepatitis, measles, and rabies (Table 17-4), or to protect travelers and health care workers who anticipate or experience exposure to pathogens for which they lack protective immunity. Antiserum also provides an antidote against the poisonous venom in some snake and insect bites. In all these instances, it is important to remember that passive immunization does not activate the host’s natural immune response. It serves as a buffer between the pathogen, or a toxin, and the host but generates no memory response, so protection is transient.
| Disease | Agent |
| Black widow spider bite | Horse antivenin |
| Botulism | Horse antitoxin |
| Cytomegalovirus | Human polyclonal Ab |
| Diphtheria | Horse antitoxin |
| Hepatitis A and B | Pooled human immunoglobulin |
| Measles | Pooled human immunoglobulin |
| Rabies | Human or horse polyclonal Ab |
| Respiratory disease | Monoclonal anti-RSV* |
| Snake bite | Horse antivenin |
| Tetanus | Pooled human immunoglobulin or horse antitoxin |
| Varicella zoster virus | Human polyclonal Ab |
Although passive immunization may be effective, it should be used with caution because certain risks are associated with the injection of preformed antibody. If the antibody was produced in another species, such as a horse (one of the most common animal sources), the recipient can mount a strong response to the isotypic determinants of the foreign antibody, or the parts of the antibody that are unique to the horse species (typically constant-region domains). This anti-isotype response can cause serious complications. Some individuals will produce IgE antibody against horse-specific determinants. High levels of these IgE–horse antibody immune complexes can induce pervasive mast-cell degranulation, leading to systemic anaphylaxis (see Chapter 15). Other individuals produce IgG or IgM antibodies specific for the foreign antibody, resulting in complement-activating immune complexes. The deposition of these complexes in the tissues can lead to type III hypersensitivity reactions. Even when purified human antiserum or human gammaglobulin is used (a mixture of IgG from many different human B cells), the recipient can generate an anti-allotype response. This recognition of foreign human immunoglobulin (within-species antigenic differences) can cause some of the same symptoms, although its intensity is usually much less than that of an anti-isotype response.
Active Immunization to Induce Immunity and Memory
The goal of active immunization is to trigger the adaptive immune response in a way that will elicit protective immunity and long-lived immunologic memory. When active immunization is successful, a subsequent exposure to the infectious agent elicits a secondary immune response that successfully eliminates the pathogen or prevents disease mediated by its products. Active immunization can be achieved by natural exposure to the infectious agent or a similar agent (e.g., cowpox exposure can protect against smallpox) or it can be acquired artificially by administration of a vaccine. An example of the former might be the “chickenpox parties” of the past, where parents invited unprotected children to come play with their pox-ridden youngster as a means to generate a natural immune response early in life. Chickenpox in adults can be more serious with more complications, so immunization while young is advisable. In active immunity, as the name implies, the immune system plays an active role—proliferation of antigen-reactive T and B cells is induced and results in the formation of protective memory cells. This is the primary goal of vaccination.
Vaccination programs have played an important role in the reduction of deaths from infectious diseases, especially among children. In the United States, vaccination of children begins at birth. The American Academy of Pediatrics sets nationwide recommendations (updated in 2017) for childhood immunizations in this country, as outlined in Table 17-5. The program recommends or requires 10 vaccines for children from birth to age 6.

|
In adolescents between the age of 11 and 12, vaccination against the sexually transmitted human papillomavirus (HPV), the primary cause of cervical cancer in women, is also recommended (see Clinical Focus Box 19-1). Vaccines against meningitis, as well as boosters for tetanus and influenza, are recommended for all on a regular schedule throughout adulthood (for the latest, see https://www.cdc.gov/vaccines/schedules/).
As illustrated in Table 17-5, children typically require boosters (repeated vaccinations or inoculations) at appropriately timed intervals to achieve protective immunity against many of the common pathogens. In the first months of life, the reason for this may be persistence of circulating maternal antibodies in the young infant. For example, passively acquired maternal antibodies can bind to epitopes on the DTaP vaccine and block adequate activation of the immune system; therefore, in order to achieve protective immunity this vaccine must be given more than once after we expect all maternal antibody has been cleared from an infant’s circulation (6–12 months). Passively acquired maternal antibody is also known to interfere with the effectiveness of the measles vaccine; for this reason, the combined measles/mumps/rubella (MMR) vaccine is not given before 12 months of age. However, in some developing countries, the measles vaccine is administered at 9 months, a little early to ensure clearance of all maternal antibodies but crucial because 30% to 50% of young children in these countries contract the disease before 15 months of age. Multiple immunizations with the polio vaccine are required to ensure that an adequate immune response is generated to each of the strains of poliovirus that make up the vaccine.
The widespread use of vaccines for common, life-threatening diseases in the United States has led to a dramatic decrease in the incidence of these diseases. As long as these immunization programs are maintained, especially in young children, the incidence of these diseases typically remains very low. However, the occurrence or even the suggestion of possible side effects to a vaccine, as well as general trends toward reduced vaccination in children, can cause a drop in vaccination rates that leads to re-emergence of the disease. For example, rare but significant side effects from the original pertussis attenuated bacterial vaccine included seizures, encephalitis, brain damage, and even death. Decreased usage of the vaccine led to an increase in the incidence of whooping cough. In 1991 and on the heels of this, an acellular pertussis vaccine (the aP in DTaP) was introduced; it was equally effective but with fewer side effects.
It is important to remember that vaccination is not 100% effective. With any vaccine, a small percentage of recipients may not respond and therefore remain (unknowingly) susceptible. In other cases, individuals may have to forgo or delay vaccination thanks to other health issues, including immune deficiency. This is not usually a serious problem if the majority of the population is immune to an infectious agent, reducing the pathogen reservoir and therefore chances of spread. In this case, the chance of a susceptible individual contacting an infected individual is very low. This phenomenon is known as herd immunity. The appearance of some recent measles epidemics, such as among college students or preschool-age children, is a testament to the power of herd immunity and likely occurred partly thanks to decreased vaccination rates in these populations. One recent example is a measles outbreak centered around an amusement park in California during the 2014-15 holiday season, highlighting the potential fallout from rising numbers of unvaccinated individuals and falling levels of herd immunity. Crowding, international travel, and reduced vaccination rates contributed to over 50 reported cases of measles amongst those who visited the park or came in contact with these individuals during this time, and may have contributed to the first U.S. death from measles in 12 years. The year 2014 saw more than three times the number of measles cases than in previous years. And with almost 50,000 cases in 2012, you would need to go all the way back to 1955 to find a worse year for pertussis (whooping cough) infections (Figure 17-9). In addition to overall sickness and hospitalization, this led to 20 deaths in U.S. children. Of note, fatality from whooping cough occurs predominantly in infants under 3 months of age, when they are either too young to receive the pertussis vaccine or before protective immunity kicks in, making them highly susceptible to transfer of an otherwise mild case from unvaccinated individuals. Despite the safety record of this vaccine and the frightening rise in this potentially deadly disease, some parents still take their chances and elect not to vaccinate their children (see Chapter 1, Clinical Focus Box 1-1).
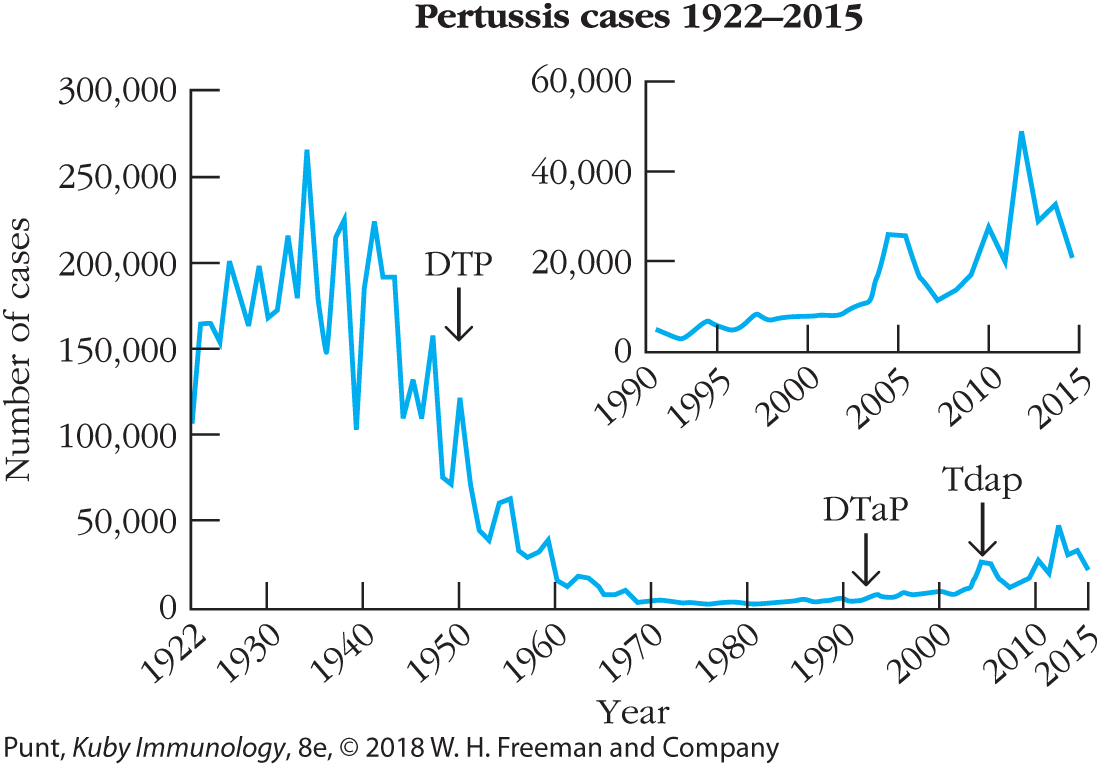
FIGURE 17-9 Introduction of the pertussis vaccine in 1950 led to a dramatic decrease in the annual incidence of this disease in the United States: pertussis cases from 1922 through 2015. Despite huge reductions in outbreaks of whooping cough (caused by pertussis) since introduction of the vaccine, the past decade (inset) has seen several outbreaks. Most are linked to travelers from other countries, where disease spreads to unvaccinated individuals, mostly young children. While many immune-competent individuals above age 1 can resolve this infection, the spread to immune-compromised individuals and babies under 6 months of age can be life-threatening and has resulted in several deaths. [Data from Centers for Disease Control and Prevention.]
Recommendations for vaccination of adults depend on the risk group. Vaccines for influenza are recommended yearly, while those that protect against meningitis and pneumonia are offered to groups living in close quarters (e.g., military recruits and incoming college students) or those with reduced immunity (e.g., the elderly). Depending on their destination, international travelers are also routinely immunized against diseases endemic to their destination, such as cholera, yellow fever, rabies and typhoid. Immunization against the deadly disease anthrax had been reserved for workers coming into close contact with infected animals or animal products. Concerns about the potential use of anthrax spores by terrorists or in biological warfare has widened use of the vaccine to military personnel and civilians in areas believed to be at risk.
There Are Several Vaccine Strategies, Each with Unique Advantages and Challenges
Three key factors must be kept in mind in the development of a successful vaccine: the vaccine must be safe, it must be effective in preventing infection, and the strategy should be reasonably achievable given the population in question. Population considerations can include geographical locale, access to the target group (which may require several vaccinations), complicating co-infections or nutritional states, and of course, cost. Critical for success is the branch or branches of the immune system that are activated, and therefore vaccine designers must target specific humoral and/or cell-mediated pathways. Protection must also reach the relevant site of infection. In some cases, mucosal surfaces are the prime candidates. An additional factor is the development of long-term immunologic memory. For example, a vaccine that induces a protective primary response may fail to induce the formation of memory cells, leaving the host unprotected after the primary response subsides. Some vaccines generate long-term memory and others, like tetanus, require regular reminders in the form of booster shots or memory responses will wane.
Before vaccines can progress from the laboratory bench to the bedside, they must go through rigorous testing in animals and humans. Most vaccines in development never progress beyond animal testing. The type of testing depends on what animal model systems are available, but frequently involves rodents and/or nonhuman primates. When these animal studies prove fruitful, follow-up clinical trials in humans can be initiated. Phase I clinical trials assess human safety; a small number of volunteers are monitored closely for adverse side effects. Only once this hurdle has been successfully passed can a trial move on to phase II, where effectiveness against the pathogen in question is evaluated. In phase II trials, volunteers are tested for measurable immune response to the immunogen. However, even when volunteers test positive for one or more of the targeted correlates of immune protection, it does not necessarily mean that a state of protective immunity has been achieved or that long-term memory is established. Again, many vaccines fail at this stage. Phase III clinical trials are run in expanded volunteer populations, where natural evidence of protection against “the real thing” is the desired outcome, and where safety, the evaluation of several measurable immune markers, and the incidence of wild-type infections with the relevant pathogen are all monitored carefully over time. Finally, phase IV trials occur after marketing and distribution, and are used to monitor safety, effectiveness, and any long-term impacts.
In some cases circulating effector cells/molecules, in addition to memory cells, are required in order to establish protection. This can depend on the incubation period of the pathogen. For influenza virus, which has a very short incubation period (1 or 2 days), disease symptoms are normally already underway by the time memory cells would be reactivated. Effective protection against disease from influenza therefore depends on maintaining high levels of neutralizing antibody via regular immunizations; those at highest risk are immunized each year, usually at the start of the flu season. For pathogens with a longer incubation period the presence of detectable neutralizing antibody at the time of infection is not always necessary. The poliovirus, for example, requires more than 3 days to begin to infect the central nervous system. An incubation period of this length gives the memory B cells time to respond by producing high levels of serum antibody. Thus, the vaccine for polio is designed to induce high levels of protective immunologic memory that can be recalled and reactivated once the virus is encountered. After immunization with the Salk vaccine (an inactivated form of polio), serum antibody levels peak within 2 weeks and then decline. However, the memory cell response continues to climb, reaching maximal levels 6 months postvaccination and persisting for many years. If an immunized individual is later exposed to the poliovirus, these memory cells will respond by differentiating into plasma cells that produce high levels of serum antibody, which defend the individual from the effects of the virus, even if they don’t block the virus from entering the body. In other words, sterilizing immunity, or the presence of immune effectors that can block infection, is not always required in order to thwart disease: poliomyelitis in this case.
In the remainder of this section, various approaches to the design of vaccines—both currently used vaccines and experimental ones—are described, with an examination of the ability of the vaccines to induce humoral and cell-mediated immunity and memory cells. As Table 17-6 indicates, the common vaccines currently in use consist of live but attenuated organisms, inactivated (killed) bacterial cells or viral particles, as well as protein or carbohydrate fragments (subunits) of the target organism. In addition, the recent Ebola vaccine relies on recombinant vector technology. The primary characteristics of each type, as well as some advantages and disadvantages, are also included.
| Vaccine type | Diseases | Advantages | Disadvantages |
| WHOLE ORGANISMS | |||
| Live attenuated | Measles Mumps Polio (Sabin vaccine) Rotavirus Rubella Tuberculosis Varicella Yellow fever |
Strong immune response; often lifelong immunity with few doses | Requires refrigerated storage; may mutate to virulent form |
| Inactivated or killed | Cholera Influenza Hepatitis A Plague Polio (Salk vaccine) Rabies Zika |
Stable; safer than live vaccines; refrigerated storage not required | Weaker immune response than live vaccines; booster shots usually required |
| PURIFIED MACROMOLECULES | |||
| Toxoid (inactivated exotoxin) | Diphtheria Tetanus |
Immune system becomes primed to recognize bacterial toxins | May require booster shots |
| Subunit | Hepatitis B Pertussis Streptococcal pneumonia |
Specific antigens lower the chance of adverse reactions | Difficult to develop |
| Conjugate | Haemophilus influenzae type b Streptococcal pneumonia |
Primes infant immune systems to recognize certain bacteria | |
| OTHER | |||
| Recombinant vector | Ebola (in clinical testing) | Mimics natural infection, resulting in strong immune response | Too early to tell |
| DNA | HPV (in clinical testing) Zika virus (in clinical testing) |
Strong humoral and cellular immune response; relatively inexpensive to manufacture | Not yet available |
Live Attenuated Vaccines
In terms of a matched exposure to the real thing, live attenuated vaccines produce the most robust response, although not without risk. For these vaccines, microorganisms are attenuated (disabled) so that they lose their ability to cause significant pathogenicity (disease) but retain their capacity for slow and transient growth within an inoculated host. This allows the immune system a taste of the real thing, but also the upper hand against a pathogen-like organism with only temporary residency. Some agents are naturally attenuated by virtue of their inability to cause disease in a given host, even while having the ability to immunize. The first vaccine used by Jenner is of this type: vaccinia virus (cowpox) inoculation of humans confers immunity to smallpox but does not cause smallpox.
Attenuation can often be achieved in the laboratory by growing a pathogenic bacterium or virus for prolonged periods under abnormal culture conditions. This selects mutants that are better suited for growth in the abnormal culture conditions than in the natural host. For example, an attenuated strain of Mycobacterium bovis, called bacillus Calmette-Guérin (BCG), was developed by growing M. bovis on a medium containing increasing concentrations of bile. After 13 years, this strain had adapted to growth in strong bile and had become sufficiently attenuated that it was suitable as a vaccine for tuberculosis. Because of variable effectiveness, relatively low prevalence, and difficulties in follow-up monitoring, BCG is not used in the United States, although it is commonly used in countries where tuberculosis is pervasive. Likewise, the Sabin form of the polio vaccine and the measles vaccine both consist of attenuated viral strains. Finally, a new weakened form of Plasmodium falciparum is being used as a vaccine for malaria (called PfSPZ). In clinical trials conducted in Mali, Africa aimed at testing this vaccine during high transmission periods, only 66% of vaccinees contracted malaria after five doses of the vaccine, compared with 93% of control subjects: a modest but significant improvement.
Attenuated vaccines have obvious advantages. Because of their capacity for growth, even transient growth, such vaccines provide prolonged immune system exposure to the epitopes (immunogens) on the attenuated organism and more closely mimic the growth patterns of the “real” pathogen. This often results in increased immunogenicity and more efficient production of highly effective memory cells. Thus, these vaccines often require only a single immunization, a major advantage in developing countries, where studies show that a significant number of individuals fail to return for boosters. This ability of attenuated vaccines to replicate within host cells thus makes them particularly suitable for inducing cell-mediated responses.
One good example of a live attenuated vaccine that has been in use for decades worldwide is the oral polio vaccine (OPV) designed by Albert Sabin. In its original form, OPV consists of three attenuated strains of poliovirus and is administered orally to children in regions where risk of polio is still relatively high. The attenuated viruses colonize the intestine and induce production of secretory IgA, an important defense against naturally acquired poliovirus. The vaccine also induces IgM and IgG classes of antibody and ultimately protective immunity to all three strains of virulent poliovirus. Unlike most other attenuated vaccines, OPV requires boosters, because the three strains of attenuated poliovirus can interfere with each other’s replication in the intestine (although there are now also mono- and divalent forms of OPV without this drawback). With the first immunization, one strain will predominate, inducing immunity to that strain. With the second immunization, the immunity generated by the previous immunization will limit the growth of the previously predominant strain in the vaccine, enabling one of the two remaining strains to colonize the intestines and induce immunity. Finally, with the third immunization, immunity to all three strains is typically achieved.
Despite these advantages, the major disadvantage of attenuated vaccines is that these live forms can sometimes mutate and revert to a more virulent form in the host—a major drawback. In the case of polio, this can therefore risk paralytic disease in the vaccinated individual, or in unprotected individuals who come in contact with these more virulent forms shed in feces. The rate of reversion of the OPV to a virulent form is extremely low: about 1 case in 2.4 million doses of vaccine. This is arguably considered an acceptable risk in areas where the danger from wild-type polio is high, so risk of paralysis is high, but maybe not in regions where the threat is minimal. This reversion can also allow pathogenic forms of the virus to find their way into the water supply and spread through a community, especially in areas where sanitation is not rigorous or wastewater is recycled. Despite less ideal immune protection, this possibility has led to the exclusive use of the safer but less effective inactivated polio vaccine in the United States since 2000 (see Table 17-5).
Attenuated vaccines also may be associated with complications similar to those seen in the natural disease. A small percentage of recipients of the measles vaccine, for example, develop postvaccination encephalitis or other complications, although the risk of vaccine-related complications is still significantly lower than risks from infection. An independent study showed that 75 million doses of measles vaccine were given between 1970 and 1993, with 48 cases of vaccine-related encephalopathy (approximately 1 per 1.5 million). This low incidence compared with the rate of infection-induced encephalopathy argues for the efficacy of the vaccine. An even more convincing argument for vaccination is the high death rate from measles—10% or more in regions where nutrition and healthcare are inadequate.
In addition to culturing methods, genetic engineering provides a means to attenuate a virus irreversibly, by selectively removing genes that are necessary for virulence or for growth in the host. This has been done with a herpesvirus vaccine for pigs, in which the thymidine kinase gene was removed. Because thymidine kinase is required for the virus to grow in certain types of cells (e.g., neurons), removal of this gene rendered the virus incapable of causing disease. A live attenuated vaccine against influenza has been developed under the name FluMist. For this, the virus was grown at lower-than-normal temperatures until a cold-adapted strain, unable to grow at human body temperature of 37°C, arose. This live attenuated virus can be administered intranasally and causes a transient infection in the upper respiratory tract, sufficient to induce a strong immune response. The virus cannot spread beyond the upper respiratory tract because of its inability to grow at the elevated temperatures of the inner body. Immunologically, this approach should provide better protection. However, changes in the formulation (going from a trivalent to a quadrivalent formula) and some resulting reports of weaker than expected responses in the United States have led to a pullback on the use of this approach, despite its continued use in the UK and Canada.
Inactivated or “Killed” Vaccines
Another common means to make a pathogen safe for use in a vaccine is by treatment with heat or chemicals. This kills the pathogen, making it incapable of replication, but still allows it to induce an immune response to at least some of the immunogens (antigens) contained within the organism. It is critically important to maintain the structure of key epitopes on surface antigens during inactivation. Heat inactivation is often unsatisfactory because it causes extensive denaturation of proteins; thus, any epitopes that depend on higher orders of protein structure are likely to be altered significantly. Chemical inactivation with formaldehyde or various alkylating agents has been more successful. The Salk inactivated polio vaccine (IPV) is produced by formaldehyde treatment of the poliovirus.
Although live attenuated vaccines generally require only one dose to induce long-lasting immunity, killed vaccines often require repeated boosters to achieve a protective immune status. Because they do not replicate in the host, killed vaccines typically induce a predominantly humoral/antibody response and are less effective than attenuated vaccines in inducing cell-mediated immunity or in eliciting a secretory IgA response, key components of many ideal protective and mucosally based responses.
Even though the pathogens they contain are killed, inactivated whole-organism vaccines still carry certain risks. A serious complication with the early Salk vaccines arose when proper inactivation procedures were not followed by some manufacturers and some of the virus in two vaccine lots were not killed, leading to paralytic polio in a significant percentage of the recipients. This led to temporary suspension of the vaccine, plus changes in all subsequent inactivation and quality control procedures for inactivated vaccines. Risk is also encountered by those who grow and inactivate the virus during vaccine manufacturing. Large quantities of the infectious agent must be handled prior to inactivation, and those exposed to the process are at risk of infection. However, in general, the safety of inactivated vaccines is greater than that of live attenuated vaccines. Inactivated vaccines are commonly used against both viral and bacterial diseases, including the classic yearly flu vaccine and vaccines for hepatitis A and cholera. In addition to their relative safety, their advantages also include stability, and ease of storage and transport.
Subunit Vaccines
Many of the risks associated with attenuated or killed whole-organism vaccines can be avoided with a strategy that uses only specific, purified macromolecules derived from the pathogen, otherwise known as a subunit approach. The three most common applications of this strategy are inactivated pathogen exotoxins (called toxoids), isolated capsular polysaccharides or surface glycoproteins, and purified key recombinant protein antigens (see Table 17-6). One limitation of some subunit vaccines, especially polysaccharide vaccines, is their inability to activate TH cells. Instead, they typically activate B cells in a thymus-independent type 2 (TI-2) manner, resulting in IgM production but little class switching, no affinity maturation, and little, if any, development of memory cells. This can be avoided in vaccines that conjugate a polysaccharide antigen to a protein carrier, which induces TH-cell responses against both the protein and polysaccharide, discussed below.
Some bacterial pathogens, including those that cause diphtheria and tetanus, produce exotoxins that account for all or most of the disease symptoms resulting from infection. Diphtheria and tetanus vaccines have been made by purifying the bacterial exotoxin and then inactivating it with formaldehyde to form a toxoid. Vaccination with the toxoid induces anti-toxoid antibodies, which are capable of binding to the toxin and neutralizing its effects. Conditions for the production of toxoid vaccines must be closely controlled and balanced to avoid excessive modification of the epitope structure while also accomplishing complete detoxification. As discussed previously, passive immunity can also be used to provide temporary protection in unvaccinated individuals exposed to organisms that produce these exotoxins, although no long-term protection is achieved in this case.
The virulence of some pathogenic bacteria depends primarily on the antiphagocytic properties of their hydrophilic polysaccharide capsule. Coating the capsule with antibodies and/or complement greatly increases the ability of macrophages and neutrophils to phagocytose such pathogens. These findings provide the rationale for vaccines consisting of purified capsular polysaccharides. The current vaccine for Streptococcus pneumoniae (the organism that causes pneumococcal pneumonia) consists of 13 antigenically distinct capsular polysaccharides (PCV13). The vaccine induces formation of opsonizing antibodies and is now on the list of vaccines recommended for all infants (see Table 17-5). The vaccine for Neisseria meningitidis, a common cause of bacterial meningitis, also consists of purified capsular polysaccharides. Surface glycoproteins on pathogens can be more difficult to detect (e.g., HIV envelope glycoprotein), and some have failed as vaccine candidates. However, a vaccine based on glycoprotein-D from HSV-2 has recently been shown to prevent genital herpes, suggesting that this may be a viable approach for future antiviral vaccines.
Theoretically, the gene encoding any immunogenic protein can be cloned and expressed in cultured cells using recombinant DNA technology, and this technique has been applied widely in the design of many types of subunit vaccines. For example, the safest way to produce sufficient quantities of the purified toxins that go into the generation of toxoid vaccines involves cloning the exotoxin genes from pathogenic organisms into easily cultured cells. A number of genes encoding surface antigens from viral, bacterial, and protozoan pathogens have also been successfully cloned into cellular expression systems for use in vaccine development. The first such recombinant antigen vaccine approved for human use is the hepatitis B vaccine, developed by cloning the gene for the major hepatitis B surface antigen (HBsAg) and expressing it in yeast cells. The recombinant yeast cells are grown in large fermenters, allowing HBsAg to accumulate in the cells. The yeast cells are harvested and disrupted, releasing the recombinant HBsAg, which is then purified by conventional biochemical techniques. Recombinant hepatitis B vaccine induces the production of protective antibodies and holds much worldwide promise for protecting against this human pathogen.
Recombinant Vector Vaccines
Recall that live attenuated vaccines prolong immunogen delivery and encourage cell-mediated responses, but have the disadvantage that they can sometimes revert to pathogenic forms. Recombinant vectors maintain the advantages of the live attenuated vaccine approach while avoiding this major disadvantage of reversion. Individual genes that encode key antigens of especially virulent pathogens can be introduced into safe attenuated viruses or bacteria that are used as live carriers. The attenuated organism serves as a vector, replicating within the vaccinated host and expressing the individual gene product/s it carries from the pathogen. Since most of the genome of the pathogen is missing, reversion potential is virtually eliminated. Recombinant vector vaccines have been prepared utilizing existing licensed live attenuated vaccines and adding to them genes encoding antigens present on newly emerging pathogens. These part old–part new chimeric virus vaccines often move more quickly through testing and approval than an entirely new product. A recent example of this is the yellow fever vaccine that was engineered to express antigens from West Nile virus. A number of organisms are under investigation as vectors in such preparations, including vaccinia virus, the canarypox virus, attenuated poliovirus, adenoviruses, attenuated strains of Salmonella, the BCG strain of Mycobacterium bovis, and certain strains of Streptococcus that normally exist in the oral cavity.
Vaccinia virus, the attenuated vaccine used to eradicate smallpox, has been widely employed as a vector for the design of new vaccines. This large, complex virus, with a genome of about 200 genes, can be engineered to carry several dozen foreign genes without impairing its capacity to infect host cells and replicate. The procedure for producing a vaccinia vector that carries a foreign gene from another pathogen is outlined in Figure 17-10. The genetically engineered vaccinia expresses high levels of the inserted gene product, which can then serve as a potent immunogen in an inoculated host. Like the smallpox vaccine, genetically engineered vaccinia vector vaccines can be administered simply by scratching the skin, causing a localized infection in host cells. If the foreign gene product expressed by the vaccinia vector is a viral envelope protein, it is inserted into the membrane of the infected host cell, inducing development of both cell-mediated and antibody-mediated immunity.

FIGURE 17-10 Production of vaccine, using a recombinant vaccinia vector. Top: The gene that encodes the desired antigen (orange) is inserted into a plasmid vector adjacent to a vaccinia promoter (pink) and flanked on either side by the vaccinia thymidine kinase (TK) gene (green). Bottom: When tissue culture cells are incubated simultaneously with vaccinia virus and the recombinant plasmid, DNA encoding the antigen is inserted into the vaccinia virus genome by homologous recombination at the site of the nonessential TK gene, resulting in a TK− recombinant virus. Cells containing the recombinant vaccinia virus are selected by addition of bromodeoxyuridine (BrdU), which kills TK+ cells.
This strategy, using the vesicular stomatitis virus (VSV) as a vaccine vector, was given a test run in 2015 during the West African Ebola outbreak. A gene for the Ebola virus surface protein was inserted into VSV, a harmless virus that infects humans and cattle. This vaccine, called rVSV-ZEBOV, was still in the early stages of development. Initial tests showed it was protective in animals and elicited an immune response in humans. The in-development vaccine was quickly marshaled for use at the tail end of the 2013-2016 West African outbreak, using a ring vaccination approach like those used successfully during the smallpox eradication campaign. To do this, individuals who came in contact with a patient with Ebola, plus their contacts, were identified and designated as one cluster, or a ring. Some ring groups were offered the experimental Ebola vaccine immediately, and others, 3 weeks after identification. It quickly became apparent that individuals in the immediate vaccination rings were protected, leading to vaccine being offered to all contacts. In an illustration of the principle of herd immunity, even contacts within a vaccine cluster that did not receive the vaccine appeared to be more protected from infection by Ebola. Final estimates from this ad hoc trial of rVSV-ZEBOV are that the vaccine is 75–100% effective. Because of the circumstances, no immune response studies were conducted in those who received the vaccine, although follow-up immunologic studies are under way in vaccinees.
Other attenuated vectors may also prove useful in vaccine preparations. A relative of vaccinia, the canarypox virus, is also large and easily engineered to carry multiple genes. Unlike vaccinia, it does not appear to be virulent, even in individuals with severe immune suppression. Another possible vector is an attenuated strain of the bacterium Salmonella typhimurium, which has been engineered with genes from the bacterium that causes cholera. The advantage of this vector is that Salmonella infects cells of the mucosal lining of the gut and therefore will induce secretory IgA production. Similar strategies are underway for organisms that enter via oral or respiratory routes, targeting bacteria that are normal flora at these sites as vectors for the addition of pathogen-specific genes. Eliciting immunity at the mucosal surface could provide excellent protection at the portal of entry for many common infectious agents, such as cholera and gonorrhea (see Advances Box 17-6).
DNA Vaccines
DNA vaccines are based on plasmid DNA encoding antigenic proteins; the plasmid DNA is injected directly into the muscle of the recipient. This strategy relies on the host cells to take up the DNA and produce the immunogenic protein in vivo, thus directing the antigen through endogenous MHC class I presentation pathways, theoretically helping to activate better CTL responses. The DNA appears either to integrate into host chromosomal DNA or to be maintained for transient periods of time in an episomal form. The hope is to deliver the DNA plasmid to antigen-presenting cells (APCs) such as dendritic cells in or near the injection area. Since muscle cells express low levels of MHC class I molecules and do not express costimulatory molecules, direct or indirect delivery to local APCs is crucial to the development of antigenic responses to these vaccines. Tests in animal models have shown that DNA vaccines are able to induce protective immunity against a number of pathogens, including influenza and rabies viruses. The addition of a follow-up booster shot with protein antigen (in combination, called a DNA prime and protein boost strategy), or inclusion of supplementary DNA sequences in the vector (sometimes called genetic adjuvants; see the Adjuvants section, below), may enhance the immune response to DNA vaccines. One sequence that has been added to some vaccines is the CpG DNA motif commonly found in many pathogens; recall that this sequence is the ligand for TLR9 (see Chapter 4).
DNA vaccines offer some potential advantages over many of the existing vaccine approaches. Since the encoded protein is expressed in the host in its natural form—there is no denaturation or modification—the immune response is directed to the antigen in a three-dimensional structure similar to that seen in the pathogen, inducing both humoral and cell-mediated immunity. Strong stimulation of both arms of the adaptive immune response typically requires immunization with a live attenuated or recombinant vector preparation, which incurs additional risk. DNA vaccines should also theoretically induce prolonged expression of the antigen, enhancing the induction of immunological memory. While in practice this has not proven true in most settings, newer technology has begun to chip away at this hurdle, producing expression in animals that can now last for months.
DNA vaccines present some practical advantages. No refrigeration of the plasmid DNA is required, eliminating long-term storage challenges. In addition, the same plasmid vector can be custom tailored to insert DNA encoding a variety of proteins, which allows the simultaneous manufacture of a variety of DNA vaccines for different pathogens, saving time and money. Methods for administering DNA vaccines include coating microscopic gold beads with the plasmid DNA and delivery via gene gun, electroporation, mucosal administration via DNA-containing liposomes, or bacterial delivery of DNA. To date, all have advantages and disadvantages, with no prevailing method that appears applicable to humans.
It has now been almost 20 years since the principle behind the use of DNA as a vaccine was proven viable. Those intervening two decades have generated many advances and multiple aborted trials but few overall successes at the end of the day. Initially, safety concerns were the primary hurdle to greater implementation, although more recently variable delivery doses, transient gene expression, and poor immunogenicity have hampered application of this strategy.
As of March 2018, there are still no DNA vaccines licensed for use in humans, although one such vaccine has been approved for veterinary use: a West Nile virus vaccine in horses. This vaccine has also been tested in humans, and after three doses, most volunteers demonstrated titers of neutralizing antibody similar to those seen in horses, as well as CD8+ and CD4+ T-cell responses against the virus. Currently, the most promising and farthest along of the human trials of a DNA vaccine are actually for the treatment of cancer (see Chapter 19); this is likely the first place we will see the licensing of this technology for human use. Since the widespread development of DNA vaccines in humans has been disappointing, the usefulness of this vaccination strategy to protect us against infectious disease is still largely unknown.
Adding a Conjugate or Multivalent Component Can Improve Vaccine Immunogenicity
One of the major drawbacks to the vaccine techniques that do not utilize a live component is that they typically induce weak immune responses due to poor immunogenicity. To address this, schemes have been developed that employ the fusing of a highly immunogenic protein (a conjugate) to these weak vaccine immunogens. Alternatively, extraneous proteins associated with strong immune activation can be added to the vaccine (multivalent) to enhance or supplement immune reactivity against a weakly immunogenic pathogen-associated antigen.
One example is the vaccine against Haemophilus influenzae type b (Hib), a major cause of bacterial meningitis and infection-induced deafness in children. A conjugate formulation of a Hib vaccine is included in the recommended childhood regimen (see Table 17-5). This consists of type b capsular polysaccharide covalently linked to a strongly immunogenic protein carrier, tetanus toxoid (Figure 17-11). Introduction of the conjugate Hib vaccines has resulted in a rapid decline in Hib cases in the United States and other countries that have introduced this vaccine. The polysaccharide-protein conjugate is considerably more immunogenic than the polysaccharide alone; and because it activates TH cells, it enables class switching from IgM to IgG. Although this type of vaccine can induce memory B cells, it cannot induce memory T cells specific for the pathogen. In the case of the Hib vaccine, it appears that the memory B cells can be activated to some degree in the absence of a population of memory TH cells, thus accounting for its efficacy.

FIGURE 17-11 A conjugate vaccine protects against Haemophilus influenzae type b (Hib). The vaccine is prepared by conjugating the surface polysaccharide of Hib (non-immunogenic) to a protein molecule such as tetanus toxoid (highly immunogenic), making the vaccine more immunogenic than either alone
A separate study used a similar technique to protect against fungal infection. Immunization with β-glucan isolated from brown alga was conjugated to diphtheria toxoid, and in animal models this vaccine induced antibodies in mice and rats that protected these animals against challenge with both Aspergillus fumigatus and Candida albicans. The protection was transferred by serum or vaginal fluid from the immunized animals, indicating that the immunity is antibody based. Infections with fungal pathogens are a serious problem for immunocompromised individuals. The availability of immunization or antibody treatment could circumvent problems with toxicity of antifungal drugs and the emergence of resistant strains, an issue that is especially important in hospital settings.
Since subunit polysaccharide or protein vaccines tend to induce humoral but not cell-mediated responses, a method is needed for constructing vaccines that contain both immunodominant B-cell and T-cell epitopes. Furthermore, if a CTL response is desired, the vaccine must be delivered intracellularly so that the peptides can be processed and presented via MHC class I molecules. One innovative means of producing a multivalent vaccine that can deliver many copies of the antigen into cells is to incorporate antigens (or DNA) into lipid vesicles called liposomes, or immunostimulating complexes (Figure 17-12a). Likewise, virus envelopes, called virosomes, can serve as a similar delivery vehicle. Protein-containing liposomes are prepared by mixing the proteins with a suspension of phospholipids under conditions that form lipid bilayer vesicles; the proteins are incorporated into the bilayer with the hydrophilic residues exposed. Immunostimulating complexes (ISCOMs) are phospholipid monolayers which likewise carry antigenic proteins. Membrane proteins from various pathogens, including influenza virus, measles virus, hepatitis B virus, and HIV, have been incorporated into liposomes and ISCOMs, and are being assessed as potential vaccines. In addition to their increased immunogenicity, liposomes and ISCOMs appear to fuse with the plasma membrane to deliver their antigens intracellularly, where it can be processed by the endogenous pathway, leading to CTL responses (Figure 17-12b).

FIGURE 17-12 Multivalent subunit vaccines. (a) Liposomes and immunostimulating complexes (ISCOMs) can all be prepared from detergent-extracted antigens or antigenic peptides. ISCOMs are composed of phospholipid monolayers and liposomes are made of phospholipid bilayers. In either case, the immunogen of interest can be inserted as a transmembrane protein or packaged inside these vesicles. (b) ISCOMs and liposomes can deliver agents inside cells, so they mimic endogenous antigens. Subsequent processing by the endogenous pathway and presentation with MHC class I molecules induces a cell-mediated response. ER = endoplasmic reticulum; TAP = transporter associated with antigen processing.
Adjuvants Are Included to Enhance the Immune Response to a Vaccine
In an ideal case, vaccines mimic most of the key immunologic events that occur during a natural infection, eliciting strong and comprehensive immune responses, but without the risks associated with “the real thing.” A discussion of vaccines would be incomplete without mentioning the importance of adjuvants. Derived from the Latin term “to help or aid,” adjuvants are substances that are added to vaccine preparations to enhance the immune response to the antigens with which they are mixed: in other words, to enhance immunogenicity. This is not completely dissimilar to the discussion above about conjugate and multivalent vaccines and can be employed for almost any vaccine strategy being employed. This is especially important to consider when the vaccine preparation is a pathogen subunit or other nonliving form of the organism, where immunogenicity is typically quite low. Nonlive vaccine preparations often also lack triggers for innate immunity, which are inherent in most live and some killed vaccines. Adjuvants primarily target these innate response elements. When mixed with the pathogen-associated antigens, these additives can also help with delivery of the vaccine to the immune system and enhance general immune responsiveness.
For almost 80 years, the only adjuvant used in human vaccines has been aluminum salts (called alum). It turns out that alum is a fairly good enhancer of TH2 responses but a weaker stimulator of TH1 pathways. Despite its long-term use and inclusion in many different vaccine preparations, the mechanism of action of alum is still poorly understood. As an adjuvant, alum is mixed in an emulsion with the immunogen and is thought to work primarily by creating slow-release delivery of the antigen at the injection site, which helps in sustained stimulation of the immune response. It may also help to recruit APCs and encourage the formation of large antigen complexes that are more likely to be phagocytosed by these cells.
In recent years, two new types of adjuvants have been licensed for use in human vaccines. One, called a virosome, is a reconstituted virus envelope containing phospholipids and virus glycoproteins but without any genetic information. This type of adjuvant is used in the Inflexal V influenza vaccine and the hepatitis A vaccine. The other, AS04, is an alum salt containing a derivative of lipopolysaccharide (LPS), found naturally on the surface of gram-negative bacteria, and a strong TLR4 agonist. Use of this adjuvant triggers PRR signaling that helps to encourage TH1 pathway responses. AS04 is currently used in vaccines against HPV and hepatitis B. All of these newer adjuvants have been found to enhance the production of antibodies as compared with unadjuvanted vaccine preparations, and may have the advantage of eliciting much greater cell-mediated immune responses than alum alone.
Some creative uses of existing adjuvants and next-generation adjuvants or immune modulators are in more basic stages of development. These include compounds designed to stimulate certain PRRs—specifically, several TLRs and at least one NOD-like receptor. Although adjuvants have improved humoral immunity, few if any actually enhance cellular immunity, especially CD8+ T-cell responses. Recently, a combination of two already licensed adjuvant components was used in mice treated with an influenza virus peptide and was found to generate protective CD8+ T-cell memory responses. This strategy is attractive, as it utilizes adjuvants with an already established track record of safety and efficacy in humans. Finally, Akiko Iwasaki and colleagues at Yale have created a novel approach aimed at protection against sexually transmitted disease and treatment of some forms of cancer caused by these infectious agents. Her strategy uses a subunit vaccine to activate the immune response, followed by local chemokine administration to recruit memory cells; this approach is called prime and pull (see Advances Box 17-6).