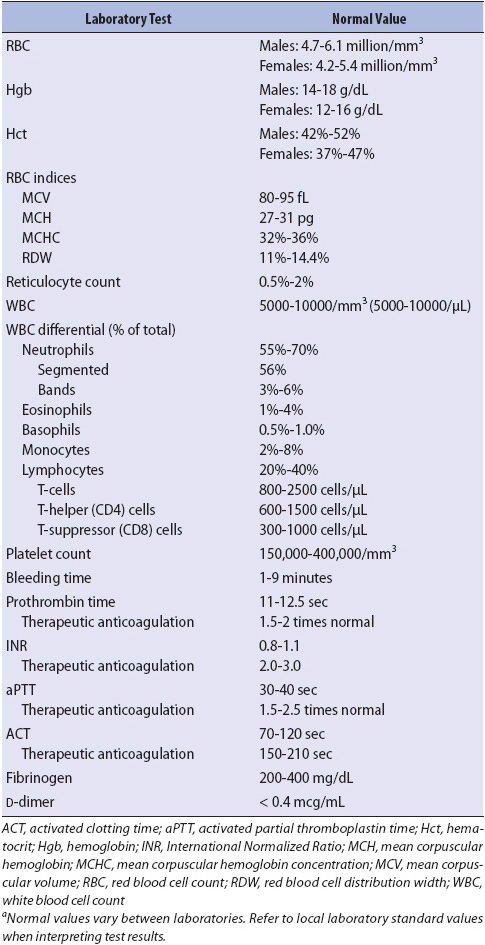
TABLE 13-1. NORMAL VALUES FOR HEMATOLOGIC AND IMMUNE SCREENING TESTSa
KNOWLEDGE COMPETENCIES
1. Analyze laboratory test results used to assess the status of the hematologic and immune systems:
• Complete blood count
• White blood cell differential
• International normalized ratio
• Activated partial thromboplastin time
• D-dimer
2. Describe the etiology, pathophysiology, clinical manifestations and collaborative management for common hematological problems in acutely ill patients.
3. Provide comprehensive management for immunosuppressed patients in the acute care setting.
The hematologic and immune systems play a major role in the body’s response to illness. Organs and tissues require a continuous supply of oxygen from the red blood cells (RBC), while the white blood cells (WBC) mount an immune response. The platelets and other coagulation components are essential for hemostasis. Assessment of these processes and treatment of hematologic and immune problems are an important part of patient management.
A complete patient assessment guides the selection of screening tests for hematologic and immune problems. Historical data are particularly important and should include family history, occupational exposures, lifestyle behaviors, diet, allergies, past medical problems, surgeries, co-morbid conditions, transfusion of blood or blood components, and current medications. Abnormal physical assessment data from each body system collectively assist in the identification of risk factors or acute abnormalities pertinent to hematologic and immune function. In addition, a variety of laboratory tests assist the clinician to evaluate problems in these systems (Table 13-1).
TABLE 13-1. NORMAL VALUES FOR HEMATOLOGIC AND IMMUNE SCREENING TESTSa
The complete blood count (CBC) is a primary assessment tool for evaluation of the hematologic and immune status. The RBC count and RBC indices, along with the hemoglobin and hematocrit levels, provide valuable information regarding the oxygen-carrying capability of the blood. The total WBC count and the WBC differential reveal the body’s ability to provide an immune response against foreign substances and to participate in the normal inflammatory process required for tissue restoration. Important information concerning hemostasis is obtained from the platelet count, with additional studies required to fully evaluate the coagulation process.
The RBC count is determined by the number of erythrocytes per cubic millimeter of blood. Normal values for men are higher than for women. A decrease in the number of RBC or in the amount of hemoglobin indicates anemia. Anemia can be due to many factors, including decreased production or increased destruction of RBC, loss of RBC by hemorrhage, vitamin B12 deficiency, and/or iron deficiency. An increase in the total number of RBC occurs as a compensatory mechanism in persons with chronic hypoxia or as an adaptation to high altitudes. Further assessment of the ability of the bone marrow to produce RBC is obtained by a reticulocyte count.
Hemoglobin is the primary carrier of oxygen to body tissues. As the number of RBCs change, so does the hemoglobin content. A decline in hemoglobin to a level as low as 7 g/dL may be well tolerated, in some patients, while in others a decline can result in significant symptoms. The rate at which the decline in hemoglobin level occurs often influences the symptoms and tolerance of the patient. A decline that occurs gradually over time is often tolerated, whereas a rapid decline frequently results in poor tolerance by the patient. Elderly patients and those with underlying cardiac or pulmonary disorders may become symptomatic with even small changes in the hemoglobin content of the blood.
Hematocrit measures the RBC mass in relationship to a volume of blood and is expressed as the percentage of cells per 100 mL of blood. Multiplying the hemoglobin value by 3 gives an estimate of hematocrit. The hematocrit is particularly sensitive to changes in the volume status of the patient. It increases with fluid losses (hemoconcentration) and decreases with increased plasma volume (hemodilution). Interpretation of hemoglobin and hematocrit results must take into account the time the values were obtained in relationship to blood volume loss, fluid loss, and/or fluid administration; for example, values obtained immediately after an acute hemorrhage may appear normal, because compensatory mechanisms have not had time to restore plasma volume. Restoration of plasma volume by compensation or crystalloid resuscitation lowers the hemoglobin and hematocrit.
The RBC indices (mean corpuscular volume, mean corpuscular hemoglobin, mean corpuscular hemoglobin concentration, and RBC distribution width) are measurements of the size, weight, and hemoglobin concentration of the individual RBCs also known as erythrocytes. These indices are useful in determining the etiology of anemia.
Leukocytes, or WBCs circulating in the blood, are measured as an indicator of the total amount of WBCs in the body. Most WBCs are not sampled in a CBC because they are marginated along capillary walls, circulating in the lymphatic system, or residing in lymph nodes and other body tissues.
Increased WBCs, or leukocytosis, is usually caused by an elevation in one type of WBC. It is most often associated with a normal immune system response to an acute infection, but is also an expected result of an inflammatory process. WBCs are known to have both positive and negative effects. Positive effects include phagocytosis of microorganisms. Potentially destructive effects include the release of oxygen-free radicals from neutrophils and excessive amounts of cytokines from macrophages. An abnormal production of leukocytes in the bone marrow occurs during leukemia.
Leukopenia refers to a decrease in the total WBC number. This occurs when bone marrow production is inhibited or during certain infections when rapid consumption of WBCs takes place. The life span of a circulating WBC is only hours to days; therefore, a constant replacement process is necessary to prevent leukopenia and immune compromise, which can result in infection and harm to the patient.
The differential is a measure of five different categories of leukocytes, with each type reported as a percentage of the total WBC count. The absolute count for each category of white cell (also referred to a cell line) is calculated by multiplying the percentage of each type of cell by the total WBC count. Increases or decreases in any one cell line help evaluate normal immune response and predict impaired immunity.
Neutrophils, or segmented neutrophils (also called “segs”), are the primary responders to infection and inflammation in the body. They also are an accurate indicator of how the immune system is functioning. With active infections the bone marrow also releases an immature form of neutrophil called a band. Bands quickly mature into segmented neutrophils with greater phagocytic properties to respond to infection. Leukocytosis is usually caused by an increased number of segmented neutrophils and is called neutrophilia. A “left shift” refers to leukocytosis with an increased percentage of bands. Neutropenia, or a decreased number of circulating neutrophils, places the body at increased risk for infection. An absolute neutrophil count (ANC = WBC × [% neutrophils + % bands]) of less than 1000 cells/mm3 severely compromises immune system response, particularly to bacterial infections.
Monocytes are large phagocytic cells that circulate briefly in the blood before maturing into macrophages. These leukocytes are important scavengers of microorganisms and other foreign material. They also activate lymphocytes by presenting antigens to T cells.
Lymphocytes are the WBCs responsible for the body’s adaptive (specific) immune responses. Subsets of T and B lymphocytes are assessed by specific cell counts. Lack of properly functioning lymphocytes or inadequate numbers of these cells places the body at risk for bacterial, viral, and fungal infections and certain malignancies. The CD4 cell is a subset of lymphocytes. It is the target of HIV infection leading to the development of acquired immunodeficiency syndrome (AIDS).
Eosinophils increase in numbers and activity during parasitic infections and allergic responses. They attach to parasites and use enzymes to kill them. Increased percentages of these cells are also seen during an allergic response. Basophils are another WBC associated with allergy. They break down during allergic reactions, releasing their intracellular contents such as heparin and histamine.
The platelet count is determined by the number of platelets per cubic millimeter of blood. Platelets are called thrombocytes because of their role in the initiation of blood coagulation at the site of damaged blood vessel walls. Two-thirds of the body’s platelets are circulating in the blood, with the remaining one-third sequestered within the spleen. Thrombocytopenia (decreased number of platelets) is associated with increased risk of spontaneous bleeding and is caused by decreased production, increased consumption, or increased destruction of platelets. Hypercoagulability of the blood can result from increased circulating platelets caused by proliferative disorders, malignancies, and inflammation. Qualitative assessment of platelet function is determined by the bleeding time.
The prothrombin time (PT) evaluates the extrinsic pathway and final common pathway of fibrin clot formation. Because of different reagents used in testing, PT values from different facilities are not standardized, so comparing results may lead to discrepancies. The International Normalized Ratio (INR) is a calculation developed to standardize interpretation of PT results. The PT and INR may be reported together, but the INR is now the recommended parameter for establishing the therapeutic range for oral anticoagulant therapy. The INR is a general test of coagulation, and will be elevated in patients with liver disease, biliary tract disease, and those who are therapeutically anticoagulated with warfarin. It is also elevated in patients with coagulopathies such as disseminated intravascular coagulation (DIC).
The activated partial thromboplastin time (aPTT) is reported in seconds and is used to evaluate fibrin clot formation stimulated by the intrinsic and common pathways of coagulation. This test is used to screen for congenital coagulation disorders and for monitoring anticoagulation with unfractionated (IV) heparin therapy. Prolonged aPTT is noted in persons with liver disease, vitamin K deficiency, and DIC.
The activated coagulation time (ACT) is reported in seconds. The test is used most commonly to monitor effects of unfractionated heparin during and following cardiovascular procedures such as cardiopulmonary bypass and percutaneous coronary interventions. It is generally performed at the point of care.
The fibrinogen level is tested during evaluation for bleeding disorders. Fibrinogen is the plasma protein that becomes the fibrin clot. Plasma levels of fibrinogen may be increased during an inflammatory response, pregnancy, or acute infection. Decreased levels are present with liver disease and DIC.
D-dimer is a very specific indicator of fibrinolysis, the natural process that breaks down fibrin clots. Levels of D-dimer are elevated in thrombotic disorders such as deep venous thrombosis (DVT) and pulmonary emboli (PE). Levels are also elevated during thrombolytic drug therapy and in DIC.
After obtaining basic laboratory screening tests, additional laboratory and diagnostic testing is necessary to identify specific etiologies for hematologic and immune function. For patients with hematologic disorders, a bone marrow aspiration, or further studies of specific clotting factor assays may be performed.
Blood, sputum, urine, and wound specimens for gram stain and culture help identify sources of infection. Molecular diagnostic techniques such as polymerase chain reaction (PCR) detect infectious agents not readily cultured, such as viruses. Noninvasive studies such as ultrasound may determine liver, spleen, or lymph node abnormalities. Radiologic procedures (radiographs, CT scans, arteriograms) may be needed to identify areas of infection or hemorrhage.
Critically and acutely ill patients often have combined abnormalities involving the hematologic and immune systems. Anemia, immune compromise, and coagulopathy are three distinct problems that may be seen together in a critical or acute care patient. Each of these problems pose major threats to the patient’s potential outcome and is evaluated separately.
Anemia is defined as a hemoglobin count less than 12 g/dL and is the most common hematologic disorder. Its etiology may be classified into disorders of RBC production, increased destruction of RBC, or acute blood loss.
A patient history gives important clues to the etiology of anemia. Decreased production may result from nutritional deficiencies in substrates necessary for RBC production such as iron, folic acid, or vitamin B12. Those at high risk for iron deficiency anemia include children, adolescents, pregnant women, elderly, and patients with malabsorption syndromes. Folic acid deficiency is common in alcoholics. Dietary vitamin B12 deficiency may occur in strict vegetarians and also occurs due to a lack of intrinsic factor (postgastrectomy or with pernicious anemia) or Crohn disease. Another common cause of anemia is chronic blood loss from the gastrointestinal (GI) tract or from heavy menstruation. Daily blood testing in hospitalized patients may also contribute to anemia, because the patient’s bone marrow cannot keep up with the loss.
Anemia may be associated with chronic illness, such as chronic inflammation, infection, cancer, hepatitis, and renal failure. Patients with renal failure experience anemia owing to the reduced production of the hormone erythropoietin. The life span of the RBC is also decreased in some chronic disease states, and the bone marrow is unable to compensate adequately, resulting in anemia. Cancer that specifically involves the bone marrow may replace normal bone marrow with malignant cells, disturb the development and maturation process of blood cells and fill the marrow with immature cells that prevent RBC generation.
Anemia can also occur in cancer patients as a result of bone marrow suppression due to their specific treatment. Here the bone marrow fails to produce cells, sometimes causing a drop in all three types of blood cells (WBC, RBC, and platelets) known as pancytopenia. Medications such as chemotherapeutic agents often suppress the bone marrow and cause anemia. Other causes of anemia include radiation therapy to marrow-producing bones such as the sternum and other long bones in the body.
Hemolytic anemia results from excessive destruction of RBCs. This can occur episodically or chronically. Abnormalities intrinsic to the RBC are usually the result of hereditary causes of hemolytic anemia, such as sickle cell disease. Extrinsic sources of hemolysis include immune destruction from a transfusion reaction, splenic disorders, damage by artificial heart valves, cardiopulmonary bypass, or use of an intra-aortic balloon pump.
Sickle cell anemia is an inherited abnormality of hemoglobin which results in chronic hemolytic anemia and occlusion of blood vessels. The problem mainly affects the African American population and can manifest itself as sickle cell trait, or the more serious sickle cell disease beginning in early childhood. During episodes of low oxygen tension, the RBCs change their shape (to a sickle rather than rounded shape) and adhere to the endothelial lining of blood vessels where they activate coagulation. This results in hemolytic anemia, blood vessel occlusion, and ischemic pain in organs and tissues. Other complications include bone disorders, injury to the spleen, and stroke. Hydroxyurea is a cytotoxic drug that may be prescribed to help prevent complications. Stress, infection, and illness can precipitate an acute exacerbation. Patient management includes hydration, blood transfusion, and pain management during acute episodes.
Acute hemorrhage also leads to anemia. Trauma, surgical blood loss, coagulopathy, GI bleeding, and bleeding from excessive anticoagulation are frequently encountered as causes of anemia in critically and acutely ill patient populations. With acute hemorrhage, both cellular components and plasma are lost simultaneously. The remaining cells are normal (normocytic, normochromic) and the main problem is an insufficient number of RBC. Until volume replacement from fluid resuscitation or mobilization of fluids from extracellular sources occurs, a drop in hematocrit may not be appreciated. Following an episode of blood loss, the reticulocyte count will generally rise as newly produced immature RBCs are released into the circulation.
Regardless of the etiology of an anemia, the critical effect of decreased RBCs and hemoglobin is a decrease in the oxygen-carrying capacity of the blood and a reduction in oxygen content. This may be tolerated if anemia develops slowly and the body can compensate, but may be life threatening if sudden blood loss occurs. Rapid loss of blood volume results in hypovolemic shock and cardiovascular instability, further reducing delivery of oxygen to body tissues.
Clinical manifestations are related to the body’s compensatory mechanisms that attempt to maintain perfusion of oxygen to vital tissues. Clinical manifestations may not be obvious until the hemoglobin level is less than 7 g/dL. As compensatory mechanisms are overwhelmed, serious signs and symptoms occur. Patients with underlying pathology involving the pulmonary and cardiovascular system have less of an ability to tolerate the effects of anemia and become symptomatic more quickly.
Cardiovascular
• Tachycardia, palpitations
• Angina
• Decreased capillary refill
• Orthostatic hypotension
• ECG abnormalities (arrhythmias, ischemic changes)
• Hypovolemic shock (hypotension, tachycardia, decreased cardiac output, increased systemic vascular resistance)
Respiratory
• Increased respiratory rate
• Dyspnea on exertion, progressing to dyspnea at rest
Skin/Musculoskeletal
• Pallor of skin and mucous membranes
• Dusky nail beds
• Decreased skin temperature
Neurologic
• Headache
• Light-headedness
• Syncope
• Irritability/agitation
• Restlessness
• Severe fatigue
Abdominal
• Enlarged liver and/or spleen
• Anorexia, nausea, vomiting
Management of the anemic patient must be guided by the severity of symptoms. The level of concern for decreases in hemoglobin and hematocrit is determined by the patient’s signs and symptoms and if active bleeding is suspected. Restoration of adequate blood to assure oxygen delivery to the tissues is a priority in critically and acutely ill patients. Identification of the etiology of anemia and resolution of the underlying cause is done simultaneously.
Oxygen delivery is a product of the amount of hemoglobin in the blood, the saturation of the hemoglobin with oxygen, and the cardiac output. Management strategies focus on optimizing each of those components.
1. Administration of supplemental oxygen can enhance oxygen saturation. Use of oxygen, particularly during activity, may minimize desaturation and dyspnea.
2. Adequate hemoglobin can be replaced in acute situations only by transfusion of RBCs. Transfusion of packed red blood cells (PRBC) is considered when blood loss is severe, the patient is actively bleeding, or when the patient is very symptomatic (Table 13-2).
3. Cardiac output can be optimized with volume replacement, including PRBCs, in situations of bleeding and hypovolemia. Other manipulations of cardiac output may be guided by hemodynamic monitoring and calculations to assess oxygen delivery and utilization.
4. Monitoring vital signs, oxygen saturation, and subjective patient data before, during, and after a change in therapy or activity identifies the patient’s ability to tolerate anemia.
5. Limiting strenuous activity and planning periods of rest are important nursing interventions for the anemic patient.
TABLE 13-2. SUMMARY OF CURRENT GUIDELINE RECOMMENDATIONS ON RED BLOOD CELL TRANSFUSION

Further diagnostic testing may be indicated to determine the etiology of anemia. Radiologic and endoscopic studies to locate sites of bleeding, particularly in the GI tract, may be necessary. Treatment of the underlying cause of anemia may include the following:
1. Administer recombinant human erythropoietin to restore bone marrow production of RBCs in chronic anemia. The response may take several weeks, so it is not appropriate in situations in which acute correction of anemia is necessary. Chronic renal failure patients and patients receiving chemotherapy may benefit from this treatment.
2. Supplemental oral ferrous sulfate or IV iron-sucrose may be indicated if iron deficiency anemia is present.
3. Vitamin B12 and folic acid–related anemia may also require supplementation.
4. Dietary consultation may be needed prior to discharge to help patients and families plan meals with foods high in iron.
1. Use small volume collection tubes and microanalysis techniques.
2. Assess the need for routine and additional blood testing to decrease diagnostic blood loss.
3. Use blood salvage systems in surgical patients.
4. Use prophylactic agents to reduce the risk of GI bleeding.
5. Screen all patients for anticoagulants and bleeding risk prior to procedures.
6. Accept normovolemic anemia in stable patients.
All critically and acutely ill patients may be considered compromised hosts because their defense mechanisms are inadequate due to a combination of factors, such as underlying disease, medical therapy, nutritional status, age, or stress. Patients in critical care units and acute or progressive care units are considered to be at high risk for infection. The term immunocompromise is applied to patients whose immune mechanisms are defective or inadequate. The patient with immunocompromise is more likely to develop an opportunistic infection. Once infection develops, it may quickly progress to systemic inflammatory response syndrome (SIRS) and sepsis.
Immune system protection from infection is categorized into three levels: natural defenses, innate (general) immunity, and adaptive (specific) immunity. Natural defenses include having intact epithelial surfaces (skin and mucous membranes) with normal chemical barriers (pH, secretions) present and all protective reflexes (blink, swallow, cough, gag, sneeze) intact. The invasive catheters and tubes used in critical and acute care units bypass these protective barriers and allow an introduction of pathogens.
The innate response to infection includes activation of the phagocytic WBC (neutrophils and monocytes) to attack the foreign microorganisms (antigens) that have entered the body, bypassing or overwhelming the natural defenses. The macrophages play a key role in processing the invading antigen and presenting it to the lymphocytes involved in the adaptive immune response.
Lymphocytes (B cells and T cells) are responsible for the orchestration of an immune response specific to each foreign protein or antigen. B lymphocytes create antigen-specific antibodies or immunoglobulins to aid in the destruction of the antigen and to protect the body from future encounters with the antigen. This is called humoral immunity. T lymphocytes have different subsets of cells created to modulate the immune system response (including CD4 helper T cells) and cells that have cytotoxic properties, the CD8 cytotoxic T cells. The immune response of the T lymphocytes is called cell-mediated immunity. Both types of lymphocytes work closely together in a specific immune response. However, humoral immunity is the primary protection against bacterial invasion and cell-mediated immunity is primarily directed against infection by viral and fungal organisms, and some malignancies. Additionally, T cells are most responsible for the rejection of foreign tissue and for delayed hypersensitivity reactions.
Deficiencies in immune system function can be categorized into primary, or congenital, immune system defects and secondary, or acquired, immune system dysfunction. Immune deficiencies may be pinpointed to a specific cell type or may involve abnormalities in multiple components of the immune system. Secondary or acquired immunodeficiencies are the most likely type encountered in critical and acute care patients. Acquired immunodeficiency may be secondary to age, malnutrition, stress, chronic disease states, drugs with immunosuppressive effects, cancer and its treatment, HIV infection, and other factors.
Today, an increased number of patients are undergoing organ transplantation and receiving immunosuppressive agents. Patients who receive organ transplants require lifelong immunosuppressive drug therapy to prevent recognition and rejection of the transplanted tissue by the immune system. Patients typically receive a combination of drugs that affect various components of the immune response. Higher doses are required during the first weeks and months after the transplant, and doses are decreased over time to minimize the risk of infection and other complications. Acute cellular rejection may be diagnosed by evidence of failure of the transplanted organ (such as elevated serum creatinine and decreased urine output in a kidney recipient) or by obtaining a biopsy diagnostic of rejection. Rejection is commonly treated by augmented immunosuppression, such as a series of doses of IV methylprednisolone. During and following treatment, these patients are at high risk for infection. Immunosuppressive drugs also may be used in the management of other disorders such as autoimmune diseases like rheumatoid arthritis and lupus.
As new regimens and new chemotherapy agents are used to treat cancer, many of these agents have the potential to produce significant bone marrow suppression. More aggressive chemotherapeutic treatment of cancer has led to higher numbers of patients with bone marrow suppression. These patients are at high risk for the development of complications, including neutropenia and risk of serious infection.
Neutropenia is a term used to describe the state where the absolute neutrophil count is less than 1000 cells/mm3 with an increased susceptibility to infection and sometimes neutropenic fever and sepsis. Many factors contribute to the susceptibility of the neutropenic patient to develop an infection. The duration of neutropenia, functional capability of the existing neutrophils, the patient’s defense mechanisms and natural barriers to infection, and endogenous and exogenous flora all are influential in the development of infection. The key to preventing sepsis and infection is early detection and intervention.
Detection of infection in the immunocompromised patient may be difficult since the body’s defense mechanisms are suppressed. Due to the lack of neutrophils, the patient may not be able to mount a vigorous inflammatory response and classic signs and symptoms of infection may be diminished or absent. Therefore redness, febrile response, or even the development of pus may not occur because purulent drainage is largely the result of dying neutrophils at the site of infection. Since the neutropenic patient can be severely infected and on the verge of becoming septic, while lacking the usual paramount signs of infection, malaise or pain may be the patient’s only complaint.
Fever in this patient population is another key sign of infection and warrants aggressive investigation. Since development of fever may not be possible, the nurse must be keenly aware of other signs of sepsis including alterations in blood pressure, pulse, and respiratory rate that are the result of compensatory mechanisms. The rapid onset of sepsis in a neutropenic patient requires diligent and meticulous assessment skills with early intervention, as these patients do not present or respond as those with a functional, normal immune system.
Human immunodeficiency virus (HIV) is another disorder which leads to immunocompromise. It primarily affects helper T cells, decreasing their number and function. This in turn has a profound effect on adaptive immunity. Following diagnosis, the CD4 cells are monitored, and a CD4 count of < 400/mm3 is associated with a poor prognosis. Viral load testing is also performed to measure the amount of viral particles per cubic millimeter of blood. Patients with HIV are susceptible to opportunistic infections and certain malignancies, and as these develop they may progress to a diagnosis of AIDS. HIV is managed with antiretroviral drugs and effective treatment of infections. Patients are surviving longer, and may develop disorders requiring hospitalization. They are most likely to require critical care if they acquire a serious opportunistic infections or experience an adverse reaction to antiretroviral therapy. They may also be admitted with conditions not directly related to HIV or AIDS.
Local Evidence of Inflammation and Infection
• Redness
• Edema
• Warmth
• Pain
• Purulent drainage
General Evidence of Infection
• Fever or hypothermia
• Rigors or shaking chills
• Fatigue and malaise
• Changes in level of consciousness
• Lymphadenopathy
• Tachycardia
• Tachypnea
Neurologic
• Headache
• Nuchal rigidity
• Changes in mental status, agitation
Respiratory
• Cough
• Change in color, amount of sputum
• Dyspnea, orthopnea
Genitourinary
• Dysuria
• Urgency
• Frequency
• Flank pain
• Abdominal pain
• Cloudy and/or bloody urine
Gastrointestinal
• Nausea
• Vomiting
• Diarrhea
• Cramping abdominal pain
• Enlarged liver or spleen
Patients with high risk for the development of infection must be identified on admission to the unit. Measures to protect and strengthen immune system function should be included in the plan of care. All healthcare team members must utilize measures to prevent the development of hospital-acquired infections. Close monitoring for signs and symptoms of a local or systemic inflammatory response is especially important to detect infection early. Identification of the source and likely organisms causing infection allows for initiation of broad-spectrum, empiric antimicrobial coverage. Culture and sensitivity reports guide the choice of drugs specific to the organisms isolated from the patient. Care is planned in a way that risks for introduction of pathogens and infection are minimized. Hand washing is the main intervention for the prevention of infection. Additionally, the number of lines, tubes, and drains is minimized when possible. Although utilization of central lines, indwelling catheters, and other devices is commonplace in critical and acute care settings, the nurse must be vigilant to constantly evaluate the ongoing need for such devices.
Immunocompromised risk factors are as follows:
1. Neonates and the elderly
2. Malnutrition
3. Use of medications with known immunosuppressive effects such as steroids, cancer chemotherapeutic agents, and transplant immunosuppressive agents
4. Recent radiation therapy to areas of the body that impact bone marrow production
5. Chronic systemic diseases such as renal or hepatic failure or diabetes
6. Diseases involving the immune system such as HIV infection
7. Loss of protective epithelial barriers through:
• Oral or nasogastric intubation
• Presence of decubitus ulcers
• Burns
• Surgical wounds
• Skin and soft tissue trauma
• Mucositis
8. Invasive catheters or prosthetic devices in place such as:
• Intravascular catheters, including peripheral, central, and arterial lines
• Indwelling urinary catheters
• Endotracheal intubation and mechanical ventilation
• Heart valve replacements
• Orthopedic hardware such as artificial joints, pins, plates, or screws
• Dialysis, apheresis catheters, shunts, fistulas, or grafts
• Cardiovascular devices such as ventricular-assist devices, pacemakers, or implantable defibrillators
• Synthetic vascular grafts
• Ventricular shunts
1. Take meticulous care of the skin and mucous membranes to prevent loss of barrier protection.
2. Use the enteral route for feeding when possible to maintain caloric intake and normal gut function.
3. Avoid the use of indwelling urinary catheters or remove them as early as possible.
4. Minimize patient stress and the release of endogenous glucocorticoids by relieving pain or using alternative methods such as guided imagery or music for relaxation, and other comfort measures (positioning, massage).
5. Administer colony-stimulating factors (G-CSF or GM-CSF) to stimulate bone marrow production of neutrophils and monocytes when appropriate.
1. All personnel and visitors are to wash their hands before and after contact with the patient. Hand washing remains the number one method to prevent hospital-acquired infection.
2. Patients at high risk should have a private room.
3. Institute respiratory hygiene/cough etiquette for patients with signs of respiratory infection and appropriate isolation for known or suspected patient infection.
4. Adhere to strict aseptic technique for all care of intravascular catheters and any invasive procedures performed at the patient’s bedside.
5. Eliminate environmental sources of infection (eg, leftover fluids used for irrigations). Clean surfaces frequently with recommended disinfectant, including bedside table, equipment, and any surfaces where contamination is likely.
6. Track the date and time fluids, tubings, and catheters and change them at the prescribed intervals.
7. Review facility protocol regarding use of filtered water, restriction of fresh fruits and vegetables, and other neutropenic precautions.
8. Encourage use of incentive spirometry, turning, deep breathing, and mobility with ambulation if possible.
1. Monitor the patient closely for signs and symptoms consistent with infection and sepsis and communicate abnormal findings to the healthcare team.
2. Initiate sepsis protocol when signs of SIRS are present.
3. Collect specimens for culture and sensitivity from potential sources of infection (eg, urine, sputum, blood, stool, wound drainage).
4. Institute antibiotic therapy as directed. See Chapter 11 Multisystem Problems, for more information on the management of sepsis.
Critically and acutely ill patients with coagulopathy may have a problem involving platelets, hemostasis, fibrinolysis, or a combination of these abnormalities. Acquired disorders of coagulation, as opposed to inherited disorders, are seen most frequently in critical and acute care units.
Platelets initiate the coagulation process at the site of blood vessel injury. Quantitative platelet disorders are associated with bleeding when the platelet count drops to less than 50,000/mm3, especially if there is tissue trauma. Spontaneous bleeding is possible at counts of < 20,000/mm3, and counts that reach 5000 to 10,000/mm3 predict high risk for hemorrhage. Four general mechanisms are responsible for thrombocytopenia: (1) decreased production of platelets by the bone marrow, (2) shortened survival due to platelet utilization and destruction, (3) sequestration of platelets in the spleen, and (4) intravascular dilution of platelets during massive transfusion.
Thrombocytopenia may also be related to immune mechanisms. Drug-induced thrombocytopenia occurs when a drug induces an antigen-antibody reaction that results in the formation of immune complexes that destroy platelets by complement-mediated lysis. There are several types of immune-related thrombocytopenia that are seen in critical and acute care. Heparin-induced thrombocytopenia (HIT) is an immune-mediated reaction to heparin that results in the formation of antiplatelet antibodies which activate platelets and form clots. This then leads to platelet consumption and a precipitous drop in the platelet count. The patient may develop intravascular clotting resulting in clinical thrombosis. Venous thrombosis is most common and may result in limb ischemia and pulmonary emboli. When this syndrome is suspected, all heparin is stopped, and confirmatory testing for HIT antibodies is performed. Treatment options include administration of direct thrombin inhibitors such as argatroban. Patients diagnosed with HIT should not receive heparin again.
A 35-year-old African American man is admitted to the progressive care unit with dyspnea and altered mental status. The patient’s history includes diabetes mellitus and chronic renal failure treated by kidney transplantation 3 months ago. Recent kidney function testing suggested possible rejection, and the patient received a course of augmented steroids. Current medications include tacrolimus, mycophenolate mofetil, prednisone, metoprolol, and insulin. Significant findings on admission include the following:

During the admission interview, the patient appears dyspneic, tachypneic, and complains of thirst. He has a dry cough, and crackles are heard over the left lung field. The physician arrives on the unit and discusses the patient with the progressive care nurse, who knows this patient from previous hospitalizations. Following analysis of the patient’s history, physical examination, and diagnostic data, a multidisciplinary care plan is developed.
Case Question 1. What are this patient’s risk factors for hematologic and immune problems?
Case Question 2. What should the nursing assessment focus on?
Case Question 3. What therapeutic interventions should be carried out immediately?
Case Question 4. Analyze the diagnostic test results and outline how the values provide diagnostic information.
Case Question 5. Analyze the assessment data and explain how it will be used to direct patient care.
Case Question 6. What are the most likely working diagnoses?
Case Question 7. What types of consultations should be ordered?
Case Question 8. What are your priority nursing interventions during these first few hours of hospitalization?
1. Risk factors include chronic immunosuppression, recently augmented immunosuppression, and diabetes.
2. Assessment should focus on the evaluation of potential sources of infection and indicators of hyperglycemia, electrolyte imbalance, and renal insufficiency.
3. Initial interventions should include IV fluids, antibiotics, supplemental oxygen, and respiratory therapy.
4. The elevated WBC and band cells indicate acute infection. The low platelet count can indicate sepsis and potential clotting problems. The serum glucose suggests poorly controlled blood sugar, and frequent testing is indicated. The elevated serum creatinine indicates renal insufficiency in the transplanted kidney. The chest x-ray suggests pneumonia.
5. The cough, respiratory distress and crackles suggest possible pneumonia. The elevated respiratory rate can be indicative of respiratory distress and diabetic ketoacidosis. The thirst may be due to fever and hyperglycemia.
6. Pneumonia, hyperglycemia, renal insufficiency.
7. An infectious disease consultation and consultation with the diabetic management team are indicated. The renal transplant team needs to be notified of the admission.
8. Nursing priorities include frequent assessment of respiratory status, blood sugar, and kidney function. Collaborative interventions such as fluids, antibiotics, and insulin are administered. Consultants are notified promptly regarding the patient’s admission and condition.
Immune thrombocytopenia purpura (ITP) is an autoimmune disorder that results in the destruction of platelets in the spleen and a platelet count of less than 20,000/mm3. This disorder was formerly called idiopathic thrombocytopenia, but was renamed when it was identified as an immune process. In adults ITP may occur as a primary disorder or may be secondary to medications or autoimmune disorders such as systemic lupus erythematosus. For some the cause may never be determined. Patients develop petechiae, purpura, and epistaxis. Splenomegaly may develop as platelets are destroyed in the spleen. Treatment options include steroids, intravenous immunoglobin, administration of monoclonal antibody treatment, and, in some cases, splenectomy.
Thrombotic thrombocytopenic purpura (TTP) is a syndrome characterized by thrombocytopenia, hemolytic anemia, renal failure, fever, and neurologic changes. The cause of this disorder is unclear, but is thought to involve dysfunction of an enzyme which leads to abnormal platelet aggregation. Patients with TTP may develop widespread vascular occlusion in organs, as well as jaundice, purpura, petechiae, and bleeding. Acutely ill individuals may be treated with plasmapheresis.
Hemolytic-uremic syndrome is characterized by thrombocytopenia, hemolytic anemia, and renal failure. It is most often the result of infectious colitis and the toxin released from E coli 0157:H7. Children are most often affected by this syndrome, and will require hospitalization for supportive care including dialysis.
Patients may have adequate numbers of platelets but still have a bleeding tendency due to qualitative platelet disorders. Drug-induced suppression of platelet function is commonly associated with use of aspirin and nonsteroidal anti-inflammatory agents (NSAIDs). In addition to many of the medications used, critically and acutely ill patients may have many factors that predispose them to the potential impairment of platelet function, including renal failure and uremia.
Disorders of hemostasis may be caused by inherited abnormalities of coagulation factors. Hemophilia types A and B are congenital deficiencies in factors VIII and IX. Von Willebrand disease represents a deficiency or dysfunction of the plasma protein of the same name. Replacement of the deficient factor keeps these chronic diseases under control. Patients with these disorders may be monitored in critical or progressive care units when undergoing routine surgical procedures or when hospitalized for other medical problems.
Acquired coagulation disorders can be associated with deficient coagulation factor production. This may be caused by a decreased intake of vitamin K, the vitamin essential for the formation of clotting factors II, VII, IX, and X. Deficiencies in vitamin K as a result of dietary deficiency, intestinal malabsorption, liver disease, use of warfarin, or antibiotic therapy are also common. Vitamin K deficiency prolongs the PT/INR. Because most coagulation factors are produced in the liver, patients with liver disease have deficiencies of fibrinogen and other factors in addition to deficiencies of the vitamin K–dependent factors.
Many of the drugs used routinely in hospitalized patients have anticoagulant and antiplatelet effects (Table 13-3). Therapeutic anticoagulation using heparin, warfarin, and other agents interferes directly with the clotting process. The intrinsic pathway and the final common pathway are affected by the administration of heparin. If bleeding from heparin is minimal, it can be controlled by decreasing the dose or temporarily stopping its administration. If bleeding is severe, the antidote to reverse heparin, protamine sulfate, may be administered intravenously. Low-molecular-weight heparin is associated with fewer bleeding and immunological complications.
TABLE 13-3. ANTICOAGULANTS COMMONLY USED IN ACUTE CARE
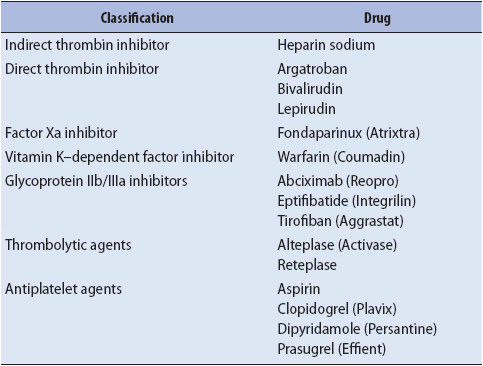
Warfarin acts by inhibiting the production of vitamin K–dependent clotting factors. Effects from warfarin take several days to be observed after initiation of the drug, but may persist for many days following administration. If significant bleeding occurs while on warfarin, replacement of vitamin K–dependent factors by use of fresh frozen plasma may be necessary. Giving replacement vitamin K may also be helpful, but its effectiveness depends on the time needed by the liver to synthesize new clotting factors.
Use of thrombolytic agents (alteplase, reteplase) to dissolve thrombi (pathologic clots) may result in patient bleeding from sites where a protective clot previously formed. These agents are used in combination with other anticoagulants, and may precipitate obvious or occult bleeding. Patients who receive anticoagulants are monitored for any sign of bleeding complications.
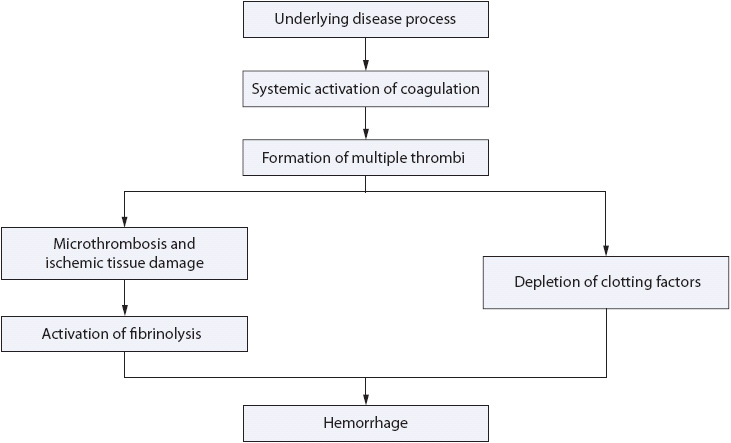
Disseminated intravascular coagulation is a complex coagulopathy which can develop in patients already critically ill from a wide variety of disorders (Table 13-4). The underlying condition triggers the release of proinflammatory cytokines, which activate the coagulation cascade and result in the formation of micro clots. The micro clots obstruct the capillaries of organs and tissues. This initiates a series of events which result in both bleeding and thrombosis (Figure 13-1). DIC can be seen in patients who are seriously ill with sepsis, traumatic injury, and extensive surgery. It is also seen in patients newly diagnosed with acute forms of leukemia and as a complication in cancer treatment.
TABLE 13-4. ETIOLOGIES OF DIC

During the process of DIC, stimulation of the clotting cascade rapidly depletes existing platelets and coagulation factors, consuming them faster than the body can replace them. Depletion of substrates of the coagulation process leaves the body at risk for spontaneous bleeding or hemorrhage from surgical sites, or even minimal trauma.
Multiple tiny clots are formed within the blood and flow to the small vessels where they are trapped. Microcirculatory thrombosis then leads to tissue ischemia, infarction, and organ dysfunction. Single or multisystem organ dysfunction may occur.
Simultaneous activation of fibrinolysis releases the enzyme plasmin. Plasmin breaks down some of the fibrin in a physiologic attempt to open the microcirculation, and this produces fibrin degradation products, including D-dimer. Anticoagulant pathways are impaired, further interfering with the balance needed for appropriate hemostasis. Clots are unable to form at new sites of injury, and existing clots are dissolved, leading to bleeding from both old and new sites. Because of the complex pathophysiology, clinical manifestations of DIC are likely to include bleeding from multiple sites and evidence of organ ischemia, including the skin, which may show ischemic changes in the hands and feet.
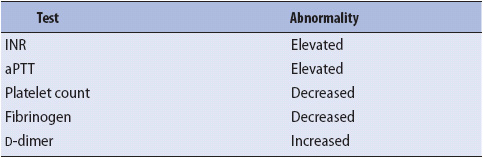
Laboratory diagnosis of DIC requires careful interpretation of coagulation panel results (Table 13-5). In many cases absolute certainty regarding a diagnosis of DIC may not be possible. With or without a clear diagnosis of DIC, a primary goal of therapy is to treat the underlying condition. In addition, supportive care is provided with volume replacement and support of vital organ systems, including ventilatory assistance. Significant bleeding is managed with blood and component therapy.
TABLE 13-5. LABORATORY RESULTS SUGGESTING DIC
Coagulopathy may be a subtle, occult process or a massive, obvious emergency. Assessment must encompass each body system, looking for evidence of abnormality in single or multiple components of the coagulation process.
Abnormal Platelet Numbers or Function
• Petechiae of skin or mucous membranes
• Spontaneous bleeding from gums or nose
• Thrombocytopenia
• Prolonged bleeding time
Abnormal Coagulation Factors
• Hemorrhage into subcutaneous tissue, muscle, or joints
• Ecchymosis, purpura
• Bleeding which responds slowly to local pressure
• Prolonged PT/INR, aPTT
• Decreased fibrinogen
• Decrease in level of specific coagulation factors
Skin/Musculoskeletal
• Oozing of blood from multiple sites, including incisions, intravascular catheters
• Petechiae
• Purpura
• Ecchymosis
• Ischemic changes in toes, fingers, nose, lips, ears
• Pain, swelling, and limited joint mobility
• Increased size of body part, increased girth
Neurologic
• Any change in level of consciousness, pupils, movement or sensation may indicate intracranial bleeding.
• Impaired vision with retinal hemorrhage.
• Headache
Gastrointestinal
• Blood in gastric aspirate
• Coffee ground emesis or gastric aspirate
• Melena or frank bloody stool
• Abdominal pain
• Enlarged liver or spleen
Genitourinary
• Hematuria
• Decreased urine output
• Vaginal bleeding
Cardiovascular
• Hypotension or labile blood pressure
• Hypovolemia and/or shock (with rapid loss of large volume of blood)
The management of coagulopathy varies with the type and severity of the disorder. The overall goal of therapy is to restore normal hemostasis and prevent/treat hypovolemic shock. Supportive care focuses on the control and prevention of further bleeding associated with activities of daily living and therapeutic interventions.
1. Treatment of quantitative platelet disorders may include transfusion of platelets. Transfusion is recommended for patients who are actively bleeding or prior to invasive procedures or surgery.
2. Destruction of platelets by immune mechanisms may be treated with steroids or IV immunoglobulin infusion. If related to use of heparin, then heparin is discontinued. Splenectomy may be performed for severe persistent problems where spleen sequestration is suspected.
3. Dysfunctional platelets may be treated by stopping the offending agent, such as aspirin or NSAIDs. Dialysis improves platelet function in patients with renal failure.
4. Acute replacement of coagulation factors can be accomplished with transfusion of fresh frozen plasma. Cryoprecipitate replaces fibrinogen, factor VIII, and von Willebrand factor. Recombinant factor VIIa may be used for persistent hemorrhage. For hemophiliac patients, factor VIII or factor IX concentrates are used to replace the specific factor deficiency.
5. Intravenous vitamin K may be used to treat warfarin-related bleeding or vitamin K deficiency.
6. Heparin therapy may be stopped, or the dosage decreased or reversed with IV protamine sulfate.
1. Modify nursing care measures to minimize trauma and prevent skin and mucous membrane breakdown:
• Provide gentle oral care.
• Use electric razor or refrain from shaving.
• Minimize use of automatic blood pressure cuffs to prevent skin trauma and subcutaneous bleeding; use manual cuffs.
• Minimize peripheral blood sampling.
• Avoid IM injections.
• Use specialty mattress, pad side rails; avoid restraint use.
• Handle patients gently when turning or moving.
• Remove adhesive dressings with care.
• Use low-suction setting to suction endotracheal tube and pharynx.
2. Modify nursing care procedures to control bleeding:
• Minimize traumatic procedures; apply direct pressure afterward for at least 5 to 10 minutes or until bleeding has stopped.
• Use ice packs on new hematomas or hemarthrosis.
• Do not dislodge or attempt to remove blood clots from areas of bleeding.
• Control environment to prevent hypothermia.
Carson JL, Grossman BJ, Kleinman S, et al. Red blood cell transfusion: a clinical practice guideline from the AABB. Ann Int Med; 2012. http://www.annals.org/content/early/2012/03/26. Accessed March 10, 2012.
Collins TA. Packed red blood cell transfusions in critically ill patients. Crit Care Nurse. 2011;31(1):25-34.
Field JJ, Vichinsky EP, DeBaun MR. Overview of the management of sickle cell disease. In: Tirnauer JS, ed. Up-To-Date. www.uptodate.com. Accessed January 30, 2013.
Gaspard KJ. Disorders of red blood cells. In: Porth CM and Matfin G, eds. Pathophysiology: concepts of altered health states, 8th ed. Philadelphia, PA: Wolters Kluwer Health; 2009.
George JN. Clinical manifestations and diagnosis of immune (idiopathic) thrombocytopenia purpura in adults. In: Tirnauer JS, ed. Up-To-Date. www.uptodate.com. Accessed January 30, 2013.
Kessler D, Shaz B, Grima K. Advances in blood transfusion. Am Nurse Today. 2012;7(3):8-12.
Kyles DM. Blood conservation and blood component replacement. In: Carlson KK, ed. Advanced Critical Care Nursing. St. Louis, MO: Saunders Elsevier; 2009.
Munro N. Hematologic complications of critical illness, anemia, neutropenia, thrombocytopenia and more. AACN Adv Crit Care. 2009;20(2):145-154.
Pagana KD, Pagana TJ. Mosby’s Manual of Diagnostic and Laboratory Tests, 4th ed. St. Louis, MO: Mosby Elsevier; 2010.
Rauen CA. Beyond the blood mess: hematologic assessment. Crit Care Nurse. 2012;32(5):42-46.
Rote NS, McCance KL. Structure and function of the hematologic system. In: Huether SE, McCance KL, eds. Pathophysiology: The Biologic Basis for Disease in Adults and Children, 6th ed. St. Louis, MO: Elsevier Mosby; 2010.
Schrier SL. Approach to the adult patient with anemia. In: Landaw SA, ed. Up-To-Date. www.uptodate.com. Accessed January 30, 2013.
Vichinsky EP. Overview of the clinical manifestations of sickle cell disease. In: Tirnauer JS, ed. Up-To-Date. www.uptodate.com. Accessed January 30, 2013.
Bonilla FA. Secondary immune deficiency due to miscellaneous causes. In: Feldweg AM, ed. Up-To-Date. www.uptodate.com. Accessed January 30, 2013.
Centers for Disease Control and Prevention. Guideline for isolation precautions: preventing transmission of infectious agents in healthcare settings. 2007. Access at www.cdc.gov.
Fishman JA. Approach to the immunocompromised patient with fever and pulmonary infiltrates. In: Thorner AR. ed. Up-To-Date. www.uptodate.com. Accessed January 30, 2013.
Freifeld AG, Bow EJ, Sepkowitz MJ, et al. Clinical practice guideline for the use of antimicrobial agents in neutropenic patients with cancer: 2010 update by the Infectious Diseases Society of America. Clin Infec Dis. 2011;52(4): e56-e93.
Kaplan JE, Benson C, Holmes KH, et al. Guidelines for the prevention and treatment of opportunistic infections in HIV-infected adults and adolescents. Recommendations from CDC, the National Institutes of Health, and the HIV Medicine Association of the Infectious Diseases Society of America. MMWR Recomm Rep. 2009;58(RR-4):1-207. Accessed 11/1/13
Porth CM. Disorders of white blood cells and lymphoid tissue. In: Porth CM, Matfin G, eds. Pathophysiology: Concepts of Altered Health States, 8th ed. Philadelphia, PA: Wolters Kluwer Health; 2009.
Relf MV, Shelton BK, Jones KM. Common immunological disorders. In: Morton PG, Fontaine DK, eds. Critical Care Nursing, 10th ed. Philadelphia, PA: Wolters Kluwer, Lippincott Williams & Wilkins, 2013.
Shelton BK. Caring for the immunocompromised patient. In: Carlson KK, ed. Advanced Critical Care Nursing. St. Louis, MO: Saunders Elsevier; 2009.
Wolff PB. Hematological and immune disorders. In: Sole ML, Klein DG, Moseley MJ. Introduction to Critical Care Nursing. 5th ed. St. Louis, MO: Saunders Elsevier; 2009.
Dressler DK. Coagulopathy in the ICU. Crit Care Nurse. 2012;32(5):48-59.
Ferraris VA, Brown JR, Despotis GJ, et al. 2011 update to the Society of Thoracic Surgeons and the Society of Cardiovascular Anesthesiologists blood conservation clinical practice guideline. Ann Thorac Surg. 2011;91(3):944-982.
Greenlaw D. Common hematological disorders. In: Morton PG, Fontaine DK, eds. Critical Care Nursing, 10th ed. Philadelphia, PA: Wolters Kluwer, Lippincott Williams & Wilkins; 2013.
James SH. Clots kill: hematologic pharmacology for ST-segment elevation myocardial infarction. Crit Care Nurse. 2012;32(6): 35-41.
Karch AM. Drugs that alter blood coagulation. Am Nurse Today. 2012;7(11):26-31.
Landaw SA, George JN. Approach to the adult patient with thrombocytopenia. In: Tirnauer JS, ed. Up-To-Date. www.uptodate.com. Accessed January 30, 2013.
Linkins LA, Dans AL, Moores LK, et al. Treatment and prevention of heparin-induced thrombocytopenia: antithrombotic therapy and prevention of thrombosis, 9th ed. American College of Chest Physicians evidence-based practice guidelines. Chest. 2012:141(2 suppl): e495S-e530S.
Mayer, B. Hematologic disorders and oncologic emergencies. In: Urden, LD, Stacy KM, Lough ME, eds. Critical Care Nursing. 6th ed. St. Louis, MO: Elsevier Mosby, 2010.
Rice TW, Wheeler AP. Coagulopathy in critically ill patients: part 1. Chest. 2009;136:1631-1643.
Warkentin TE. Heparin-induced thrombocytopenia in critically ill patients. Crit Care Clin. 2011;27(4):805-823.
Wheeler AP, Rice TW. Coagulopathy in critically ill patients: part 2. Chest. 2010;137:185-194.
*As noted earlier, immune-compromised patients may not show any of these clinical signs and symptoms. They may have some, they may have none. The neutropenic patient may have very subtle, suppressed signs of sepsis; thus heightened vigilance is necessary so that essential treatment is provided.