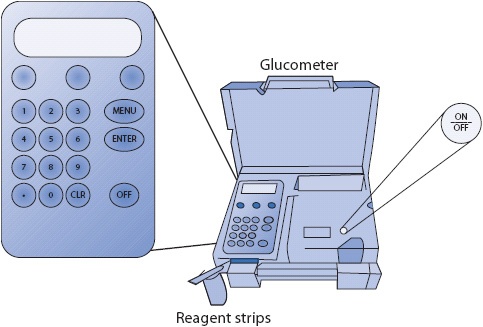
Figure 16-1. Reagent strip and glucometer for bedside testing of blood glucose levels.
KNOWLEDGE COMPETENCIES
1. Outline the nursing management of patients receiving blood glucose monitoring.
2. Describe the etiology, pathophysiology, clinical presentation, patient needs, and principles of management for:
• Hyperglycemic states
• Diabetic ketoacidosis
• Hyperosmolar hyperglycemic states
• Acute hypoglycemia
• Syndrome of inappropriate antidiuretic hormone secretion
• Diabetes insipidus
Good glycemic control is paramount to improved morbidity and mortality in critically and acutely ill patients. Frequent assessments of blood glucose levels in these patients are commonly performed at the bedside using small quantities of blood obtained from finger sticks, or via arterial lines or central venous catheters. A drop of blood is placed onto a chemical reagent strip and inserted into a portable glucometer (Figure 16-1). This point-of-care (POC) glucometer bedside analysis allows for more rapid interventions of glycemic disorders than is possible from laboratory glucose analysis. Newer technologies have greatly enhanced both the usability and accuracy of bedside glucometers.
Figure 16-1. Reagent strip and glucometer for bedside testing of blood glucose levels.
Despite the obvious benefits of glucometers and improved technology, inaccuracies of glucose measurements can occur. However, recent studies have found discrepancies of 10% to 36% between glucometer values and laboratory glucose values. While nationally an acceptable discrepancy between glucometer values and laboratory values is +/– 20%, such a variance range may be detrimental. Any large discrepancies between laboratory and the bedside glucometers should be investigated. This is particularly true when blood glucose values are in the lower ranges. In this instance, a blood glucose reading of 70 mg/dL may actually be closer to 54 mg/dL—a range that may require immediate action. There are many reasons for these discrepancies, but one important reason is that these glucometers were not originally developed or intended for use in critically ill and/or unstable patients. The FDA is currently weighing new, stricter industry guidelines for glucometer efficacy and an acceptable range for POC and laboratory glucose value discrepancies.
While POC glucometers were not specifically designed for potentially unstable patients, they do provide timely and reasonably accurate glucose monitoring. One common source of errors in glucose measurement is the incorrect operation of the glucometer device. Other causes may include the use of expired glucose reagent strips or insufficient blood application on the strip. Exogenous glucose contamination of blood samples for testing can also impair testing accuracy. This can happen as a result of an incorrectly withdrawn blood sample from arterial or central venous line. Some researchers have found that arterial sampling may overestimate glucose levels. Also, laboratory glucose analysis errors may occur if venous sampling is obtained at a site above an intravenous (IV) infusion of a glucose-containing solution. If any doubt exists about the accuracy of the glucometer value, a repeat measurement should be done or a laboratory analysis obtained. Tips for glucometer use are reviewed in Table 16-1.
TABLE 16-1. TIPS FOR BLOOD GLUCOMETER (BGM) USE

Several clinical conditions may influence POC glucose measurements. Shock or hypotensive states, along with vasopressor use, can lead to inadequate tissue perfusion in fingers thereby increasing inaccuracies of fingerstick glucometer analysis (usually overestimation of the blood glucose). Hematocrit levels may also adversely affect glucose levels. A hematocrit value of less than 34% can result in overestimation of blood glucose, while hematocrit values of greater than 55% may lead to underestimation of the blood glucose. Other naturally occurring blood substances can also interfere with glucose measurement accuracy, such as high triglycerides, which are associated with higher glucose levels. Oxygenation and uric acid levels may result in erroneously low blood glucose levels. Finally, patients receiving certain drugs, such as acetaminophen, L-dopa, and ascorbic acid, may yield erroneous results because the drug may chemically affect some reagent strips. Manufacturers of reagent strips are currently endeavoring to develop strips impervious to drug interference. Clinical situations that may affect the accuracy of POC glucose monitoring are listed in Table 16-2.
TABLE 16-2. CLINICAL SITUATIONS THAT MAY AFFECT THE ACCURACY OF POC BLOOD GLUCOSE MEASUREMENTS

Several options may help provide greater accuracy in bedside glucose monitoring. Studies suggest that glucose levels obtained from standing unit-based blood gas and chemistry analyzers, are more accurate than those from glucometers. Because these unit analyzers provide essential chemistry values such as potassium simultaneously with glucose values, they may quickly alert the nurse to critical values; for example, insulin induces a shift of potassium from the extracellular to the intracellular compartment, which can lead to hypokalemia and subsequently life-threatening arrhythmias. Knowing the potassium in conjunction with the glucose level may potentially avert serious patient decline. In cases of instability and/or rapidly changing interventions and treatments, it may be especially important to use a standing POC unit versus a standalone bedside glucometer.
It is possible that continuous glucose monitoring (CGM) devices that are currently used by millions of people with insulin-dependent diabetes in the outpatient setting may one day be accurately used in critically and acutely ill patient populations as well. These devices use subcutaneous glucose sensors and have demonstrated that they optimize insulin therapy, metabolic control, and safety in the outpatient setting. Data from the CGM can be downloaded to a computer for a visual display of the patient’s continuous glucose levels, as well as daily and weekly glucose trends. These devices also provide safety benefits as they come with hypo- and hyperglycemia alarms. Currently, only the subcutaneous CGM is FDA-approved; however, in future, intravenous glucose sensors are likely to be developed and available for use in hospitalized patients as well.
Prior to hospital discharge, patients requiring ongoing glucose monitoring should be evaluated for competency using the glucometer. It is important to first determine the patient’s fasting glycemic goal. Underlying patient morbidities, cognitive skills, frailty, and age affect glycemic target goals. In a relatively healthy outpatient, a target fasting glucose between 85 and 140 mg/dL is usually acceptable. Two-hour postprandial, blood glucose levels should be kept less than 180 mg/dL whenever possible. These goals are achieved through the use of oral hypoglycemic agents, insulin, and in outpatients, injectable incretins (ie, GI hormones that increase the release of insulin from beta cells and enhance proper glucose metabolism). Accurate glucometer measurements are essential to safely achieve glycemic targets.
Ideally, patients should test their blood glucose levels before each meal and at bedtime to evaluate the effectiveness of their therapy, especially if on insulin therapy. This can also improve safety if ongoing insulin dose adjustments are necessary. However, frequent glucose-monitoring schedules may not be feasible, and some patients may struggle with adherence to rigid self-monitoring schedules. In these instances, patients are encouraged to test at least once in a day at alternating times to track glucose patterns. Fasting and preprandial glucose levels, as well as bedtime, postexercise, and 2-hour postprandial glucose measurements are recommended. At the very least, “paired testing” performed once or twice a week can effectively trend blood glucose levels. Paired tests are performed by checking blood glucose just prior to a meal and repeated 2 hours after the meal.
Diabetes is the fourth most common comorbidity plaguing hospitalized patients. This disease, along with the specter of hyperglycemia, carries a legacy of vascular compromise and is associated with a three- to fourfold increase in hospital morbidity and mortality. A more troubling fact is that 12% of patients, without a history of diabetes, will develop hyperglycemia during hospitalization. Unfortunately, these patients have a nearly 18-fold increased risk of in-hospital mortality—far greater than those with known diabetes. This increase in mortality is believed to be related to the patients’ exaggerated response to physiologic stress and lack of physiologic resiliency.
Hyperglycemia occurs in hospitalized patients due to natural metabolic responses to acute injury and stress. During acute illness, the liver produces and releases glucose in response to glucocorticoids, catecholamines, growth hormone, and various cytokines (interleukin-6 [1L-6], interleukin-1a [1L-1a], and tumor necrosis factor-alpha). As a result, fat and protein are catabolized and blood glucose surges. Conditions such as myocardial infarction, stroke, surgery, trauma, pain, and sepsis may cause the release of these biological mediators and counter-regulatory hormones. In essence, the greater the stress response, the higher the blood glucose will be. To help minimize the adverse outcomes associated with hyperglycemia, rigorous glucose monitoring and effective management of blood glucose is essential. This is usually accomplished in critically ill patients utilizing frequent blood glucose testing paired with continuous insulin infusion. Standard infusion protocols, or standing order sets, are often used to maintain glucose values in the targeted range.
A great deal of controversy exists related to how tightly glucose should be controlled in the hospitalized patient. Some studies have shown that tight glucose control using insulin infusions (pre- and postprandial blood glucose target near 110 mg/dL) can improve morbidity and mortality and reduce infections in the critically ill, postsurgical cardiovascular patient population, despite an increased risk of hypoglycemia. Unfortunately, this mortality benefit has not been demonstrated in medical ICU patients or patients in the general wards or progressive care units. In fact, intensive insulin therapy in medical ICU population has been found to slightly increase mortality due to the associated increase in hypoglycemia. Based on these recent findings, the American Diabetes Association and American Association of Clinical Endocrinologists (AACE) have jointly recommended a revised glucose target of 140 to 180 mg/dL in the ICU setting, and between 100 and 180 mg/dL for most patients admitted to general medical-surgical units. In the medical-surgical population, it is advised that preprandial sugars be less than 140 mg/dL and random or postprandial sugars be less than 180 mg/dL.
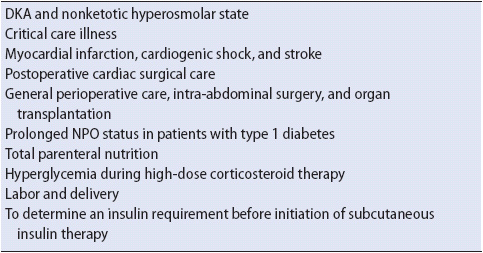
An insulin infusion is preferable in all hyperglycemic, critically and acutely ill patients, not just those experiencing diabetic ketoacidosis (DKA) and hyperosmolar hyperglycemic states (HHS). Patients at greatest risk are those undergoing major cardiovascular surgery and organ transplants, those with decompensated diabetes (such as DKA and HHS), those in cardiogenic shock or renal failure, and in those receiving high-steroid doses (Table 16-3). These patients often have increased hepatic glucose production, impaired insulin release and sensitivity, and widely fluctuating insulin needs.
TABLE 16-3. COMMON INDICATIONS FOR IV INSULIN INFUSIONS
IV insulin infusions are also preferred over subcutaneous insulin injections due to erratic tissue absorption in the presence of hypotension, generalized edema, and use of vasopressors. Many hospitals have adopted insulin infusion protocols for use in critical and acute care areas. An effective insulin infusion protocol should incorporate an algorithm that easily adapts to individual patient responses, attains the glucose goal quickly with minimal hypoglycemic risk, and may be used hospital wide. Infusion rates should be increased, decreased, or stopped temporarily based on blood glucose readings and the prescribed algorithm. Whatever protocol is used, it is important to consider the degree of insulin resistance. Patients who are highly insulin resistant may require a much higher hourly infusion rate.
Along with an insulin infusion, hyperglycemic patients will require a tandem infusion of 0.9% normal saline or 5% dextrose and 0.45% normal saline, at a rate commensurate with the patients’ fluid requirements. A dextrose solution is always preferred in patients with type 1 diabetes. Most patients will also require simultaneous infusion of potassium as insulin is known to drive potassium into cells, especially into liver and muscle cells, which may increase the risk of hypokalemia.
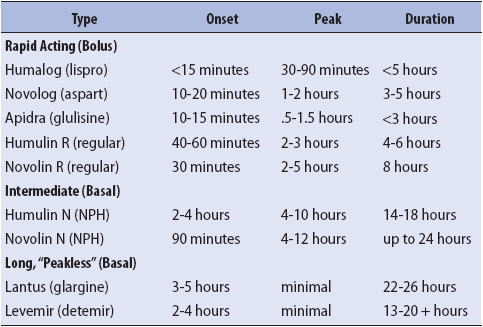
When the infusion is discontinued, subcutaneous insulin is often started using a basal insulin to cover glucose produced endogenously by the liver. Bolus insulin, also called correctional insulin, is used to cover food intake and intermittent surges of blood glucose. It is essential that all type 1 diabetics receive basal insulin or DKA will ensue. If the patient is to receive subcutaneous, multi-dose insulin (MDI) following the infusion, then both basal and bolus insulin should be provided a minimum of 30 minutes prior to discontinuing the IV as IV insulin disappears from the bloodstream within 5 minutes after discontinuation of IV insulin therapy. It is important to note that the effects of long-acting, basal insulin, such as glargine and detemir, do not appear for several hours after injection and should be administered 2 hours prior to discontinuing the insulin infusion. Insulin actions by type of insulin are listed in Table 16-4. After discontinuation of the insulin infusion POC blood glucose testing should continue before meals and at bedtime in patients who are eating, or every 4-6 hours in patients who are NPO or receiving continuous enteral feeding.
TABLE 16-4. INSULIN ACTION CHART BY TYPE
Diabetes ketoacidosis (DKA) and hyperglycemic hyperosmolar (HHS) are two extremes in the spectrum of decompensated diabetes. The incidence of DKA is defined as acute hyperglycemia with acidosis, and HHS is classified as acute hyperglycemia without acidosis (nonketotic).
Diabetes is a metabolic disease that results in inadequate uptake of glucose by cells, resulting in hyperglycemia. There are 13 forms of diabetes mellitus (DM) that fall within one or both main diabetes classifications: types 1 and 2 diabetes. Type 1 DM is an autoimmune disorder and often has a juvenile or early adulthood onset, although it can occur at any age. When it occurs in middle or old age, type 1 DM is referred to as latent autoimmune diabetes in adults (LADA). The key disorder in type 1 DM is minimal or absent insulin secretion by the pancreatic beta islet cells. Type 2 diabetes usually occurs in older adults, but can be seen in youth, and is associated with impaired insulin receptor sensitivity. Insulin production in type 2 diabetes may initially be high or normal, then falls dramatically as the disease progresses. Although hyperglycemia is a shared feature, the etiology, risk factors, pathophysiology, and management priorities vary considerably for each classification of diabetes.
Insulin is normally released from the pancreas by beta islet cells (Islets of Langerhans) in response to an increase in blood glucose. Insulin is necessary for cellular uptake of glucose by most cells in the body (except brain and liver cells). Without insulin, the glucose fails to enter cells and accumulates in the blood, resulting in hyperglycemia and a vascular inflammatory state. Cells deprived of glucose begin to starve, triggering a mobilization of stored glucose via the breakdown of protein and fat (gluconeogensis) and release of stored glucose from the liver (glycogenolysis). This triggers a complex series of physiologic processes that account for the major signs and symptoms associated with DKA and HHS.
The most common scenarios associated with DKA are underlying or concomitant infection (40%), missed insulin treatments (25%), and newly diagnosed, previously unknown diabetes (15%). Other associated causes make up roughly 20% in the various series. Among the other causes are myocardial infarction, stroke, trauma, and pancreatitis. Although DKA is primarily a complication of type 1 diabetes, it can occur (rarely) in some forms of type 2 diabetes under conditions of extreme stress, including “ketone-prone” type 2 diabetes a disorder found in African American males (Table 16-5).
TABLE 16-5. CAUSES OF DKA

In general, DKA consists of the biochemical triad of hyperglycemia, ketonemia, and metabolic acidosis (with a large anion gap). This disorder is typically characterized by hyperglycemia (> 300 mg/dL), low bicarbonate level (< 15 mEq/L), and acidosis (pH < 7.30) with ketonemia and ketonuria. While definitions vary, moderate DKA can be categorized by pH of less than 7.2 and serum bicarbonate less than 10 mEq/L, whereas severe DKA has pH of less than 7.1 and bicarbonate less than 5 mEq/L.
DKA can develop in less than 24 hours. The initiating event in DKA is an insufficient or absent level of circulating insulin. This insulin deficiency results in increased fatty acid metabolism, increased liver gluconeogenesis (formation of glucose from amino acids and proteins), and increased secretion of counterregulatory hormones, including glucagon and the stress hormones (catecholamines, cortisol, and growth hormone). These hormones counteract the glucose-lowering effects of insulin and are released in response to stress and other stimuli. The pathophysiology of DKA can be organized into two main components: fluid volume deficit and acid-base imbalance (Figure 16-2).
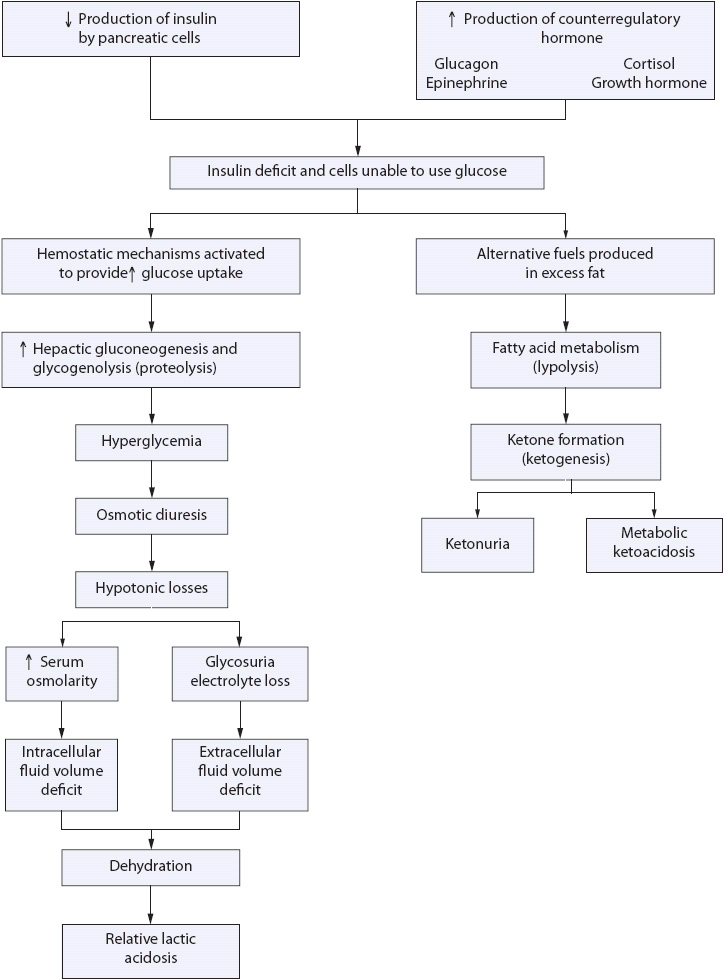
Figure 16-2. Pathogenesis of DKA.
Because of the insulin deficiency, there is both hyperglycemia and increased amino acid release from cells. The stress response in the body leads to metabolic decompensation, and stress hormones further trigger a rise in plasma glucose and ketones. The hyperglycemia causes an osmotic diuresis and hypotonic losses leading to fluid volume deficits (intracellular and extracellular) and electrolyte losses. As serum glucose exceeds the renal threshold, glycosuria results. In the absence of insulin, protein stores are also broken down by the liver into amino acids and then into glucose for energy. This further increases serum blood glucose, increases urine glucose, and worsens the osmotic diuresis and ketonemia. Urinary losses of water, sodium, magnesium, calcium, and phosphorus cause an increase in serum osmolality and decreased electrolyte levels. Potassium levels may be increased or decreased, depending on the amount of nausea and vomiting, acid-base balance, and fluid status of the patient. This hyperosmolality causes additional fluid shifts from the intracellular to the extracellular space, increasing dehydration. Hypovolemic shock can result from severe fluid losses in DKA. Volume depletion decreases glomerular filtration of glucose and creates a cycle of progressive hyperglycemia. The increase in serum osmolarity also is thought to further impair insulin secretion and promote insulin resistance. The altered neurologic status frequently seen in these patients is due primarily to brain cell dehydration and serum hyperosmolarity.
Cells without glucose starve and begin to use existing stores of fat and protein to provide energy for body processes (gluconeogenesis). Fats are broken down faster than they can be metabolized in the liver, which results in an accumulation of ketone acids. These ketone acids are usually cleared in peripheral tissues. If the ketogenic pathway is overwhelmed ketone acids accumulate in the blood stream where hydrogen ions (H+) dissociate, causing a profound metabolic acidosis. Acetone is formed during this process and is responsible for the “fruity breath” found in these patients.
Metabolic acidosis may be worsened with severe fluid volume deficits because hypovolemia results in tissue hypoperfusion and production of lactic acids from anaerobic metabolism. Excess lactic acid results in what is called increased anion gap (increased body acids). Sodium, potassium, chloride, and bicarbonate are responsible for maintaining a normal anion gap in the body which is normally less than 12 to 14 mEq/L (Table 16-6). The anion gap represents the difference between the cations (Na+, K+) and anions (Cl–, HCO3–). Ketone accumulation, a by-product of gluconeogenesis, causes an increase in the anion gap more than 14 mEq/L.
TABLE 16-6. CALCULATION OF ANION GAP (NORMAL <12 mEq/L)a

The normal physiologic response to metabolic acidosis is to produce bicarbonate to buffer the ketones and H+ ions. The patient with DKA often has diminished bicarbonate levels because of the osmotic diuresis. The respiratory system attempts to compensate by blowing off carbon dioxide to restore normal blood pH. This explains the deep rapid breathing, called “Kussmaul respirations,” often seen in these patients.
Metabolic acidosis also results in potentially life-threatening electrolyte imbalances. Serum potassium is elevated initially in DKA probably due to potassium shifts from the intracellular to the extracellular space because of the acidosis. Later, hypokalemia is common because of insulin-induced transfer of plasma potassium into cells and increased urinary excretion of potassium with the osmotic diuresis.
An 18-year-old woman was admitted to the MICU with a diagnosis of DKA. She was stressed about numerous school examinations, had run out of her basal insulin, glargine, and was only taking random, short-acting insulin to cover her meals, as she neglected to check her blood sugars. During the past 2 days she had been experiencing flu-like symptoms (vomiting, abdominal cramping). On arrival in the ED, she was flushed and vaguely confused, clutching an extralarge cup of diet soda. Significant findings on her admission profile were:
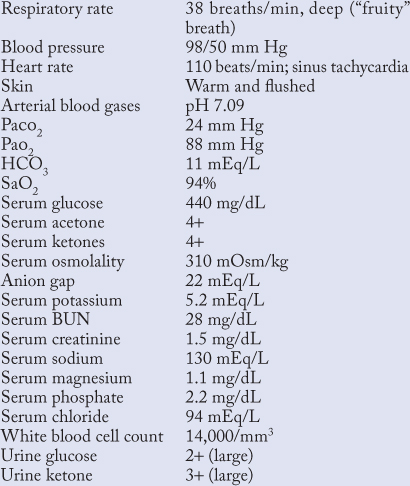
Case Question 1. True or False–Intravenous hydration should be given prior to insulin administration.
Case Question 2. True or False–Sodium bicarbonate IV is indicated in this case.
Case Question 3. True or False–Despite a mildly-elevated serum potassium, a tandem IV with potassium should still be added after hydration and an insulin infusion have been initiated.
1. True
2. False (used if pH is < 7.0)
3. True
The pathogenesis of HHS is similar to the pathogenesis of DKA with the following differences. HHS is classified as hyperglycemia with profound dehydration in the absence of ketosis. The onset of hyperglycemia in HHS is progressive. Many of these patients have a history of type 2 DM with some circulating insulin levels. The extremely severe hyperglycemia in HHS results in profound extracellular fluid volume contraction, marked intracellular dehydration, and excessive loss of electrolytes. In addition, because there is some insulin, secretion lipolysis is suppressed. Therefore, there is no overproduction of ketones and no specific physical signs and symptoms of ketosis (no Kussmaul respiration, renal excretion of ketones, abdominal pain, nausea, vomiting, or anorexia). The lack of these emergent signs and symptoms may cause these patients to not seek early treatment. Sustained osmotic diuresis results, leading to massive volume losses, electrolyte imbalance, and central nervous system (CNS) dysfunction. Mortality rates, therefore, are higher with HHS, because of the severe volume loss and because it occurs more frequently in a chronically ill patients. Death results from CNS depression of vital body functions (cardiac and respiratory centers in the brain are depressed), cerebral edema, cardiovascular collapse, renal shutdown, and vascular embolism.
A 72-year-old man was admitted to the MICU with a diagnosis of hyperglycemic crisis. He lives alone with his small dog. His daughter dialed 911 after finding her father unresponsive at his home. She reported that he had complained of flulike symptoms 3 weeks earlier. His history is significant for heart failure and type 2 DM. His daily medications include carvedilol 6.25 mg orally twice a day, lipitor 40 mg once a day, lisinopril 20 mg orally once a day, furosemide (Lasix) 20 mg orally twice a day, KCl 20 mEq/d orally, and glipizide 10 mg orally twice a day. On arrival in the ED he was comatose. Significant findings on his admission profile were:

Case Question 1. True or False–Because of the extremely high blood sugar, insulin administration should precede IV hydration.
Case Question 2. True or False–The relative metabolic acidosis is related to ketone acid accumulation.
Case Question 3. True or False–The mortality of HHS exceeds that of DKA.
1. False (Hydration more effectively lowers blood sugar and reduces stroke risk)
2. False (It is due to lactic acid accumulation related to dehydration)
3. True

The management of the patient in acute DKA and HHS revolves around six primary areas: fluid replacement, treatment of hyperglycemia, electrolyte replacement, treatment of any underlying disorders, prevention and management of complications, and patient/family teaching.
Treatment of intracellular and extracellular fluid volume deficits is a priority for both DKA and HHS to restore intravascular volume and prevent cardiovascular collapse. Initial volume replacement is based on assessment of vascular status.
1. Administer normal saline (0.9%). The choice of IV fluid depends on the initial blood pressure readings and the serum sodium level. The presence of hyperglycemia and dehydration masks the true serum sodium level, requiring a correction of serum sodium levels prior to IV fluid selection (Table 16-7). IV fluids are generally infused at rapid rates (1000 to 2000 mL in the first hour, 1000 mL in the second hour, and then at 500 mL/h) until fluid volume is restored or initially around 15 to 20 mL/kg/h.
2. Titrate the rate of infusion based on urine output, mean arterial blood pressure, and central venous pressure measurements. Typically, the patient with HHS has more profound fluid volume deficits, but because the patient is older and often has other underlying medical problems, the rate of fluid replacement needs to be carefully titrated. Serum glucose falls with initiation of fluids alone. It is critical that insulin therapy not be started without simultaneously correcting the fluid deficit. Otherwise, the result is an acute loss of vascular volume, worsening of the hypernatremia, shock, and increased risk of mortality.
3. Change IV fluid to 5% dextrose with 0.45 NaCl at 150 to 200 mL/h when serum glucose reaches 250 mg/dL. Maintain insulin therapy.
TABLE 16-7. CORRECTION OF SERUM SODIUM LEVELS IN THE PRESENCE OF HYPERGLYCEMIA

In both DKA and HHS some insulin replacement is needed, although the requirements in DKA are typically lower than HHS.
1. Regular insulin 0.15 U/kg as IV bolus.
2. Initiate low-dose IV insulin at a rate of 0.1 U/kg/h. If serum glucose does not fall by 50 to 70 mg/dL in the first hour, double insulin infusion on an hourly basis until glucose falls by 50 to 70 mg/dL.
3. Monitor serum glucose levels closely and titrate insulin infusion accordingly. Once the serum glucose reaches 250 mg/dL, the insulin infusion should be decreased to a rate of 2 to 4 U/h and the IV fluids changed to half normal saline with glucose (D5-1/2NS). This ensures that hypoglycemia does not occur during ongoing treatment of the acute condition. It is essential that insulin infusion continues in the patient with DKA until the serum pH is corrected to avoid intracellular hypokalemia. Additional glucose may be needed to achieve this outcome. Glucose-containing solution should also be started in the patient with HHS when serum glucose reaches 250 to 300 mg/dL to protect against cerebral edema.
Electrolyte deficits are usually present in both DKA and HHS due to the osmotic diuresis. Hypokalemia may be masked by acidosis. Potassium levels rise 0.6 mEq/L for every 0.1 drop in pH.
1. Administer potassium supplements according to serum levels:
• If serum K+ is < 3.3 mEq/L, hold insulin and administer 40 mEq K+/h (2/3 KCl and 1/3 KPO4–) until K+ is > 3.3 mEq/L.
• If serum K+ is > 5.0, hold K+ and check K+ every 2 hours.
• If serum K+ is > 3.3 mEq/L or < 5.0 mEq/L give 20 to 30 mEq K+ in each liter of volume replacement.
Replacement of potassium is a priority during the correction of hyperglycemia to avoid hypokalemia during rehydration, when potassium moves into the cell along with glucose. To avoid cardiac arrhythmias associated with hypokalemia, delay insulin administration until serum potassium levels are greater than 3.3 mEq/L. The rate of potassium chloride infusion should be adjusted according to frequently monitored serum potassium levels and the urine output.
2. Monitor magnesium, calcium, and phosphate levels every 2 hours during rehydration. Hemodilution may further decrease serum levels of these electrolytes. Magnesium and calcium replacements are given based on serum levels. Total body phosphorous levels are depleted due to osmotic diuresis. This may result in impaired cardiac and respiratory functions. Phosphate deficiencies are usually corrected with volume replacement. If needed, the administration of potassium phosphate 20 mEq/L is the best method of phosphate replacement as it replaces both potassium and phosphate simultaneously. Phosphate replacements should not be administered in patients with renal failure. If hypokalemia is refractory to potassium replacement, magnesium replacement should be considered.
3. Assess need for bicarbonate therapy:
• If pH is < 6.9, dilute NaHCO3– (100 mmol) in 400 mL H2O. Infuse at 200 mL/h.
• If pH is 6.9 to 7.0, dilute NaHCO3– (50 mmol) in 200 mL H2O. Infuse at 200 mL/h.
• If pH is > 7.0, hold NaHCO3–.
Repeat HCO3 administration every 2 hours until pH is > 7.0. Monitor serum K+ closely.
The precipitating cause for the hyperglycemic emergency needs to be determined. Underlying infection is a common precipitating factor in both DKA and HHS.
1. Investigate precipitating factors utilizing the following tests: urinalysis, complete blood count, ECG, chest x-ray, and appropriate cultures. Administer antibiotics as appropriate if infection is suspected.
2. Obtain history from patient and family about the possibility of missed insulin doses.
1. Monitor serum glucose, electrolytes (sodium and potassium), and arterial blood gases every 1 to 2 hours until normal levels are attained.
2. Measure serum phosphate and magnesium initially and repeat as necessary.
3. Monitor temperature, blood pressure, pulse, respiratory rate, pulse oximetry, urinary output, and central venous pressure at frequent intervals.
4. Evaluate neurologic status at frequent intervals. Institute seizure precautions if cerebral edema is suspected. Institute measures to avoid aspiration in patients with altered mental status. Administer dexamethasone and mannitol if appropriate.
5. Titrate fluid replacement carefully to prevent heart failure. Auscultate lung sounds frequently during fluid replacement.
6. Administer anticoagulants as ordered. Hyperosmolar patients are at great risk for developing thrombosis.
Particularly in type I DM, the key to prevention of recurrent DKA is adequate patient education regarding diabetes management. Contact the diabetes educator (if available) to help teach the skills needed to manage diabetes once the patient is stable and ready to receive information. Table 16-8 outlines the required skills for diabetic management. Return demonstrations by the patient or designated caregiver are essential. Instruction regarding the need for routine medical follow-up and the availability of hospital and community resources is also an important component of the diabetes management plan. The patient is typically discharged on the inpatient insulin doses (via multiple dose insulin or insulin pump). Sometimes a patient who was on pre-admission insulin may resume the pre-admission doses unless an adjusted dose is required due to weight loss, decreased renal function, or marked increase in exercise (typically less insulin is required with those conditions).
TABLE 16-8. SKILLS FOR DIABETIC MANAGEMENT

Hypoglycemia is a blood glucose level less than 60 mg/dL and is a common endocrine emergency. Hypoglycemia results from the imbalance between glucose production and glucose utilization. Of the acute complications, hypoglycemia is most common in insulin-dependent (types 1 and 2) diabetics. It also can occur with type 2 diabetics who are treated with oral hypoglycemic agents, especially sulfonylureas like glipizide, glyburide, and glimepiride.
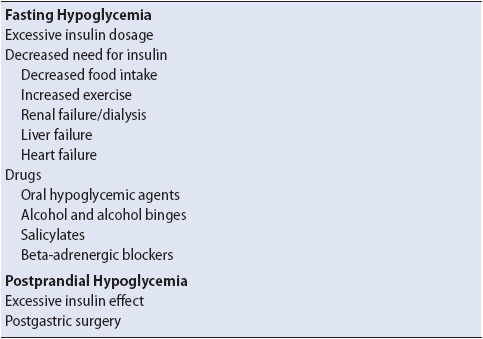
Hypoglycemia can be divided into two categories: fasting hypoglycemia (> 5 hours after a meal) and postprandial hypoglycemia (1-2 hours after a meal) (Table 16-9). Fasting hypoglycemia occurs when the normal physiologic response (gluconeogenesis and glycogenolyisis) to a falling glucose level is altered and there is an imbalance in glucose production and utilization. Hypoglycemia in a diabetic person is most commonly caused by excessive insulin or oral hypoglycemic agent, too much exercise, or not enough caloric intake. Exercise can lead to increased insulin sensitivity immediately or hours later as sugar moves readily into muscles. One cause of postprandial hypoglycemia is gastric bypass surgery because surgical alterations cause smaller amounts of foods to be ingested and this food passes more rapidly through the small intestine. In addition, the resultant rapid weight loss decreases insulin resistance further, increasing the risk of hypoglycemia.
TABLE 16-9. CAUSES OF HYPOGLYCEMIA (PARTIAL LISTING)
Glucose is the obligate fuel for the brain and CNS. The brain is unable to synthesize or store glucose and must rely on circulating plasma blood glucose levels for survival. As blood glucose declines rapidly, epinephrine, glucagon, glucocorticoids, and growth hormones are released. Patients exhibit adrenergic symptoms—tachycardia, anxiety, sweating, trembling, and hunger. These symptoms can occur even if the blood glucose is normal but there is a sudden acute decline (ie, blood glucose level rapidly decreases to 80-90 mg/dL). In moderate to severe hypoglycemic reactions, the CNS is affected, signifying that the brain is being deprived of the glucose it needs.
Hypoglycemic unawareness is an autonomic neuropathy with potentially serious consequences. Hypoglycemic unawareness is defined as the loss of adrenergic symptoms of hypoglycemia that prompt a patient to act to prevent the progression of severe hypoglycemia and it results from altered counterregulation systems as described. Both type 1 and 2 diabetics may have deficiencies in counterregulation systems.
• Mild hypoglycemic symptoms (adrenergic response)
– Diaphoresis (most common)
– Tremors
– Shakiness
– Tachycardia
– Paresthesias
– Pallor
– Excessive hunger
– Anxiety
• Moderate to severe hypoglycemic symptoms (CNS or neuroglycopenic symptoms)
– Headache
– Inability to concentrate
– Mood changes
– Drowsiness
– Irritability
– Confusion
– Impaired judgment
– Slurred speech
– Staggering gait
– Double or blurred vision
– Morning headaches
– Nightmares
– Psychosis (late)
– Seizures
– Coma
• Serum blood glucose level for fingerstick glucose < 60 mg/dL
The management of the patient with acute hypoglycemia depends on the severity of the reaction. Principles of management include normalization of blood glucose concentrations and patient teaching.
Treatment of the hypoglycemia depends on its severity as described below.
1. Administer 10- to 15-g carbohydrate (Table 16-10). Follow in 10 minutes with another 10 to 15 g if the condition does not improve.
2. Obtain a blood glucose measurement.
3. If the next meal is more than 2 hours away, provide the patient with a complex carbohydrate (ie, 4-oz milk).
4. If patient is not alert enough to swallow or unable to do so, inject 1 to 2 mg glucagon. If the patient cannot swallow and has a feeding tube, administer a liquid source of glucose (regular, non-diet soda).
TABLE 16-10. EXAMPLES OF FOODS WITH 10 TO 15 G OF CARBOHYDRATE EQUIVALENTS FOR TREATMENT OF MILD HYPOGLYCEMIC REACTIONS

1. Administer IV glucose. The initial bolus is 50% dextrose (equivalent of 25-g glucose) followed by a continuous IV infusion until oral replacement is possible.
2. Glucagon Hcl 1 to 2 mg IV/IM/SC may be given in the hospital or at home and repeated every few hours.
3. Provide for patient rest.
4. Monitor glucose levels frequently for several hours.
The best treatment for hypoglycemia is prevention.
1. Teach the early signs and symptoms of hypoglycemia. Instruct the patient to always carry a source of fast-acting carbohydrate (see Table 16-10).
2. Advise the patient not to skip or delay meals and to limit alcohol to no more than 2-oz hard liquor, 8-oz wine, or 24-oz beer per day. It is advisable to never drink on an empty stomach. When an alcoholic beverage is consumed, it is prudent to ingest some protein-rich calories.
3. Evaluate the patient’s pattern of blood glucose self-monitoring.
4. Teach the patient and family or friends how to give glucagon for severe reactions.
5. Stress the importance of wearing visible diabetes identification.
6. Assess the patient’s pattern of activity and alert the patient to the risk of hypoglycemia within minutes to five hours following exercise.
Antidiuretic hormone (ADH), also known as arginine vasopressin (AVP), is produced by the hypothalamus and is stored in the posterior pituitary gland. ADH exerts its primary effects in the distal collecting tubules of the kidneys where it decreases water excretion, conserving body water, thereby increasing urine concentration (osmolality) and hemodilution. Osmoreceptors in the hypothalamus monitor changes in blood osmolality. An increase of osmolality by 2% leads to ADH release by the posterior pituitary. In high concentration, usually via exogenous administration, ADH has potent vasopressor effects along with pro-coagulant (platelet aggregation) properties. The syndrome of inappropriate antidiuretic hormone (SIADH) and diabetes insipidus (DI) are the most common disorders associated with ADH secretion in the critically ill.
The syndrome of inappropriate antidiuretic hormone is characterized by excessive release of ADH unrelated to the plasma osmolality, or the concentration of electrolytes and other osmotically active particles. Normal mechanisms that control ADH secretion fail, causing impaired water excretion and profound hyponatremia. SIADH is a syndrome of water intoxication.
There are many causes of SIADH (Table 16-11). Vasopressin can be produced by a variety of malignancies, most commonly oat cell carcinoma of the lung. Therefore, patients who develop “idiopathic” SIADH are screened for malignant tumors. SIADH is also commonly associated with pulmonary conditions, metabolic and traumatic CNS disorders, and drugs, particularly chlorpropamide, thiazide diuretics, opiates, and barbiturates. Surgical patients are also at risk because of increased vasopressin secretion due to perioperative surgical stress and the use of opiate analgesics such as morphine.
TABLE 16-11. ETIOLOGIES OF SIADH (PARTIAL LISTING)

Clinically, SIADH is distinguished by hyponatremia and water retention that progresses to water intoxication. The seriousness of the patient’s signs and symptoms depends on how fast the serum sodium falls. As water intoxication progresses and the serum becomes more hypotonic, brain cells swell, causing neurologic impairment. Without treatment, irreversible brain damage and death can occur.
EARLY
• Urine volume decreased and concentrated
• Nausea
• Vomiting
• Headache
• Impaired taste
• Dulled sensorium
• Muscle weakness and cramps
• Anorexia
• Weight gain
• Crackles
• Dyspnea
• Increased CVP, PCWP
• Weakness/fatigue
LATE
• Confusion
• Hostility
• Aberrant respirations
• Hypothermia
• Coma
• Convulsions
DIAGNOSTIC TESTS
• Serum Na+ < 130 mEq/L
• Serum osmolality < 280 mOsm/kg
• Increased urine osmolality > 500 mOsm/kg
• Urine sodium > 20 mEq/L
• Blood urea nitrogen and creatinine decreased (hemodilution)
Principles of management depend on the severity and duration of the hyponatremia. Recognition of early clinical manifestations of SIADH is key to prevent life-threatening complications. Continued assessment of neuromuscular, cardiac, gastrointestinal, and renal systems is important. Generally, treatment focuses on restricting fluids, replenishing sodium deficits, and in severe cases of hyponatremia, inhibiting antidiuretic actions. Treatment of the underlying disorder is also a priority.
Fluid restriction is the mainstay of treatment and, to be effective, a negative water balance must be achieved.
1. Treatment of mild hyponatremia (sodium level > 125 and < 135 mEq/L) includes fluid restriction of 800 to 1000 mL/d. This allows sodium level to correct over 3 to 10 days. If fluid restriction alone is not effective, demeclocycline (Declomycin) can be administered. Demeclocycline allows excretion of water because it inhibits the effect of ADH on the renal tubules.
2. If severe neurologic symptoms of SIADH are present along with severe hyponatremia (< 125 mEq/L), administer 3% saline infusion over 2-3 hours. Furosemide is also given to increase urinary water excretion.
3. Assess cardiovascular and respiratory functions closely to evaluate the effects of the excess volume on these systems. Right and left ventricular volumes may increase, causing heart failure. Tachypnea, reports of shortness of breath, and fine crackles are indicators of fluid overload and impending heart failure.
4. Provide for patient comfort with limited fluid intake. Provide for frequent mouth care. Explain why fluid is being restricted and allow the patient to develop the schedule for allotted fluid intake. If the patient complains of nausea, administer an antiemetic prior to meals.
1. In severe symptomatic hyponatremia, infuse 3% saline at a rate of 0.1 mL/kg/min for 2 hours to raise plasma sodium. Monitor closely for signs of hypernatremia, fluid overload, and heart failure because this treatment causes a transient increase in the serum sodium.
2. Monitor neurologic status closely and protect the patient from harm. Institute seizure precautions as necessary. Monitor respiratory status closely.
In cases where SIADH does not resolve within 1 to 2 weeks, drugs that interfere with the renal effect of vasopressin, such as demeclocycline, may be ordered. The full effect of these drugs makes them unsuitable for acute management of the syndrome.
Diabetes insipidus (DI) results from a group of disorders in which there is an absolute or relative deficiency of ADH (called central DI) or an insensitivity to its effects on the renal tubules (called nephrogenic DI) (Figure 16-3). Diabetes insipidus may complicate the course of critically and acutely ill patients and can result in acute fluid and electrolyte disturbances.
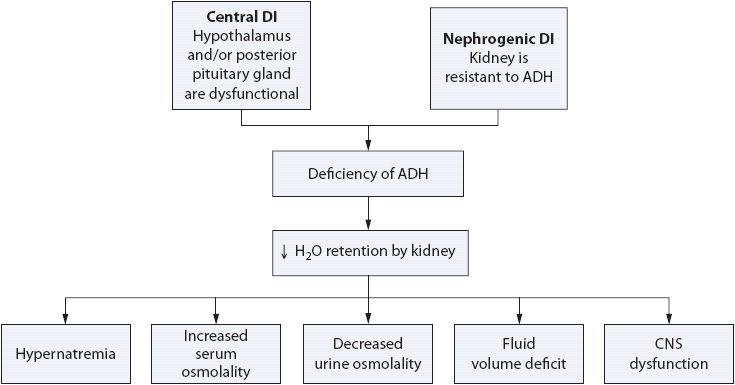
Figure 16-3. Pathogenesis of DI.
There are many causes of DI (Table 16-12). Central or neurogenic DI results from damage to the hypothalamic/pituitary system. An absolute deficiency of ADH results in an impaired ability to concentrate urine, polyuria, and a subsequent risk for dehydration. Patients with head trauma or those who have had neurosurgery must be watched closely for at least 7 to 10 days after the injury for evidence of DI as DI does not present for at least 48 to 72 hours after the initial hypothalamic or hypophyseal (system of blood vessels that link the hypothalamus and the anterior pituitary) trauma.
TABLE 16-12. CAUSES OF DI

Nephrogenic DI is characterized by renal tubule insensitivity to ADH and develops because of structural or functional changes in the kidney. This results in impaired urine-concentrating ability and free water conservation. Nephrogenic DI is less dramatic than neurogenic DI in its onset and appearance.
Regardless of the etiology, in DI the ability of the body to increase ADH secretion or respond to ADH is impaired. A persistent output of dilute urine and increasing hemoconcentration is the hallmark of DI. Signs and symptoms of dehydration are present in those patients in whom the thirst mechanism has been impaired (neurogenic DI) or in whom there is inadequate fluid replacement. In addition, if a hyperosmolar state exists, intracellular brain volume depletion occurs as water moves from within the brain cells to the plasma. Typically, symptoms manifest when serum sodium levels exceed 155 mEq/L.
• Polydipsia (if alert)
• Polyuria (5-20 L in 24 hours)
• Orthostatic hypotension
• Weight loss
• Tachycardia
• Decreased CVP, PCWP
• Poor skin turgor
• Dry mucous membranes
• Confusion
• Restlessness
• Lethargy
• Irritability
• Seizures
• Coma
• Water deprivation test
• Serum sodium > 155 mEq/L
• Serum osmolality > 295 mOsm/kg/L
• Urine osmolality inappropriately low with high serum osmolality (< 150 mOsm/kg/L)
• Urine specific gravity decreased
• BUN and creatinine increased (hemoconcentration)
The management of the patient in DI is directed at correcting the profound fluid volume deficit and electrolyte imbalances associated with this condition. If fluid losses are not replaced, hypovolemic shock can rapidly develop (see Chapter 9, Cardiovascular System). In some cases of DI, vasopressin or agents that simulate ADH release and renal response to ADH are prescribed to treat the disorder. As with other disorders, diagnosis and treatment of the cause of DI are priorities.
If the patient is alert and the thirst mechanism is not impaired, allow the patient to drink water to maintain normal serum osmolality. In many critically ill patients, this is not possible.
1. Administer hypotonic volume, such as dextrose 5% in water, quarter-strength or half-strength saline, IV as prescribed to restore the hypotonic fluid lost through osmotic diuresis. The administration of normal saline to replace volume is usually contraindicated because it presents an added renal load, promoting osmotic diuresis and worsening dehydration. In severe DI, where large amounts of fluid replacement are required, the IV intake is usually titrated to urine output; for example, 400 mL of urine output for 1 hour is replaced with 400 mL IV fluid the next hour. Hypotonic saline solutions are preferred (quarter-strength or half-strength saline). A good rule of thumb is to reduce serum sodium by 0.5 mEq/L every hour but no more than 12 mEq/L per day.
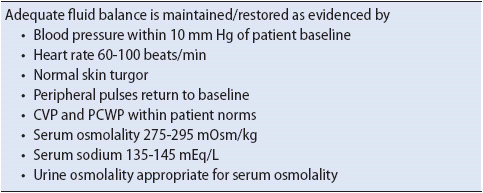
2. Monitor fluid status: Hourly urine outputs along with measurements of urine specific gravity every 1-2 hours should be done along with daily weight and strict intake and output. Monitor for signs of continuing fluid volume deficit. If the serum Na is > 155 mEq/L, rehydration should occur over 48 hours. A serum Na + > 170 necessitates ICU care. Expected outcomes for the patient with DI are listed in Table 16-13.
3. Monitor neurologic status continuously. An altered level of consciousness indicates intracellular dehydration of the brain and hypovolemia.
4. Frequent electrolyte monitoring is recommended during the initial phase of treatment.
TABLE 16-13. EXPECTED OUTCOMES FOR THE PATIENT WITH DI
In central DI, desmopressin (DDAVP), an ADH analogue, is the drug of choice and is available in subcutaneous, IV, intranasal, and oral preparations. Desmopressin acts on the distal tubules and collecting ducts of the kidney to increase water reabsorption and has very specific actions with little or no ADH-like activity elsewhere in the body, most notably vasopressor effects that are prominent in another vasopressin analogue, aqueous (arginine) vasopressin (Pitressin). Vasopressin is only given intramuscularly or IV and is useful only for very short duration in unconscious patients in whom recovery of ADH secretion is expected. However, aqueous vasopressin is less specific than DDAVP and can cause profound vasoconstriction in splanchnic, portal, coronary, cerebral, peripheral, pulmonary, and intrahepatic vessels—thus it is not preferred for DI therapy in critically and acutely ill patients unless DDAVP is ineffective. Adjunctive therapy to enhance ADH release includes nonhormonal agents such as chlorpropamide, carbamazepine, thiazides, and nonsteroidal anti-inflammatory drugs (NSAIDs).
1. If the patient is unconscious, injectable DDAVP is given IV or IM 1 to 4 μg every 12 hours until therapeutic goals are achieved, such as a urine output of 2-3 ml/kg/hour, urine specific gravity 1.010-1.020 and serum sodium 140-145 mEq/l. In conscious patients, the nasal replacement route is given 10 to 20 μg by spray 2 to 3 times a day. It is important that DDAVP or other ADH analogues not be administrated unless serum sodium is at least above 145 mmol/L, as serious hyponatremia may result. Oral formulations of ADH have a slower onset and duration of action and are not useful in acute situations. Major side effects to watch for include headache, abdominal cramps, or allergic reactions such as facial flushing. Monitor for overmedication, which may precipitate hypervolemia. Signs and symptoms of fluid volume excess include dyspnea, hypertension, weight gain, and angina. Hyponatremia is another serious consequence and if it develops rapidly can cause extreme cerebral edema and osmotic demyelination syndrome. Therefore, close monitoring of serum sodium is necessary.
2. For nephrogenic DI, DDAVP intranasal or oral agents may be adminstered or alternative drugs such as chlorpropamide or NSAIDs (such as indomethacin) may be given. Volume excess remains a risk from treatment. Chlorpropamide, an older antidiabetic agent, may also result in hypoglycemia.
American Diabetes Association. Clinical practice recommendations. Diabetes Care. 2013;36:S1-S110.
Endocrine Society. Management of hyperglycemia in hospitalized patients in non-critical care setting: an endocrine society clinical practice guideline. January 2012. Accessed February 18, 2013.
Karon BS, Gandhi GY, Nuttall GA, Bryant SC. Accuracy of Roche Accu-Chek from whole blood capillary, arterial, and venous glucose values in patients receiving intensive intravenous insulin therapy after cardiac surgery. Am J Clin Pathol. 2007;127(6):919-926.
Klonoff DC. The Food and Drug Administration is now preparing to establish tighter performance requirements for blood glucose monitors. J Diabetes Sci Technol. 2010;4(3):499-504.
Malone B. Blood glucose meters: is FDA ready to tighten up accuracy standards? Clin Lab News. 2010;36(5):1-4.
Vaddiraju S, Burgess DJ, Tomazos I, Jain FC, Papadimitrakopoulos F. Technologies for continuous glucose monitoring: current problems and future promises. J Diabetes Sci Technol. 2010;4(6):1540-1562.
American Diabetes Association. Standards of medical care in diabetes 2012 (Position Statement). Diabetes Care. 2012;35 (suppl 1): S11-S63.
Blouin D. Too much of a good thing: management of diabetic ketoacidosis in adults. Can Fam Physician. January 1, 2012;58:55-57.
Inzucchi SE, Siegel MD. Glycemic control in the ICU—how tight is too tight? N Engl J Med. 2009;360:1346-1349.
Management of Hyperglycemia in Hospitalized Patients in Non-Critical Care Setting: An Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab. January 1, 2012;97:16-38.
Moghissi ES, Korytkowski MT, DiNardo M, et al. American Association of Clinical Endocrinologists and American Diabetes Association Consensus Statement on Inpatient Glycemic Control. Endocrine Practice;2009;15:1-17 and Diabetes Care. 2009;Jun;32(6):1119-1131.
Rajesh G, Hurwitz S. Hypoglycemia, with or without insulin therapy, is associated with increased mortality among hospitalized patients. Diabetes Care. December 17, 2012, doi: 10.2337/dc12-1296. Accessed February 18, 2012.
Sato H, Carvalho G, Sato T, et al. The Association of Preoperative Glycemic Control, Intraoperative Insulin Sensitivity, and Outcomes after Cardiac Surgery. J Clin Endocrinol Metab. September 2010;95(9):4338-4344.
Bui H, To T, Stein R, Fung K, Daneman D. Is diabetic ketoacidosis at disease onset a result of missed diagnosis? J Pediatr. 2010;Mar;156(3):472-477.
The NICE-SUGAR Study Investigators. Intensive versus conventional glucose control in critically ill patients. N Engl J Med. 2009;360:1283-1297.
The NICE-SUGAR Study Investigators. Hypoglycemia and risk of death in critically ill patients. N Engl J Med. 2012; 367:1108-1118.
Umpierrez G. Randomized study of basal-bolus insulin therapy in the inpatient management of patients with type 2 diabetes undergoing general surgery (RABBIT 2 surgery). Diabetes Care. 2011;34:256-261.
Van den Berghe G, Wilmer A, Hermans G, et al. Intensive insulin therapy in the medical ICU. N Engl J Med. 2006;354:449-461.
Van den Berghe G, Wouters P, Weekers F, et al. Intensive insulin therapy in the critically ill patients. N Engl J Med. 2001;345:1359-1367.
Crawford A, Harris H. Waterworld, part 2: understanding diabetes insipidus in adults. Nurs Crit Care. 2012;7(1):12-16.
Ellison DH, Berl T. Clinical practice. The syndrome of inappropriate antidiuresis. N Engl J Med. 2007;356:2064-2072.
Gross P. Clinical management of SIADH. Ther Adv in Endo and Metab. 2012;3(2):61-73.
Loh JA, Verbalis JG. Disorders of water and salt metabolism associated with pituitary disease. Endocrinol Metab Clin North Am. Mar 2008;37(1):213-234.
Mavrakis AN, Tritos NA. Diabetes insipidus with deficient thirst: report of a patient and review of the literature. Am J Kidney Dis. May 2008;51(5):851-859.
Thornton SN. Thirst and hydration: physiology and consequences of dysfunction. Physiol Behav. 2010;100:15-20.
Tomky D. Detection, prevention, and treatment of hypoglycemia in the hospital. Diabetes Spectr. 2005;18(1):39-44.