TABLE 9-1. CHEST PAIN ASSESSMENT
KNOWLEDGE COMPETENCIES
1. Identify indications for, complications of, and nursing management of patients undergoing coronary angiography and percutaneous coronary interventions.
2. Describe the etiology, pathophysiology, clinical presentation, patient needs, and principles of management of patients with ischemic heart disease.
3. Discuss the indications for, complications of, and management of patients undergoing electrophysiology studies.
4. Discuss the etiology, pathophysiology, clinical presentation, patient needs, and principles of management of patients in shock, heart failure, and hypertensive crisis.
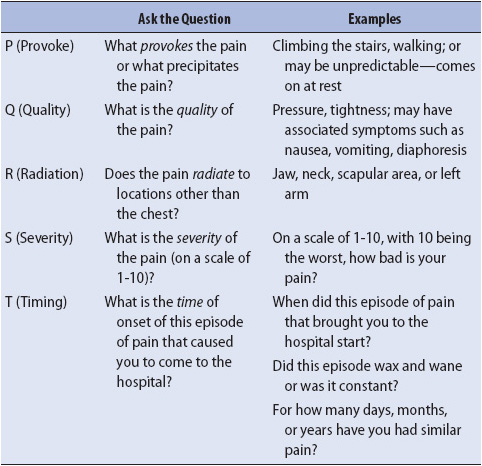
Obtaining an accurate assessment of chest pain history is an important aspect of differentiating cardiac chest pain from other sources of pain (eg, musculoskeletal, respiratory, anxiety). Ischemic chest pain, caused by lack of oxygen to the myocardium, must be quickly identified for therapeutic interventions to be effective. The most important descriptors of ischemic pain include precursors of pain onset, quality of the pain, pain radiation, severity of the pain, what relieves the pain, and timing of onset of the current episode of pain that brought the patient to the hospital. Each of these descriptors can be assessed using the “PQRST” nomogram (Table 9-1). This nomogram prompts the clinician to ask a series of questions which help clarify the characteristics of the cardiac pain.
TABLE 9-1. CHEST PAIN ASSESSMENT
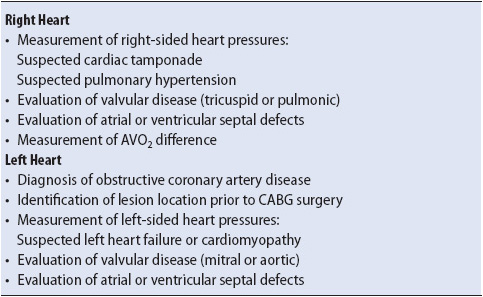
Coronary angiography is a common and effective method for visualizing the anatomy and patency of the coronary arteries. This procedure, also known as cardiac catheterization, is used to diagnose atherosclerotic lesions or thrombus in the coronary vessels. Cardiac catheterization is also used for evaluation of valvular heart disease, including stenosis or insufficiency, atrial or ventricular septal defects, congenital anomalies, and cardiac wall motion abnormalities (Table 9-2).
TABLE 9-2. INDICATIONS FOR CARDIAC CATHETERIZATION
Prior to cardiac catheterization, the patient should be NPO for at least 6 to 12 hours, in the event that emergency intubation is required during the procedure. NPO may indicate everything except medications which should be taken with small sips of water the day of the procedure. Typically, if the patient is on insulin or taking oral hypoglycemics the doses may need to be adjusted or held the day of the procedure. There are other medications that may need to be held. Benadryl may be administered prior to beginning the procedure as a precautionary measure against allergic reaction to the dye. Unfractionated heparin and platelet inhibitor agents (including aspirin, glycoprotein IIb/IIIa receptor inhibitors, and/or clopidogrel) may be administered to prevent catheter-induced platelet aggregation during the procedure. Typically, patients remain awake during the procedure, allowing them to facilitate the catheterization process by controlling respiratory patterns (eg, breath holding during injection of radiopaque dye to improve the quality of the image). An anxiolytic agent, such as diazepam, is frequently administered during the procedure to decrease anxiety or restlessness.
An intracoronary catheter is inserted through a “sheath” or vascular introducer placed in a large artery, most commonly the femoral artery (Figure 9-1A). In recent years there has been an increase in the use of the radial artery as catheters have been made smaller, allowing easier access to the vessel. If inserted via the femoral artery the catheter is then advanced into the ascending abdominal aorta, across the aortic arch, and into the coronary artery orifice located at the base of the aorta (Figure 9-1B). Ionic dye, visible to the observer or operator under fluoroscopy (x-ray), is then injected into the coronary arterial tree by the catheter. If the cardiac valves, septa, or ventricular wall motion is being evaluated, the catheter is advanced directly into the left ventricle, followed by injection of dye (Figure 9-1C). During a right heart catheterization, the catheter is inserted into the venous system through the inferior vena cava, passed through the right ventricle, and advanced into the pulmonary artery.

Figure 9-1. Coronary angiography. (A) Insertion of the coronary catheter into the femoral artery through a percutaneously inserted introducer sheath. (B) Coronary catheter advancement into the aorta and the left coronary artery. (C) Catheter advancement into the left ventricle.
The coronary vascular tree consists of a left and a right system (Figure 9-2). The left system consists of two main branches, the left anterior descending (LAD) artery and the left circumflex (LCx) artery. The right system has one main branch, the right coronary artery (RCA). Both systems have a number of smaller vessels that branch off these three primary arterial vessels. A clinically significant stenosis is considered to be an obstruction of 75% or greater in a major coronary artery or one of its major branches. If there is significant disease in only one of the major arteries, the patient is said to have single-vessel disease. If two major vessels are affected, the patient has two vessel disease. If significant disease exists in all three major coronary arteries then the patient has three-vessel disease. Frequently, the microvasculature, or smaller vessels branching off the major coronary artery, may also have blockages. It is common to refer to these multiple lesions as diffuse disease.
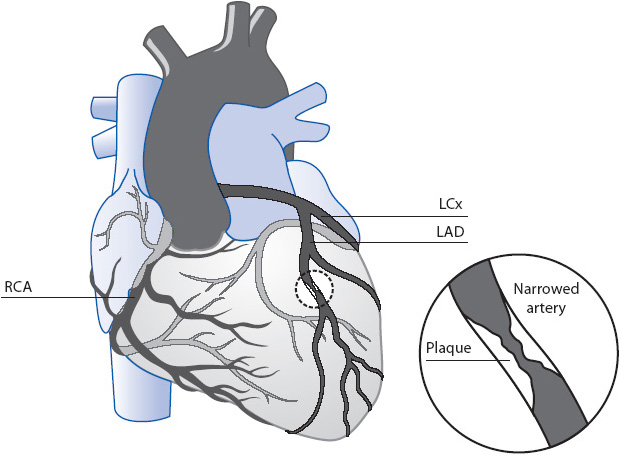
Figure 9-2. Coronary artery circulation with a coronary vessel narrowed with plaque formation.
A cineventriculogram is obtained by radiographic imaging during the injection of dye after advancing the catheter from the aorta, through the aortic valve, and into the left ventricle (see Figure 9-1C). The cineventriculogram provides information on ventricular wall motion, ejection fraction, and the presence and severity of mitral regurgitation and aortic regurgitation. Ejection fraction, or the percentage of blood volume ejected from the left ventricle with each contraction, is the gold standard for determining left ventricular function and is helpful in selecting treatment strategies. A left ventricular ejection fractions (LVEF) normal value is 55% to 60%. The LVEF is one of the most important predictors of long-term outcome following acute myocardial infarction (AMI). Patients with ejection fractions less than 20% have nearly 50% 1-year mortality. Another important measurement is the pressure in the left ventricle at the end of diastole. This is called “left ventricular end-diastolic pressure (LVEDP).” It, too, is an important determinant of ventricular function and is considered to be a predictor of morbidity and mortality in patients with heart failure (HF) and those undergoing cardiac surgery. The normal LVEDP is 6-12 mm Hg.
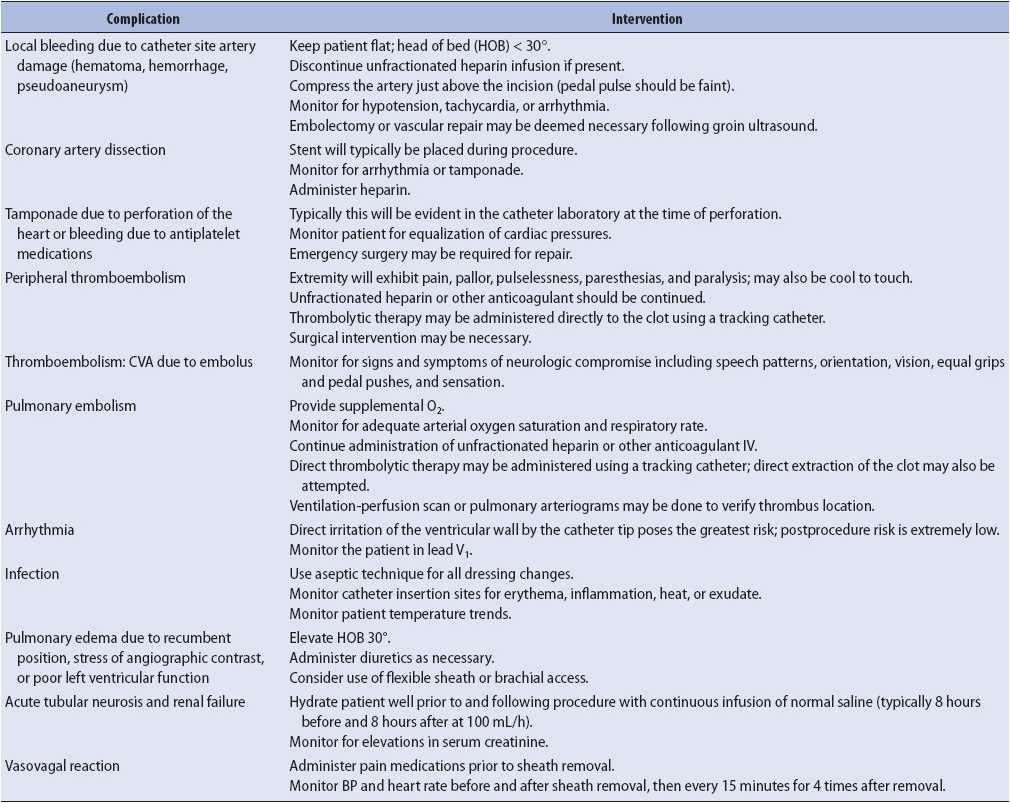
During cardiac catheterization, a number of complications may occur, including arrhythmia; coronary vasospasm; coronary dissection; allergic reaction to the dye; atrial or ventricular perforation resulting in pericardial tamponade; embolus to an extremity, a lung, or, rarely, the brain; acute closure of the left main coronary; myocardial infarction (MI); or death. Common management and prevention strategies for catheterization complications are summarized in Table 9-3.
Percutaneous coronary interventions (PCIs) include percutaneous transluminal coronary angioplasty (PTCA), insertion of one or more stents, and coronary atherectomy. PTCA, also termed angioplasty or balloon angioplasty, is a cardiac catheterization with the addition of a balloon apparatus on the tip of the catheter for revascularizing the myocardium (Figure 9-3). The catheter tip is advanced, generally over a guidewire, into the coronary artery until the balloon is positioned across the atherosclerotic lesion in the vessel. Once properly positioned, the balloon is inflated to stretch the vessel wall, resulting in fracture and compression of the atherosclerotic plaque improving blood flow through the vessel. As a result, the degree of stenosis is reduced. This allows a higher rate and volume of blood flow through the vessel, which translates clinically into fewer symptoms of angina and better exercise tolerance.
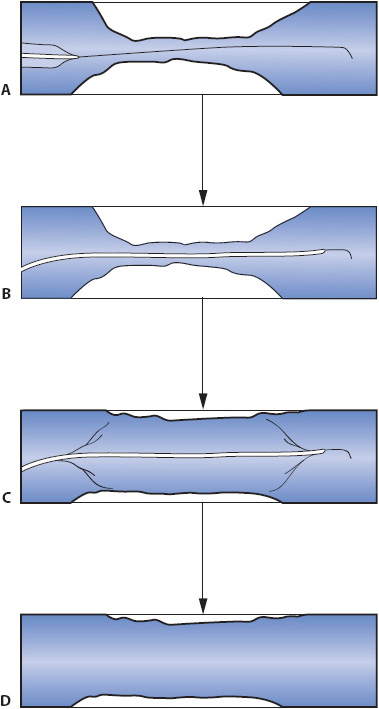
Figure 9-3. Percutaneous transluminal coronary angioplasty (PTCA). (A) PTCA catheter being advanced into the narrowed coronary artery over a guidewire. (B) Catheter position prior to balloon inflation. (C) Balloon inflation. (D) Coronary vessel following catheter removal.
Angioplasty is associated with the same complications found during cardiac catheterization. In addition, complications related to manipulation of the coronary artery itself may also occur. The most common serious complications include a 2% to 10% incidence of complete occlusion of the vessel (abrupt closure), AMI (1%-5% incidence), and the need for emergency coronary artery bypass surgery (1%-2% incidence). The most important predictor of complications of MI and abrupt vessel closure is reduced coronary flow through the lesion prior to the procedure. A universal scale, the TIMI Scale, is used to quantify this rate of coronary flow. The scale rates coronary blood flow as follows: no perfusion, penetration without perfusion, partial perfusion, and complete perfusion.
In addition to routine balloon angioplasty, a number of other devices are now commonly used for percutaneous coronary revascularization. Intracoronary stents are small metallic mesh tubes placed across the stenotic area and expanded with an angioplasty balloon (Figure 9-4). Once expanded, the tube is permanently anchored in the vessel wall. Stents are effective in decreasing the rate of abrupt vessel closure seen with traditional PTCA. Some stents are coated with a drug that is bonded to a material on the stent causing the drug to be released directly onto the arterial wall over several months to years. These drug-coated stents have been shown to significantly reduce the restenosis rate associated with metal stents. Atherectomy catheters and lasers are used infrequently; however, patient outcomes are not significantly better than those achieved with traditional balloon catheters and stent deployment and may result in higher rates of complication, including AMI. Each of these devices may offer advantages over traditional balloon angioplasty catheters in situations involving specific vascular anatomy (eg, ostial lesions) or lesion morphology (eg, high degree of calcified plaque).
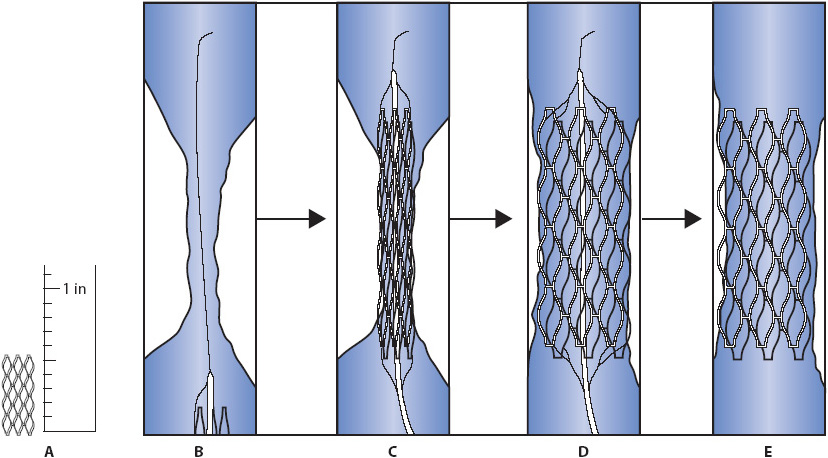
Figure 9-4. Intracoronary stent. (A) Size of stent device when fully deployed. (B) Insertion of stent into a narrowed area of a coronary artery on a balloon-inflatable catheter. (C) Inflation of the balloon catheter to expand the stent. (D) Inflation complete with stent fully expanded. (E) Stent following removal of balloon catheter.
Myocardial ischemia is the lack of adequate blood supply to the heart, resulting in an insufficient supply of oxygen to meet the demands of the heart muscle. This supply-demand mismatch, known as ischemia, is most often caused by thrombus formation at a site of atherosclerotic plaque rupture within a coronary artery. Decreased oxygen supply to myocardial tissue may cause a variety of symptoms such as chest discomfort (angina), shortness of breath, diaphoresis, and nausea. Unstable angina, defined as angina that is of new onset, increasing in frequency, or occurring at rest, and AMI are referred to as the acute coronary syndromes (ACS), which form the spectrum of acute ischemic heart disease.
Intracoronary thrombus formation, and the resulting obstruction of coronary blood flow, is the pathophysiologic mechanism of acute ischemic heart disease. Preexisting atherosclerosis and spasm of the smooth muscle wall of the coronary arteries, termed fixed obstructions, may also contribute to reduced flow. In some situations, coronary artery spasm may play a major role, unrelated to underlying atherosclerosis, causing MI. These occurrences are sometimes associated with cocaine abuse seen in MI in young patients.
The formation of a thrombus in coronary arteries is initiated by the fissuring and rupture of atherosclerotic plaque in the vessel wall of the coronary artery (Figure 9-5). A continuous, dynamic process occurs whereby plaque may become unstable during periods of active accumulation of more lipid into the core of the plaque. The plaque then ruptures, dispelling its contents into the lumen of the coronary artery and causing activation of clotting factors at the site of plaque rupture. The rupture of plaque and resultant thrombus formation may eventually occlude the coronary artery.
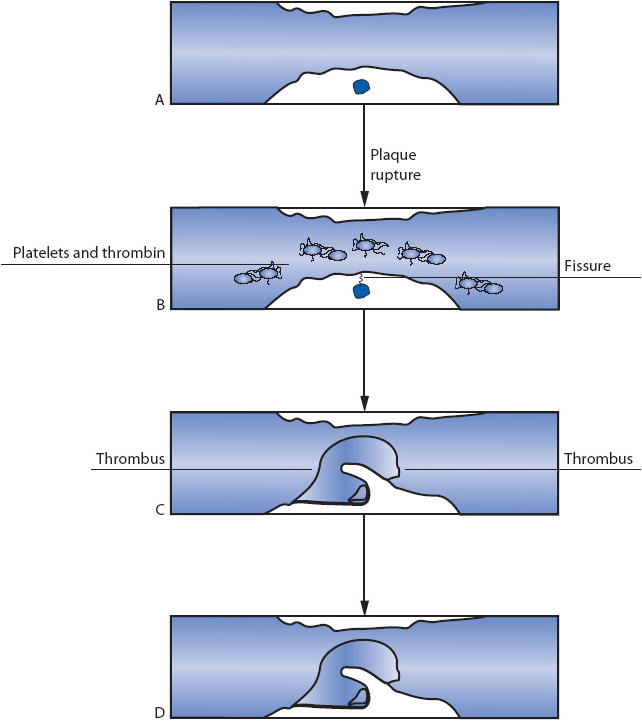
Figure 9-5. Atherosclerotic plaque formation. (A) Stable plaque. (B) Plaque with cap disruption. (C) Moderate amount of layered thrombus. (D) Occlusive thrombus.
Although most people have some degree of atherosclerotic plaque formation by age 30, the vast majority of these plaques are considered “stable.” They are covered by smooth fibrous caps allowing adequate blood flow through the coronary arteries, and are not prone to development of unstable angina or MI. In young, growing plaques, the fibrous cap may become thin and rupture, resulting in unstable angina, ischemia, or MI.
A variety of factors predispose a plaque to fissure and rupture. Characteristics of plaque at increased risk for rupture include:
• Location of the lesion in the vascular tree: Areas of greater turbulence of flow and dynamic activity during the cardiac cycle are at higher risk.
• Size of the lipid pool within the plaque: A large amount of lipid inside the plaque core is more likely to be associated with plaque disruption.
• Infiltration of the plaque with macrophages: Macrophages are thought to weaken the integrity of the fibrous cap of the plaque, making it more susceptible to rupture.
A 62-year-old man presents to the emergency department (ED) with complaints of pain in his chest and jaw. The pain, originally occurring only with exertion and resolving with rest, became increasingly persistent over the past 2 to 3 days. On the evening of his arrival, the patient experienced a 15-minute episode of severe pain while watching television. This episode he characterized as a “tight, burning feeling in my chest, and an aching in my jaw” that did not vary with respiratory effort and was accompanied by diaphoresis, nausea, and shortness of breath.
On arrival to the ED, his pain and nausea had resolved, pulse oximetry showed oxygen saturation of 98% on room air, and his vital signs were:
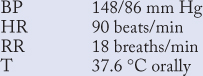
On physical examination, heart sounds were normal, without S3, S4, or murmurs. Initial diagnostic tests revealed:
• ECG: Normal sinus rhythm with nonspecific ST-T wave changes
• Chest x-ray: Normal cardiac silhouette, clear lungs
A more detailed assessment of his history revealed increasing dyspnea on exertion and fatigue for the previous 6 months. Despite these symptoms, he had continued his daily 2.5-mile walking routine, sometimes experiencing shortness of breath several times during the walk. The patient reported smoking cigarettes in the past, one pack per day for 20 years, but quit 25 years ago. No ankle swelling, nocturnal dyspnea, or orthopnea were reported, nor was he aware of any family history of cardiac problems, coronary artery disease, diabetes, or hypertension.
He was started on aspirin based on his history and the likelihood of underlying coronary artery disease. He was then admitted for observation and evaluation of cardiac enzymes. (See section on cardiac enzymes I section on MI below.)
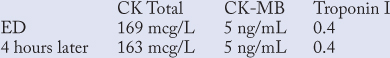
Six hours after presenting to the ED, the patient had recurrent tightness in his chest. An ECG showed T-wave inversion in the anterior leads. Sublingual nitroglycerin 0.4 mg was administered every 5 minutes with complete relief of the pressure following the second tablet. An unfractionated heparin drip was started. Subsequent cardiac enzymes showed:
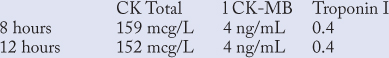
Other laboratory results were normal with the exception of elevated cholesterol and triglycerides on the lipid panel. Following receipt of these results, he was scheduled for an exercise tolerance test.
The ECG recorded a heart rate of 118 beats/min after 6 minutes of exercise. Onset of chest tightness during the last minute of exercise was described as similar to that which brought him to the hospital and correlated with 1.5-mm ST depression in leads V4 to V6. A cardiac catheterization was scheduled.
Coronary angiography showed a 75% obstruction of the LAD artery and 90% obstruction of the diagonal branch of the same artery. LVEF was 55%. A coronary angioplasty (PTCA) was performed on both lesions.
Case Question 1. While in the ED, an important aspect of his care would be to:
(A) Obtain repeat ECGs intermittently every 4 hours
(B) Monitor the patient’s ECG continuously with continuous ST-segment monitoring
(C) Monitor platelet levels every 6 hours
(D) Assess breath sounds every 2 hours
Case Question 2. The ST-segment depression and T-wave inversion would be indicative of a:
(A) Non–ST-segment elevation MI
(B) ST-segment elevation MI
(C) Coronary spasm
(D) Pericarditis
Case Question 3. Following the PTCA, which of the following would be a clear sign of acute closure of one or both target vessels?
(A) Increased heart rate of 115 beats/min
(B) Hypotension
(C) 4 mm ST-segment elevation in leads V3-V4
(D) All of the above
Answers: 1: B; 2: A; 3: C
Although these characteristics determine the likelihood of plaque rupture, they are not easily identified by clinical assessment, stress testing, or cardiac catheterization. Plaque rupture may be caused by a number of environmental or hormonal factors, known as triggers (Table 9-4). These triggers may disrupt the plaque and precipitate an acute coronary event. Some of the triggers for atherosclerotic plaque rupture can be manipulated or controlled, such as blood pressure (BP), blood glucose level, and stress. In the clinical setting, management of these variables may decrease the risk for AMI, reinfarction, and reocclusion. They should be closely monitored.
TABLE 9-4. HORMONAL AND ENVIRONMENTAL TRIGGERS OF PLAQUE RUPTURE
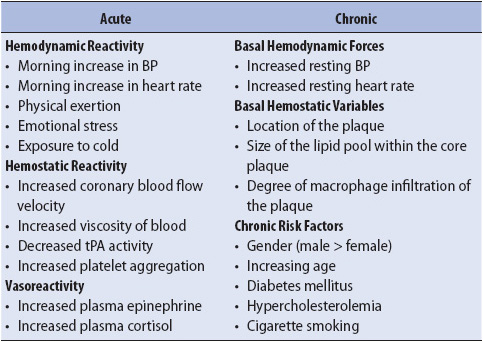
When these triggers combine to cause plaque rupture, the lipid pool is exposed and a rough surface on the intima of the vessel wall occurs, stimulating the local effects of hormonal and immune factors and initiating thrombus formation. At the same time, the fibrinolytic system is stimulated, creating a dynamic process of simultaneous attempts to form and dissolve the clot. Because of the dynamic nature of the clotting process, the thrombus may be completely or only partially obstructive, or may fluctuate intermittently between the two stages. Regardless of the maturity of the clot, the process of thrombus formation may lead to obstruction of blood flow, diminishing oxygen delivery to distal myocardium and creating a mismatch between the supply of and demand for oxygen.
Because the underlying pathology of the ischemia-related diagnoses is the same (plaque rupture and thrombus formation), ischemic heart disease encompasses the entire spectrum of ischemic coronary events that are referred to as the ACS. ACS represents a continuum of clinical events that may result from the supply-demand mismatch including unstable angina, non–ST-segment elevation MI (NSTEMI), or ST-segment elevation MI (STEMI) (Figure 9-6).
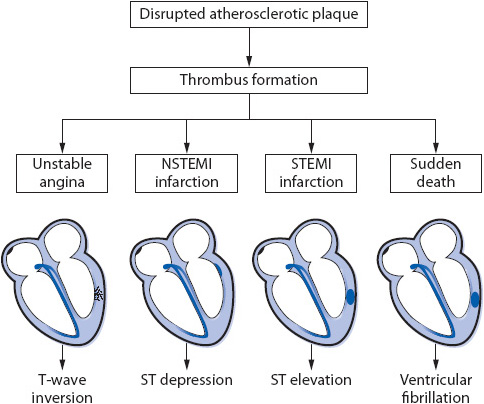
Figure 9-6. Pathophysiologic steps leading to acute coronary events.
Following a decrease in oxygen supply to the myocardium, the cell membranes lose their integrity and fluid moves into the cell. The cell is no longer able to regulate its internal and external environment. The cell dies, releasing cytotoxic substances into the bloodstream. When they die, cardiac myocytes release significant amounts of myoglobin, troponin I and T, as well as cardiac-specific creatine kinase (CK-MB), causing elevation in these laboratory values and confirming the MI diagnosis.
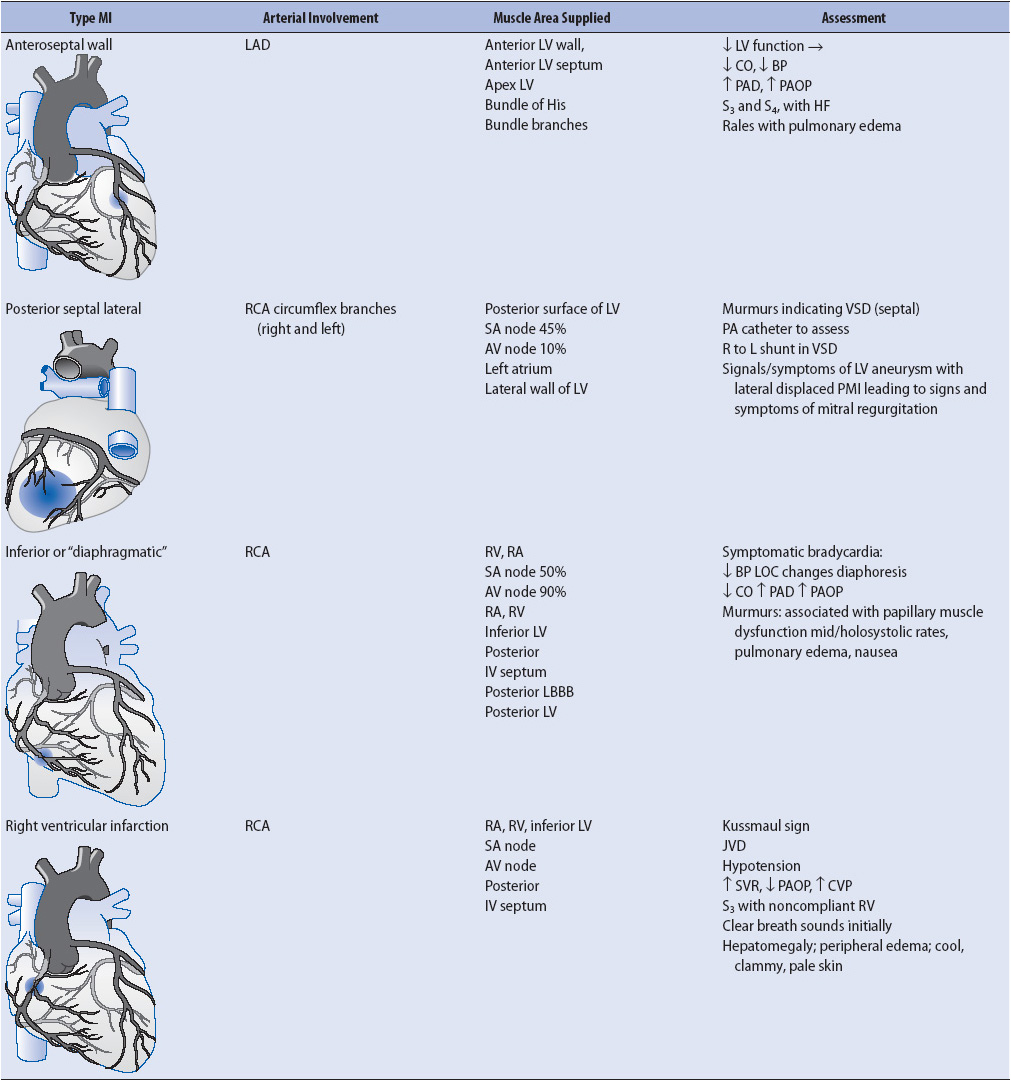
Clinical presentation across the spectrum of ACS is similar, with slight differences depending on the involved vessels (Table 9-5).
1. Pain or discomfort, usually in the chest (see Table 9-1)
• Pressure or tightness in the chest
• Jaw or neck pain
• Left arm ache or pain
• Epigastric discomfort
• Scapular back pain
2. Nausea/vomiting
3. Hemodynamic instability
• Hypotension (systolic BP < 90 mm Hg or 20 mm Hg below baseline)
• Cardiac index (CI) (< 2.0 L/min/m2)
• Elevated pulmonary artery diastolic (PAD) and/or pulmonary artery occlusion pressure (PAOP)
• Skin cool, clammy, diaphoretic
4. Dyspnea
5. Dysrhythmia/Conduction Defects
• Left bundle branch block (LBBB)
• Tachycardia/bradycardia
• Frequent premature ventricular contractions
• Ventricular fibrillation
6. Anxiety, sense of impending catastrophe
7. Denial
TABLE 9-5. CLINICAL PRESENTATION OF MYOCARDIAL ISCHEMIA AND INFARCTION

Some patient populations are predictably different in their description of chest discomfort, such as women and diabetics. Women frequently present with symptoms that are more vague such as feeling tired, short of breath, and lack of energy. Women may be prone to deny their symptoms for longer periods of time than men, delaying their arrival to the ED and often rendering them ineligible for thrombolytic therapy. In addition, women are typically postmenopausal when signs and symptoms of atherosclerotic disease become apparent. This predominantly older patient population may pose problems of its own such as anxiety, fear of the inability to care for oneself following MI, and other concerns to geriatric patient populations, which must be considered.
Diabetics are another patient population with atypical differences in symptoms when experiencing an MI. Diabetics have atypical pain secondary to neuropathies, and early development of atherosclerotic disease. Coronary artery disease (CAD) in this patient population is diffuse, and poor distal vascular anatomy is common. Lesion morphology in diabetic patients is also more difficult to revascularize, either using percutaneous or surgical methods.
1. 12-lead electrocardiogram (ECG): Transient changes may occur and resolve; most commonly T-wave inversion or ST-segment depression.
2. Cardiac enzymes (Troponin [I or T], myoglobin, and CK-MB): Normal (Figure 9-7).
3. Cardiac catheterization: Not recommended in the acute setting, except in the case of continued pain/discomfort without relief from nitroglycerin. Catheterization results may be normal, or with visible atherosclerotic disease, but without a complete occlusion or thrombus.
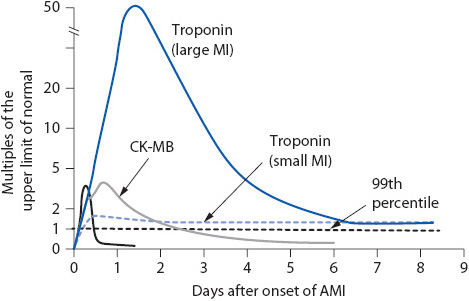
Figure 9-7. Timing and levels of biomarkers associated with heart injury. (Modified from Antman EM: Decision making with cardiac troponin tests. N Engl J Med 346:2079, 2002; and Jaffe AS, Babiun L, Apple FS: Biomarkers in acute cardiac disease: The present and the future. J Am Coll Cardiol 48:1, 2006.)
1. 12-lead ECG: Thirty-five percent of patients with AMI have ST-segment elevation (see Chapter 18, Advanced ECG Concepts). Approximately 65% of those with AMI have no ECG or other diagnostic changes.
2. Creatine kinase (CK and CK-MB): (see Figure 9-7).
• Total CK > 150 to 180 mcg/L
• MB band > 10 ng/mL or 0.3% of total.
• Peaks at 12 hours after symptom onset.
• CK-MB isoforms have better sensitivity and specificity for detecting MI within the first 6 hours.
3. Troponin T: > 0.1 to 0.2 ng/mL.
• Begins to increase 3 to 5 hours after symptom onset
• Remains elevated for 14 to 21 days
4. Troponin I: > 0.4 ng/mL.
• Begins to increase 3 hours after onset of myocardial ischemia
• Peaks at 14 to 18 hours
• Remains elevated for 5 to 7 days
5. Myoglobin: Present in serum, 17.4 to 105.7 ng/mL
• Released from myocardium within 2 hours of coronary occlusion
• Peaks in 6 to 7 hours
• Better marker for early detection of MI; better negative indicator if negative
6. Cardiac catheterization: Ventricular wall motion abnormalities (also may be seen by echocardiography); total occlusion of one or more coronary arteries.
Because most complications of acute ischemic heart disease directly result from reduced coronary flow, a primary objective in patient management is to optimize blood flow to the myocardium. Additional goals are to prevent complications of ischemia and infarction, alleviate chest discomfort/pain, and reduce anxiety.
Regardless of whether a patient presents with unstable angina or AMI, restoration and maintenance of coronary blood flow is important to improve patient outcomes. Interventions to optimize blood flow to the myocardium include pharmacologic measures, such as antiplatelet or antithrombin agents, and mechanical measures, such as percutaneous coronary revascularization (eg, angioplasty, stent, or other) or coronary artery bypass grafting (CABG). Refer to Table 9-6 for evidence-based guidelines for AMI. The intervention selected and the optimal timing of the intervention depends on whether the occlusion of the artery is total or partial. This determination must be made as accurately and as quickly as possible, as a totally occluded artery will soon result in tissue necrosis or MI (Figure 9-8 for algorithm on acute chest pain management). All unstable arteries benefit from the following interventions which stabilize the artery and optimize coronary arterial flow.
TABLE 9-6. EVIDENCE-BASED PRACTICE: ACS—ST ELEVATION MI AND NON–ST-ELEVATION MI

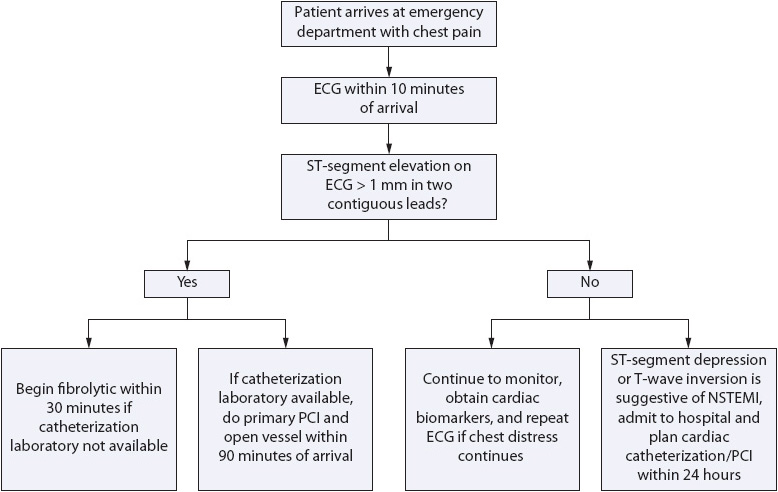
Figure 9-8. Algorithm for management of acute chest pain.
1. Decrease activity of coagulation system with pharmacologic therapy (Figure 9-9).
• Antiplatelet agents: Aspirin, GP IIb/IIIa receptor blocking agents (eg, abciximab [Reopro®], eptifibatide [Integrilin®], and tirofiban [Aggrastat®]), thienopyridine agents (eg, clopidogrel [Plavix®])
• Antithrombin agents: Indirect (eg, unfractionated heparin, low-molecular-weight heparin), direct (eg, bivalirudin [Angiomax®])
2. Increase ventricular filling time (decrease heart rate).
• Beta-blockers
• Bed rest for 24 hours
3. Decrease preload.
• Nitrates
• Diuretics
• Morphine sulfate
4. Decrease afterload.
• Angiotensin-converting enzyme (ACE) inhibitors or angiotensin receptor blockers (ARBs)
• Hydralazine
5. Decrease myocardial oxygen consumption (MVO2).
• Beta-blockers
• Bed rest for 24 hours
6. Reduce risk for sudden cardiac death due to ventricular tachycardia/ventricular fibrillation.
• Beta blockers
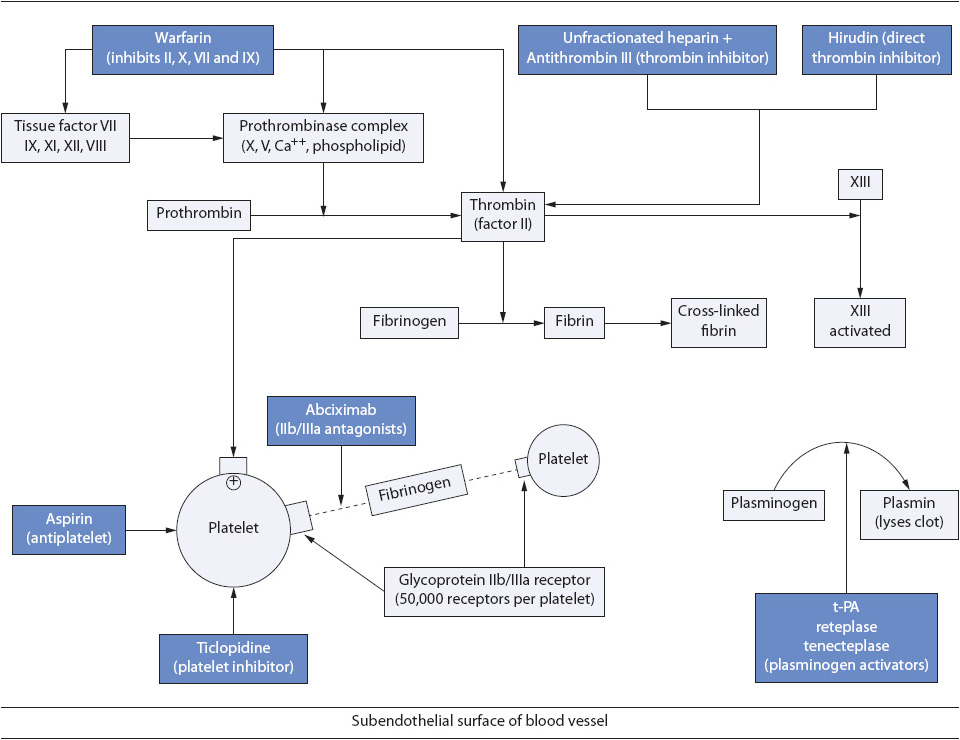
Figure 9-9. Coagulation sequence and site of antithrombotic/antiplatelet drug activity.
Totally occluded arteries require, in addition to the above pharmacologic interventions, further reperfusion therapy, such as fibrinolysis, angioplasty, or CABG, to effectively restore blood flow to the coronary artery. In the event of left main coronary artery stenosis or three-vessel disease, urgent or emergent CABG is usually considered. In the acute setting, for ST-segment elevated MI (STEMI) fibrinolytic therapy is often the fastest, most universally available method for reperfusion if a catheterization laboratory is not available or operational 24 hours a day. The indications, contraindications, and common complications of fibrinolytic therapy are listed in Tables 9-7 and 9-8. In those settings where the catheterization laboratory is operational 24 h/day, primary PCI is indicated. Studies have indicated that primary PCI may be associated with better outcomes and fewer complications than with the use of fibrinolytic agents.
TABLE 9-7. INDICATIONS AND CONTRAINDICATIONS FOR THROMBOLYTIC THERAPY
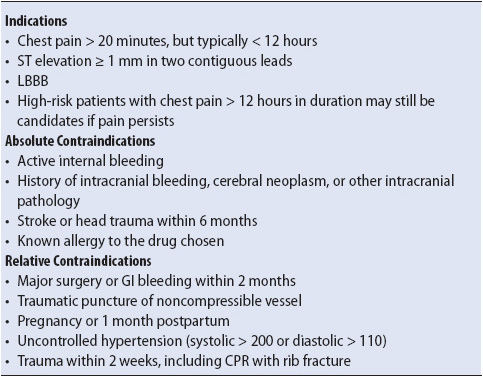
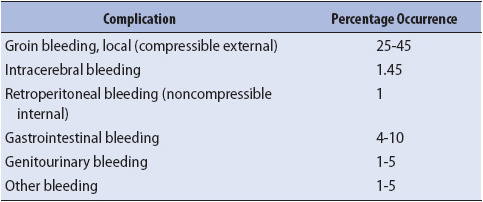
TABLE 9-8. COMPLICATIONS OF FIBROLYTIC THERAPY
Coronary artery bypass grafting is one method of revascularization generally used in patients with atherosclerosis of three or more coronary vessels or in the case of significant left main CAD. CABG is performed both electively, as well as emergently, and may be performed either prior to or following an MI. The CABG procedure requires induction with general anesthesia, and possible initiation of cardiopulmonary bypass (blood is diverted outside of the body to a pump that mechanically oxygenates the blood before returning it to the arterial circulation) and placement of a graft into the coronary arterial tree (Figure 9-10). Technological advances have resulted in the development of stabilizer devices permitting CABG to be performed without placing the patient on cardiopulmonary bypass. The heart continues to beat while the surgeon places a device over the coronary artery site where the bypass graft is to be anastomosed, which stabilizes the small area allowing for suturing to occur. This is often referred to as “beating heart surgery” or “off pump” coronary artery bypass (OPCAB). The graft, generally a leg vein, left internal mammary artery, or radial artery, is inserted past the distal end of the blockage in the coronary artery and, in the case of a leg vein graft and radial artery graft, anastomosed to the aorta. Multiple grafts may be inserted based on the number of blockages present and the availability of viable insertion sites in the patient’s native coronary tree.
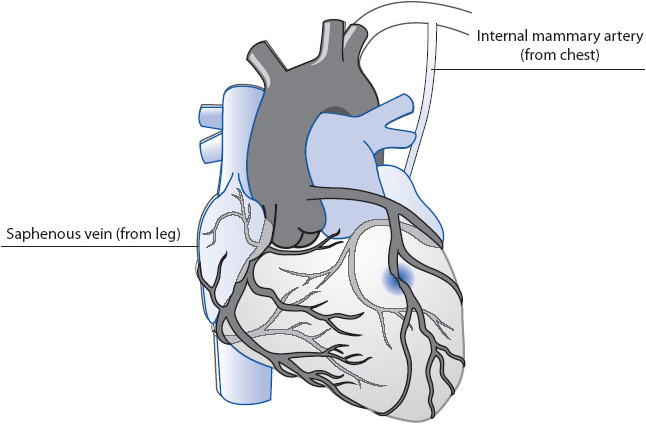
Figure 9-10. Coronary artery bypass grafting (CABG).
The indications for CABG and long-term patient outcome following this procedure have been intensively reviewed over the past decade. In general, patients with three-vessel disease, poor LVEF (< 35%), or significant disease in the left main coronary artery have lower long-term morbidity and mortality with surgical revascularization (CABG) compared to medical therapy or percutaneous interventions such as angioplasty or stent. Diabetics with multivessel disease have also been found to fare better following CABG than following percutaneous interventions including drug-eluting stents. CABG may also be indicated as an emergent “rescue” procedure in patients whose coronary artery severely dissects or fractures during an attempted percutaneous procedure.
Several populations of patients may be considered poor candidates for coronary bypass, including the very elderly, debilitated patients, patients with severely diseased distal coronary vasculature (eg, some diabetics), and patients with extremely low LVEF (eg, < 5%-15%). Patients with low ejection fractions often have difficulty being weaned from cardiopulmonary bypass following the procedure. Other contraindications are those related to general anesthesia risk, including severe chronic obstructive pulmonary disease, pulmonary edema, or pulmonary hypertension.
The following is a general overview of the early postoperative management of CABG patients:
1. Maintain hemodynamic stability: A variety of cardiac drugs are administered to maintain hemodynamic stability in the first 24 hours postoperatively. The following hemodynamic values may serve as guides for inotropic and vasopressor administration along with intravascular fluid therapy. In general, values greater or lower than the following require intervention:
• Mean arterial pressure: 70 to 80 mm Hg
• CI: 2.0 to 3.5 L/min/m2
• PAD/PAOP: 10 to 12 mm Hg (used primarily to evaluate need for volume replacement)
• Central venous pressure (CVP): 5 to 10 mm Hg (used primarily to evaluate need for volume replacement)
• HR: Intrinsic or paced rhythm in range of 80 to 100 beats/min to keep CI ≥ 2.0
• If radial artery graft used, monitor for arterial spasm. Prophylactic nitroglycerin drip and nitro paste.
2. Maintain ventilation and oxygenation: Ventilation and oxygenation are maximized in the early postoperative period with mechanical ventilation. Within 2 to 12 hours, most patients have recovered from the anesthesia effects and are sufficiently stable to allow weaning from mechanical ventilation and extubation. Individuals with preexisting pulmonary problems may require longer periods of intubation until weaning can be successfully accomplished. Following weaning and extubation, supplemental O2 therapy usually is required for 1 to 2 days to maintain PaO2 or SaO2 in normal ranges. Postoperative atelectasis and pleural effusions are a common occurrence after cardiopulmonary bypass, requiring frequent pulmonary interventions (eg, coughing and deep breathing, incentive spirometry, ambulation) to maintain ventilation and oxygenation.
3. Prevention of postoperative complications:
• Bleeding from vascular graft anastomosis sites: Frequent monitoring of mediastinal tube drainage, hematocrit, and coagulation status; avoidance of even brief periods of hypertension.
• Cardiac tamponade: Frequent assessment for signs and symptoms of tamponade which include tachycardia, SOB, anxiety/decreased LOC, paradoxus, sinus tachycardia, decreased mediastinal tube drainage, increased CVP, PAD, and PAOP (note: these are often within 2 to 3 mm Hg of each other, which is called equalization of pressures or diastolic plateau), muffled heart sounds, decreased BP, and cardiac output.
• Infection: Antibiotics may be used prophylactically for 48 hours; temperature spike within 24 hours postoperatively is not abnormal (may be related to pulmonary atelectasis).
• Cardiac arrhythmias: ECG and continuous ST-segment monitoring, treat unstable rhythms, maintain K+ and Mg+ within normal limits with IV replacement.
• Relief of postoperative pain and anxiety: Analgesic administration is typically required to ensure pain relief, especially to facilitate ambulation, coughing, and deep breathing.
• If median sternotomy performed ensure sternal precautions are implemented, eg, avoid hyperextension of chest (arms and shoulders pulled posteriorly).
Complications associated with acute ischemic syndromes include recurrent ischemia, infarction or reinfarction, onset of HF, and arrhythmias.
1. Prevent recurrent ischemia, infarction, or reinfarction: Continue pharmacologic interventions to inhibit prothrombotic events, including ischemia and infarction (eg, antiplatelet and antithrombin agents). Assess for recurrent angina with frequent chest pain assessment and serial 12-lead ECG and continuous ST-segment ischemia monitoring. (See AACN Practice Alert: ST-Segment Monitoring.)
2. Continuously monitor for arrhythmias: Monitor, if possible, for 24 to 72 hours following an ischemic episode.
3. Minimize potential for HF: Minimize myocardial oxygen consumption with the administration of beta-blockers, limit physical activity (bed rest), and avoid increases in metabolic rate (eg, fever). Reduce left ventricular afterload with the administration of ACE inhibitors or ARBs and hydralazine.
Pain relief improves coronary flow by decreasing the level of circulating catecholamines, thereby decreasing BP (after-load) and heart rate (myocardial oxygen consumption). Nitrates typically relieve anginal pain by dilating coronary arteries and increasing blood flow, thereby improving myocardial oxygenation and directly treating the source of the pain. Another pharmacologic intervention commonly used to relieve pain in ischemia is morphine sulfate. Although morphine is a potent narcotic that has been criticized for masking cardiac pain, it is also a potent vasodilator and effectively vasodilates coronary as well as peripheral arteries, resulting in mild afterload reduction. Severe pain, unable to be relieved with nitrates or a combination of nitrates and morphine, is typically an indication for immediate PCI if available or transfers to a referring institution for emergency PCI.
The reduction of anxiety in ischemic heart disease is important for a number of reasons. The most important physiologically is the reduction of catecholamine secretion and decrease in sympathetic tone following relaxation in the anxious patient. This effect has been shown to decrease the incidence of dysrhythmia and promote vasodilation and afterload reduction. Decreasing anxiety may also increase the patient’s ability to process new information regarding his or her diagnosis, and to better understand instructions for tests or procedures that will be done.
Relief of pain typically is most effective in reducing patient anxiety. In the event that pain is not relieved with nitroglycerin, or fibrinolytics in the initial treatment of ischemia, pain relievers such as morphine sulfate or anxiolytics such as midazolam or lorazepam (short- or intermediate-acting benzodiazepines respectively) are usually effective.
A number of interventions may be done at the bedside to promote relaxation, including specific relaxation and imagery techniques, meditation, music therapy, and the use of relaxation tapes. Providing the patient and family with adequate information regarding unfamiliar surroundings, when the physician may be available to speak with them, possible “unknowns” such as tests or procedures, and important expectations such as visitation guidelines helps provide a sense of security and facilitates relaxation by increasing the patient’s level of comfort with the situation. Anxiety can also be decreased by offering the patient opportunities for control in the acute setting. Examples include the timing of simple activities such as visitor presence, bathing, and eating.
In the past 25 years, the cardiology subspecialty of electro-physiology (EP) has grown significantly and continues to expand. An electro-physiology study involves the insertion of several catheters with multiple electrodes on each into the right atrium and ventricle, the coronary sinus and, in some cases across the intra-atrial septum (trans-septal) into the left atrium. The multiple electrode locations (Figure 9-11) are used to record the impulse initiative and/or the conduction of the electrical waveform (depolarization process) through the myocardial tissue. The purpose of the EP may be for a variety of reasons such as: (1) to identify the site of origin for a dysrhythmia like atrial fibrillation or ventricular tachycardia; (2) to map the conduction pathway to identify the location of a conduction disturbance; (3) to identify the presence of an anomalous pathway which may be seen with Wolf-Parkinson-White Syndrome; (4) to determine the need for an implantable device for instance a pacemaker, or cardiovertor defibrillator (ICD or PCD). If the study identifies the site of origin for a dysrhythmia or abnormal pathway, the EP physician is able to use radiofrequency (electricity) ablation or cryoablation (extreme cold) to destroy the site or the abnormal pathway. Following the procedure, the patient needs to be monitored for similar groin complications as seen with cardiac catheterization. Additional monitoring includes observing for the onset of AV blocks as the AV node and/or Bundle of His tissue might have been injured during ablation of the anomalous pathway. If there is evidence of the AV block during the EP study, a pacemaker will be implanted at that time.
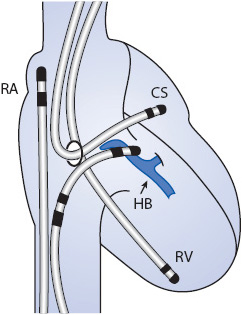
Figure 9-11. Electrophysiology study: Multiple catheters with electrodes are threaded into the right side of the heart. This allows for local monitoring and recording of the electrical activity as the heart depolarizes and repolarizes. The recordings help to locate the origin of the arrhythmia or conduction abnormality across the anomalpous pathway. (Source: Medtronic, Inc. Minneapolis, MN.) Abbreviations: RA, right atrium; RV, right ventricle; HB, his bundle; CS, coronary sinus.
Heart failure is a broad term referring to the inability of the heart to eject an adequate cardiac output to meet the oxygen and metabolic requirements of the body. A number of underlying disease processes may contribute to this “weak pump” syndrome, with coronary atherosclerosis, valvular heart disease, hypertension, and cardiomyopathy as the most common causes. Although the underlying causes are diverse, the progressive process which occurs in response to one of these initiating events is the same.
Although HF may result from a number of underlying etiologies, those causing left ventricular systolic dysfunction are the most common contributors. The pathophysiology of HF is a three-stage process, beginning with an initial insult to the myocardium (phase I), followed by a response phase (phase II), and resulting in the clinical syndrome known as HF, characterized by exhaustion of compensatory mechanisms (phase III) (Figure 9-12). Regardless of the precipitating event, the physiologic progression of the syndrome, once initiated, is the same.
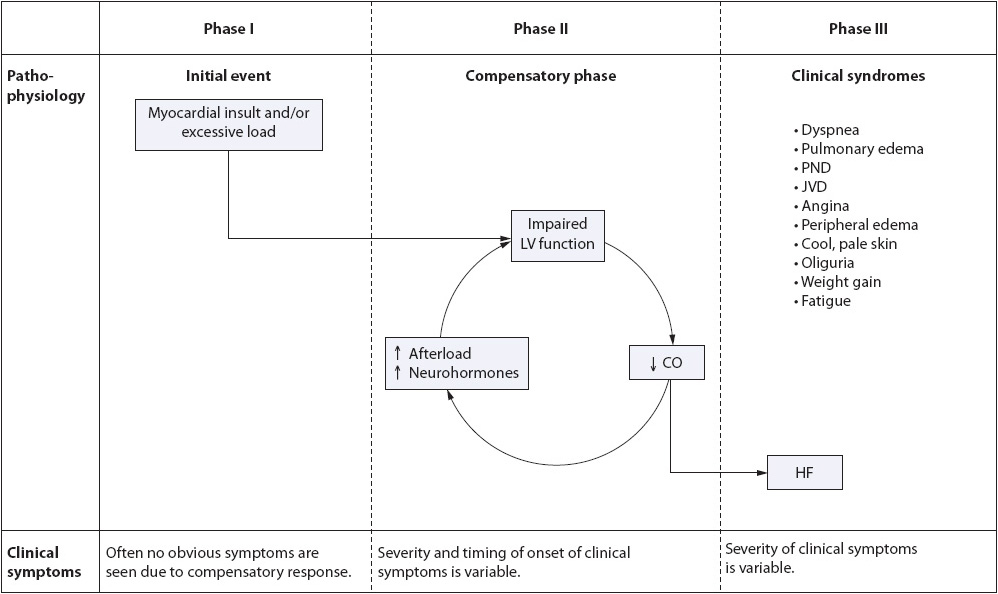
Figure 9-12. Pathophysiology of heart failure during phases I, II, and III.
Phase I of HF is characterized by an initiating event (eg, MI, viral infection, chemotherapeutic agents, valvular heart disease, hypertension, idiopathic cardiomyopathy), which causes loss of myocytes. This cell loss or permanent damage to the myocytes can be either localized or diffuse, resulting in compromised ventricular function. To date, over 700 initiating factors, such as acute ischemic damage, viruses, and toxins, have been isolated as contributors to myocardial insult and HF.
• Result of phase I: Decreased stroke volume secondary to an initial insult to the myocardium.
A number of adaptive mechanisms occur in response to the initial insult in an effort to maintain adequate cardiac output to meet the body’s needs. This phase is sometimes referred to as the compensatory phase (Figures 9-12 and 9-13). These compensatory mechanisms or responses include the Frank-Starling response, myocardial remodeling, and the neurohormonal response.
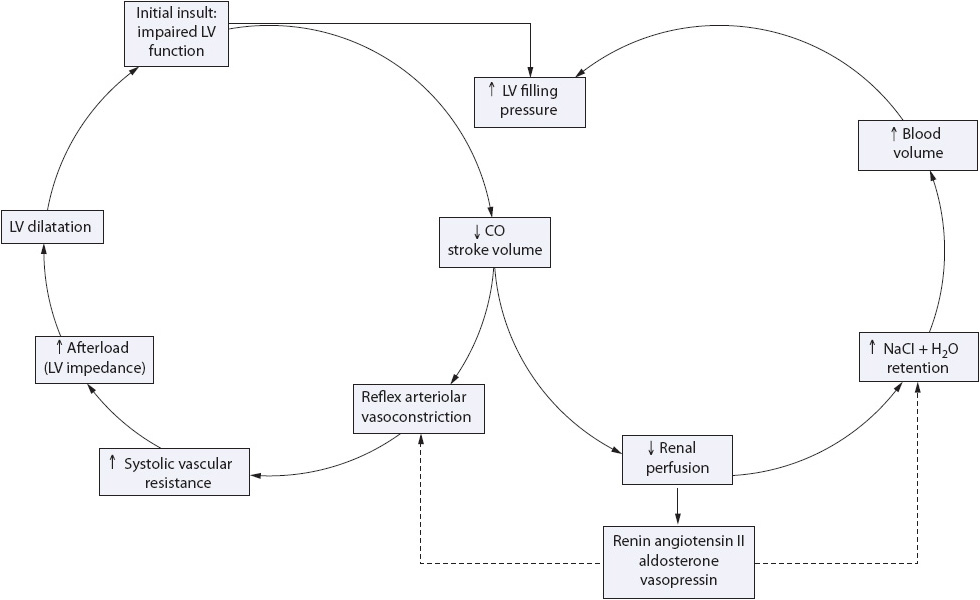
Figure 9-13. Compensatory mechanisms of heart failure.
As cardiac output decreases and the sympathetic nervous system is activated, alpha-1 receptors are stimulated, resulting in arteriolar and venous vasoconstriction. This adaptive response initially results in increased venous return to the ventricle, increased ventricular end-diastolic volume, stretching of the ventricular myocytes, and improved stroke volume. Later, as overstretching of the ventricle occurs, this compensatory mechanism is lost, resulting in left ventricular decompensation and myocardial hypertrophy (Figure 9-14). Additionally, there is increased expression of granules in the left ventricle causing an increased release of brain natriuretic peptide (BNP).
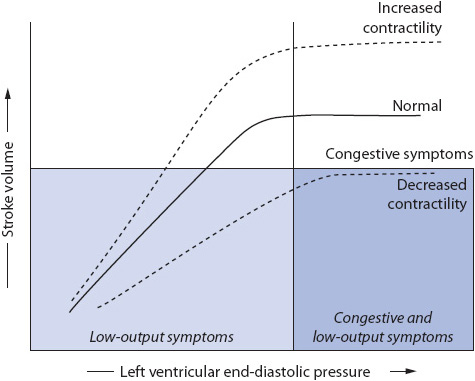
Figure 9-14. Frank-Starling curve.
Increased BNP levels in the serum are used as markers of severity of ventricular failure.
In response to increased vascular volume and decreased myocardial function (loss of the Frank-Starling response), the left ventricle dilates and hypertrophies. This distortion of the normal left ventricular anatomy causes mitral regurgitation and further left ventricular dilatation. Angiotensin II, a by-product of the renin-angiotensin system activation, directly induces myocyte hypertrophy as well. The result of these factors is decreased left ventricular reserve (stretch), increased preload (high residual volume in the ventricle following systole), and further mitral regurgitation.
In response to decreased stroke volume and decreased renal perfusion, several neurohormonal systems are activated, each of which acts to compensate for the decrease in stroke volume. These include:
1. Adrenergic nervous system: Adrenergic nervous system activity is heightened in the setting of impaired ventricular function as a direct result of baroreceptor stimulation. These baroreceptors mediate the sympathetic nervous system, which in turn stimulates the beta-1 receptors. This results in an increase in heart rate and contractility.
2. Renin-angiotensin-aldosterone system: Decreased renal perfusion stimulates the release of renin, increasing the production of angiotensin I and II and the release of aldosterone. This causes arteriolar vasoconstriction, decreased cardiac output, increased arterial BP and peripheral resistance, increased ventricular filling pressures, sodium and potassium retention (imbalance), increased volume overload, increased left ventricular wall stress, increased ventricular dilation and hypertrophy, and increased sympathetic nervous system arousal.
3. Arginine vasopressin (AVP) system: AVP is a potent vasoconstrictor that is normally inhibited by stretch receptors in the atria during atrial distension. In HF, these receptors are less sensitive, causing a decrease in AVP inhibition. This results in systemic vasoconstriction, further increasing afterload (the pressure the ventricle must work against to eject blood out to the system). Increases in AVP availability also lead to an inability to excrete free water, hypoosmolarity, and, in general, inability to autoregulate further AVP production.
4. Atrial natriuretic peptide (ANP): ANP is a counter-regulatory hormone that opposes all three of the above systems, resulting in vasodilation and sodium excretion. ANP is produced in response to atrial distension and results in decreased formation of renin, decreased effects of angiotensin II, decreased release of aldosterone and vasopressin, and enhanced renal excretion of sodium and water. In chronic HF, the levels of ANP remain elevated, but are less so than in the acute decompensation phase (phase III).
The effects of the compensatory mechanisms in phase II lead to an increase in circulating volume and perfusion to vital organs. Eventually, these mechanisms are self-limiting and a vicious cycle of increased afterload and volume overload results. The neurohormonal response is no longer beneficial in the chronic state but, as seen in phase III, becomes detrimental leading to changes in the myocyte DNA, resulting in programmed cell death (apoptosis) and further loss of myocytes.
• Result of phase II: Ventricular hypertrophy, weakened myocytes, increased arteriolar resistance, increased vascular volume, and increased ventricular wall stress occur in an effort to maintain adequate cardiac output.
A 75-year-old man presents to the ED with diaphoresis and severe dyspnea. Initial assessment revealed the following:

A pulse oximeter revealed 83% oxygen saturation.
Laboratory work, including an arterial blood gas sample, was done with the following results:

Oxygen was initiated at 4 L/min via nasal cannula. An ECG was done and showed left ventricular hypertrophy, and left bundle branch block. His chest x-ray showed an enlarged cardiac silhouette and bilateral infiltrates. A pulmonary artery catheter was placed and the following parameters were found (refer Chapter 3 for hemodynamic parameters):

A dobutamine drip was started at 2.5 mcg/kg/min, and furosemide 40 mg IV was given. Cardiac catheterization was performed the next morning with the following findings:
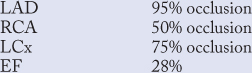
Severe asyneresis
Case Question 1. The purpose of starting the dobutamine infusion and administering the furosemide is to:
(A) Increase myocardial contractility and reduce ventricular preload
(B) Increase myocardial contractility and reduce ventricular afterload
(C) Reduce myocardial contractility and increase ventricular preload
(D) Increase myocardial contractility and increase ventricular afterload
Case Question 2. Following the initiation of dobutamine and administration of furosemide, you would expect which of the following to occur?
(A) HR 120 beats/min; PA 40/38, PAOP unchanged
(B) HR 110 beats/min; PA 32/25 mm Hg; PAOP 24 mm Hg
(C) HR 95 beats/min; PA unchanged; RA 14 mm Hg
(D) BP 105/80 mm Hg, PA 49/38 mm Hg; RA 14 mm Hg
Case Question 3. After reviewing the cardiac catheterization results, you would anticipate the patient having one of the following procedures:
(A) Implantation of a HeartMate II LVAD as destination therapy
(B) Ventricular aneurysmectomy/reconstruction surgery
(C) Mitral valve repair
(D) Insertion of a dual chamber biventricular pacemaker
Answers: 1: A; 2: B; 3: D
When the adaptive mechanisms of phase II fail, the clinical syndrome of HF follows. This third phase of HF is extremely variable in onset and presentation. The clinical expression and course of the disease is determined by the extent of the initial insult and myocyte damage, the severity of hemodynamic burden (volume overload), and the patient’s individual neurohormonal response to these changes. Phase III is characterized by a progressive deterioration of cardiovascular functioning due to the relationship between compromised left ventricular function and excessive cardiac afterload (Figure 9-15). As ventricular dilation occurs, brain natriuretic peptide (BNP) levels in the serum increase.
• Result of phase III: Clinical signs and symptoms of HF are evident, resulting in decreased functional status and activity intolerance for the patient.
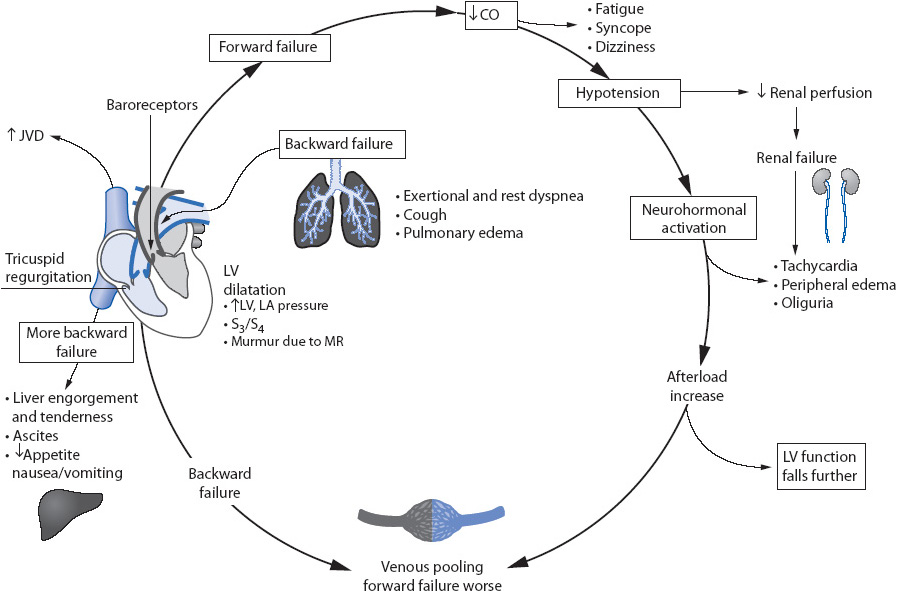
Figure 9-15. Clinical features of heart failure.
Regardless of the underlying cause of the weak pump, patients with HF present with clinical signs and symptoms of intravascular and interstitial volume overload, as well as manifestations of inadequate tissue perfusion. Common findings in HF include:
• Dyspnea (especially with exertion, commonly severe in the acute setting)
• Paroxysmal nocturnal dyspnea
• Pulmonary edema (pronounced crackles)
• Jugular venous distention (JVD)
• Chest discomfort or tightness
• Peripheral edema
• Cool, pale, cyanotic skin
• Oliguria
• Reported weight gain
• Fatigue
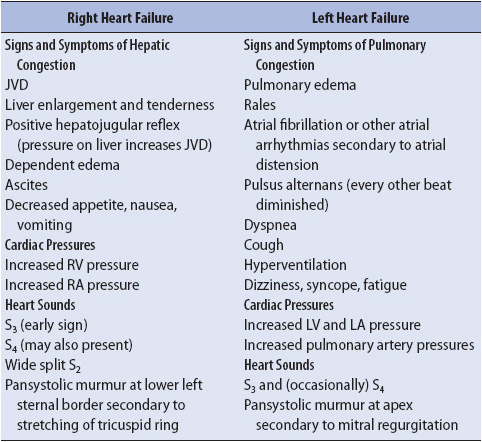
More specific physical signs and symptoms may vary in individuals depending on the ventricle which is primarily involved. A summary of clinical findings specific to left and right ventricular failure is presented in Table 9-9.
TABLE 9-9. CLINICAL SIGNS AND SYMPTOMS SPECIFIC TO RIGHT-AND LEFT-SIDED HEART FAILURE
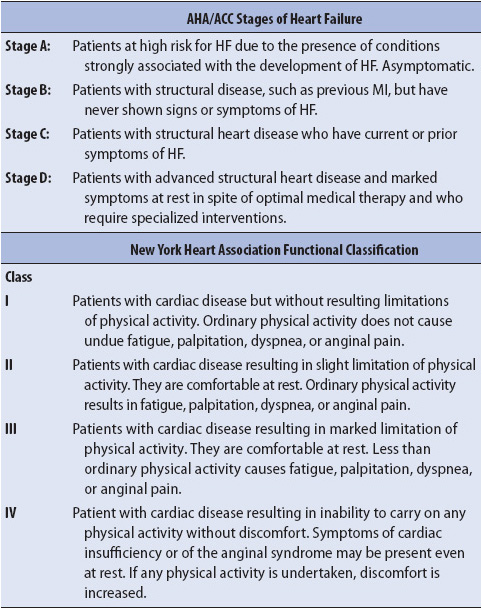
Because subjective assessment of symptoms and their severity may vary from clinician to clinician, classification systems have been developed to standardize symptom severity as well as the evolution and progression of HF. The American College of Cardiology and the American Heart Association developed a staging system that addresses the evolution and progression of HF. A second system, known as the New York Heart Association (NYHA) Functional Classification System, is used to provide systematic assessment of patient status and to benchmark improvement or deterioration from initial evaluation (Table 9-10).
TABLE 9-10. CLASSIFICATION OF CARDIOVASCULAR DISABILITY
A number of conditions, both cardiac and noncardiac, are similar to HF in their clinical presentation and should be ruled out as possible diagnoses in the initial assessment. These conditions include MI, pulmonary disease, dysrhythmias, anemia, renal failure, nephrotic syndrome, and thyroid disease.
• 12-lead ECG: Acute ST-T wave changes, low voltage, left ventricular hypertrophy, atrial fibrillation or other tachyarrhythmias, bradyarrhythmias, Q waves from previous MI, LBBB
• Chest x-ray: Cardiomegaly, cardiothoracic ratio 0.5
• Complete blood count: Low red cell count (anemia)
• Urinalysis: Proteinuria, red blood cells, or casts
• Creatinine: Elevated
• Albumin: Decreased
• Serum sodium and potassium: Decreased
• PAP: Elevated
• CI: < 2.0 L/min/m2
• Brain natriuretic peptide: Elevated
• Echocardiography: Dilated left ventricle, right ventricle, or right atria; hypertrophied left ventricle; AV valve incompetence; diffuse or segmental hypocontractility; atrial thrombus; pericardial effusion; LVEF < 40%
• Radionuclide ventriculography: More precise measure of right ventricular dysfunction and LVEF
Acute management of HF has changed dramatically over the past decade, from an emphasis on the micromanagement of hemodynamic parameters, primarily using positive inotropes, to an emphasis on functional capacity and long-term survival with the use of neurohormonal blocking agents. This shift is due to a better understanding of the neurohormonal response and the dependence of the body on these mechanisms for compensation in low-output states. Goals of patient management in HF revolve around four general principles: (1) treatment of the underlying cause (eg, ischemia, valvular dysfunction), (2) management of fluid volume overload, (3) improvement of ventricular function, and (4) patient and family education.
The most effective, but often the most difficult, management strategy for HF is to limit the damage done by the initial insult. This limitation of myocardial damage and cell loss maximizes the amount of viable ventricular muscle, myocardial contractility, and overall ventricular function.
• Administer fibrinolytic therapy as soon as possible for eligible patients in the setting of AMI or facilitate immediate transfer to the cardiac catheterization laboratory for primary PCI (see the previous section on acute ischemic heart disease).
• Revascularization may be warranted in patients with persistent ischemia as a preventive measure against eventual tissue necrosis.
• Valve replacement or repair or other surgical corrections (ventricular reconstruction surgery) should be undertaken as soon as possible to prevent prolonged overstretching of the ventricular myocardium.
Decrease preload by the use of diuretic therapy, limitation of dietary sodium, and restriction of free water.
• Diuretics should be initiated according to the severity of the patient’s signs and symptoms. More severe symptoms require intravenous therapy and loop diuretics, and less severe symptoms may be managed adequately on loop diuretics. Thiazide diuretics may be added later if the patient does not respond to the loop diuretics. Caution must be taken not to diurese the patient too fast as rapid loss of fluid can lead to activation of the renin-angiotensin system.
• Sodium and fluid restriction should be monitored carefully, with sodium intake not exceeding 2 g/day and free water not exceeding 1500 mL in a 24-hour period. Obtain nutrition consult to reinforce sodium and water restrictions.
• Serum sodium and potassium should be monitored on a regular basis to prevent inadvertent electrolyte imbalances (each day or two in the acute setting, depending on the aggressiveness of therapy).
Improvement in left ventricular function is accomplished by decreasing the workload on the heart with preload and after-load reduction and by augmenting ventricular contractility. Ventricular function may be measured directly in the acute setting by monitoring CI using a noninvasive cardiac output monitor. As has been demonstrated by a number of large clinical trials, traditional micromanagement of hemodynamic variables, such as CI with inotropic drugs, may be detrimental to long-term patient outcome. Current recommendations do not advocate this as an initial management strategy.
• Decrease preload (see above).
• Decrease afterload by administration of pharmacologic therapy, including ACE inhibitors and vasodilators. ACE inhibitors are recommended in all HF patients with a LVEF < 40% unless otherwise contraindicated or not tolerated. Contraindications to ACE inhibitor therapy include previous intolerance, potassium > 5.5 mEq/L, hypotension with systolic blood pressure less than 90 mm Hg, and serum creatinine greater than 3.0 mg/dL. Cautious initiation of low-dose therapy in patients with contraindications may still be considered. If the patient is ACE inhibitor intolerant (eg, experiences a cough), an ARB may be administered. Vasodilators may also be used in conjunction with diuretics and ACE inhibitors if further afterload reduction is necessary. Nitrates are often used concomitantly with ACE inhibitors and diuretics to augment afterload reduction, especially in the case of underlying atherosclerotic disease, still the largest single contributor to HF. Angiotensin receptor blockers may be used if the patient does not tolerate the side effects of an ACE inhibitor (eg, cough).
• ACE inhibitors and beta-blockers are considered cornerstone therapy for HF in an effort to reverse the remodeling of the left ventricle. Aldosterone antagonists may be used as add on therapy. Lastly isosorbide, dinitrate, and hydralazine are used for special populations. Digoxin has been shown to improve symptoms but is no longer considered to be first-line therapy unless paroxysmal atrial fibrillation or atrial flutter is present. Digoxin may be used to control the ventricular rate in this situation.
• Beta-blockers are also used to reduce the incidence of ventricular tachycardia and ventricular fibrillation, the most common cause of death in HF patients. Recommended beta-blockers for the management of HF include carvedilol, metoprolol, and bisoprolol. Caution should be taken when initiating a beta-blocker in a patient with reactive airway disease.
• BNP (nesiritide [Natrecor®]) has been another recent addition to the management of decompensated HF. Nesiritide’s effects include promoting diuresis and vasodilation, thereby decreasing ventricular preload and afterload. The agent may also inhibit angiotensin II as well as some of the other neuroendocrine compensatory mechanisms associated with HF. Nesiritide is recommended for acutely decompensated ventricular failure.
• Dual-chamber biventricular pacemaker/implantable cardioverter defibrillator (ICD): Approximately 60% of patients with dilated cardiomyopathy develop LBBB. In the presence of LBBB, the right and left ventricles no longer contract simultaneously but in a series causing the intraventricular septum to shift inappropriately, interfering with the aortic and mitral valve functioning. There have been several studies demonstrating significantly improved outcomes (quality of life, survival rates, etc) with the use of a dual chamber biventricular pacemaker. This technology stimulates both ventricles simultaneously, causing both to contract at the same time resulting in a narrowing of the QRS complex and improved myocardial contractility and cardiac output. Often the pacing technology is combined with an ICD because sudden cardiac death related to ventricular tachycardia/fibrillation is the most common cause of death in these patients.
• Cardiac assist devices (left ventricular, right ventricular, or both) can provide temporary maintenance or preservation of ventricular function, especially as a bridge to recovery, bridge to cardiac transplantation, or as destination therapy (discharge to home). These devices may be inserted percutaneously via the femoral artery or femoral vein or surgically using the medial sternotomy or thoracotomy approach (see Chapter 19, Advanced Cardiovascular Concepts). Left ventricular apical cannulation allows ambulation and physical rehabilitation. Technological developments have contributed to the development of small axial flow pumps allowing many to be implanted with the drive line (power source) exiting the skin. Risks related to insertion of these devices include infection, peripheral embolization including stroke, and, for some, long-term weaning difficulties in the event that an organ donor is not available. Presently, the Heart Mate II® is approved for destination therapy (a replacement for heart transplant).
1. Intra-aortic balloon pump (IABP): Femoral artery cannulation with the IABP allows for ventricular support, but restricts the patient to bed rest and compromises arterial flow to the cannulated limb. These patients will be in the intensive care unit (ICU).
2. Minimally invasive catheter-based micro-axial flow ventricular assist devices: These are frequently used to reduce ventricular afterload and myocardial work. They may be inserted through the femoral artery across the aortic valve into the left ventricle or introduced via the femoral vein into the right atrium and through an atrial septostomy, and positioned in the left atrium. As with the IABP, the patient is restricted to bedrest.
3. Ventricular reconstruction: Many patients with end-stage HF have a previous history of CAD and MI resulting in the development of a ventricular aneurysm on the anterior wall of the left ventricle. A surgical procedure can be performed removing the aneurysm, reducing the size of the ventricle resulting in increased contractility and cardiac output. Studies have shown that some patients experience improvement in physical functioning and NYHA Functional Class following this procedure.
4. Extracoporeal membrane oxygenation (ECMO): In recent years the use of ECMO has become more common for severely decompensated HF patients who are in cardiogenic shock. This is used as a bridge to recovery, a link to a ventricular assist device (VAD), or a bridge to transplant. The use of this technology is limited to the ICU.
Patients who present with HF to the progressive care unit have high acuity levels, require more pharmacological interventions, and have an increased need for emotional support surrounding the serious nature of the hospital admission. Previous admissions for HF make patients more aware of the serious nature of acute episodes. Patient education, which is appropriately addressed in the acute-care setting, includes the following:
1. Both patient and family may require crisis interventions. The nurse may help by encouraging the verbalization of fears related to role adaptations or changes in family responsibility, lifestyle alterations and limitations, and death and dying. The completion of advanced directives and living wills should be initiated if not previously addressed.
2. Family involvement in the acute-care phase should be strongly encouraged, including assistance with activities of daily living such as bathing, and “patterning” of daily activities to allow for frequent periods of rest and spacing of exertional activity. In addition, family involvement in reading or other leisure activity with the patient is often restful and relaxing, and may be useful as a diversional activity. If possible, the family should also be present for reinforcement of patient teaching regarding the medical regimen, the importance of fluid and sodium restriction, and the need for daily weights.
Shock is the inability of the circulatory system to deliver enough blood to meet the oxygen and metabolic requirements of body tissues. This clinical syndrome may result from ineffective pumping of the heart (cardiogenic shock), insufficient volume of circulating blood (hypovolemic shock), or massive vasodilation of the vascular bed causing maldistribution of blood (distributive shock). Although the specific definition of shock and strategies for patient management vary according to the underlying pathophysiology, the principle of ineffective or insufficient oxygen delivery to meet the needs of body tissues remains consistent.
The ineffective delivery of oxygen to the tissues leads to cellular dysfunction, rapidly progressing to organ failure and finally to total body system failure. The cause of the initial onset of the shock syndrome may be from any number of underlying problems, including heart problems, fluid loss, and trauma. Because the body responds in the same way, differences between cardiogenic, hypovolemic, and distributive shock are obvious to the clinician only after the initial assessment has provided key information about the patient’s acute illness. Given the history, the clinician can classify shock into one of three major pathologic groups and proceed to further determine the patient’s needs with the help of diagnostic testing. Because interventions for patient management are directed at the cause, it is essential for the underlying pathophysiology to be clearly understood.
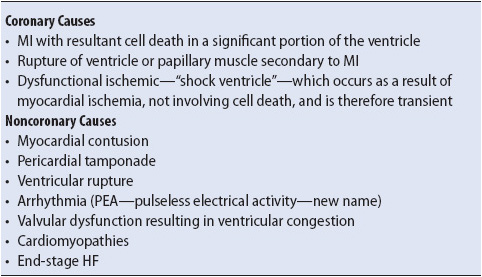
In cardiogenic shock, the heart is unable to pump enough blood to meet the oxygen and metabolic needs of the body. Pump failure is caused by a variety of factors, the most common being CAD. A number of other factors may cause pump failure, however, and are typically categorized as coronary or noncoronary causes (Table 9-11).
TABLE 9-11. CAUSES OF CARDIOGENIC SHOCK
A 49-year-old man was found slumped in his living room chair, cool and clammy but still breathing. His wife phoned emergency medical services, which arranged air transportation to the local emergency room. On arrival, his vital signs were as follows:

Oxygen at 60% by face mask had been initiated in flight, as well as intravenous normal saline running wide open, 450 mL having already infused. Dopamine was started at a rate of 5 mcg/kg/min. A stat ECG showed “tombstone” ST elevation in anterior leads (V2, V3, V4), with reciprocal changes in leads II, III, and aVF. The patient was taken for immediate PTCA. In the laboratory, cardiac catheterization findings were as follows:

On return to the progressive care unit, the nurse obtained hemodynamic parameters as follows:

Case Question 1. Given the patient’s history, ECG changes, and hemodynamic profile, you know the type of shock this patient is experiencing is:
(A) Hypovolemic
(B) Distributive
(C) Cardiogenic
(D) Neurogenic
Case Question 2. Following the interventional procedure the primary goal for this patient is to:
(A) Reduce myocardial workload
(B) Dilate the pulmonary vascular bed
(C) Administer a diuretic
(D) Intubate the patient to improve oxygen delivery
Case Question 3. You anticipate the next intervention for this patient will be:
(A) Initiate a vasodilator to reduce afterload
(B) Give volume to improve preload
(C) Titrate the dopamine infusion up to 7.5 mcg/kg/min
(D) Insertion of an intra-aortic balloon
Answers: 1: C; 2: A; 3: D
In all cardiogenic shock cases, the heart ceases to function effectively as a pump, resulting in decreases in stroke volume and cardiac output. This leads to a decrease in blood pressure and tissue perfusion. The inadequate emptying of the ventricle increases left atrial pressure, which then increases pulmonary venous pressure. As a result, pulmonary capillary pressure increases, resulting in pulmonary edema.
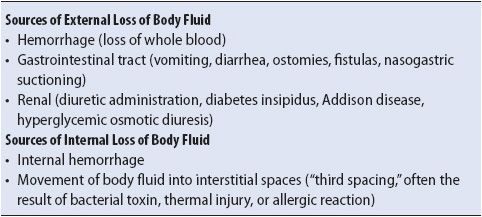
Hypovolemic shock occurs when there is inadequate volume in the vascular space. This volume depletion may be caused by blood loss, either internal or external, or by the vascular fluid volume shifting out of the vascular space into other body fluid spaces (Table 9-12). The loss of vascular volume results in insufficient circulating blood to maintain tissue perfusion.
TABLE 9-12. CAUSES OF HYPOVOLEMIC SHOCK
The pathophysiology of hypovolemic shock is related directly to a decreased circulating blood volume. When an insufficient amount of blood is circulating, the venous blood returning to the heart is insufficient. As a result, right and left ventricular filling pressures are insufficient, decreasing stroke volume and cardiac output. As in cardiogenic shock, when cardiac output is decreased, BP is low and tissue perfusion is poor.
Distributive shock is characterized by an abnormal placement or distribution of vascular volume, occurring in three situations: (1) sepsis, (2) neurologic damage, and (3) anaphylaxis. In each of these situations, the pumping function of the heart and the total blood volume are normal, but the blood is not appropriately distributed throughout the vascular bed. Massive vasodilation occurs in each of these situations for various reasons, causing the vascular bed to be much larger than normal. In this enlarged vascular bed, the usual volume of circulating blood (approximately 5 L) is no longer sufficient to fill the vascular space, causing a decrease in blood pressure and inadequate tissue perfusion. For this reason distributive shock is also referred to as relative hypovolemic shock.
Of the distributive shock syndromes, septic shock is most commonly seen in the critical care setting. In the field or emergency department setting, anaphylaxis and neurogenic shock are also common and typically result from allergic reactions and trauma-related spinal cord injury.
Regardless of underlying etiology, all three types of shock (cardiogenic, hypovolemic, distributive) activate the sympathetic nervous system, which in turn initiates neural, hormonal, and chemical compensatory mechanisms in an attempt to improve tissue perfusion (Figure 9-16). Cellular changes that occur as a result of these compensatory mechanisms are similar in all types of shock. Progression of these cellular changes follows a predictable, four-stage course.
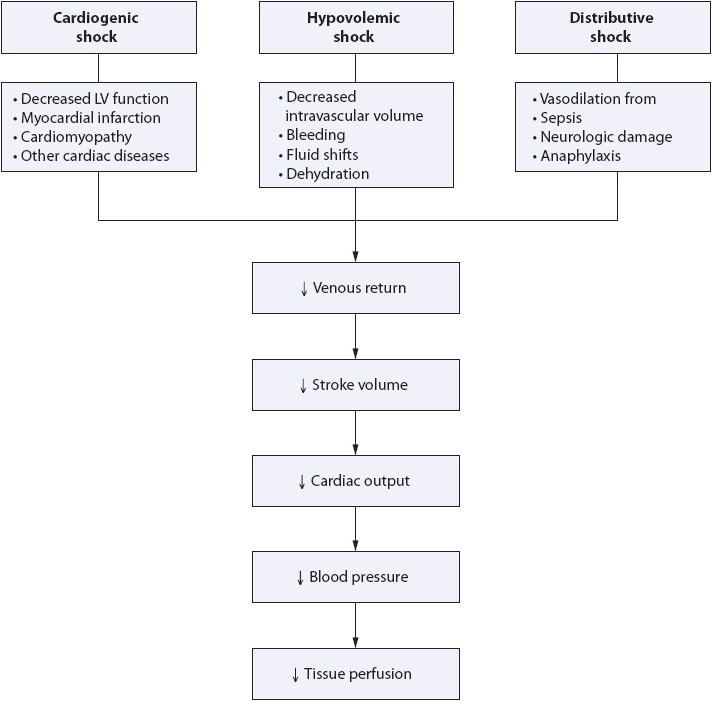
Figure 9-16. Pathophysiology of shock.
The initial stage of shock represents the first cellular changes resulting from the decrease in oxygen delivery to the tissue. These changes include decreased aerobic and increased anaerobic metabolism, leading to increases in serum lactic acid. No obvious clinical signs and symptoms are apparent during this stage of shock.
The compensatory stage is composed of a number of physiologic events that represent an attempt to compensate for decreases in cardiac output and restore adequate oxygen and nutrient delivery to the tissues (Figure 9-17). These events can be organized into neural, hormonal, and chemical responses. Neural responses involve the baroreceptors in the aortic arch and carotid arteries, detecting changes in the arterial BP, and responding by activating the vasomotor center of the medulla. Hypovolemia and resultant hypotension lead to activation of the sympathetic nervous system. The sympathetic nervous system initiates neural, hormonal, and chemical compensatory mechanisms causing peripheral vasoconstriction and elevation of the BP. The sympathetic nervous system activation produces vasoconstriction of the peripheral circulation, shunting blood to vital organs (autoregulation). Vasoconstriction of the peripheral circulation shunts blood to the vital organs, reducing renal blood flow, which activates the hormonal response.
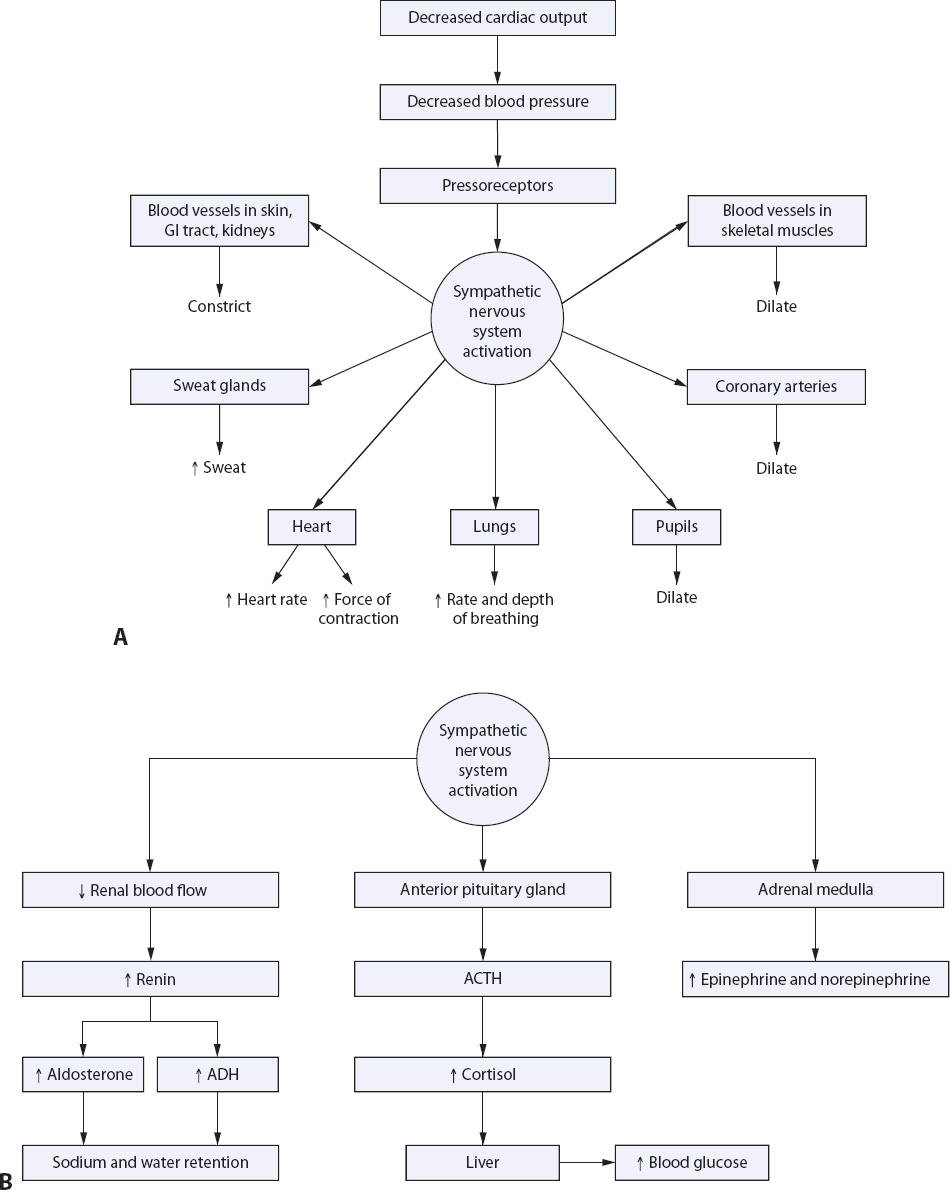
Figure 9-17. Compensatory response to shock. (A) Neural compensation. (B) Hormonal compensation.
Hormonal responses include increased production of catecholamines and adrenocorticotropic hormone (ACTH) and activation of the renin-angiotensin-aldosterone system. As a direct result of decreased renal blood flow, renin is released from the juxtaglomerular cells in the kidney, combining with angiotensinogen from the liver resulting in the production of angiotensin I. Angiotensin I, circulating in the blood, is converted to angiotensin II in the lungs. As was discussed in more detail in the HF section, this hormonal response results in direct peripheral vasoconstriction, as well as release of aldosterone from the adrenal cortex and antidiuretic hormone (ADH) from the pituitary gland. Sodium and potassium retention, in conjunction with increased ADH, ACTH, and circulating catecholamines, effectively increases intravascular volume, heart rate, and blood pressure, and decreases urine output.
Chemical responses during the compensatory stage are related to the respiratory ventilation-perfusion imbalance, which occurs as a result of sympathetic stimulation, redistribution of blood flow, and decreased pulmonary perfusion. A respiratory alkalosis ensues, adversely affecting the patient’s level of consciousness, and causing restlessness, and agitation.
These compensatory mechanisms are effective for finite periods of time, which may vary depending on the individual and presence of comorbidities. The younger and healthier the patients prior to the shock episode, the more likely they are to survive a prolonged episode of shock. In the absence of vascular volume replacement, these intrinsic vasopressors eventually fail as a compensatory mechanism, and the patient enters the progressive, and finally refractory, stages of shock, usually resulting in death.
The progressive stage is characterized by end-organ failure due to cellular damage from prolonged compensatory changes. The compensatory changes, which were effective in supporting blood pressure and therefore tissue perfusion, are no longer effective and severe hypoperfusion ensues. Impaired oxygen delivery to the tissues results in multiple organ system dysfunction (MODS)—typically beginning with gastrointestinal and renal failure—followed by respiratory and/or cardiac failure and loss of liver and cerebral function. See Chapter 11 for more on sepsis and MODS.
The refractory stage, as its name implies, is the irreversible stage of shock. At this stage, cell death has progressed to such a point as to be irreparable, and death is imminent.
Clinical signs and symptoms vary depending on the underlying cause of shock and the stage of shock in which the patient presents.
• Initial stage: No visible signs and symptoms evident from ongoing cellular changes
• Compensatory stage
• Consciousness: Restless, agitated, confused
• Blood pressure: Normal or slightly low
• Respiratory rate: Increased (> 20 breaths/min)
• Skin: Cool, clammy, may be cyanotic
• Peripheral pulses: Weak and thready
• Urine output: Concentrated and scant (< 30 mL/h)
• Bowel sounds: Hypoactive, possible abdominal distension
• Laboratory results
• Glucose: Increased
• Sodium: Increased
• PaO2: Decreased
• PaCO2: Decreased
• pH: Increased
• Progressive stage
• Consciousness: Unresponsive to verbal stimuli
• Blood pressure: Inadequate (< 90 mm Hg systolic)
• Heart rate: Increased > 90/min
• Respiratory rate: Increased, shallow
• Skin: Cold, cyanotic, mottled
• Peripheral pulses: Weak and thready, may be absent
• Urine output: Scant (< 20 mL/h) and concentrated
• Bowel sounds: Absent
• Laboratory results
• Amylase: Increased
• Lipase: Increased
• SGPT/SGOT: Increased
• Lactate: Increased
• CPK: Increased
• Creatinine: Increased
• Blood urea nitrogen: Increased
• PaO2: Decreased
• PaCO2: Increased
• pH: Decreased
• HCO3: Decreased
• ECG: Tachycardia
• Pulmonary arterial pressure: PAD/PAOP high (> 12 mm Hg), RAP high (> 8 mm Hg)
• Echocardiogram: Ventricular wall motion abnormalities, cardiac tamponade, ventricular rupture
• Hypovolemic
• Pulmonary arterial pressure: PAD/PAOP low (< 8 mm Hg), RAP low (< 5 mm Hg), RVEDVI low
• Ultrasound: Groin or retroperitoneal hemorrhage
• Distributive
• Septic: WBC ≥ 12,000 or ≤ 4,000 > 10% neutrophils, serum lactate > 4 mmol/L, positive blood cultures (in 50% of patients).
• Anaphylactic: Arterial blood gas shows inadequate oxygenation.
• Neurogenic: Computed tomography (CT) scan and magnetic resonance imaging (MRI) shows spinal cord damage.
Differences in the underlying cause of shock lead to some variation in the principles of management. The basic goals of therapy for all forms of shock, however, include the need to correct the underlying cause of shock, improvement of oxygenation, and restoration of adequate tissue perfusion. Generally a patient in shock will be transferred to the critical care unit for management of their shock state.
• Cardiogenic: Remove coronary obstruction or correct tamponade, if present, and support ventricular contractility to increase cardiac output.
• Hypovolemic: Identify source and stop bleeding if possible; correct fluid shunting or third spacing with electrolyte management.
• Distributive
• Anaphylactic: Intubate for oxygenation and treat the underlying allergic reaction using antidote or steroid therapy.
• Septic: Implement 3 hour “bundle” including obtaining blood cultures and serum lactate; administer broad spectrum antibiotics and 30 mL/kg crystalloid for hypotension; implement early goal-directed therapy protocol, removal of infected tissue or device, refer to AACN Practice Alert: Severe Sepsis for evidence-based practices for the management of severe sepsis and septic shock. (See Chapter 11 for more on sepsis.)
• Neurogenic: Severing of the cord may be irreversible; however, intubation provides respiratory support while the underlying cause is identified.
• Assess for patent airway and intubate if necessary.
• Administer oxygen at 100% or as necessary until PaO2 is adequate (> 60 to 70 mm Hg).
• Administer fluid volume expanders (normal saline, lactated Ringer solution, or plasmanate) in large rapid boluses. Type and cross-match for blood type and administer blood as necessary for hypovolemic shock.
• Initiate vasoactive drug therapy.
Hypertension is typically a chronic disease of BP elevation that is often masked, especially in the early years of onset, by lack of warning signs or symptoms. Hypertensive crisis is an acute episode or exacerbation, occurring infrequently in a small percentage of hypertensive patients and characterized by the pivotal effect the particular episode and its treatment may have on the patient’s long-term outcome. In most cases, the numerical or absolute value of the arterial BP is less important than its impact on the individual’s underlying risk of target organ damage, specifically cerebrovascular, coronary, and renal diseases.
Although a number of clinical syndromes commonly are associated with hypertension and many underlying etiologies may contribute to the progression of hypertensive disease, the pathophysiology of hypertension is similar regardless of the underlying cause.
An acute hypertensive crisis begins with elevation of the systolic or diastolic BP causing a threat, direct or indirect, to an organ or body system. Acute, severe increases in pressure may cause serious, life-threatening cerebrovascular and cardiovascular compromise. Prolonged hypoperfusion of an organ system leads to ischemia, necrosis, and organ system failure.
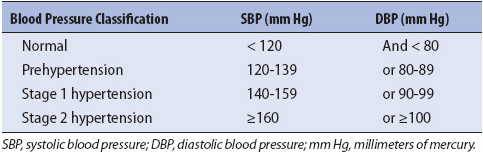
Because of the increased risk of such events in all hypertensive patients, morbidity and mortality directly related to hypertension is high, and long-term, consistent therapy in all stages of hypertension is necessary. Hypertension can be described in stages as described below or classified according to the value of the blood pressure. Refer Table 9-13 for the classification of blood pressure for adults.
• Stage 1 hypertension: Benign hypertension is characterized by slightly elevated blood pressure (140-160 mm Hg systolic/90 mm Hg diastolic, in adults) for long periods of time, with little if any end-organ damage. Stage 1 hypertension does not tend to cause acute problems or complications, unless other comorbid conditions, such as atherosclerotic disease, are present. The pressure does not typically exacerbate or precipitate an acute emergent event (generally not > 140-160 mm Hg systolic/90 mm Hg diastolic, in adults).
• Accelerated hypertension: Often used interchangeably with malignant hypertension, the stage known as accelerated hypertension is generally considered a precursor to malignant hypertension, and is characterized by an increase in the patient’s baseline BP.
• Malignant hypertension: Hypertension typically is a chronic disease in which elevation in BP occurs slowly, over a period of several years. Because of its gradual onset, the body adapts to increased pressures in the vascular bed and the patient frequently is asymptomatic for years, eventually able to tolerate pressures of up to 200/120 mm Hg without experiencing significant symptoms or clinical events. This type of presentation often is identified “accidentally,” secondary to hospitalization for another problem. Generally patients with malignant hypertension are at risk for significant end-organ damage because of the severity of high pressure in the vascular bed and inability of the circulatory system to further adapt or compensate in the event of additional stressors.
• Hypertensive crisis: Hypertensive crisis is characterized by a severe elevation in BP, relative to the individual’s baseline BP, which causes risk of end-organ damage and poor long-term outcome due to permanent organ system damage if the immediate episode is not treated quickly and aggressively.
• Special populations: In pregnant women and in children a less severe elevation in blood pressure may result in significant end-organ damage and is therefore considered to be a “hypertensive crisis” at values much lower than would be expected to be problematic in the average adult. The absolute value of the blood pressure varies significantly depending on the situation and the individual involved; for example, pre-eclampsia, considered to be a hypertensive crisis in pregnancy, may occur at pressures as low as 130/100 to 160/100 mm Hg.
Diagnosis of hypertensive crisis is not based on the absolute value of the blood pressure, but rather on the following combined criteria:
• Rapidity of the rise of the BP
• Duration of prior hypertension
• Clinical determination of the immediate threat to vital organ function
• Headache
• Blurred vision
• Nosebleed
• Dizziness or vertigo
• Transient ischemic attack
• Diminished peripheral pulses or bruits
• Carotid or abdominal bruit
• Heart sounds with S3 and/or S4
• Systolic and/or diastolic murmurs
• Gastrointestinal bleeding
• Pulmonary edema
• Shortness of breath
• Fatigue
• Malaise
• Weakness
• Nausea and vomiting
• Hematuria
• Dysuria
• Funduscopic findings: Arteriovenous thickening, arteriolar narrowing, hemorrhage, papilledema, or exudates
• Chest x-ray: Myocardial hypertrophy, pulmonary infiltrates
• Computed tomography: Arteriolar narrowing and arteriovenous thickening
• Specific tests to target organ damage
• Renal angiography
• Coronary angiography
• Carotid/cerebral angiography
• Magnetic resonance imaging: Cerebral vascular malperfusion
Management of the patient with acute exacerbation of hypertension, or hypertensive crisis, revolves around three primary objectives: reduction of arterial pressure, evaluation and treatment of target organ damage, and preparation and planning for continuous and consistent outpatient follow-up.
Ascertain correct arterial blood pressure. Verify arterial blood pressure, being sure to assess bilateral measurements with the correct cuff size if using sphygmomanometry, as well as orthostatic pressures if possible (lying and sitting up, if standing is not possible). Each measurement should be 2 minutes apart and both right and left measurements should be documented. If differences between the right and left measurements are greater than 10 mm Hg, the higher reading should be used to gauge therapy. In most acute situations, priority should be given to establishing a stable arterial access site for direct, invasive monitoring of BP.
Initiate pharmacologic intervention. For acute high arterial pressure, intravenous pharmacologic intervention is the fastest, most effective means of reducing arterial blood pressure. A number of agents are used in the acute setting for management of hypertensive crisis (Table 9-14). Aggressiveness of pharmacologic intervention should be based on the severity of blood pressure elevation (immediate risk of stroke), the immediate risk of irreversible target organ damage (renal and hepatic function related to drug metabolism and clearance also should be considered), and any confounding conditions or risk factors which are present (for example, the fetus in pre-eclampsia). In general, acute, severe (accelerated malignant/stages 3 and 4) hypertension should be treated as quickly and aggressively as can be tolerated by the patient in order to prevent the immediate risk of hypertensive encephalopathy, dissecting aortic aneurysm, MI, or intracranial hemorrhage. Maintenance of cerebral perfusion pressure is imperative during treatment, and overly aggressive pharmacologic management poses the threat of cerebrovascular compromise due to a sudden drop in arterial pressure and inability of the autoregulatory mechanism to adjust. Other organ systems dependent on higher pressure for perfusion include the renal and coronary systems. A sudden, severe drop in systemic arterial pressure may result in ischemic episodes or acute renal failure.
TABLE 9-14. COMMON DRUGS USED TO MANAGE ACUTE HYPERTENSIVE EPISODES

For non-acute hypertension, dietary alteration and relaxation, or biofeedback techniques may be used in addition to pharmacologic measures to reduce the morbidity and mortality of hypertension. Although these measures are most effective when employed long term as part of a cohesive outpatient follow-up program, initiating these strategies in the acute setting may help emphasize their importance.
Concomitant to initiation of pharmacologic intervention, the assessment and prevention of target organ disease is important to avoid irreversible damage. Target organs typically at risk include the brain, heart, kidneys, and eyes. Strategies to prevent damage to these organ systems during hypertensive crisis include the following:
• Brain: Reduce diastolic pressure by one-third (not to go < 95 mm Hg) using aggressive pharmacologic measures (see Table 9-14).
• Heart: Reduce diastolic and systolic pressure by one-third; administer combination therapy if possible (vasodilator and beta-blocker) or ACE inhibitor for afterload reduction; monitor for ischemic changes on ECG.
• Kidneys: Reduce systolic and diastolic BP using pharmacologic measures; monitor serum creatinine and urine specific gravity as well as proteinuria and hematuria; for patients with severe existing renal impairment, use of ACE inhibitors may exacerbate their renal compromise and is therefore contraindicated in patients with bilateral renal artery stenosis; administer diuretics to maintain serum sodium and adequate diuresis.
• Eyes: Reduce systolic and diastolic BP; observe retina for evidence of hemorrhage, exudate, or papilledema; instruct the patient with blurring of vision regarding his or her environment, especially location of the call bell.
You are taking care of a patient, 4 days post anterior MI, who is just transferred to the progressive care unit from a medicine floor with severe shortness of breath. Your initial assessment reveals the following:

Case Question 1. What is your initial intervention?
(A) Obtain an arterial blood gas measurement
(B) Initiate a nesiritide infusion
(C) Prepare to intubate the patient
(D) Call a Code Blue
Case Question 2. What is the most likely underlying cause for this patient’s respiratory compromise?
(A) Acute decompensated HF with pulmonary edema
(B) Abrupt onset of septic shock with ARDS
(C) Acute anxiety attack
(D) Hypovolemic shock
Case Question 3. Management of this condition would most likely include what interventions?
1. Administration of a diuretic to reduce preload
2. Initiation of a dobutamine infusion at 5 mcg/kg/min
3. Consideration for mechanical support
4. Consideration for intubation and ventilator support
(A) 4
(B) 1 and 2
(C) 3 and 4
(D) All of the above
Answers: 1: C; 2: B; 3: D
Following control of hypertension in the acute phase, patient education should be initiated regarding the serious and chronic nature of the disease. Often, the clinician may have an opportunity in the acute stage to make an impact regarding the seriousness of uncontrolled hypertension and its potentially debilitating effects. Prior to beginning the educational process, assessment should include:
1. Family history of hypertension, cardiovascular disease, coronary artery disease, stroke, diabetes mellitus, and hyperlipidemia
2. Lifestyle history including weight gain, exercise, and smoking habits
3. Dietary patterns including high sodium, alcohol, and dietary fat intake or low potassium intake
4. Knowledge of hypertension and impact of previous medical therapy for hypertension (compliance, side effects, results, or efficacy)
Bonow RO, Mann DL, Zipes DP, Libby P, eds. Braunwald’s Heart Disease: A Textbook of Cardiovascular Medicine. 9th ed. Philadelphia, PA: WB Saunders; 2012.
Moser DK, Riegel B. Cardiac Nursing: A Companion to Braunwald’s Heart Disease. Canada: Saunders; 2008.
Woods SL, Froelicher ESS, Motzer SA, Bridges EJ. Cardiac Nursing. 6th ed. Philadelphia, PA: Lippincott, Williams & Wilkins; 2010.
Hardin S, Kaplow R. Cardiac Surgery Essentials for Critical Care Nursing. Dudbury, MA: Jones & Bartlett Publishing; in press; 2010.
South T. Coronary artery bypass surgery. Crit Care Nurs Clin NA. 2011;23(4):573-586.
Todd BA. Cardiothoracic Surgical Nursing Secrets. St Louis, MO: Mosby-Year Book, Inc; 2005.
Cahoon W, Flattery MP. ACC/AHA non-ST elevation myocardial infarction guidelines revision: 2007: implications for nursing practice. Prog Cardiovasc Nurs. 2008;23(1):53-56.
Naples RM, Harris JW, Ghaemmaghami CA. Critical care aspects in the management of patients with acute coronary syndromes. Emerg Med Clin N Am. 2008;26:685-702.
Thygesen K, Alpert JS, Jaffe AS, et al. Third universal definition of myocardial infarction. Circulation. 2012;126:2020-2035.
Albert NM. Fluid management strategies in heart failure. Crit Care Nurs. 2012;32(2):20-33.
Albert NM. Heart failure with preserved systolic function: giving well-deserved attention to the “other” heart failure. Crit Care Nurs Q. 2007;30(4):287-296.
Daleiden-Burns A, ed. Heart failure. Crit Care Nurs Q. 2007;30(4):285-286.
English MA. Advanced concepts in heart failure. Crit Care Nurs Q. 1995;18(1):56-64.
Fara-Erny A. Heart failure: challenges and outcomes. J Cardiovasc Nurs. 2000;14(4):v-vii.
Litton KA. Demystifying ventricular assist devices. Crit Care Nurs Q. 2011;34(2):200-207.
Bridges EJ, Dukes S. Cardiovascular aspects of septic shock. Crit Care Nurs. 2005;25(2):14-42.
Cheng JM, den Ull CA, Hoeks SE, et al. Percutaneous left ventricular assist devices vs intra-aortic balloon pump counterpulsation for treatment of cardiogenic shock: a meta-analysis on controlled trials. Eur Heart J. 2009;30(17):2101-2108.
Kelley DM. Hypovolemic shock: an overview. Crit Care Nurs Q. 2005;28(1):2-19.
McAtee ME. Cardiogenic shock. Crit Care Nurs Clin of NA. 2011;23(4):607-616.
Reynolds HR, Hochman JS. Cardiogenic shock: current concepts and improving outcomes. Circulation. 2008;117(5):686-697.
Rivers E, Nguyen B, Havstad S, et al. Early goal-directed therapy in the treatment of severe sepsis and septic shock. N Eng J Med. 2001;345(19):1368-1377.
Topalian S, Ginsberg F, Parillo JE. Cardiogenic shock. Crit Care Med. 2008;36(suppl 1):S66-S74.
Schulenburg M. Management of hypertensive emergencies: implications for the critical care nurse. Crit Care Nurs Q. 2007;30(2):80-93.
Smithburger PL, Kane-Gill SL, Seybert AL. Recent advances in the treatment of hypertensive emergencies. Crit Care Nurs. 2010;30(5).
Whelton PK, Appel LJ, Sacco RL, et al. Sodium, blood pressure, and cardiovascular disease: further evidence supporting the American Heart Association sodium reduction recommendations. Circulation. 2012;126:2880-2889.
Adams D, Bridges CR, Casey DE, et al. 2012 ACCF/AHA focused update of the guideline for the management of patients with unstable angina/non-ST-elevation myocardial infarcton: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Circulation. 2012;126:875-910.
American Association of Critical-Care Nurses. AACN Practice Alert: Noninvasive Blood Pressure Monitoring. Aliso Viejo, CA: American Association of Critical-Care Nurses; 2010, June.
American Association of Critical-Care Nurses. AACN Practice Alert: Severe Sepsis. Aliso Viejo, CA: American Association of Critical-Care Nurses; 2006, April.
American Association of Critical-Care Nurses. AACN Practice Alert: ST-Segment Monitoring. Aliso Viejo, CA: American Association of Critical-Care Nurses; 2008, April.
Dellinger RP, Carlet JM, Masur H, Gerlach H. The surviving sepsis guidelines for the management of severe sepsis and septic shock: background, recommendations, and discussion from an evidence-based review. Crit Care Med. 2004;32(suppl 11).
Dellinger RP, Levy MM, Rhodes A, et al. Surviving sepsis campaign: international guidelines for management of severe sepsis and septic shock: 2012. Crit Care Med. 2013;41(2):580-637.
Drew BJ, Calif RM, Funk M, et al. Practice standards for electrocardiographic monitoring in hospital settings. Circulation. 2004;110:2721-2746.
Hillis LD, Smith PK, Anderson JL, et al. 2011 ACCF/AHA guideline for coronary artery bypass surgery: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Circulation. 2011;124:e652-e735.
Levine GN, Bates ER, Blankenship JC, et al. 20100 ACCF/AHA/SCAI Guidelines for percutaneous coronary intervention: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines and the Society for Cardiovascular Angiography and Interventions. Circulation. 2011;124:e574-e651.
Lindenfeld J, Albert NM, Boehmer JP, et al. Executive summary: HSFA 2010 comprehensive heart failure practice guideline. J Card Fail. 2010;16(6):475-539.
Masoudi FA, Bonow RO, Brindis RG, et al. ACC/AHA 2008 statement on performance measurement and reperfusion therapy: a report of the ACC/AHA Task Force on Performance Measures. Circulation. 2008;118:2649-2661.
National Institute of Health. The seventh report of the Joint National Committee on Prevention, Detection, Evaluation and Treatment of High Blood Pressure. U.S. Department of Health and Human Services. [NIH Publication No. 04-5230]. Bethesda, MD: NIH: August 2004.
O’Gara PT, Kushner FG, Ascheim DD, et al. 2013 ACCF/AHA Guideline for the management of ST-elevation myocardial infarction: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Circulation. 2013;127:529-555.
Peura JL, Colvin-Adams M, Francis GS, et al. Recommendations for the use of mechanical circulatory support: device strategies and patient selection: a scientific statement from the American Heart Association. Circulation. 2012;126:2648-2667.