Pediatric Pharmacogenetics, Pharmacogenomics, and Pharmacoproteomics
Jonathan B. Wagner, Matthew J. McLaughlin, J. Steven Leeder
Interindividual variability in the response to similar doses of a given medication is an inherent characteristic of both adult and pediatric populations. Pharmacogenetics , the role of genetic factors in drug disposition and response, has resulted in many examples of how variations in human genes can lead to interindividual differences in pharmacokinetics and drug response at the level of individual patients. Pharmacogenetic variability contributes to the broad range of drug responses observed in children at any given age or developmental stage. Therefore, it is expected that children will benefit from the promise of personalized medicine —identifying the right drug for the right patient at the right time (Fig. 72.1 ). However, pediatricians are keenly aware that children are not merely small adults. Numerous maturational processes occur from birth through adolescence such that utilization of information resulting from the Human Genome Project and related initiatives must take into account the changing patterns of gene expression that occur over development to improve pharmacotherapeutics in children.
Definition of Terms
The terms pharmacogenomics and pharmacogenetics tend to be used interchangeably, and precise, consensus definitions are often difficult to determine. Pharmacogenetics classically is defined as the study or clinical testing of genetic variations that give rise to interindividual response to drugs. Examples of pharmacogenetic traits include specific adverse drug reactions, such as unusually prolonged respiratory muscle paralysis due to succinylcholine, hemolysis associated with antimalarial therapy, and isoniazid-induced neurotoxicity, all of which were found to be a consequence of inherited variations in enzyme activity. The importance of pharmacogenetic differences has become better understood and is exemplified by the half-life of several drugs being more similar in monozygotic twins than in dizygotic twins. However, it is important to note that in addition to pharmacogenetic differences, environmental factors (diet, smoking status, concomitant drug or toxicant exposure), physiologic variables (age, sex, disease, pregnancy), and patient adherence all contribute to variations in drug metabolism and response. Likewise, ethnicity is another potential genetic determinant of drug variability. Chinese patients who are HLA-B*1502 positive have an increased risk of carbamazepine-induced Stevens-Johnson syndrome; white patients who are HLA-B*5701 positive have an increased risk of hypersensitivity to abacavir (Table 72.1 ).
Table 72.1
Examples of Effects of Gene Polymorphisms on Drug Response
| GENE | ENZYME/TARGET | DRUG | CLINICAL RESPONSE |
|---|---|---|---|
| BCHE | Butyrylcholinesterase | Succinylcholine | Prolonged paralysis |
| CYP2C9 | Cytochrome P450 2C9 | Warfarin | Individuals having ≥1 reduced function alleles require lower doses of warfarin for optimal anticoagulation, especially initial anticoagulant control. |
| CYP2C19 | Cytochrome P450 2C19 | Clopidogrel | Individuals having ≥1 loss-of-function alleles have reduced capacity to form pharmacologically active metabolite of clopidogrel and reduced antiplatelet effect. |
| CYP2D6 | Cytochrome P450 2D6 | Codeine |
Poor metabolizers—individuals with 2 loss-of-function alleles—do not metabolize codeine to morphine and thus experience no analgesic effect. Ultrarapid metabolizers—individuals with ≥3 functional alleles—may experience morphine toxicity. |
| G6PD | Glucose-6-phosphate dehydrogenase | Primaquine (others) | Hemolysis |
| HLA-A*3101 | Human leukocyte antigen A31 | Carbamazepine | Carriers of HLA-A*3101 allele have increased risk of SJS and TEN from carbamazepine. |
| HLA-B*1502 | Human leukocyte antigen B15 | Allopurinol | Han Chinese carriers of HLA-B*1502 allele have increased risk of SJS and TEN from carbamazepine. |
| HLA-B*5701 | Human leukocyte antigen B57 |
Abacavir Flucloxacillin |
Carriers of HLA-B*5701 allele have increased risk of hypersensitivity reactions to abacavir- and flucloxacillin-induced liver injury. |
| HLA-B*5801 | Human leukocyte antigen B58 | Allopurinol | Carriers of HLA-B*5801 allele have increased risk of severe cutaneous adverse reactions to allopurinol, including hypersensitivity reactions, SJS, and TEN. |
| NAT2 | N -Acetyltransferase 2 | Isoniazid, hydralazine | Individuals homozygous for “slow acetylation” polymorphisms are more susceptible to isoniazid toxicity, or hydralazine-induced systemic lupus erythematosus. |
| SLCO1B1 | Organic anion–transporting protein (OATP) 1B1 | Simvastatin | Carriers of the SLCO1B1*5 allele are at increased risk for musculoskeletal side effects from simvastatin. |
| TPMT | Thiopurine S -methyltransferase |
Azathioprine 6-Mercaptopurine |
Individuals homozygous for an inactivating mutation have severe toxicity if treated with standard doses of azathioprine or 6-mercaptopurine; rapid metabolism causes undertreatment. |
| UGT1A1 | Uridine diphospho-glucuronosyltransferase 1A1 | Irinotecan | UGT1A1*28 allele is associated with decreased glucuronidation of SN-38, the active metabolite of irinotecan, and increased risk of neutropenia. |
| VKORC1 | Vitamin K oxidoreductase complex 1 | Warfarin | Individuals with a haplotype associated with reduced expression of VKORC1 protein (therapeutic target of warfarin) require lower doses of the drug for stable anticoagulation. |
SJS, Stevens-Johnson syndrome; TEN, toxic epidermal necrolysis.
Pharmacogenomics represents the marriage of pharmacology and genomics and can be defined as the broader application of genome-wide technologies and strategies to identify both disease processes that represent new targets for drug development and factors predictive of efficacy and risk of adverse drug reactions.
Pharmacokinetics describes what the body does to a drug. It is often studied in conjunction with pharmacodynamics , which explores what a drug does to the body. The pharmacokinetic properties of a drug are determined by the genes that control the drug's disposition in the body (absorption, distribution, metabolism, excretion). Drug-metabolizing enzymes and drug transporters play a particularly important role in this process (Table 72.2 ), and the functional consequences of genetic variations in many drug-metabolizing enzymes have been described between individuals of both similar and different ethnic groups. The most common clinical manifestation of pharmacogenetic variability in drug biotransformation is an increased risk of concentration-dependent toxicity caused by reduced clearance and consequent drug accumulation. However, an equally important manifestation of this variability is lack of efficacy caused by variations in metabolism of prodrugs that require biotransformation to be converted into a pharmacologically active form of a medication. The pharmacogenetics of drug receptors and other target proteins involved in signal transduction or disease pathogenesis can also be expected to contribute significantly to interindividual variability in drug disposition and response.
Table 72.2
Some Important Relationships Between Drugs and Cytochrome P450 (CYP) Enzymes* and P-Glycoprotein Transporter
| ENZYME | DRUG SUBSTRATES | INHIBITORS | INDUCERS |
|---|---|---|---|
| CYP1A2 | Caffeine, clomipramine (Anafranil † ), clozapine (Clozaril † ), theophylline | Cimetidine (Tagamet † ) | Omeprazole (Prilosec † ) |
| Fluvoxamine (Luvox † ) | Tobacco | ||
| Ciprofloxacin (Cipro ) | |||
| CYP2C9 | Diclofenac (Voltaren † ), ibuprofen (Motrin † ), piroxicam (Feldene † ), Losartan (Cozaar ), irbesartan (Avapro ), celecoxib (Celebrex ), tolbutamide (Orinase † ), warfarin (Coumadin † ), phenytoin (Dilantin ) | Fluconazole (Diflucan ) | Rifampin (Rifadin † ) |
| Fluvastatin (Lescol ) | |||
| Amiodarone (Cordarone ) | |||
| Zafirlukast (Accolate ) | |||
| CYP2C19 | Omeprazole, lansoprazole (Prevacid ), pantoprazole (Protonix ), (S)-mephenytoin, (S) -citalopram (Lexapro ); nelfinavir (Viracept ), diazepam (Valium † ), voriconazole (Vfend ) | Cimetidine | Rifampin |
| Fluvoxamine | |||
| CYP2D6 | CNS-active agents: Atomoxetine (Strattera ), amitriptyline (Elavil † ), desipramine (Norpramin † ), imipramine (Tofranil † ), paroxetine (Paxil ), haloperidol (Haldol † ), risperidone (Risperdal ), thioridazine (Mellaril † ) |
Fluoxetine (Prozac † ) Paroxetine (Paxil ) |
|
| Antiarrhythmic agents: Mexiletine (Mexitil ), propafenone (Rythmol ) |
Amiodarone (Cordarone † ) Quinidine (Quinidex † ) |
||
| β-Blockers: Propranolol (Inderal † ), metoprolol (Lopressor † ), timolol (Blocadren † ) | Terbinafine | ||
| Narcotics: Codeine, dextromethorphan, hydrocodone (Vicodin † ) | |||
| Others: Tamoxifen (Nolvadex ) |
Cimetidine Ritonavir |
||
| CYP3A4 | Calcium channel blockers: Diltiazem (Cardizem † ), felodipine (Plendil ), nimodipine (Nimotop ), nifedipine (Adalat † ), nisoldipine (Sular ), nitrendipine, verapamil (Calan † ) | Amiodarone | Barbiturates |
|
Carbamazepine (Tegretol † ) Phenytoin (Dilantin † ) |
|||
| Immunosuppressive agents: Cyclosporine A (Sandimmune , Neoral † ), tacrolimus (Prograf ) | Efavirenz (Sustiva ) | ||
| Corticosteroids: Budesonide (Pulmicort ), cortisol, 17β-estradiol, progesterone, testosterone |
Fluconazole Ketoconazole (Nizoral † ) Itraconazole (Sporanox ) |
Nevirapine (Viramune ) | |
| Macrolide antibiotics: Clarithromycin (Biaxin ), erythromycin (Erythrocin † ), troleandomycin (TAO ) |
Clarithromycin Erythromycin Troleandomycin |
||
| Anticancer agents: Cyclophosphamide (Cytoxan † ), gefitinib (Iressa ), ifosfamide (Ifex ), tamoxifen, vincristine (Oncovin † ), vinblastine (Velban † ), | Imatinib | Rifampin | |
| Ritonavir ‡ | |||
| Benzodiazepines: Alprazolam (Xanax † ), midazolam (Versed † ), triazolam (Halcion † ) | St. John's wort | ||
| Opioids: Alfentanil (Alfenta † ), fentanyl (Sublimaze † ), sufentanil (Sufenta † ) | |||
| HMG-CoA reductase inhibitors: Lovastatin (Mevacor ) † , simvastatin (Zocor ), atorvastatin (Lipitor ) | |||
| HIV protease inhibitors: Indinavir (Crixivan ), nelfinavir, ritonavir (Norvir ), saquinavir (Invirase, Fortovase ), amprenavir (Agenerase ) |
Ritonavir ‡ Indinavir |
||
| Others: Quinidine (Quinidex † ), sildenafil (Viagra ), eletriptan (Relpax ), ziprasidone (Geodon ) |
Grapefruit juice Nefazodone (Serzone ) |
||
| P-glycoprotein | Aldosterone, amprenavir, atorvastatin, cyclosporine, dexamethasone (Decadron † ), digoxin (Lanoxin † ), diltiazem, domperidone (Motilium ), doxorubicin (Adriamycin † ), erythromycin, etoposide (VePesid ), fexofenadine (Allegra ), hydrocortisone, indinavir, ivermectin (Stromectol ), lovastatin, loperamide (Imodium † ), nelfinavir, ondansetron (Zofran ), paclitaxel (Taxol ), quinidine, saquinavir, simvastatin, verapamil, vinblastine, vincristine | Amiodarone | Amprenavir |
| Carvedilol (Coreg ) | Clotrimazole (Mycelex † ) | ||
| Clarithromycin | Phenothiazine | ||
| Cyclosporine | Rifampin | ||
| Erythromycin | Ritonavir ‡ | ||
| Itraconazole | St. John's wort | ||
| Ketoconazole | |||
| Quinidine | |||
| Ritonavir ‡ | |||
| Tamoxifen | |||
| Verapamil |
† Also available generically.
‡ Can be both an inhibitor and an inducer.
CNS, Central nervous system.
From Med Lett 2003;45:47.
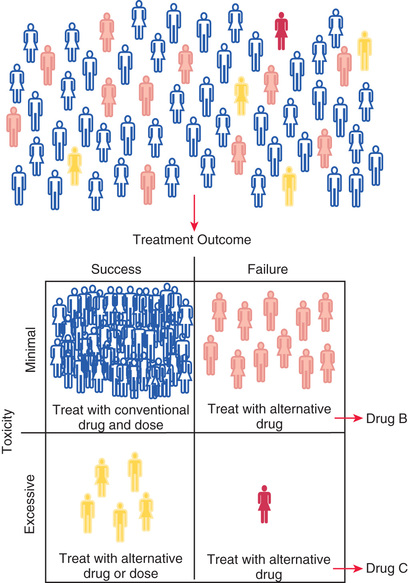
Therapeutic drug monitoring (TDM) programs recognize that all patients are unique and that the serum concentration-time data for an individual patient theoretically can be used to optimize pharmacotherapy. TDM programs have been the earliest application of personalized medicine; however, routine TDM does not necessarily translate to improved patient outcome in all situations.
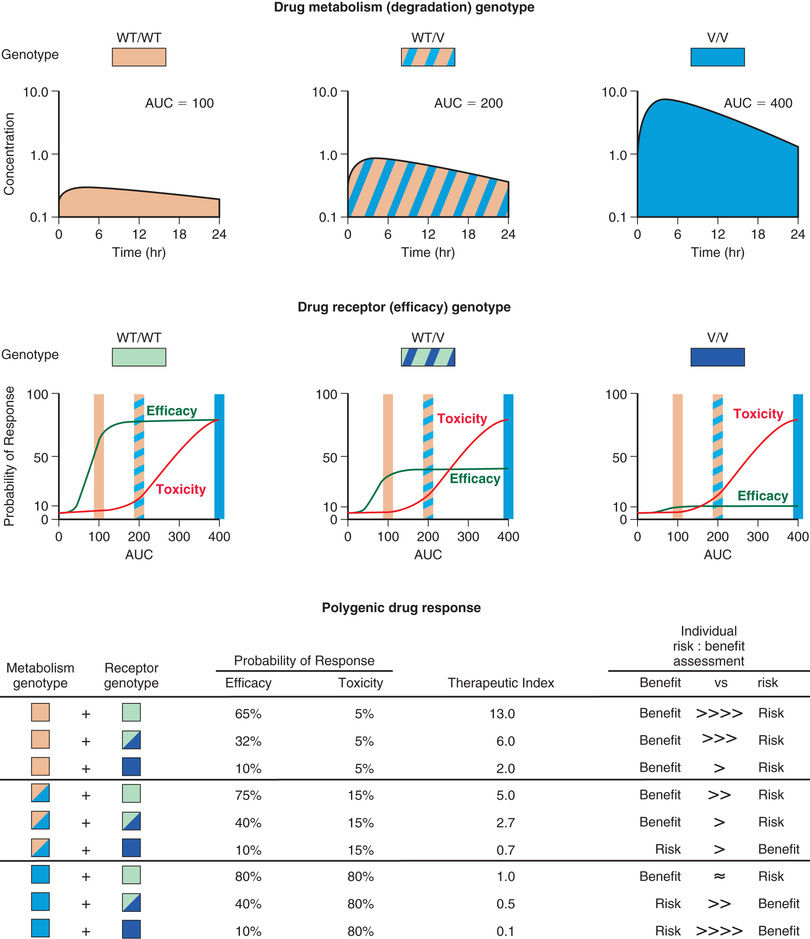
The concept of personalized medicine is based on the premise that the information explosion accompanying the application of genomic technologies to patient-related problems will allow (1) stratification of patient populations according to their response to a particular medication (e.g., lack of drug efficacy or excessive toxicity) and (2) stratification of diseases into specific subtypes that are categorized according to genomic criteria and by response to particular treatments. Personalized medicine has become supplanted by individualized medicine , which takes into consideration the vast amount of information that can be collected from an individual patient and applied to inform decisions for that patient. Precision medicine is an emerging approach for disease treatment and prevention that considers individual variability in genes, environment, and lifestyle for each person; it reflects the progression in delivery of care for more accurately diagnosing or treating a patient at an individual level. As the amount of data specific to an individual patient increases (e.g., genomic data, electronic health records), precision medicine can be further divided into precision diagnosis and precision therapeutics ; pharmacokinetics, pharmacodynamics, and pharmacogenomics all represent tools that can be applied to implement precision therapeutics for children.
Genetic polymorphisms (variations ) result when copies of a specific gene present within a population do not have identical nucleotide sequences. The term allele refers to one of a series of alternative DNA sequences for a particular gene. In humans, there are 2 copies of every gene. An individual's genotype for a given gene is determined by the set of alleles that the individual possesses. The most common form of genetic variation involves a single base change at a given location, referred to as a single nucleotide polymorphism (SNP) (see Chapter 95 ). At the other end of the spectrum are copy number variations (CNVs) , which refer to the deletion or duplication of identical or near-identical DNA sequences that may be thousands to millions of bases in size. CNVs occur less frequently than SNPs, but may constitute 0.5–1% of an individual's genome and thereby contribute significantly to phenotypic variation. Haplotypes are collections of SNPs and other allelic variations that are located close to each other; when inherited together, these create a catalog of haplotypes, or HapMap . When the alleles at a particular gene locus on both chromosomes are identical, a homozygous state exists, whereas the term heterozygous refers to the situation in which different alleles are present at the same gene locus. The term genotype refers to an individual's genetic constitution, whereas the observable characteristics or physical manifestations constitute the phenotype , which is the net consequence of genetic and environmental effects (see Chapters 94 –101 ).
Pharmacogenetics focuses on the phenotypical consequences of allelic variation in single genes. Pharmacogenetic polymorphisms are monogenic traits that are functionally relevant to drug disposition and action and are caused by the presence (within one population) of >1 allele (at the same gene locus) and >1 phenotype with regard to drug interaction with the organism. The key elements of pharmacogenetic polymorphisms are heritability, the involvement of a single gene locus, functional relevance, and the fact that distinct phenotypes are observed within the population only after drug challenge.
Developmental or Pediatric Pharmacogenetics and Pharmacogenomics
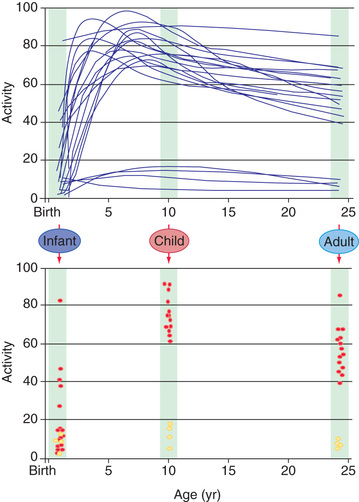
Our current understanding of pharmacogenetic principles involves enzymes responsible for drug biotransformation . Individuals are classified as being “fast,” “rapid,” or “extensive” metabolizers at one end and “slow” or “poor” metabolizers at the other end of the continuum. This may or may not also include an “intermediate” metabolizer group, depending on the particular enzyme. With regard to biotransformation, children are more complex than adults; fetuses and newborns may be phenotypically “slow” or “poor” metabolizers for certain drug-metabolizing pathways because of their stage of development and may acquire a phenotype consistent with their genotype at some point later in the developmental process as they mature. Examples of drug-metabolizing pathways that are significantly affected by ontogeny include glucuronidation and some of the cytochrome P450 (CYP) activities. It is also apparent that not all infants acquire drug metabolism activity at the same rate, a result of interactions between genetics and environmental factors. Interindividual variability in the trajectory (i.e., rate and extent) of acquired drug biotransformation capacity may be considered a developmental phenotype (Fig. 72.2 ). This helps to explain the considerable variability in some CYP activities observed immediately after birth.
In contrast to pharmacogenetic studies that typically target single genes, pharmacogenomic analyses are considerably broader in scope and focus on complex and highly variable drug-related phenotypes with targeting of many genes. Genome-wide genotyping technologies and massively parallel “next-generation” sequencing platforms for genomic analyses continue to evolve and allow evaluation of genetic variation at more than 1 million sites throughout an individual genome for SNP and CNV analyses. Genome-wide association studies (GWAS) have been conducted in several pediatric settings, in part to identify novel genes involved in disease pathogenesis that can lead to new therapeutic targets. GWAS are also being applied to identify genetic associations with response to drugs, such as warfarin and clopidogrel, and risk for drug-induced toxicity, including statin-induced myopathy and flucloxacillin hepatotoxicity. The “Manhattan plot,” a form of data presentation for GWAS, is becoming more common in many medical journals (Fig. 72.3A ). Whole genome and exome sequencing have been applied in a diagnostic setting to identify disease-causing genetic variation, usually in the context of rare, undiagnosed diseases that would otherwise require a “diagnostic odyssey” lasting several years before a definitive diagnosis is made (and thereby delaying therapeutic intervention). Contained within this genome sequence is the pharmacogenome , and an area of intense interest is the development of bioinformatics tools to determine a patient's drug metabolism and response genotype from whole genome sequence data.
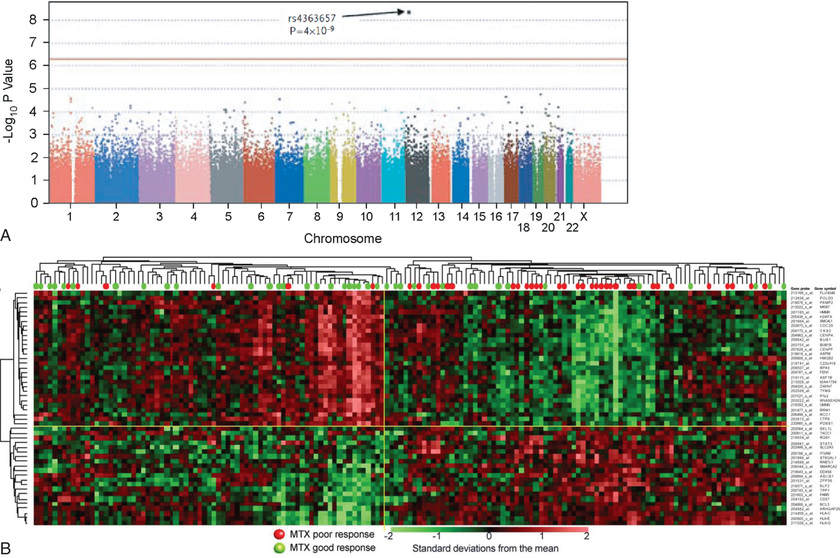
Investigating differential gene expression before and after drug exposure has the potential to correlate gene expression with variable drug responses and uncover the mechanisms of tissue-specific drug toxicities. These types of studies use microarray technology to monitor global changes in expression of thousands of genes (the transcriptome ) simultaneously. Genomic sequencing technologies can also be applied to RNA (RNA-Seq) and result in a more complete and quantitative assessment of the transcriptome. Gene expression profiling data from microarrays or RNA-Seq analyses are used to improve disease classification and risk stratification and are common in oncology. This approach has been widely used to address treatment resistance in acute lymphoblastic leukemia and has provided clinically relevant insights into the mechanistic basis of drug resistance and the genomic basis of interindividual variability in drug response. Subsets of transcripts, or gene expression “signatures,” are being investigated as potential prognostic indicators for identifying patients at risk for treatment failure (Fig. 72.3B ).
Pharmacoproteomic and Metabolomic Tools
Proteomic studies use many different techniques to detect, quantify, and identify proteins in a sample (expression proteomics) and to characterize protein function in terms of activity and protein-protein or protein–nucleic acid interactions (functional proteomics ). Mass spectrometry–based analyses are able to provide quantitative data regarding protein abundance, and several studies have been applied to pediatric liver samples, for example, to generate more accurate developmental trajectories for several drug-metabolizing enzymes and transporters.
Metabolomics and metabonomics utilize sophisticated analytical platforms, such as nuclear magnetic resonance (NMR) spectroscopy and liquid or gas chromatography coupled with mass spectral detection, to measure the concentrations of all small molecules present in a sample. Metabolomics refers to the complete set of low-molecular-weight molecules (metabolites) present in a living system (cell, tissue, organ or organism) at a particular developmental or pathologic state. Metabonomics is defined as the study of how the metabolic profile of biologic systems change in response to alterations caused by pathophysiologic stimuli, toxic exposures, or dietary changes. Pharmacometabonomics involves prediction of the outcome, efficacy, or toxicity of a drug or xenobiotic intervention in an individual patient based on a mathematical model of preintervention metabolite signatures.
Drug Biotransformation: Applications to Pediatric Therapy
The major consequence of pharmacogenetic polymorphisms in drug-metabolizing enzymes is concentration-dependent toxicity caused by impaired drug clearance. In certain cases, reduced conversion of prodrug to therapeutically active compounds is also of clinical importance (see Table 72.2 ). Chemical modification of drugs by biotransformation reactions generally results in termination of biologic activity through decreased affinity for receptors or other cellular targets as well as more rapid elimination from the body. The process of drug biotransformation can be very complex but is characterized by 3 important features: (1) the concept of broad substrate specificity , in which a single isozyme may metabolize a large variety of chemically diverse compounds; (2) many different enzymes may be involved in the biotransformation of a single drug (enzyme multiplicity ); and (3) a given drug may undergo several different types of reactions. One example of this product multiplicity occurs with racemic warfarin, in which at least 7 different hydroxylated metabolites are produced by different CYP isoforms.
Drug biotransformation reactions are conveniently classified into 2 main types, which occur sequentially and serve to terminate biologic activity and enhance elimination (see Chapter 73 ). Phase I reactions introduce or reveal (through oxidation, reduction, or hydrolysis) a functional group within the substrate drug molecule that serves as a site for a phase II conjugation reaction. Phase II reactions involve conjugation with endogenous substrates, such as acetate, glucuronic acid, glutathione, glycine, and sulfate. These reactions further increase the polarity of an intermediate metabolite, make the compound more water soluble, and thereby enhance its renal excretion. Interindividual variability in drug biotransformation activity (for both phase I and phase II reactions) is a consequence of the complex interplay among genetic (genotype, sex, race or ethnic background) and environmental (diet, disease, concurrent medication, other xenobiotic exposure) factors. The pathway and rate of a given compound's biotransformation are a function of each individual's unique phenotype with respect to the forms and amounts of drug-metabolizing enzymes expressed.
The CYP enzymes (CYPs) are quantitatively the most important of the phase I enzymes . These heme-containing proteins catalyze the metabolism of many lipophilic endogenous substances (steroids, fatty acids, fat-soluble vitamins, prostaglandins, leukotrienes, thromboxanes) as well as exogenous compounds, including a multitude of drugs and environment toxins. CYP nomenclature is based on evolutionary considerations and uses the root symbol CYP for cytochrome P450. CYPs that share at least 40% homology are grouped into families denoted by an Arabic number after the CYP root. Subfamilies, designated by a letter, appear to represent clusters of highly related genes. Members of the human CYP2 family, for example, have >67% amino acid sequence homology. Individual P450s in a subfamily are numbered sequentially (e.g., CYP3A4, CYP3A5). CYPs that have been identified as being important in human drug metabolism are predominantly found in the CYP1, CYP2, and CYP3 gene families. Importantly, enzyme activity may be induced or inhibited by various agents (see Table 72.2 ).
Phase II enzymes include arylamine N -acetyltransferases (NAT1, NAT2), uridine diphospho-glucuronosyltransferases (UGTs), epoxide hydrolase, glutathione S -transferases (GSTs), sulfotransferases (SULTs), and methyltransferases (catechol O -methyltransferase, thiopurine S -methyltransferase, several N -methyltransferases). As with the CYPs, UGTs, SULTs, and GSTs are gene families with multiple individual isoforms, each having its own preferred substrates, mode of regulation, and tissue-specific pattern of expression.
For most CYPs, genotype-phenotype relationships are influenced by development in that fetal expression is limited (with the exception of CYP3A7) and functional activity is acquired postnatally in isoform-specific patterns. Clearance of some compounds appears to be greater in children relative to adults, and the correlation between genotype and phenotype in neonatal life through adolescence may be obscured.
CYP2D6
The CYP2D6 gene locus is highly polymorphic, with >110 allelic variants identified to date (http://www.imm.ki.se/CYPalleles/cyp2d6.htm ; see Table 72.2 ). Individual alleles are designated by the gene name (CYP2D6) followed by an asterisk, and an Arabic number. By convention, CYP2D6*1 designates the fully functional wild-type allele. Allelic variants are the consequence of point mutations, single–base pair deletions or additions, gene rearrangements, or deletion of the entire gene, resulting in a reduction or complete loss of activity. Inheritance of 2 recessive, nonfunctional or “null' alleles results in the poor-metabolizer (PM) phenotype , which is found in approximately 5–10% of whites and approximately 1–2% of Asians. In whites the *3, *4, *5, and *6 alleles are the most common loss-of-function alleles and account for approximately 98% of PM phenotypes. In contrast, CYP2D6 activity on a population basis tends to be lower in Asian and African American populations because of a lower frequency of nonfunctional alleles (*3, *4, *5, and *6) and a relatively high frequency of population-selective alleles associated with decreased activity (“reduced function” alleles) relative to the wild-type CYP2D6*1 allele. The CYP2D6*10 allele occurs at a frequency of approximately 50% in Asians, whereas CYP2D6*17 and CYP2D6*29 occur at relatively high frequencies in persons of black African origin.
In addition to nonfunctional and partial-function alleles, the presence of gene duplication and multiplication events (≥3 copies of CYP2D6 gene in tandem on a single chromosome) further complicates the prediction of phenotype from genotype information. The concept of “activity score” has been developed to simplify translation of CYP2D6 genotype information into a predicted phenotype of CYP2D6 activity for a particular patient. Fully functional alleles (*1, *2, *35, etc.) are assigned a value of “1”, reduced-function alleles (*9, *10, *17, *29 ) are assigned a value of “0.5”, and nonfunctional alleles (*3-*6, etc.) are assigned a value of “0”; for duplications/multiplication events, the allele score is multiplied by the number of copies detected (*10 × 2 = 0.5 × 2 = “1”). The activity score for an individual is the sum of the scores for each chromosome, with poor metabolizers (PMs) defined by a score of “0”, whereas a score of “0.5” indicates an intermediate-metabolizer (IM) phenotype , and a score >2 indicating an ultrarapid-metabolizer (UM) phenotype ; scores of 1 to 2 are referred to as extensive metabolizers (EMs) . The activity score classification system has been adopted by the Clinical Pharmacogenetics Implementation Consortium (CPIC; see below). In the past, individuals with an activity score of “1” have been referred to as “IMs,” and any reference to IM status in literature before 2012 likely refers to a genotype with the equivalent of 1 functional allele, in contrast to the current definition (0.5).
CYP2D6 is involved in the biotransformation of >40 therapeutic entities, including several β-receptor antagonists, antiarrhythmics, antidepressants, antipsychotics, and morphine derivatives † (see Table 72.2 ). CYP2D6 substrates commonly encountered in pediatrics include selective serotonin reuptake inhibitors (SSRIs; fluoxetine, paroxetine), risperidone, atomoxetine, promethazine, tramadol, and codeine. Furthermore, over-the-counter cold remedies (e.g., dextromethorphan, diphenhydramine, chlorpheniramine) are also CYP2D6 substrates. An analysis of CYP2D6 ontogeny in vitro that utilized a relatively large number of samples revealed that CYP2D6 protein and activity remain relatively constant after 1 wk of age up to 18 yr. Similarly, results from an in vivo longitudinal phenotyping study involving >100 infants over the 1st year of life demonstrated considerable interindividual variability in CYP2D6 activity, but no relationship between CYP2D6 activity and postnatal age between 2 wk and 12 mo. Furthermore, a cross-sectional study involving 586 children reported that the distribution of CYP2D6 phenotypes in children was comparable to that observed in adults by at least 10 yr of age. Thus, both available in vitro and in vivo data, although based on phenotype data rather than information on drug clearance from pharmacokinetic studies, imply that genetic variation is more important than developmental factors as a determinant of CYP2D6 variability in children.
One consequence of CYP2D6 developmental pharmacogenetics may be the syndrome of irritability, tachypnea, tremors, jitteriness, increased muscle tone, and temperature instability in neonates born to mothers receiving SSRIs during pregnancy. Controversy exists as to whether these symptoms reflect a neonatal withdrawal (hyposerotonergic) state or represent manifestations of serotonin toxicity analogous to the hyperserotonergic state associated with the SSRI-induced serotonin syndrome in adults. Delayed expression of CYP2D6 (and CYP3A4) in the 1st few weeks of life is consistent with a hyperserotonergic state caused by delayed clearance of paroxetine and fluoxetine (CYP2D6) or sertraline (CYP3A4) in neonates exposed to these compounds during pregnancy. Furthermore, decreases in plasma SSRI concentrations and resolution of symptoms would be expected with increasing postnatal age and maturation of these pathways. Given that treatment of a “withdrawal” reaction may include administration of an SSRI, there is considerable potential for increased toxicity in affected neonates. Resolution of the question whether symptoms are caused by withdrawal vs a hyperserotonergic state is essential for appropriate management of SSRI-induced neonatal adaptation syndromes. Until further data are available, it would be prudent to consider newborns and infants <28 days of age as CYP2D6 PMs.
In older children, drug accumulation and resultant concentration-dependent toxicities in CYP2D6 genotypic poor metabolizers should be anticipated in the same way that they are in adults due to the risk of significant morbidity and mortality. Although a fluoxetine-related death has been reported in a 9 yr old child with a CYP2D6 PM genotype, experience with paroxetine indicates that the risk of drug accumulation may also occur, under certain conditions, in individuals at the opposite end of the activity spectrum. For example, chronic dosing of paroxetine may lead to greater-than-anticipated drug accumulation in children classified as CYP2D6 EMs. In fact, the largest decreases in paroxetine clearance observed with ascending doses are seen in patients who have the greatest clearance at the initial dose level (10 mg/day) and are predicted to have the greatest CYP2D6 activity based on CYP2D6 genotype. This seemingly paradoxical effect appears to involve oxidation of paroxetine within the CYP2D6 active site to form a reactive intermediate that is associated with irreversible modification of the CYP2D6 protein in or near the active site and loss of enzyme activity. As a consequence, CYP2D6 activity progressively declines such that drug accumulation may occur over time, placing CYP2D6 EM patients also at increased risk of concentration-dependent toxicity.
Theoretically, younger children may experience decreased efficacy or therapeutic failure with drugs such as codeine and tramadol that are dependent on functional CYP2D6 activity for conversion to the pharmacologically active species. CYP2D6 catalyzes the O -demethylation of codeine to morphine. Infants and children appear capable of converting codeine to morphine and achieving morphine:codeine ratios comparable to those of adults. However, in one study, morphine and its metabolites were not detected in 36% of children receiving codeine making the level of analgesia from codeine unreliable in the studied pediatric population. Interestingly, levels of morphine and its metabolites were not related to CYP2D6 phenotype. Finally, ultrarapid CYP2D6 metabolism of codeine may result in opiate intoxication, including maternal ultrarapid metabolism of codeine, which can result in high serum and breast milk concentrations of morphine and may have adverse effects in the breastfed neonate.
Rapid metabolism and clearance of CYP2D6 substrates may also contribute to poor therapeutic response because of an inability to achieve adequate plasma concentrations, even when medications are dosed at the maximum approved dose level. The product label for atomoxetine (Strattera) indicates that CYP2D6 PMs have a systemic exposure to the drug (e.g., amount of drug in body over time as determined by area under plasma concentration-time curve) that is 10 times greater than in typical individuals (EMs), and yet the same starting dose of 0.5 mg/kg is recommended for all patients. A genotype-stratified pharmacokinetic study of atomoxetine in children and adolescents with attention-deficit/hyperactivity disorder (ADHD) confirmed an 11-14-fold difference in average systemic exposure between PM and EM groups. However, the most informative finding was the 50-fold range in exposure (30-fold, if exposure corrected for actual mg dose administered) between the PM participant with the highest exposure and the UM participant (3 functional alleles) with the lowest exposure. Using the results of this single-dose study to simulate atomoxetine exposure at steady state for each study participant revealed that even at the maximum recommended dose of atomoxetine, exposure was likely to be subtherapeutic for the majority of patients with ≥1 functional CYP2D6 alleles.
Avoiding ineffective treatment at one end of the spectrum and excessive toxicity at the other are potential benefits of individualizing doses based on genomic information for medications dependent on a polymorphic clearance pathway, such as CYP2D6. The CPIC has published several guidelines that include CYP2D6 substrates, such as the CPIC guideline for codeine,* SSRIs, † and tricyclic antidepressants. ‡ Although pediatric data are sparse, these links serve as valuable sources of information regarding the effect of genotype on the dose-exposure relationship for several CYP2D6 substrates.
CYP2C9
Although several clinically useful compounds are substrates for CYP2C9 § (see Table 72.2 ), the effects of allelic variation are most profound for drugs with a narrow therapeutic index, such as phenytoin, warfarin, and tolbutamide. In vitro studies show a progressive increase in CYP2C9 expression from 1–2% of mature levels in the 1st trimester to approximately 30% at term. Considerable variability (approximately 35-fold) in expression is apparent over the 1st 5 mo of life, with about half the samples studied exhibiting values equivalent to those observed in adults. One interpretation of these data is that broad interindividual variability exists in the rate at which CYP2C9 expression is acquired after birth, and in general, the ontogeny of CYP2C9 activity in vivo, as inferred from pharmacokinetic studies of phenytoin in newborns, is consistent with the in vitro results. The apparent half-life of phenytoin is prolonged (approximately 75 hours) in preterm infants, but decreases to approximately 20 hr in term newborns. By 2 wk of age, the half-life has further declined to 8 hr. The appearance of concentration-dependent (saturable) metabolism of phenytoin, reflecting the functional acquisition of CYP2C9 activity, does not appear until approximately 10 days of age. The maximal velocity of phenytoin metabolism has been reported to decrease from an average of 14 mg/kg/day in infants to 8 mg/kg/day in adolescents, which may reflect changes in the ratio of liver mass to total body mass observed over this period of development, as has been observed for warfarin.
Several allelic variants of CYP2C9 have been reported, but not all have been evaluated for their functional consequences. The CYP2C9*2 allele is associated with approximately 5.5-fold decreased intrinsic clearance for S -warfarin relative to the wild-type enzyme. Allelic variations resulting in amino acid changes within the enzyme active site, such as the CYP2C9*3, CYP2C9*4, and CYP2C9*5 alleles, are associated with activities that are approximately 5% of the wild-type protein. Approximately one third of the white population carries a variant CYP2C9 allele (typically *2 and *3 alleles), whereas the *2 and *3 alleles are virtually nonexistent in African American, Chinese, Japanese, or Korean populations. In contrast, the *5 allele has been detected in blacks but not in whites. The risk of bleeding complications in patients treated with warfarin and with concentration-dependent toxicity in patients treated with phenytoin is most pronounced for individuals with a CYP2C9*3/*3 genotype. Although the relationship between the CYP2C9 genotype and warfarin dosing and pharmacokinetics has not been as extensively studied in children, consequences of allelic variation can be expected to be similar to those observed in adults. In adults, CYP2C9 and VKORC1 genotype and patient age, sex, and weight can account for 50–60% of the variation in warfarin dose requirements. A large part of the variation is still unknown, but may be at least partially attributed to interactions with other drugs and foods.
CYP2C19
In vitro, CYP2C19 protein and catalytic activity can be detected at levels representing 12–15% of mature values by 8 wk of gestation and remain essentially unchanged throughout gestation and at birth. Over the 1st 5 mo of postnatal age, CYP2C19 activity increases linearly. Adult levels are achieved by 10 yr of age, although variability in expression is estimated to be approximately 21-fold between 5 mo and 10 yr of age. The major source of this variability is likely pharmacogenetic in nature. The CYP2C19 PM phenotype (also known as mephenytoin hydroxylase deficiency ) is present in 3–5% of the white population and 20–25% of Asians. Although 25 variant alleles have been reported to date, the 2 most common variant alleles, CYP2C19*2 and CYP2C19*3, result from single-base substitutions that introduce premature stop codons and, consequently, truncated polypeptide chains that possess no functional activity. Despite consistent increases in CYP2C19 activity observed in vitro over the 1st 5 months of life, the results of an in vivo phenotyping study with omeprazole in Mexican children revealed a broad range of activity and implied that 17% of infants <4 mo of age could be classified as PMs (no PMs were detected beyond that point). In contrast, 20% of children 3–9 mo old were classified as ultrarapid metabolizers (UMs) compared with 6% of infants 1-3 mo of age. For omeprazole, pharmacokinetic parameters comparable to those observed in adults are achieved by age 2 yr.
CYP2C19 also plays an important role in the metabolism of lansoprazole. In Japanese adults treated with lansoprazole, amoxicillin, and clarithromycin for Helicobacter pylori infection, the eradication rate for CYP2C19 PMs (97.8%) and heterozygous EMs (1 functional CYP2C19 allele; 92.1%) was significantly greater than that observed in homozygous EMs (72.7%). Initial treatment did not eradicate H. pylori in 35 patients, 34 of whom had at least 1 functional CYP2C19 allele, and eradication could be achieved with higher lansoprazole doses in almost all cases. Given that the frequency of the functional CYP2C19*1 allele is considerably greater in whites (0.84 [84%]) than Japanese (0.55 [55%]), eradication failure can be expected to occur more frequently in whites. Because proton pump inhibitors are widely used in children, pharmacogenetic as well as developmental considerations should guide pediatric dosing strategies.
CYP3A4, CYP3A5, and CYP3A7
The CYP3A subfamily consists of four members in humans (CYPs 3A4, 3A5, 3A7, and 3A43) and is quantitatively the most important group of CYPs in terms of human hepatic drug biotransformation. These isoforms catalyze the oxidation of many different therapeutic entities, several of which are of potential importance to pediatric practice † (see Table 72.2 ). CYP3A7 is the predominant CYP isoform in fetal liver and can be detected in embryonic liver as early as 50–60 days' gestation. CYP3A4, the major CYP3A isoform in adults, is essentially absent in fetal liver, but increases gradually throughout childhood. Over the 1st 6 mo of life, CYP3A7 expression exceeds that of CYP3A4, although its catalytic activity toward most CYP3A substrates is rather limited compared with CYP3A4. CYP3A4 is also abundantly expressed in intestine, where it contributes significantly to the first-pass metabolism of oral drugs, which are substrates (e.g., midazolam). CYP3A5 is polymorphically expressed and is present in approximately 25% of adult liver samples studied in vitro.
Several methods have been proposed to measure CYP3A activity. Using these various phenotyping probes, CYP3A4 activity has been reported to vary widely (up to 50-fold) among individuals, but the population distributions of activity are essentially unimodal and evidence for polymorphic activity has been elusive. Although 20 allelic variants have been identified (http://www.imm.ki.se/CYPalleles/cyp3a4.htm ), most occur relatively infrequently and do not appear to be of clinical importance. Of interest to pediatrics is the CYP3A4*1B allele present in the CYP3A4 promoter region. The clinical significance of this allelic variant appears limited with respect to drug biotransformation activity, despite in vitro assays showing 2-fold increased activity over the wild-type CYP3A4*1 allele. Although no association appears to exist between the CYP3A4*1B allele and age of menarche, a significant relationship does exist between the number of CYP3A4*1B alleles and the age at onset of puberty, as defined by Tanner breast score. In one study, 90% of 9 yr old girls with a CYP3A4*1B/*1B genotype had a Tanner breast score of ≥2 vs 56% of CYP3A4*1A/*1B heterozygotes and 40% of girls homozygous for the CYP3A4*1A allele. Because CYP3A4 plays an important role in testosterone catabolism, it was proposed that the estradiol:testosterone ratio may be shifted toward higher values in the presence of the CYP3A4*1B allele and might trigger the hormonal cascade that accompanies puberty. Intestinal CYP3A4 activity is inhibited by grapefruit juice and may result in higher levels of the many drugs metabolized by this enzyme; very large quantities of grapefruit juice may also inhibit the hepatic CYP3A4.
Polymorphic CYP3A5 expression is largely caused by an SNP in intron 3 that creates a cryptic splice site and gives rise to messenger RNA splice variants that retain part of intron 3 with a premature stop codon. The truncated mRNA transcripts associated with this allele, CYP3A5*3, cannot be translated into a functional protein. Individuals with at least one wild-type CYP3A5*1 allele express functional CYP3A5 protein, whereas those homozygous for CYP3A5*3 (CYP3A5*3/*3) do not express appreciable amounts of functional protein. Approximately 60% of African Americans show functional hepatic CYP3A5 activity, compared with only 33% of European Americans.
Clinically important consequences of CYP3A5 allelic variation have been reported in children. In pediatric heart transplant patients with a CYP3A5*1/*3 genotype, tacrolimus concentrations were approximately 25% of those observed in patients with CYP3A5*3/*3 genotypes, when corrected for dose, in the highly vulnerable period immediately after transplant (≤2 wk), and 50% less at 3, 6, and 12 mo after transplant. Thus, larger doses of tacrolimus are required in patients with functional CYP3A5 protein to achieve comparable blood levels and to minimize the risk of rejection. Of concern, <15% of tacrolimus concentrations in the immediate posttransplant period were within the therapeutic target range, highlighting the need for prospective, precision-guided tacrolimus trials in the pediatric population. In addition to CYP3A5 expressor genotype, younger age was associated with lower tacrolimus concentrations. The same age and genotype relationship is observed for renal transplantation. Conversely, the same age- and genotype-tailored treatment is more challenging in liver transplantation unless the donor CYP3A5 is known. In pediatric liver transplant recipients, CYP3A5 expressor genotype was not associated with tacrolimus concentrations and dosing. This implies that hepatic metabolism, from the donor liver and genotype status, plays a larger role in tacrolimus concentrations than intestinal metabolism or the recipient's CYP3A5 genotype status. Collectively, these pediatric tacrolimus datasets have informed the CPIC to recommend a 1.5-2-fold increase in tacrolimus dosing, followed by close plasma drug monitoring, in children and adolescents with at least one CYP3A5*1 allele (https://cpicpgx.org/guidelines/guideline-for-tacrolimus-and-cyp3a5/ ).
Glucuronosyl Transferases (UGTs)
The UGT gene superfamily catalyzes the conjugation (with glucuronic acid) of several drugs used clinically in pediatrics, including morphine, acetaminophen, nonsteroidal antiinflammatory drugs, and benzodiazepines. The effect of development on glucuronidation capacity has been well described and is illustrated by hyperbilirubinemia, gray baby syndrome (cardiovascular collapse associated with high doses of chloramphenicol in newborns), and the 3.5-fold increase in morphine clearance observed in premature neonates at 24-39 wk postconception age. As with the CYPs, there are multiple UGT isoforms, and the acquisition of functional UGT activity appears to be isoform and substrate specific.
UGT1A1 is the major UGT gene product responsible for bilirubin glucuronidation, and >100 genetic alterations have been reported (Table 72.3 ), most of which are rare and are more properly considered mutations rather than gene polymorphisms. Inheritance of 2 defective alleles is associated with reduced bilirubin-conjugating activity and gives rise to clinical conditions such as Crigler-Najjar and Gilbert syndromes. More frequently occurring polymorphisms involve a dinucleotide (TA) repeat in the atypical TATA box of the UGT1A1 promoter. The wild-type UGT1A1*1 allele has 6 repeats (TA6 ), and the TA5 (UGT1A1*33), TA7 (UGT1A1*28), and TA8 (UGT1A1*34) variants are all associated with reduced activity. UGT1A1*28, the most frequent variant, is a contributory factor to prolonged neonatal jaundice. This variant is also associated with impaired glucuronidation and thus toxicity of the active metabolite SN-38 of the chemotherapeutic agent irinotecan. Allelic variations in UGT1A7 and UGT1A9 have also been associated with irinotecan toxicity in adults with colorectal cancer.
Table 72.3
Internet Resources for Pharmacogenetics and Pharmacogenomics*
* All sites were accessible on July 14, 2017.
The consequences of allelic variation in the UGT2B family are less certain. The predominant routes of morphine elimination include biotransformation to the pharmacologically active 6-glucuronide (M6G) and the inactive 3-glucuronide (M3G). M6G formation is almost exclusively catalyzed by UGT2B7, whereas several UGTs in the UGT1A subfamily and UGT2B7, both contribute to M3G formation. Increased M6G:morphine ratios have been reported in individuals homozygous for the SNPs constituting the UGT2B7*2 allele. Although individuals genotyped as UGT2B7*2/*2 may produce higher-than-anticipated concentrations of pharmacologically active morphine and its metabolites, prospective studies addressing phenotype-genotype correlations and the consequences of morphine analgesia have had conflicting results.
Thiopurine S -Methyltransferase
Thiopurine S -methyltransferase (TPMT ) is a cytosolic enzyme that catalyses the S -methylation of aromatic and heterocyclic sulfur-containing compounds, such as 6-mercaptopurine (6MP), azathioprine, and 6-thioguanine, used in the treatment of acute lymphoblastic leukemia (ALL), inflammatory bowel disease (IBD), and juvenile idiopathic arthritis and the prevention of renal allograft rejection. To exert its cytotoxic effects, 6MP requires metabolism to thioguanine nucleotides by a multistep process initiated by hypoxanthine guanine phosphoribosyltransferase. TPMT prevents thioguanine nucleotide production by methylating 6MP (Fig. 72.4A ). TPMT activity is usually measured in erythrocytes, with erythrocyte activity reflecting what is found in other tissues, including liver and leukemic blasts. Although approximately 89% of whites and blacks have high TPMT activity and 11% have intermediate activity, 1 in 300 individuals inherits TPMT deficiency as an autosomal recessive trait (Fig. 72.4B ). In newborn infants, peripheral blood TPMT activity is 50% greater than in race-matched adults and shows a distribution of activity consistent with the polymorphism characterized in adults. No data currently indicate how long this higher activity is maintained, although TPMT activities were comparable to previously reported adult values in a population of Korean schoolchildren age 7–9 yr. In patients with intermediate or low activity, more drug is shunted toward production of cytotoxic thioguanine nucleotides. TPMT can also methylate 6-thioinosine 5′-monophosphate to generate a methylated metabolite capable of inhibiting de novo purine synthesis (Fig. 72.4C ).
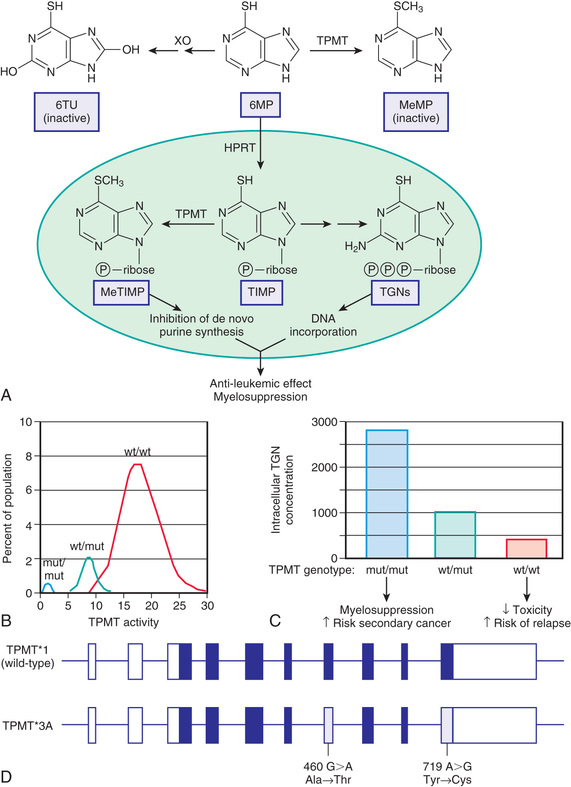
Multiple SNP variants have been identified in the TPMT gene, and a GWAS from 2 independent pediatric ALL cohorts confirmed that TPMT activity is a monogenic pharmacogenetics trait; 3 variants (TPMT*2, *3A, *3C ) account for 98% of whites with low activity and have high predictive capacity for TPMT phenotype. TPMT*3A is the most common and is characterized by 2 nucleotide transition mutations, G460A and A719G, that lead to 2 amino acid substitutions, Ala154Thr and Tyr240Cys (Fig. 72.4D ). The TPMT*3A allele occurs more frequently in white (9.5%) and Hispanic (7.0%) patients and is absent in black patients. In contrast, TPMT*3C is reported to be the predominant variant allele in black patients (12.2%), and only rarely observed in white or Hispanic patients; overall, black patients have lower TPMT activities than nonblack patients. The *3A and *3C variants each result in loss of functional activity through the production of unstable proteins that are subject to accelerated proteolytic degradation.
The relatively few patients with low to absent TPMT activity (0.3%) are at increased risk for severe myelosuppression if treated with routine doses of thiopurines; thus they require a 10-15-fold reduction in dose to minimize this risk. Furthermore, if not dosed properly, patients may be at increased risk for relapse as a result of inadequate or a lack of treatment with thiopurines. Given the expanding use of 6MP and azathioprine in pediatrics to treat IBD and juvenile arthritis and to prevent renal allograft rejection, TPMT pharmacogenetics is not trivial, and a CPIC guideline assists with genotype-guided dosing. † However, TPMT genotype is not the only determinant of intolerance to thiopurines. Multiple studies have also implicated genetic variation in NUDT15, a nucleotide diphosphatase that converts thioguanine triphosphate to thioguanine monophosphate, thereby reducing incorporation of thioguanine into DNA; reduction or loss of this activity results in greater-than-expected thioguanine incorporation into DNA and thus increased cytotoxicity. Reduced-function NUDT15 alleles are more common in Hispanic patients and those with Asian ancestry, and patients who have inherited 2 reduced-function alleles tolerate thiopurine doses that are much lower (10%) than normal. Thus it is reasonable to expect that both TPMT and NUDT15 genotype will need to be considered for individualized thiopurine treatment.
Pharmacogenetics of Drug Transporters
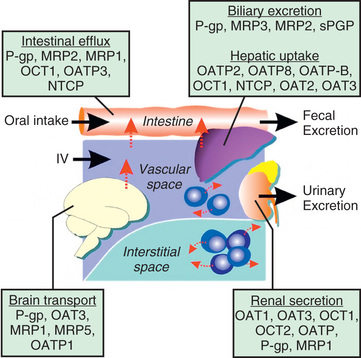
There are several major types of membrane transporters, including organic anion transporters (OATs), organic anion–transporting polypeptides (OATPs), organic cation transporters (OCTs), and the adenosine triphosphate–binding cassette (ABC) transporters, such as P-glycoprotein and the multidrug-resistant proteins. Membrane transporters are heavily involved in drug disposition and actively transport substrate drugs between organs and tissues. Drug transporters are expressed at numerous epithelial barriers, such as intestinal epithelial cells, hepatocytes, renal tubular cells, and the blood-brain barrier (BBB) (Fig. 72.5 ). Transporters often are also determinants of drug resistance, and many drugs work by affecting the function of transporters. As such, polymorphisms in the genes encoding these proteins may have a significant effect on the absorption, distribution, metabolism, and excretion as well as the pharmacodynamic effect of a wide variety of compounds.
Adenosine Triphosphate–Binding Cassette Superfamily
The ATP-binding cassette (ABC) transporters belong to the largest known transporter gene family and translocate a variety of substrates, including chemotherapy agents. ABC multidrug transporter expression has been implicated in tumor cell resistance to anticancer therapy, altered disposition of chemotherapy drugs, and toxic side effects associated with chemotherapy. More recently, the genetic heterogeneity of several ABC transporter genes has been described. Apart from having at least one ATP-binding domain, these transporters are characterized by a signature sequence of amino acid residues within the domain. In humans the ABC transporters function as efflux pumps, which together with detoxification enzymes, constitute a complex, integrated, “chemoimmunologic defense” system against drugs and other foreign chemicals. A variety of epithelial barriers, including the kidney, liver, and BBB have abundant expression of ABC transporters, such as P-glycoprotein (P-gp; also known as MDR1 ), and multidrug-resistant proteins (MRPs) 1, 2, and 3. Powered by ATP, these transporters actively extrude substrates from the respective cell and organ.
Considerable genetic variation has been reported in the superfamily of ABC transporter genes. Many studies have investigated the relationship between ABCB1 genotype or haplotype and P-gp expression, activity, or drug response, yielding inconsistent results, largely due to methodological limitations. No association between genotype and drug disposition or response would be expected if the drug of interest were not substrate for P-gp. However, even when drugs are tested for transport by P-gp using in vitro systems, the results are not necessarily conclusive, as is the case for carbamazepine. On the other hand, an association between ABCB1 genotype and drug response was observed in patients receiving antidepressants that were ABC substrates (e.g., citalopram, paroxetine, amitriptyline, venlafaxine)., but not in drugs that were not substrates (e.g., mirtazapine).
Studies conducted in children need to also consider the ontogeny of P-gp expression. Based on studies using human lymphocytes, it appears that P-gp activity is high at birth, decreases between 0 and 6 mo, and stabilizes between 6 mo and 2 yr of age. In contrast, P-gp can be detected in human neural stem/progenitor cells and decreases with differentiation. Furthermore, P-gp has been proposed as an endothelial marker for development of the BBB, and expression increases with postnatal age as the BBB matures. Proteomic analysis of the ontogeny of hepatic P-gp has demonstrated that P-gp expression increases through infancy, achieving 50% of adult expression at approximately 3 yr and reaching a plateau during adolescence. Thus the developmental patterns of P-gp expression likely are tissue specific, but data still are sparse in this regard. Nevertheless, expression of P-gp at a young age in gut and liver likely represents a protective mechanism in which both endogenous and exogenous toxins are efficiently excreted from the body. However, developmental patterns of expression in tissues of drug response, such as lymphocytes and tumors, may also affect the efficacy of intracellular drugs. For example, polymorphisms in the gene have been shown to be predictive of the ability to wean corticosteroids after heart transplantation, as well as the susceptibility to and clinical outcome of treatment for pediatric ALL. On the other hand, immaturity of P-gp expression in the developing BBB may contribute to discrete periods of increased susceptibility to drug toxicity in the central nervous system. However, for most other drugs, including immunosuppressants and protease inhibitors, studies investigating the effect of ABCB1 polymorphisms in drug disposition and response have yielded conflicting results. In one study investigating the relationship between ABCB1 genotype and cyclosporin pharmacokinetics, an effect of genotype on oral availability was only apparent in children >8 yr of age. Although these results require further replication, the implication is that a better understanding of transporter ontogeny is required to properly design and interpret pharmacogenetic studies of ABCB1 in pediatric populations.
Organic Anion–Transporting Polypeptides
Organic anion–transporting polypeptides (OATPs) in the solute carrier organic anion transporter (SLCO) are a family of glycoprotein transporters with 12 transmembrane-spanning domains expressed in various epithelial cells. There are 11 OATPs in humans, some of which are ubiquitously expressed and others whose expression is restricted to specific tissues. Typical substrates include bile salts, hormones and their conjugates, toxins, and various drugs. The solute carrier, human OATP 1A2 (OATP1A2, OATP-A, OATP1, and OATP) is highly expressed in the intestine, kidney, cholangiocytes, and BBB and may be important in the absorption, distribution, and excretion of a broad array of clinically important drugs. Several nonsynonymous polymorphisms have been identified in the gene encoding OATP1A2, SLCO1A2 (SLC21A3), with some of these variants demonstrating functional changes in the transport of OATP1A2 substrates.
OATP1B1 (SLCO1B1) and OATP1B3 (SLCO1B3) are liver-specific transporters and promote the cellular uptake of endogenous substrates, such as bilirubin, bile acids, DHEA-sulfate, and leukotriene C4, as well as various drugs, including several statins, methotrexate, and enalapril. Allelic variation in OATP1B1 (specifically the SLCO1B1*5 allele) results in reduced clearance and increased systemic exposure of several statin drugs (atorvastatin, pravastatin, simvastatin) and has been associated with an increased risk of musculoskeletal side effects from simvastatin. The expression of OATP1B1 in human pediatric liver tissue was independent of age in all samples, but age dependency was demonstrated in samples homozygous for the SLCO1B1 reference sequence (i.e., SLCO1B1*1A/*1A genotype). Therefore, not only genotype, but also growth and development, may influence OATP1B1 protein expression in the developing child. To date, only one study has investigated the effect of SLCO1B1 genotype on statin disposition in children, reporting a genotype-phenotype relationship for pravastatin that was discordant with the relationship observed in adults. However, data with simvastatin in dyslipidemic children and adolescents (LDL >130 mg/dL) suggest that the genotype-phenotype relationships observed in adults are also present in this population, but the magnitude of the genetic effect may be greater in pediatric patients.
Several studies have confirmed that the 2 SNPs determining the most common SLCO1B1 haplotypes (*1a, *1b, *5, and *15 ), rs4149056 and rs2306283, are associated with decreased clearance of high-dose methotrexate in children with ALL. Genotyping for SLCO1B1 may be helpful in identifying patients at increased risk of toxicity from reduced clearance or increased accumulation of methotrexate. In the pediatric liver proteomic analysis, OATP1B3 expression was age dependent, with a 3-fold difference observed between neonates and adults. Similar to P-gp, expression steadily increased during childhood; however, 50% of adult level expression was much earlier (6 mo) compared with P-gp.
Organic Cation Transporters
Organic cation transporters (OCTs) in the SCL22A subfamily are primarily expressed on the basolateral membrane of polarized epithelia and mediate the renal secretion of small organic cations. Originally, OCT1 (also known as SLC22A1) was thought to be primarily expressed in liver, but recent studies have also localized its expression to the apical side of proximal and distal renal tubules. Hepatic OCT1 expression was found to be age dependent with almost a 5-fold difference between neonates and adults. OCT2 (SLC22A2) is predominantly expressed on the basolateral surface of proximal renal tubules. In adults, allelic variation in OCT1 and OCT2 is associated with increased renal clearance of metformin. The role of genetic variation of OCT1 and OCT2 has not been studied in children, but developmental factors appear to be operative. Neonates possess very limited ability to eliminate organic cations, but this function increases rapidly during the 1st few months of life, and when standardized for body weight or surface area, it tends to exceed adult levels during the toddler stage.
Polymorphisms in Drug Receptors
Receptors are the targets for drugs and endogenous transmitters because of their inherent molecular recognition sites. Drugs and transmitters bind to the receptor to produce a pharmacologic effect. Variability in the receptor protein or the ion channel may determine the magnitude of the pharmacologic response. Polymorphisms of the β2 -adrenergic receptor gene (ADRB2) are associated with variable responses to bronchodilator drugs.
Drug responses are seldom monogenic events because multiple genes are involved in both drug binding to the pharmacologic target and the subsequent downstream signal transduction events that ultimately manifest collectively as a therapeutic effect. Although genotypes at a particular locus may show a statistically significant effect on the outcome of interest, they may account for only a relatively small amount of the overall population variability for that outcome. A particular group of SNPs in the corticotropin-releasing hormone receptor 1 gene (CRHR1) is associated with a statistically significant improvement in forced expiratory volume in 1 second (FEV1 ), but accounts for only 6% of the overall variability in response to inhaled corticosteroids. A series of subsequent studies has determined that allelic variation in several genes in the steroid pathway contributes to overall response to this form of therapy.
The listing and classification of receptors is a major initiative of the International Union of Pharmacology (IUPHAR). The list of receptors and voltage-gated ion channels is available on the IUPHAR website (http://www.iuphar-db.org ). The effect of growth and development on the activities and binding affinities of these receptors, effectors, and ion channels has been studied in animals to some extent but remains to be elucidated in humans.
Current and Future Applications in Pediatrics
Progress in the treatment of acute lymphoblastic leukemia shows how the application of pharmacogenomic principles can improve pediatric drug therapy (see Chapter 522.1 ). Despite improved understanding of the genetic determinants of drug response, however, many complexities remain to be resolved. Patients with ALL who have 1 wild-type allele and intermediate TPMT activity tend to have a better response to 6MP therapy than patients with 2 wild-type alleles and full activity. Reduced TPMT activity also places patients at risk for irradiation-induced secondary brain tumors and etoposide-induced acute myeloid leukemia. Pharmacogenetic polymorphisms of several additional genes, such as NUDT15, also have the potential to influence successful treatment of ALL. Multiple genetic and treatment-related factors interact to create patient subgroups with varying degrees of risk. These represent an opportunity for pharmacogenomic approaches to identify subgroups of patients who will benefit from specific treatment regimens and those who will be at risk for short-term and long-term toxicities (Fig. 72.6 ).