Poisoning
Jillian L. Theobald, Mark A. Kostic
Poisoning is the leading cause of injury-related death in the United States, surpassing that from motor vehicle crashes. Most these deaths are unintentional (i.e., not suicide). In adolescents, poisoning is the 3rd leading cause of injury-related death. Of the >2 million human poisoning exposures reported annually to the National Poison Data Systems (NPDS) of the American Association of Poison Control Centers (AAPCC), approximately 50% occur in children <6 yr old, with the highest number of exposures occurring in 1 and 2 yr olds. Almost all these exposures are unintentional and reflect the propensity for young children to put virtually anything in their mouth. Fortunately, children <6 yr old account for <2% of all poisoning fatalities reported to NPDS.
More than 90% of toxic exposures in children occur in the home, and most involve a single substance. Ingestion accounts for the majority of exposures, with a minority occurring by the dermal, inhalational, and ophthalmic routes. Approximately 40% of cases involve nondrug substances, such as cosmetics, personal care items, cleaning solutions, plants, and foreign bodies. Pharmaceutical preparations account for the remainder of exposures, and analgesics, topical preparations, vitamins, and antihistamines are the most commonly reported categories.
The majority of poisoning exposures in children <6 yr old can be managed without direct medical intervention beyond a call to the regional poison control center (PCC) . This is because the product involved is not inherently toxic or the quantity of the material is not sufficient to produce clinically relevant toxic effects. However, a number of substances can be highly toxic to toddlers in small doses (Table 77.1 ). In 2015, carbon monoxide (CO), batteries, and analgesics (mainly opioids) were the leading causes of poison-related fatalities in young children (<6 yr). In addition, stimulants/street drugs, cardiovascular (CV) drugs, and aliphatic hydrocarbons were significant causes of mortality.
Table 77.1
| SUBSTANCE | TOXICITY |
|---|---|
| Aliphatic hydrocarbons (e.g., gasoline, kerosene, lamp oil) | Acute lung injury |
| Antimalarials (chloroquine, quinine) | Seizures, dysrhythmias |
| Benzocaine | Methemoglobinemia |
| β-Adrenergic receptor blockers † | Bradycardia, hypotension |
| Calcium channel blockers | Bradycardia, hypotension, hyperglycemia |
| Camphor | Seizures |
| Caustics (pH <2 or >12) | Airway, esophageal and gastric burns |
| Clonidine | Lethargy, bradycardia, hypotension |
| Diphenoxylate and atropine (Lomotil) | CNS depression, respiratory depression |
| Hypoglycemics, oral (sulfonylureas and meglitinides) | Hypoglycemia, seizures |
| Laundry detergent packets (pods) | Airway issues, respiratory distress, altered mental status |
| Lindane | Seizures |
| Monoamine oxidase inhibitors | Hypertension followed by delayed cardiovascular collapse |
| Methyl salicylate | Tachypnea, metabolic acidosis, seizures |
| Opioids (especially methadone, buprenorphine) | CNS depression, respiratory depression |
| Organophosphate pesticides | Cholinergic crisis |
| Phenothiazines (especially chlorpromazine, thioridazine) | Seizures, dysrhythmias |
| Theophylline | Seizures, dysrhythmias |
| Tricyclic antidepressants | CNS depression, seizures, dysrhythmias, hypotension |
* ”Small dose” typically implies 1 or 2 pills or 5 mL.
† Lipid-soluble β-blockers (e.g., propranolol) are more toxic than water-soluble β-blockers (e.g., atenolol).
CNS, Central nervous system.
Poison prevention education should be an integral part of all well-child visits, starting at the 6 mo visit. Counseling parents and other caregivers about potential poisoning risks, poison-proofing a child's environment, and actions in the event of an ingestion diminishes the likelihood of serious morbidity or mortality. Poison prevention education materials are available from the American Academy of Pediatrics (AAP) and regional PCCs. Through a U.S. network of PCCs, anyone at any time can contact a regional poison center by calling the toll-free number 1-800-222-1222 . Parents should be encouraged to share this number with grandparents, relatives, babysitters, and any other caregivers.
Product safety measures, poison prevention education, early recognition of exposures, and around-the-clock access to regionally based PCCs all contribute to the favorable exposure outcomes in young children. Poisoning exposures in children 6-12 yr are much less common, involving only approximately 10% of all reported pediatric exposures. A 2nd peak in pediatric exposures occurs in adolescence. Exposures in the adolescent age-group are primarily intentional (suicide or abuse or misuse of substances) and thus often result in more severe toxicity (see Chapter 140 ). Families should be informed and given anticipatory guidance that nonprescription and prescription medications, and even household products (e.g., inhalants), are common sources of adolescent exposures. Although adolescents (age 13-19 yr) account for only about 12% of exposures, they constituted a much larger proportion of deaths. Of the 90 poison-related pediatric deaths in 2015 reported to NPDS, 58 were adolescents (5% of all fatalities called in to poison centers). Pediatricians should be aware of the signs of drug abuse or suicidal ideation in adolescents and should aggressively intervene (see Chapter 40 ).
Prevention
Deaths caused by unintentional poisoning among younger children have decreased dramatically over the past 2 decades, particularly among children <5 yr old. In 1970, when the U.S. Poison Packaging Prevention Act was passed, 226 poisoning deaths of children <5 yr old occurred, compared with only 24 in 2015. Poisoning prevention demonstrates the effectiveness of passive strategies, including the use of child-resistant packaging and limited doses per container. Difficulty using child-resistant containers by adults is an important cause of poisoning in young children today. In 18.5% of households in which poisoning occurred in children <5 yr old, the child-resistant closure was replaced, and 65% of the packaging used did not work properly. Almost 20% of ingestions occur from drugs belonging to grandparents, who have difficulty using traditional child-resistant containers and often put their medications in pill organizers that are not childproof.
Even though there has been success in preventing poisoning in young children, there has been a remarkable rise in adolescent poison-related death over the past 20 years. This has mirrored the increasing rate of antidepressant prescriptions written by healthcare providers and the epidemic increase in opioid-related fatalities.
Approach to the Poisoned Patient
The initial approach to the patient with a witnessed or suspected poisoning should be no different than that in any other sick child, starting with stabilization and rapid assessment of the airway, breathing, circulation (pulse, blood pressure), and mental state, including Glasgow Coma Scale score and laryngeal reflexes (see Chapters 80 and 81 ). In any patient with altered mental status, a serum dextrose concentration should be obtained early, and naloxone administration should be considered. A targeted history and physical examination serves as the foundation for a thoughtful differential diagnosis, which can then be further refined through laboratory testing and other diagnostic studies.
History
Obtaining an accurate problem-oriented history is of paramount importance. Intentional poisonings (suicide attempts, drug abuse/misuse) are typically more severe than unintentional, exploratory ingestions. In patients without a witnessed exposure, historical features such as age of the child (toddler or adolescent), acute onset of symptoms without prodrome, multisystem organ dysfunction, or high levels of household stress should suggest a possible diagnosis of poisoning. In patients with a witnessed exposure, determining exactly what the child was exposed to and the circumstances surrounding the exposure is crucial to initiating directed therapy quickly. For household and workplace products, names (brand, generic, chemical) and specific ingredients, along with their concentrations, can often be obtained from the labels. PCC specialists can also help to identify possible ingredients and review the potential toxicities of each component. Poison center specialists can also help identify pills based on markings, shape, and color. If referred to the hospital for evaluation, parents should be instructed to bring the products, pills, and/or containers with them to assist with identifying and quantifying the exposure. If a child is found with an unknown pill, a list of all medications in the child's environment, including medications that grandparents, parents, siblings, caregivers, or other visitors might have brought into the house, must be obtained. In the case of an unknown exposure, clarifying where the child was found (e.g., garage, kitchen, laundry room, bathroom, backyard, workplace) can help to generate a list of potential toxins.
Next, it is important to clarify the timing of the ingestion and to obtain some estimate of how much of the substance was ingested. It is better to overestimate the amount ingested to prepare for the worst-case scenario. Counting pills or measuring the remaining volume of a liquid ingested can sometimes be useful in generating estimates. For inhalational, ocular, or dermal exposures, the concentration of the agent and the length of contact time with the material should be determined, if possible.
Symptoms
Obtaining a description of symptoms experienced after ingestion, including their timing of onset relative to the time of ingestion and their progression, can generate a list of potential toxins and help anticipate the severity of the ingestion. Coupled with physical exam findings, reported symptoms assist practitioners in identifying toxidromes, or recognized poisoning syndromes, suggestive of toxicity from specific substances or classes of substances (Tables 77.2 to 77.4 ).
Table 77.2
Selected Historical and Physical Findings in Poisoning
| SIGN | TOXIN |
|---|---|
| ODOR | |
| Bitter almonds | Cyanide |
| Acetone | Isopropyl alcohol, methanol, paraldehyde, salicylates |
| Rotten eggs | Hydrogen sulfide, sulfur dioxide, methyl mercaptans (additive to natural gas) |
| Wintergreen | Methyl salicylate |
| Garlic | Arsenic, thallium, organophosphates, selenium |
| OCULAR SIGNS | |
| Miosis | Opioids (except propoxyphene, meperidine, and pentazocine), organophosphates and other cholinergics, clonidine, phenothiazines, sedative-hypnotics, olanzapine |
| Mydriasis | Anticholinergics (e.g., antihistamines, TCAs, atropine), sympathomimetics (cocaine, amphetamines, PCP), post–anoxic encephalopathy, opiate withdrawal, cathinones, MDMA |
| Nystagmus | Anticonvulsants, sedative-hypnotics, alcohols, PCP, ketamine, dextromethorphan |
| Lacrimation | Organophosphates, irritant gas or vapors |
| Retinal hyperemia | Methanol |
| CUTANEOUS SIGNS | |
| Diaphoresis | Cholinergics (organophosphates), sympathomimetics, withdrawal syndromes |
| Alopecia | Thallium, arsenic |
| Erythema | Boric acid, elemental mercury, cyanide, carbon monoxide, disulfiram, scombroid, anticholinergics, vancomycin |
| Cyanosis (unresponsive to oxygen) | Methemoglobinemia (e.g., benzocaine, dapsone, nitrites, phenazopyridine), amiodarone, silver |
| ORAL SIGNS | |
| Salivation | Organophosphates, salicylates, corrosives, ketamine, PCP, strychnine |
| Oral burns | Corrosives, oxalate-containing plants |
| Gum lines | Lead, mercury, arsenic, bismuth |
| GASTROINTESTINAL SIGNS | |
| Diarrhea | Antimicrobials, arsenic, iron, boric acid, cholinergics, colchicine, opioid withdrawal |
| Hematemesis | Arsenic, iron, caustics, NSAIDs, salicylates |
| Constipation | Lead |
| CARDIAC SIGNS | |
| Tachycardia | Sympathomimetics, anticholinergics, antidepressants, antipsychotics, methylxanthines (theophylline, caffeine), salicylates, cellular asphyxiants (cyanide, carbon monoxide, hydrogen sulfide), withdrawal (ethanol, sedatives, clonidine, opioids), serotonin syndrome, neuroleptic malignant syndrome, MDMA, cathinones |
| Bradycardia | β-Blockers, calcium channel blockers, digoxin, clonidine, organophosphates, opioids, sedative-hypnotics |
| Hypertension | Sympathomimetics, anticholinergics, monoamine oxidase inhibitors, serotonin syndrome, neuroleptic malignant syndrome, clonidine withdrawal |
| Hypotension | β-Blockers, calcium channel blockers, cyclic antidepressants, iron, antipsychotics, barbiturates, clonidine, opioids, arsenic, amatoxin mushrooms, cellular asphyxiants (cyanide, carbon monoxide, hydrogen sulfide), snake envenomation |
| RESPIRATORY SIGNS | |
| Depressed respirations | Opioids, sedative-hypnotics, alcohol, clonidine, barbiturates |
| Tachypnea | Salicylates, sympathomimetics, caffeine, metabolic acidosis, carbon monoxide, hydrocarbon aspiration |
| CENTRAL NERVOUS SYSTEM SIGNS | |
| Ataxia | Alcohols, anticonvulsants, sedative-hypnotics, lithium, dextromethorphan, carbon monoxide, inhalants |
| Coma | Opioids, sedative-hypnotics, anticonvulsants, antidepressants, antipsychotics, ethanol, anticholinergics, clonidine, GHB, alcohols, salicylates, barbiturates |
| Seizures | Sympathomimetics, anticholinergics, antidepressants (especially TCAs, bupropion, venlafaxine), cholinergics (organophosphates), isoniazid, camphor, lindane, salicylates, lead, nicotine, tramadol, water hemlock, withdrawal |
| Delirium/psychosis | Sympathomimetics, anticholinergics, LSD, PCP, hallucinogens, lithium, dextromethorphan, steroids, withdrawal, MDMA, cathinones |
| Peripheral neuropathy | Lead, arsenic, mercury, organophosphates, nicotine |
GHB, γ-Hydroxybutyrate; LSD, lysergic acid diethylamide; MDMA, 3,4-methylenedioxymethamphetamine (Ecstasy); NSAIDs, nonsteroidal antiinflammatory drugs; PCP, phencyclidine; TCAs, tricyclic antidepressants.
Table 77.3
Recognizable Poison Syndromes (“Toxidromes”)
| TOXIDROME | SIGNS | POSSIBLE TOXINS | |||||
|---|---|---|---|---|---|---|---|
| Vital Signs | Mental Status | Pupils | Skin | Bowel Sounds | Other | ||
| Sympathomimetic | Hypertension, tachycardia, hyperthermia | Agitation, psychosis, delirium, violence | Dilated | Diaphoretic | Normal to increased | Amphetamines, cocaine, PCP, bath salts (cathinones), ADHD medication | |
| Anticholinergic | Hypertension, tachycardia, hyperthermia | Agitated, delirium, coma, seizures | Dilated | Dry, hot | Diminished | Ileus urinary retention | Antihistamines, TCAs, atropine, jimsonweed |
| Cholinergic | Bradycardia, BP, and temp typically normal | Confusion, coma, fasciculations | Small | Diaphoretic | Hyperactive | Diarrhea, urination, bronchorrhea, bronchospasm, emesis, lacrimation, salivation | Organophosphates (insecticides, nerve agents), carbamates (physostigmine, neostigmine, pyridostigmine) Alzheimer medications, myasthenia treatments |
| Opioids | Respiratory depression bradycardia, hypotension, hypothermia | Depression, coma, euphoria | Pinpoint | Normal | Normal to decreased | Methadone, buprenorphine, morphine, oxycodone, heroin, etc. | |
| Sedative-hypnotics | Respiratory depression, HR normal to decreased, BP normal to decreased, temp normal to decreased | Somnolence, coma | Small or normal | Normal | Normal | Barbiturates, benzodiazepines, ethanol | |
| Serotonin syndrome (similar findings with neuroleptic malignant syndrome) | Hyperthermia, tachycardia, hypertension or hypotension (autonomic instability) | Agitation, confusion, coma | Dilated | Diaphoretic | Increased | Neuromuscular hyperexcitability: clonus, hyperreflexia (lower > upper extremities) | SSRIs, lithium, MAOIs, linezolid, tramadol, meperidine, dextromethorphan |
| Salicylates | Tachypnea, hyperpnea, tachycardia, hyperthermia | Agitation, confusion, coma | Normal | Diaphoretic | Normal | Nausea, vomiting, tinnitus, ABGs with primary respiratory alkalosis and primary metabolic acidosis; tinnitus or difficulty hearing | Aspirin and aspirin-containing products, methyl salicylate |
| Withdrawal (sedative-hypnotic) | Tachycardia, tachypnea, hyperthermia | Agitation, tremor, seizure, hallucinosis, delirium tremens | Dilated | Diaphoretic | Increased | Lack of access to ethanol, benzodiazepines, barbiturates, GHB, or excessive use of flumazenil | |
| Withdrawal (opioid) | Tachycardia | Restlessness, anxiety | Dilated | diaphoretic | Hyperactive | Nausea, vomiting, diarrhea | Lack of access to opioids or excessive use of naloxone |
ABGs, Arterial blood gases; ADHD, attention-deficit/hyperactivity disorder; BP, blood pressure; GHB, γ-hydroxybutyrate; HR, heart rate; MAOIs, monoamine oxidase inhibitors; PCP, phencyclidine; SSRIs, selective serotonin reuptake inhibitors; temp, temperature; TCAs, tricyclic antidepressants.
Table 77.4
| TOXIDROME | SYMPTOMS AND SIGNS | EXAMPLES |
|---|---|---|
| α1 -Adrenergic receptor antagonists | CNS depression, tachycardia, miosis | Chlorpromazine, quetiapine, clozapine, olanzapine, risperidone |
| α2 -Adrenergic receptor agonist | CNS depression, bradycardia, hypertension (early), hypotension (late), miosis | Clonidine, oxymetazoline, tetrahydrozoline, tizanidine, dexmedetomidine |
| Clonus/myoclonus | CNS depression, myoclonic jerks, clonus, hyperreflexia | Carisoprodol, lithium, serotonergic agents, bismuth, organic lead, organic mercury, serotonin or neuroleptic malignant syndrome |
| Sodium channel blockers | CNS toxicity, wide QRS | Cyclic antidepressants and structurally related agents, propoxyphene, quinidine/quinine, amantadine, antihistamines, bupropion, cocaine |
| Potassium channel blockers | CNS toxicity, long QT interval | Antipsychotics, methadone, phenothiazines |
| Cathinones, synthetic cannabinoids | Hyperthermia, tachycardia, delirium, agitation, mydriases | See Chapter 140 . |
CNS, Central nervous system.
From Ruha AM, Levine M: Central nervous system toxicity. Emerg Med Clin North Am 32(1):205–221, 2014, p 208.
Past Medical and Developmental History
Underlying diseases can make a child more susceptible to the effects of a toxin. Concurrent drug therapy can also increase toxicity because certain drugs may interact with the toxin. A history of psychiatric illness can make patients more prone to substance abuse, misuse, intentional ingestions, and polypharmacy complications. Pregnancy is a common precipitating factor in adolescent suicide attempts and can influence both evaluation of the patient and subsequent treatment. A developmental history is important to ensure that the exposure history provided is appropriate for the child's developmental stage (e.g., report of 6 mo old picking up a large container of laundry detergent and drinking it should indicate urgent need for treatment, or indicate a severe condition, or “red flag”).
Social History
Understanding the child's social environment helps to identify potential sources of exposures (caregivers, visitors, grandparents, recent parties or social gatherings) and social circumstances (new baby, parent's illness, financial stress) that might have contributed to the ingestion (suicide or unintentional). Unfortunately, some poisonings occur in the setting of serious neglect or intentional abuse.
Physical Examination
A targeted physical examination is important to identifying the potential toxin and assessing the severity of the exposure. Initial efforts should be directed toward assessing and stabilizing the airway, breathing, circulation, and mental status. Once the airway is secure and the patient is stable from a cardiopulmonary standpoint, a more extensive physical exam can help to identify characteristic findings of specific toxins or classes of toxins.
In the poisoned patient, key features of the physical exam are vital signs, mental status, pupils (size, reactivity), nystagmus, skin, bowel sounds, and muscle tone. These findings might suggest a toxidrome, which can then guide the differential diagnosis and management.
Laboratory Evaluation
A basic chemistry panel (electrolytes, renal function, glucose) is necessary for all poisoned or potentially poisoned patients. Any patient with acidosis (low serum bicarbonate level on serum chemistry panel) must have an anion gap calculated because of the more specific differential diagnoses associated with an elevated anion gap metabolic acidosis (Table 77.5 ). Patients with a known overdose of acetaminophen should have liver transaminases (ALT, AST) assessed, as well as an international normalized ratio (INR). A serum creatinine kinase level is indicated on any patient with a prolonged “down time” to evaluate for rhabdomyolysis . Serum osmolality is only helpful as a surrogate marker for a toxic alcohol exposure if a serum concentration of the alcohol cannot be obtained in a reasonable time frame. A urine pregnancy test is mandatory for all postpubertal female patients. Based on the clinical presentation and the presumed poison, additional lab tests may also be helpful. Acetaminophen is a widely available medication and a commonly detected co-ingestant with the potential for severe toxicity. There is an effective antidote to acetaminophen poisoning that is time dependent. Given that patients might initially be asymptomatic and might not report or be aware of acetaminophen ingestion, an acetaminophen level should be checked in all patients who present after an intentional exposure or ingestion.
For select intoxications (e.g., salicylates, some anticonvulsants, acetaminophen, iron, digoxin, methanol, ethanol, lithium, ethylene glycol, theophylline, CO, lead), quantitative blood concentrations are integral to confirming the diagnosis and formulating a treatment plan. However, for most exposures, quantitative measurement is not readily available and is not likely to alter management. All intoxicant levels must be interpreted in conjunction with the history. For example, a methanol level of 20 mg/dL 1 hr after ingestion may be nontoxic, whereas a similar level 24 hr after ingestion implies a significant poisoning. In general, patients with multiple or chronic exposures to a drug or other chemical will be more symptomatic at lower drug levels than those with a single exposure.
Both the rapid urine drug-of-abuse screens and the more comprehensive drug screens vary widely in their ability to detect toxins and generally add little information to the clinical assessment. This is particularly true if the agent is known and the patient's symptoms are consistent with that agent. If a drug screen is ordered, it is important to know that the components screened for, and the lower limits of detection, vary from laboratory to laboratory. In addition, the interpretation of most drug screens is hampered by many false-positive and false-negative results. Many opiate toxicology screens poorly detect hydrocodone, and do not detect the fully synthetic opioids at all (e.g., methadone, buprenorphine, fentanyl). Several common benzodiazepines may not be detected, as may not synthetic cannabinoids or “bath salts.” The amphetamine screen, on the other hand, is typically overly sensitive and often is triggered by prescription amphetamines and some over-the-counter cold preparations. As such, the urine drug-of-abuse screen is typically of limited utility for medical clearance, but may serve a useful function for psychiatrists in their evaluation of the adolescent patient. Besides its psychiatric usefulness, urine drug-of-abuse screens are potentially helpful in patients with altered mental status of unknown etiology, persistent unexplained tachycardia, and acute myocardial ischemia or stroke at a young age. These screens can also be useful in the assessment of a neglected or abused child. Consultation with a medical toxicologist can be helpful in interpreting drug screens and directing which specific drug levels or other lab analyses might aid in patient management.
In the case of a neglected or allegedly abused child, a positive toxicology screen can add substantial weight to a claim of abuse or neglect. In these cases and any case with medicolegal implications, any positive screen mus t be confirmed with gas chromatography/mass spectroscopy, which is considered the gold standard measurement for legal purposes.
Additional Diagnostic Testing
An electrocardiogram (ECG) is a quick and noninvasive bedside test that can yield important clues to diagnosis and prognosis. Particular attention should be paid to the ECG intervals (Table 77.6 ). A widened QRS interval, putting the patient at risk for monomorphic ventricular tachycardia, suggests blockade of fast sodium channels. A widened QTc interval suggests effects at the potassium rectifier channels and portends a risk of torsades de pointes (polymorphic ventricular tachycardia).
Chest radiography may reveal signs of pneumonitis (e.g., hydrocarbon aspiration), noncardiogenic pulmonary edema (e.g., salicylate toxicity), or a foreign body. Abdominal radiography is most helpful in screening for the presence of lead paint chips or other foreign bodies. It may detect a bezoar (concretion), demonstrate radiopaque tablets, or reveal drug packets in a “body packer.” Further diagnostic testing is based on the differential diagnosis and pattern of presentation.
Principles of Management
The principles of management of the poisoned patient are supportive care, decontamination, directed therapy (antidotes, ILE), and enhanced elimination. Few patients meet criteria for all these interventions, although clinicians should consider each option in every poisoned patient so as not to miss a potentially lifesaving intervention. Antidotes are available for relatively few poisons (Tables 77.7 and 77.8 ), thus emphasizing the importance of meticulous supportive care and close clinical monitoring.
Table 77.7
Common Antidotes for Poisoning
| POISON | ANTIDOTE | DOSAGE | ROUTE | ADVERSE EFFECTS, WARNINGS, COMMENTS |
|---|---|---|---|---|
| Acetaminophen | N -Acetylcysteine (Mucomyst) | 140 mg/kg loading, followed by 70 mg/kg q4h | PO | Vomiting (patient-tailored regimens are the norm) |
| N -Acetylcysteine (Acetadote) | 150 mg/kg over 1 hr, followed by 50 mg/kg over 4 hr, followed by 100 mg/kg over 16 hr | IV |
Anaphylactoid reactions (most commonly seen with loading dose) (Higher doses of the infusion are often recommended depending on acetaminophen level or degree of injury) |
|
| Anticholinergics | Physostigmine | 0.02 mg/kg over 5 min; may repeat q5-10 min to 2 mg max | IV/IM |
Bradycardia, seizures, bronchospasm Note: Do not use if conduction delays on ECG. |
| Benzodiazepines | Flumazenil | 0.2 mg over 30 sec; if response is inadequate, repeat q1min to 1 mg max | IV |
Agitation, seizures from precipitated withdrawal (doses over 1 mg) Do not use for unknown or polypharmacy ingestions. |
| β-Blockers | Glucagon | 0.15 mg/kg bolus followed by infusion of 0.05-0.15 mg/kg/hr | IV | Vomiting, relative lack of efficacy |
| Calcium channel blockers | Insulin | 1 unit/kg bolus followed by infusion of 0.5-1 unit/kg/hr | IV |
Hypoglycemia Follow serum potassium and glucose closely. |
| Calcium salts | Dose depends on the specific calcium salt | IV | ||
| Carbon monoxide | Oxygen | 100% FIO 2 by non-rebreather mask (or ET if intubated) | Inhalation | Some patients may benefit from hyperbaric oxygen (see text). |
| Cyanide | Hydroxocobalamin (Cyanokit) | 70 mg/kg (adults: 5 g) given over 15 min | IV | Flushing/erythema, nausea, rash, chromaturia, hypertension, headache |
| Digitalis | Digoxin-specific Fab antibodies (Digibind, DigiFab) |
1 vial binds 0.6 mg of digitalis glycoside; #vials = digitalis level × weight in kg/100 |
IV | Allergic reactions (rare), return of condition being treated with digitalis glycoside |
| Ethylene glycol, methanol | Fomepizole | 15 mg/kg load; 10 mg/kg q12h × 4 doses; 15 mg/kg q12h until ethylene glycol level is <20 mg/dL | IV |
Infuse slowly over 30 min. If fomepizole is not available, can treat with oral ethanol (80 proof) |
| Iron | Deferoxamine | Infusion of 5-15 mg/kg/hr (max: 6 g/24 hr) | IV | Hypotension (minimized by avoiding rapid infusion rates) |
| Isoniazid (INH) | Pyridoxine |
Empirical dosing: 70 mg/kg (max dose = 5 g) If ingested dose is known: 1 g per gram of INH |
IV | May also be used for Gyromitra mushroom ingestions |
| Lead and other heavy metals (e.g., arsenic, inorganic mercury) | BAL (dimercaprol) | 3-5 mg/kg/dose q4h, for the 1st day; subsequent dosing depends on the toxin | Deep IM |
Local injection site pain and sterile abscess, vomiting, fever, salivation, nephrotoxicity Caution: prepared in peanut oil; contraindicated in patients with peanut allergy |
| Calcium disodium EDTA | 35-50 mg/kg/day × 5 days; may be given as a continuous infusion or 2 divided doses/day | IV | Vomiting, fever, hypertension, arthralgias, allergic reactions, local inflammation, nephrotoxicity (maintain adequate hydration; follow UA and renal function) | |
| Dimercaptosuccinic acid (succimer, DMSA, Chemet) | 10 mg/kg/dose q8h × 5 days, then 10 mg/kg q12h × 14 days | PO | Vomiting, hepatic transaminase elevation, rash | |
| Methemoglobinemia | Methylene blue, 1% solution | 0.1-0.2 mL/kg (1-2 mg/kg) over 5-10 min; may be repeated q30-60 min | IV | Vomiting, headache, dizziness, blue discoloration of urine |
| Opioids | Naloxone |
1 mg if patient not likely to be addicted. 0.04-0.4 mg if possibly addicted; repeated as needed; may need continuous infusion |
IV, intranasal, IO, IM, nebulized |
Acute withdrawal symptoms if given to addicted patients May also be useful for clonidine ingestions (typically at higher doses) |
| Organophosphates | Atropine | 0.05-0.1 mg/kg repeated q5-10 min as needed | IV/ET | Tachycardia, dry mouth, blurred vision, urinary retention |
| Pralidoxime (2-PAM) | 25-50 mg/kg over 5-10 min (max: 200 mg/min); can be repeated after 1-2 hr, then q10-12h as needed | IV/IM | Nausea, dizziness, headache, tachycardia, muscle rigidity, bronchospasm (rapid administration) | |
| Salicylates | Sodium bicarbonate | Bolus 1-2 mEq/kg followed by continuous infusion | IV |
Follow potassium closely and replace as necessary. Goal urine pH: 7.5-8.0 |
| Sulfonylureas | Octreotide and dextrose | 1-2 µg/kg/dose (adults 50-100 µg) q6-8h | IV/SC | |
| Tricyclic antidepressants | Sodium bicarbonate | Bolus 1-2 mEq/kg; repeated bolus dosing as needed to keep QRS <110 msec | IV | Indications: QRS widening (≥110 msec), hemodynamic instability; follow potassium. |
BAL, British antilewisite; DMSA, dimercaptosuccinic acid; ECG, electrocardiogram; FIO 2 , fraction of inspired oxygen; EDTA, ethylenediaminetetraacetic acid; ET, endotracheal tube; IO, intraosseous; max, maximum; UA, urinalysis.
Table 77.8
Poison control center personnel are specifically trained to provide expertise in the management of poisoning exposures. Parents should be instructed to call the poison control center (1-800-222-1222 ) for any concerning exposure. PCC specialists can assist parents in assessing the potential toxicity and severity of the exposure. They can further determine which children can be safely monitored at home and which children should be referred to the emergency department for further evaluation and care. Although up to one third of calls to PCCs involve hospitalized patients, and 90% of all calls for exposures in children <6 yr old are managed at home. The AAPCC has generated consensus statements for out-of-hospital management of common ingestions (e.g., acetaminophen, iron, calcium channel blockers) that serve to guide poison center recommendations.
Supportive Care
Careful attention is paid first to the “ABCs” of airway, breathing, and circulation; there should be a low threshold to aggressively manage the airway of a poisoned patient because of the patient's propensity to quickly become comatose. In fact, endotracheal intubation is often the only significant intervention needed in many poisoned patients. An important caveat is the tachypneic patient with a clear lung examination and normal oxygen saturation. This should alert the clinician to the likelihood that the patient is compensating for an acidemia. Paralyzing such a patient and underventilating might prove fatal. If intubation is absolutely necessary for airway protection or a tiring patient, a good rule of thumb is to match the ventilatory settings to the patient's preintubation minute ventilation.
Hypotensive patients often are not hypovolemic but are poisoned, and aggressive fluid resuscitation may lead to fluid overload. If hypotension persists after 1 or 2 standard boluses of crystalloid, infusion of a direct-acting vasopressor, such as norepinephrine or epinephrine, is preferred. Dysrhythmias are managed in the standard manner, except for those caused by agents that block fast sodium channels of the heart, for which boluses of sodium bicarbonate are given.
Seizures should primarily be managed with agents that potentiate the γ-aminobutyric acid (GABA) complex, such as benzodiazepines or barbiturates. The goal of supportive therapy is to support the patient's vital functions until the patient can eliminate the toxin. Patients with an elevated creatine phosphokinase (CPK) should be aggressively hydrated with crystalloid, with a goal urine output of 1-2 mL/kg/hr and close monitoring of CPK trend.
Decontamination
The majority of poisonings in children are from ingestion, although exposures can also occur by inhalational, dermal, and ocular routes. The goal of decontamination is to minimize absorption of the toxic substance. The specific method employed depends on the properties of the toxin itself and the route of exposure. Regardless of the decontamination method used, the efficacy of the intervention decreases with increasing time since exposure. Decontamination should not be routinely employed for every poisoned patient. Instead, careful decisions regarding the utility of decontamination should be made for each patient and should include consideration of the toxicity and pharmacologic properties of the exposure, route of the exposure, time since the exposure, and risks vs benefits of the decontamination method.
Dermal and ocular decontamination begins with removal of any contaminated clothing and particulate matter, followed by flushing of the affected area with tepid water or normal saline (NS). Treating clinicians should wear proper protective gear when performing irrigation. Flushing for a minimum of 10-20 min is recommended for most exposures, although some chemicals (e.g., alkaline corrosives) require much longer periods of flushing. Dermal decontamination, especially after exposure to adherent or lipophilic (e.g., organophosphates) agents, should include thorough cleansing with soap and water. Water should not be used for decontamination after exposure to highly reactive agents, such as elemental sodium, phosphorus, calcium oxide, and titanium tetrachloride. After an inhalational exposure, decontamination involves moving the patient to fresh air and administering supplemental oxygen if indicated.
Gastrointestinal (GI) decontamination strategies are most likely to be effective in the 1 or 2 hours after an acute ingestion . GI absorption may be delayed after ingestion of agents that slow GI motility (anticholinergic medications, opioids), massive amounts of pills, sustained-release (SR) preparations, and agents that can form pharmacologic bezoars (e.g., enteric-coated salicylates). GI decontamination more than 2 hr after ingestion may be considered in patients who ingest toxic substances with these properties. However, even rapid institution of GI decontamination with activated charcoal will, at best, bind only approximately 30% of the ingested substance. GI decontamination should never supplant excellent supportive care and should not be employed in an unstable or persistently vomiting patient. Described methods of GI decontamination include induced emesis with ipecac, gastric lavage, cathartics, activated charcoal, and whole-bowel irrigation (WBI). Of these, only activated charcoal and WBI are of potential benefit.
Syrup of Ipecac
Syrup of ipecac contains 2 emetic alkaloids that work in both the central nervous system (CNS) and locally in the GI tract to produce vomiting. Many studies have failed to document a significant clinical impact from the use of ipecac and have documented multiple adverse events from its use. The AAP, the American Academy of Clinical Toxicology (AACT), and the AAPCC have all published statements in favor of abandoning the use of ipecac.
Gastric Lavage
Gastric lavage involves placing a large tube orally into the stomach to aspirate contents, followed by flushing with aliquots of fluid, usually water or NS. Although gastric lavage was used routinely for many years, objective data do not document or support clinically relevant efficacy. This is particularly true in children, in whom only small-bore tubes can be used. Lavage is time-consuming and painful and can induce bradycardia through a vagal response to tube placement. It can delay administration of more definitive treatment (activated charcoal) and under the best circumstances, only removes a fraction of gastric contents. Thus, in most clinical scenarios, the use of gastric lavage is no longer recommended.
Single-Dose Activated Charcoal
Activated charcoal is a potentially useful method of GI decontamination. Charcoal is “activated” by heating to extreme temperatures, creating an extensive network of pores that provides a very large adsorptive surface area that many (but not all) toxins will bind to, preventing absorption from the GI tract. Charged molecules (i.e., heavy metals, lithium, iron) and liquids do not bind well to activated charcoal (Table 77.9 ). Charcoal is most likely to be effective when given within 1 hr of ingestion. Administration should also be avoided after ingestion of a caustic substance, as it can impede subsequent endoscopic evaluation. A repeat dose of activated charcoal may be warranted in the cases of ingestion of an extended-release product or, more frequently, with a significant salicylate poisoning as a result of its delayed and erratic absorption pattern.
The dose of activated charcoal, with or without sorbitol, is 1 g/kg in children or 50-100 g in adolescents and adults. Before administering charcoal, one must ensure that the patient's airway is intact or protected and that the patient has a benign abdominal examination. In the awake, uncooperative adolescent or child who refuses to drink the activated charcoal, there is little utility and potential morbidity associated with forcing activated charcoal down a nasogastric (NG) tube, and such practice should be avoided. In young children, practitioners can attempt to improve palatability by adding flavorings (chocolate or cherry syrup) or giving the mixture over ice cream. Approximately 20% of children vomit after receiving a dose of charcoal, emphasizing the importance of an intact airway and avoiding administration of charcoal after ingestion of substances that are particularly toxic when aspirated (e.g., hydrocarbons). If charcoal is given through a gastric tube in an intubated patient, placement of the tube should be carefully confirmed before activated charcoal is given. Instillation of charcoal directly into the lungs can have disastrous effects. Constipation is another common side effect of activated charcoal, and in rare cases, bowel perforation has been reported.
Cathartics (sorbitol, magnesium sulfate, magnesium citrate) have been used in conjunction with activated charcoal to prevent constipation and accelerate evacuation of the charcoal-toxin complex. There are no data demonstrating their value and numerous reports of adverse effects from cathartics, such as dehydration and electrolyte imbalance.
Whole-Bowel Irrigation
Whole-bowel irrigation (WBI) involves instilling large volumes (35 mL/kg/hr in children or 1-2 L/hr in adolescents) of a polyethylene glycol electrolyte solution (e.g., GoLYTELY) to “wash out” the entire GI tract. This technique may have some success for the ingestion of SR preparations, substances not well adsorbed by charcoal (e.g., lithium, iron), transdermal patches, foreign bodies, and drug packets. In children, WBI is most frequently administered to decontaminate the gut of a child whose abdominal radiograph demonstrates multiple lead paint chips. Careful attention should be paid to assessment of the airway and abdominal exam before initiating WBI. WBI should never be given to a patient with signs of obstruction or ileus or with a compromised airway. Given the rate of administration and volume needed to flush the system, WBI is typically administered by NG tube. WBI is continued until the rectal effluent is clear. If the WBI is for a child with ingested paint chips, the end-point will be clearing of the chips from the bowel based on repeat radiographs. Complications of WBI include vomiting, abdominal pain, and abdominal distention. Bezoar formation might respond to WBI but may also require endoscopy or surgery.
Directed Therapy
Antidotal Therapy
Antidotes are available for relatively few toxins (Tables 77.7 and 77.8 ), but early and appropriate use of an antidote is a key element in managing the poisoned patient.
Intralipid Emulsion Therapy
Intralipid emulsion (ILE) therapy is a potentially lifesaving intervention. ILE therapy sequesters fat-soluble drugs, decreasing their impact at target organs. It also enhances cardiac function by supplying an alternative energy source to a depressed myocardium and acting on calcium channels in the heart, increasing myocardial calcium and thus cardiac function. Intralipid is most effective as a reversal agent for toxicity from inadvertent intravenous (IV) injection of bupivacaine. Using the same 20% Intralipid used for total parenteral nutrition (TPN), a bolus dose of 1.5 mL/kg is given over 3 min, followed by an infusion of 0.25 mL/kg/min until recovery or until a total of 10 mL/kg has been infused. Lipophilic drugs, those in which the logarithm of the coefficient describing the partition between 2 solvents (hydrophobic phase and hydrophilic phase) is >2, have the most potential to be bound by ILE. These include, but are not limited to, calcium channel blockers (verapamil, diltiazem), bupropion, and tricyclic antidepressants.
Enhanced Elimination
Enhancing elimination results in increased clearance of a poison that has already been absorbed. It is only useful for a few toxins and in these cases is a potentially lifesaving intervention. Methods of enhanced elimination include urinary alkalinization, hemodialysis, and multidose activated charcoal.
Urinary Alkalinization
Urinary alkalinization enhances the elimination of drugs that are weak acids by forming charged molecules, which then become trapped in the renal tubules. Charged molecules, being polar and hydrophilic, do not easily cross cellular membranes, thus they remain in the renal tubules and are excreted. Urinary alkalinization is accomplished by a continuous infusion of sodium bicarbonate–containing IV fluids, with a goal urine pH of 7.5-8. Alkalinization of the urine is most useful in managing salicylate and methotrexate toxicity. Complications of urinary alkalinization include electrolyte derangements (e.g., hypokalemia, hypocalcemia), fluid overload, and excessive serum alkalinization. Serum pH should be closely monitored and not exceed a pH >7.55. Patients typically unable to tolerate the volumes required for alkalinization are those with heart failure, kidney failure, pulmonary edema, or cerebral edema.
Hemodialysis
Few drugs or toxins are removed by dialysis in amounts sufficient to justify the risks and difficulty of dialysis. Toxins amenable to dialysis have the following properties: low volume of distribution (<1 L/kg) with a high degree of water solubility, low molecular weight, and low degree of protein binding. Hemodialysis may be useful for toxicity from methanol, ethylene glycol, salicylates, theophylline, bromide, lithium, and valproic acid. Hemodialysis is also used to correct severe electrolyte disturbances and acid-base derangements resulting from the ingestion (e.g., severe metformin-associated lactic acidosis).
Multidose Activated Charcoal
Whereas single-dose activated charcoal is used as a method of decontamination, multidose activated charcoal (MDAC ) can help to enhance the elimination of certain toxins. MDAC is typically given as 0.5 g/kg every 4-6 hr (for 4 doses). MDAC enhances elimination by 2 proposed mechanisms: interruption of enterohepatic recirculation and “GI dialysis.” The concept of GI dialysis involves using the intestinal mucosa as a dialysis membrane and pulling toxins from the bloodstream back into the intraluminal space, where they are adsorbed to the charcoal. The AACT/European Association of Poisons Centres and Clinical Toxicologists position statement recommends MDAC in managing significant ingestions of carbamazepine, dapsone, phenobarbital, quinine, and theophylline. As with single-dose activated charcoal, contraindications to use of MDAC include an unprotected airway and a concerning abdominal examination (e.g., ileus, distention, peritoneal signs). Thus the airway and abdominal exam should be assessed before each dose. A cathartic (e.g., sorbitol) may be given with the 1st dose, but it should not be used with subsequent doses because of the risk of dehydration and electrolyte derangements. Although MDAC reduces the serum level of an intoxicant quicker than without MDAC, it has not been shown to have a significant impact on outcome.
Select Compounds in Pediatric Poisoning
See other chapters for herbal medicines (Chapter 78 ), drugs of abuse (Chapter 140 ), and environmental health hazards (Chapters 735-741 ).
Pharmaceuticals
Analgesics
Acetaminophen.
Acetaminophen (APAP) is the most widely used analgesic and antipyretic in pediatrics, available in multiple formulations, strengths, and combinations. Consequently, APAP is commonly available in the home, where it can be unintentionally ingested by young children, taken in an intentional overdose by adolescents and adults, or inappropriately dosed in all ages. In the United States, APAP toxicity remains the most common cause of acute liver failure and is the leading cause of intentional poisoning death.
Pathophysiology.
APAP toxicity results from the formation of a highly reactive intermediate metabolite, N -acetyl-p -benzoquinone imine (NAPQI). In therapeutic use, only a small percentage of a dose (approximately 5%) is metabolized by the hepatic cytochrome P450 enzyme CYP2E1 to NAPQI, which is then immediately joined with glutathione to form a nontoxic mercapturic acid conjugate. In overdose, glutathione stores are overwhelmed, and free NAPQI is able to combine with hepatic macromolecules to produce hepatocellular necrosis. The single acute toxic dose of APAP is generally considered to be >200 mg/kg in children and >7.5-10 g in adolescents and adults. Repeated administration of APAP at supratherapeutic doses (>90 mg/kg/day for consecutive days) can lead to hepatic injury or failure in some children, especially in the setting of fever, dehydration, poor nutrition, and other conditions that serve to reduce glutathione stores.
Any child with a history of acute ingestion of >200 mg/kg (unusual in children <6 yr) or with an acute intentional ingestion of any amount should be referred to a healthcare facility for clinical assessment and measurement of a serum APAP level.
Clinical and Laboratory Manifestations.
Classically, 4 general stages of APAP toxicity have been described (Table 77.10 ). The initial signs are nonspecific (i.e., nausea and vomiting) and may not be present. Thus the diagnosis of APAP toxicity cannot be based on clinical symptoms alone, but instead requires consideration of the combination of the patient's history, symptoms, and laboratory findings.
Table 77.10
If a toxic ingestion is suspected, a serum APAP level should be measured 4 hr after the reported time of ingestion. For patients who present to medical care more than 4 hr after ingestion, a stat APAP level should be obtained. APAP levels obtained <4 hr after ingestion, unless “nondetectable,” are difficult to interpret and cannot be used to estimate the potential for toxicity. Other important baseline lab tests include hepatic transaminases, renal function tests, and coagulation parameters.
Treatment.
When considering the treatment of a patient poisoned or potentially poisoned with APAP, and after assessment of the ABCs, it is helpful to place the patient into one of the following four categories.
1 Prophylactic.
By definition, these patients have a normal aspartate transaminase (AST). If the APAP level is known and the ingestion is within 24 hr of the level being drawn, treatment decisions are based on where the level falls on the Rumack-Matthew nomogram (Fig. 77.1 ). Any patient with a serum APAP level in the possible or probable hepatotoxicity range per the nomogram should be treated with N -acetylcysteine (NAC). This nomogram is only intended for use in patients who present within 24 hr of a single acute APAP ingestion with a known time of ingestion. If treatment is recommended, they should receive NAC as either oral Mucomyst or IV Acetadote for 24 or 21 hr, respectively. Repeat AST and APAP concentration drawn toward the end of that interval should be obtained. If the AST remains normal and the APAP becomes nondetectable, treatment may be discontinued. If the AST becomes elevated, the patient moves into the next category of treatment (injury). If APAP is still present, treatment should be continued until the level is nondetectable. In the case of a patient with a documented APAP level, normal AST, and an unknown time of ingestion, treatment should ensue until the level is nondetectable, with normal transaminases.
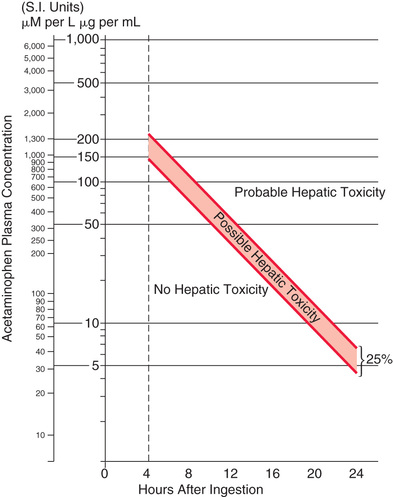
The importance of instituting therapy with either IV or oral NAC no later than 8 hr from the time of ingestion cannot be overemphasized. No patient, regardless of the size of the ingestion, who receives NAC within 8 hr of overdose should die from liver failure. The longer from the 8 hr mark the initiation of therapy is delayed, the greater the risk of acute liver failure. Any patient presenting close to or beyond the 8 hr mark after an APAP overdose should be empirically started on NAC pending laboratory results.
2 Hepatic Injury.
These patients are exhibiting evidence of hepatocellular necrosis, manifested first as elevated liver transaminases (usually AST first, then alanine transaminase [ALT]), followed by a rise in the INR. Any patient in this category requires therapy with NAC (IV or oral). When to discontinue therapy in the clinically well patient remains controversial, but in general the transaminases and INR have peaked and fallen significantly “toward” normal (they do not need to be normal). Most patients' liver enzymes will peak 3 or 4 days after their ingestion.
3 Acute Liver Failure.
The King's College criteria are used to determine which patients should be referred for consideration of liver transplant. These criteria include acidemia (serum pH <7.3) after adequate fluid resuscitation, coagulopathy (INR >6), renal dysfunction (creatinine >3.4 mg/dL), and grade III or IV hepatic encephalopathy (see Chapter 391 ). A serum lactic acid >3 mmol/L (after IV fluids) adds to both sensitivity and specificity of the criteria to predict death without liver transplant. The degree of transaminase elevation does not factor in to this decision-making process.
4 Repeated Supratherapeutic Ingestion.
APAP is particularly prone to unintentional overdose through the ingestion of multiple medications containing the drug or simply because people assume it to be safe at any dose. Ingestion of amounts significantly greater than the recommended daily dose for several days or more puts one at risk for liver injury. Because the Rumack-Matthew nomogram is not helpful in this scenario, a conservative approach is taken. In the asymptomatic patient, if the AST is normal and the APAP is <10 µg/mL, no therapy is indicated. A normal AST and an elevated APAP warrants NAC dosing for at least long enough for the drug to metabolize while the AST remains normal. An elevated AST puts the patient in the “hepatic injury” category previously described. A patient presenting with symptoms (i.e., right upper quadrant pain, vomiting, jaundice) should be empirically started on NAC pending lab results.
NAC is available in oral and IV forms, and both are considered equally efficacious (see Table 77.7 for the dosing regimens of the oral vs IV form). The IV form is used in patients with intractable vomiting, those with evidence of hepatic failure, and pregnant patients. Oral NAC has an unpleasant taste and smell and can be mixed in soft drink or fruit juice or given by NG tube to improve tolerability of the oral regimen. Administration of IV NAC (as a standard 3% solution to avoid administering excess free water, typically in 5% dextrose), especially the initial loading dose, is associated in some patients with the development of anaphylactoid reactions (non–immunoglobulin E mediated). These reactions are typically managed by stopping the infusion; treating with diphenhydramine, albuterol, and/or epinephrine as indicated; and restarting the infusion at a slower rate once symptoms have resolved. IV NAC is also associated with mild elevation in measured INR (range: 1.2-1.5) because of laboratory interference. IV dosing, however, delivers less medication to the liver compared with the oral regimen. As a result, many toxicologists now recommend higher doses of the IV formulation in patients with large overdoses. Transaminases, synthetic function, and renal function should be followed daily while the patient is being treated with NAC. Patients with worsening hepatic function or clinical status might benefit from more frequent lab monitoring. A patient-tailored approach is now the norm for when to stop NAC therapy, for deciding whom to refer for transplantation evaluation, and often for the dose of IV NAC in patients with either very high APAP levels or signs of injury. Consultation with the regional PCC and medical toxicologist can help streamline the care of these patients, ultimately shortening their length of stay with potentially improved outcomes.
Salicylates.
The incidence of salicylate poisoning in young children has declined dramatically since APAP and ibuprofen replaced aspirin as the most commonly used analgesics and antipyretics in pediatrics. However, salicylates remain widely available, not only in aspirin-containing products but also in antidiarrheal medications, topical agents (e.g., keratolytics, sports creams), oil of wintergreen, and some herbal products. Oil of wintergreen contains 5 g of salicylate in 1 teaspoon (5 mL), meaning ingestion of very small volumes of this product has the potential to cause severe toxicity.
Pathophysiology.
Salicylates lead to toxicity by interacting with a wide array of physiologic processes, including direct stimulation of the respiratory center, uncoupling of oxidative phosphorylation, inhibition of the tricarboxylic acid cycle, and stimulation of glycolysis and gluconeogenesis. The acute toxic dose of salicylates is generally considered to be >150 mg/kg. More significant toxicity is seen after ingestions of >300 mg/kg, and severe, potentially fatal, toxicity is described after ingestions of >500 mg/kg.
Clinical and Laboratory Manifestations.
Salicylate ingestions are classified as acute or chronic, and acute toxicity is much more common in pediatric patients. Early signs of acute salicylism include nausea, vomiting, diaphoresis, and tinnitus. Moderate salicylate toxicity can manifest as tachypnea and hyperpnea, tachycardia, and altered mental status. The tachycardia largely results from marked insensible losses from vomiting, tachypnea, diaphoresis, and uncoupling of oxidative phosphorylation. Thus, careful attention should be paid to volume status and early volume resuscitation in the significantly poisoned patient. Signs of severe salicylate toxicity include mild hyperthermia, coma, and seizures. Chronic salicylism can have a more insidious presentation, and patients can show marked toxicity (e.g. altered mental status, noncardiogenic pulmonary edema, acidemia) at significantly lower salicylate levels than in acute toxicity.
Classically, laboratory values from a patient poisoned with salicylates reveal a primary respiratory alkalosis and a primary, elevated anion gap metabolic acidosis. Early in the course of acute salicylism, respiratory alkalosis dominates and the patient is alkalemic. As the respiratory stimulation diminishes, the patient will move toward acidemia. Hyperglycemia (early) and hypoglycemia (late) have been described. Abnormal coagulation studies and acute kidney injury may be seen but are not common.
Serial serum salicylate levels should be closely monitored (every 2-4 hr initially) until they are consistently downtrending. Salicylate absorption in overdose is unpredictable and erratic, especially with an enteric-coated product, and levels can rapidly increase into the highly toxic range, even many hours after the ingestion. The Done nomogram is of poor value and should not be used. Serum and urine pH and electrolytes should be followed closely. An APAP level should be checked in any patient who intentionally overdoses on salicylates, because APAP is a common co-ingestant, and people often confuse or combine their nonprescription analgesic medications. Salicylate toxicity can cause a noncardiogenic pulmonary edema, especially in chronic overdose; consequently, a chest radiograph is recommended in any patient in respiratory distress.
Treatment.
For the patient who presents soon after an acute ingestion, initial treatment should include gastric decontamination with activated charcoal. Salicylate pills occasionally form bezoars, which should be suspected if serum salicylate concentrations continue to rise many hours after ingestion or are persistently elevated despite appropriate management. Gastric decontamination is typically not useful after chronic exposure.
Initial therapy focuses on aggressive volume resuscitation and prompt initiation of sodium bicarbonate therapy in the symptomatic patient, even before obtaining serum salicylate levels. Therapeutic salicylate levels are 10-20 mg/dL, and levels >25 or 30 mg/dL warrant treatment.
The primary mode of therapy for salicylate toxicity is urinary alkalinization . Urinary alkalinization enhances the elimination of salicylates by converting salicylate to its ionized form, “trapping” it in the renal tubules, thus enhancing elimination. In addition, maintaining an alkalemic serum pH decreases CNS penetration of salicylates because charged particles are less able to cross the blood-brain barrier. Alkalinization is achieved by administration of a sodium bicarbonate infusion at approximately 2 times maintenance fluid rates. The goals of therapy include a urine pH of 7.5-8, a serum pH of 7.45-7.55, and decreasing serum salicylate levels. In general, in the presence of an acidosis, an aspirin-poisoned patient's status can be directly related to the patient's serum pH: the lower the pH, the greater the relative amount of salicylate in the uncharged, nonpolar form and the greater the penetration of the blood-brain barrier by the drug. Careful attention should also be paid to serial potassium levels in any patient on a bicarbonate infusion, since potassium will be driven intracellularly and hypokalemia impairs alkalinization of the urine. For these reasons, potassium is often added to the bicarbonate drip. Repeat doses of charcoal may be beneficial because of the often delayed and erratic absorption of aspirin. Parenteral glucose should be provided to any salicylate-poisoned patients with altered mental status because they may have CNS hypoglycemia (i.e., neuroglycopenia) not seen in a peripheral serum glucose test.
In patients with severe toxicity, hemodialysis may be required. Indications for dialysis include severe acid-base abnormalities (specifically severe acidosis and acidemia), a rising salicylate level (despite adequate decontamination and properly alkalinized urine), pulmonary edema, cerebral edema, seizures, and renal failure. Serum salicylate concentrations alone are not a clear indicator of the need for dialysis and should always be interpreted along with the clinical status of the patient.
Ibuprofen and Other Nonsteroidal Antiinflammatory Drugs (NSAIDs).
Ibuprofen and other NSAIDs are often involved in unintentional and intentional overdoses because of their widespread availability and common use as analgesics and antipyretics. Fortunately, serious effects after acute NSAID overdose are rare because of their wide therapeutic index.
Pathophysiology.
NSAIDs inhibit prostaglandin synthesis by reversibly inhibiting the activity of cyclooxygenase (COX), the primary enzyme responsible for the biosynthesis of prostaglandins. In therapeutic use, side effects include GI irritation, reduced renal blood flow, and platelet dysfunction. To minimize these side effects, NSAID analogs have been developed that are more specific for the inducible form of COX (the COX-2 isoform) than the constitutive form, COX-1. However, overdose of the more selective COX-2 inhibitors (e.g., celecoxib [Celebrex]) is treated the same as overdose of nonspecific COX inhibitors (e.g., ibuprofen) because at higher doses, COX-2–selective agents lose their COX inhibitory selectivity.
Ibuprofen, the primary NSAID used in pediatrics, is well tolerated, even in overdose. In children, acute doses of <200 mg/kg rarely cause toxicity, but ingestions of >400 mg/kg can produce more serious effects, including altered mental status and metabolic acidosis.
Clinical and Laboratory Manifestations.
Symptoms usually develop within 4-6 hr of ingestion and resolve within 24 hr. If toxicity does develop, it is typically manifested as nausea, vomiting, and abdominal pain. Although GI bleeding and ulcers have been described with chronic use, they are rare in the setting of acute ingestion. After massive ingestions, patients can develop marked CNS depression, anion gap metabolic acidosis, renal insufficiency, and (rarely) respiratory depression. Seizures have also been described, especially after overdose of mefenamic acid. Specific drug levels are not readily available, nor do they inform management decisions. Renal function studies, acid-base balance, complete blood count (CBC), and coagulation parameters should be monitored after very large ingestions. Co-ingestants, especially APAP, should be ruled out after any intentional ingestion.
Treatment.
Supportive care, including use of antiemetics and acid blockade as indicated, is the primary therapy for NSAID toxicity. Decontamination with activated charcoal should be considered if a patient presents within 1-2 hr of a potentially toxic ingestion. There is no specific antidote for this class of drugs. Given the high degree of protein binding and excretion pattern of NSAIDs, none of the modalities used to enhance elimination is particularly useful in managing these overdoses. Unlike in patients with salicylate toxicity, urinary alkalinization is not helpful for NSAID toxicity. Patients who develop significant clinical signs of toxicity should be admitted to the hospital for ongoing supportive care and monitoring. Patients who remain asymptomatic for 4-6 hr after ingestion may be considered medically cleared.
Prescription Opioids.
Opioids are a frequently abused class of medications in both IV and oral forms. The opioid epidemic gripping the United States and other countries is discussed in Chapter 140 . Two specific oral opioids, buprenorphine and methadone, merit mention because of potential life-threatening toxicity in toddlers with ingestion of even 1 pill. Both agents are used in managing opioid dependence, although buprenorphine is the drug of choice. Methadone is also widely used in the treatment of chronic pain, meaning multiday prescriptions can be filled. Both drugs are readily available for illicit purchase and potential abuse. Both drugs are of great potential toxicity to a toddler, especially buprenorphine because of its long half-life and high potency.
Pathophysiology.
Methadone is a lipophilic synthetic opioid with potent agonist effects at µ-opioid receptors, leading to both its desired analgesic effects and undesired side effects, including sedation, respiratory depression, and impaired GI motility. Methadone is thought to cause QTc interval prolongation through interactions with the human ether-a-go-go–related gene (hERG)-encoded potassium rectifier channel. Its duration of effect for pain control averages only about 8 hr, whereas the dangerous side effects can occur up to 24 hr from the last dose and longer after overdose. Methadone has an average half-life >25 hr, which may be extended to >50 hr in overdose.
Suboxone is a combination of buprenorphine, a potent opioid with partial agonism at µ-opioid receptors and weak antagonism at κ-opioid receptors, and naloxone. Naloxone has poor oral bioavailability but is included in the formulation to discourage diversion for IV use, during which it can precipitate withdrawal. Suboxone is formulated for buccal or sublingual administration; consequently, toddlers can absorb significant amounts of drug even by sucking on a tablet. Buprenorphine has an average half-life of 37 hr.
Clinical and Laboratory Manifestations.
In children, methadone and buprenorphine ingestions can manifest with the classic opioid toxidrome of respiratory depression, sedation, and miosis. Signs of more severe toxicity can include bradycardia, hypotension, and hypothermia. Even in therapeutic use, methadone is associated with a prolonged QTc interval and risk of torsades de pointes. Accordingly, an ECG should be part of the initial evaluation after ingestion of methadone or any unknown opioid. Neither drug is detected on routine urine opiate screens, although some centers have added a separate urine methadone screen. Levels of both drugs can be measured, although this is rarely done clinically and is seldom helpful in the acute setting. An exception may be in the cases involving concerns about neglect or abuse, at which point urine for gas chromatography/mass spectroscopy, the legal gold standard, should be sent to confirm and document the presence of the drug.
Treatment.
Patients with significant respiratory depression or CNS depression should be treated with the opioid antidote naloxone (see Table 77.7 ). In pediatric patients who are not chronically taking opioids, the full reversal dose of 1-2 mg should be used. In contrast, opioid-dependent patients should be treated with smaller initial doses (0.04-0.4 mg), which can then be repeated as needed to achieve the desired clinical response, avoiding abrupt induction of withdrawal. Because the half-life of methadone and buprenorphine is much longer than that of naloxone, patients can require multiple doses of naloxone. These patients may benefit from a continuous infusion of naloxone, typically started at two thirds of the reversal dose per hour and titrated to maintain an adequate respiratory rate and level of consciousness. Patients who have ingested methadone should be placed on a cardiac monitor and have serial ECGs to monitor for the development of a prolonged QTc interval. If a patient does develop a prolonged QTc, management includes close cardiac monitoring, repletion of electrolytes (potassium, calcium, and magnesium), and having a defibrillator readily available should the patient develop torsades de pointes.
Given the potential for clinically significant and prolonged toxicity, any toddler who has ingested methadone, even if asymptomatic, should be admitted to the hospital for at least 24 hr of monitoring. Some experts advocate a similar approach to management of buprenorphine ingestions, even in the asymptomatic patient. All such cases should be discussed with a PCC or medical toxicologist before determining disposition.
Cardiovascular Medications
β-Adrenergic Receptor Blockers.
β-Blockers competitively inhibit the action of catecholamines at the β-adrenergic receptor. Therapeutically, β-blockers are used for a variety of conditions, including hypertension, coronary artery disease, tachydysrhythmias, anxiety disorders, migraines, essential tremor, and hyperthyroidism. Because of its lipophilicity and blockade of fast sodium channels, propranolol is considered to be the most toxic member of the β-blocker class. Overdoses of water-soluble β-blockers (e.g., atenolol) are associated with milder symptoms.
Pathophysiology.
In overdose, β-blockers decrease chronotropy and inotropy in addition to slowing conduction through atrioventricular nodal tissue. Clinically, these effects are manifested as bradycardia, hypotension, and heart block. Patients with reactive airways disease can experience bronchospasm as a result of blockade of β2 -mediated bronchodilation. β2 -Blockers interfere with glycogenolysis and gluconeogenesis, which can sometimes lead to hypoglycemia, especially in patients with poor glycogen stores (e.g., toddlers).
Clinical and Laboratory Manifestations.
Toxicity typically develops within 6 hr of ingestion, although it may be delayed after ingestion of sotalol or slow-release (SR) preparations. The most common features of severe poisoning are bradycardia and hypotension. Lipophilic agents, including propranolol, can enter the CNS and cause altered mental status, coma, and seizures. Overdose of β-blockers with membrane-stabilizing properties (e.g., propranolol) can cause QRS interval widening and ventricular dysrhythmias.
Evaluation after β-blocker overdose should include an ECG, frequent reassessments of hemodynamic status, and blood glucose. Serum levels of β-blockers are not readily available for routine clinical use and are not useful in management of the poisoned patient.
Treatment.
In addition to supportive care and GI decontamination as indicated, glucagon is theoretically the preferred antidote of choice for β-blocker toxicity (see Table 77.7 ). Glucagon stimulates adenyl cyclase and increases levels of cyclic adenosine monophosphate (cAMP) independent of the β-receptor. Glucagon is typically given as a bolus and, if this is effective, followed by a continuous infusion. In practice, glucagon is often only marginally effective, limited by its proemetic effects, especially at the high doses typically required. Other potentially useful interventions include calcium, vasopressors, and high-dose insulin. Seizures are managed with benzodiazepines, and QRS widening should be treated with sodium bicarbonate. Children who ingest 1 or 2 water-soluble β-blockers are unlikely to develop toxicity and can typically be discharged to home if they remain asymptomatic over a 6 hr observation period. Children who ingest SR products, highly lipid-soluble agents, and sotalol can require longer periods of observation before safe discharge. Any symptomatic child should be admitted for ongoing monitoring and directed therapy.
Calcium Channel Blockers.
Calcium channel blockers (CCBs) are used for a variety of therapeutic indications and have the potential to cause severe toxicity, even after exploratory ingestions. Specific agents include verapamil, diltiazem, and the dihydropyridines (e.g., amlodipine, nifedipine). Of these, diltiazem and verapamil are the most dangerous in overdose because of their higher lipophilicity and direct cardiac suppressant effects.
Pathophysiology.
CCBs antagonize L -type calcium channels, inhibiting calcium influx into myocardial and vascular smooth muscle cells. Verapamil works primarily by slowing inotropy and chronotropy and has no effect on systemic vascular resistance (SVR). Diltiazem has effects both on the heart and the peripheral vasculature. The dihydropyridines exclusively diminish SVR. Verapamil and diltiazem can significantly diminish myocardial contractility and conduction, with diltiazem also lowering SVR. By contrast, dihydropyridines will decrease the SVR, leading to vasodilation and reflex tachycardia, although this receptor selectivity may be lost after a large overdose. Because the same L -type calcium channels blocked by CCBs are also on the pancreatic islet cells, any patient significantly poisoned with a CCB usually is hyperglycemic.
Clinical and Laboratory Manifestations.
The onset of symptoms typically is soon after ingestion, although it may be delayed with ingestions of SR products. Overdoses of CCBs lead to hypotension, accompanied by bradycardia, normal heart rate, or even tachycardia, depending on the agent. A common feature of CCB overdose is the patient exhibiting profound hypotension with preserved consciousness.
Initial evaluation should include an ECG, continuous and careful hemodynamic monitoring, and rapid measurement of serum glucose levels. Both the absolute degree of hyperglycemia and the percentage increase in serum glucose have been correlated with the severity of CCB toxicity in adults. The development of hyperglycemia can even precede the development of hemodynamic instability. Blood levels of CCBs are not readily available and are not useful in guiding therapy.
Treatment.
Once initial supportive care has been instituted, GI decontamination should begin with activated charcoal as appropriate. WBI may be beneficial in a stable patient after ingestion of an SR product. Calcium channel blockade in the smooth muscles of the GI tract can lead to greatly diminished motility; thus any form of GI decontamination should be undertaken with careful attention to serial abdominal tests.
Calcium salts, administered through a peripheral IV line as calcium gluconate or a central line as calcium chloride, help to overcome blocked calcium channels. High-dose insulin euglycemia therapy is considered the antidote of choice for CCB toxicity. An initial bolus of 1 unit/kg of regular insulin is followed by an infusion at 0.5-1 unit/kg/hr (see Table 77.7 ). The main mechanism of high-dose insulin euglycemia is to improve the metabolic efficiency of a poisoned heart that is in need of carbohydrates for energy (instead of the usual free fatty acids), but has minimal circulating insulin. Blood glucose levels should be closely monitored, and supplemental glucose may be given to maintain euglycemia, although this is rarely necessary in the severely poisoned patient.
Additional therapies include judicious IV fluid boluses and vasopressors (often in very high doses). Cardiac pacing is rarely of value. Lipid emulsion therapy (discussed earlier) is a potentially lifesaving intervention, especially for patients poisoned with the more lipid-soluble CCBs, verapamil and diltiazem. In extreme cases an intraaortic balloon pump or extracorporeal membrane oxygenation (ECMO) are potential rescue devices. Given the potential for profound and sometimes delayed toxicity in toddlers after ingestion of 1 or 2 CCB tablets, hospital admission and 12-24 hr of monitoring for all of these patients is strongly recommended.
Clonidine.
Although originally intended for use as an antihypertensive, the number of clonidine prescriptions in the pediatric population has greatly increased because of its reported efficacy in the management of attention-deficit/hyperactivity disorder (ADHD), tic disorders, and other behavioral disorders. With this increased use has come a significant rise in pediatric ingestions and therapeutic misadventures. Clonidine is available in pill and transdermal patch forms.
Pathophysiology.
Clonidine, along with the closely related agent guanfacine , is a centrally acting α2 -adrenergic receptor agonist with a very narrow therapeutic index. Agonism at central α2 receptors decreases sympathetic outflow, producing lethargy, bradycardia, hypotension, and apnea. Toxicity can develop after ingestion of only 1 pill or after sucking on or swallowing a discarded transdermal patch. Even a “used” transdermal patch might contain as much as one-third to one-half the original amount of drug.
Clinical and Laboratory Manifestations.
The most common clinical manifestations of clonidine toxicity are lethargy, miosis, and bradycardia. Hypotension, respiratory depression, and apnea may be seen in severe cases. Very early after ingestion, patients may be hypertensive in the setting of agonism at peripheral α-receptors and resulting vasoconstriction. Symptoms develop relatively soon after ingestion and typically resolve within 24 hr. Serum clonidine concentrations are not readily available and are of no clinical value in the acute setting. Although signs of clinical toxicity are common after clonidine overdose, death from clonidine alone is extremely unusual.
Treatment.
Given the potential for significant toxicity, most young children warrant referral to a healthcare facility for evaluation after unintentional ingestions of clonidine. Gastric decontamination is usually of minimal value because of the small quantities ingested and the rapid onset of serious symptoms. Aggressive supportive care is imperative and is the cornerstone of management. Naloxone, often in high doses, has shown variable efficacy in treating clonidine toxicity. Other potentially useful therapies include atropine, IV fluid boluses, and vasopressors. Symptomatic children should be admitted to the hospital for close cardiovascular and neurologic monitoring. Also, in a patient receiving chronic clonidine or guanfacine therapy, rapid discontinuation of the drug, or even missing 1 or 2 doses, could lead to potentially dangerous elevations in blood pressure.
Digoxin.
Digoxin is a cardiac glycoside extracted from the leaves of Digitalis lanata . Other natural sources of cardiac glycosides include Digitalis purpura (foxglove), Nerium oleander (oleander), Convallaria majalis (lily of the valley), Siberian ginseng, and the Bufo marinus toad. Therapeutically, digoxin is used in the management of heart failure and some supraventricular tachydysrhythmias. Acute overdose can occur in the setting of dosing errors (especially in younger children), unintentional or intentional medication ingestion, or exposure to plant material containing digitalis glycosides. Regarding exposure to such plants, toxicity is unusual unless the poison is concentrated in the form of a tea. Chronic toxicity can result from alteration of the digoxin dose, alteration in digoxin clearance as a result of renal impairment, or drug interactions.
Pathophysiology.
Digoxin blocks the sodium-potassium adenosine triphosphatase (Na+ ,K+ -ATPase) pump, leading to intracellular loss of K+ and gain of Na+ and calcium (Ca2+ ). This resulting rise in Ca2+ available to the contractile myocardium improves inotropy. An increase in myocardial automaticity leads to subsequent atrial, nodal, and ventricular ectopy. Digoxin also affects nodal conduction, leading to a prolonged refractory period, decreased sinus node firing, and slowed conduction through the atrioventricular node. Impaired Na+ /K+ exchange results in dangerously high levels of serum K+ . Overall, digoxin overdose manifests as a combination of slowed or blocked conduction and increased ectopy.
Clinical and Laboratory Manifestations.
Nausea and vomiting are common initial symptoms of acute digoxin toxicity, manifesting within 6 hr of overdose. Cardiovascular manifestations include bradycardia, heart block, and a wide variety of dysrhythmias. CNS manifestations consist of lethargy, confusion, and weakness. Chronic toxicity is more insidious and may also manifest as altered mental status and visual disturbances (rare).
Initial assessment should include an ECG, serum digoxin level, serum potassium, and kidney function tests. The serum digoxin level should be assessed at least 6 hr after ingestion and carefully interpreted in the setting of clinical symptoms, because the digoxin level alone does not entirely reflect the severity of intoxication. In acute ingestions, serum potassium is an independent marker of morbidity and mortality, with levels >5.5 mEq/L predicting poor outcomes. In chronic toxicity, serum K+ concentration is less useful as a prognostic marker and may be altered from concomitant use of diuretics.
Digoxin has a very narrow therapeutic index. Therapeutic plasma digoxin concentrations are 0.5-2.0 ng/mL; a level >2 ng/mL is considered toxic and >6 ng/mL is considered potentially fatal (in chronic poisonings). As with all serum levels of intoxicants, one must be careful to interpret the number in the context of the scenario of the poisoning and the status of the patient. An acutely poisoned patient may have a very high serum level and minimal to no symptoms, whereas a patient with a chronic or acute on chronic poisoning will usually be sicker with a lower serum level.
Numerous drug interactions affect plasma digoxin concentrations. Medications known to increase serum digoxin concentrations include the macrolides, erythromycin and clarithromycin, spironolactone, verapamil, amiodarone, and itraconazole.
Treatment.
Initial treatment includes good general supportive care and gastric decontamination with activated charcoal if the ingestion was recent. An antidote for digoxin, digoxin-specific antibody fragments (Fab: Digibind or DigiFab) is available (see Table 77.7 ). Fab fragments bind free digoxin in both the intravascular and the interstitial spaces to form a pharmacologically inactive complex that is subsequently eliminated renally. Indications for Fab fragments include life-threatening dysrhythmias, K+ value >5-5.5 mEq/L, serum digoxin level >15 ng/mL at any time or >10 ng/mL 6 hr after ingestion, clinically significant hypotension or other CV instability, altered mental state, and renal failure. Atropine is potentially useful in managing symptomatic bradycardia. Although dogma states that patients on digoxin with severe hyperkalemia and QRS widening on the ECG should not receive calcium salts, this has not been supported in the literature. Once stabilized, consultation with a cardiologist is recommended in the management of patients receiving chronic digoxin therapy, because administration of Fab fragments can lead to recurrence of the patient's underlying dysrhythmias or dysfunction.
Iron.
Historically, iron was a common cause of childhood poisoning deaths. However, preventive measures such as childproof packaging have significantly decreased the rates of serious iron toxicity in young children. Iron-containing products remain widely available, with the most potentially toxic being adult iron preparations and prenatal vitamins. The severity of an exposure is related to the amount of elemental iron ingested. Ferrous sulfate contains 20% elemental iron, ferrous gluconate 12%, and ferrous fumarate 33%. Multivitamin preparations and children's vitamins rarely contain enough elemental iron to cause significant toxicity. Furthermore, nonionic forms of iron, carbonyl iron and iron polysaccharide also do not cause significant toxicity.
Pathophysiology.
Iron is directly corrosive to the GI mucosa, leading to hematemesis, melena, ulceration, infarction, and potential perforation. Early iron-induced hypotension is caused by massive volume losses, increased permeability of capillary membranes, and vasodilation mediated by free iron. Iron accumulates in tissues, including the Kupffer cells of the liver and myocardial cells, leading to hepatotoxicity, coagulopathy, and cardiac dysfunction. Metabolic acidosis develops in the setting of hypotension, hypovolemia, and iron's direct interference with oxidative phosphorylation and the Krebs cycle. Pediatric patients who ingest >40 mg/kg of elemental iron should be referred to medical care for evaluation, although moderate to severe toxicity is typically seen with ingestions >60 mg/kg.
Clinical and Laboratory Manifestations.
Iron toxicity is described in 5 often-overlapping stages. The 1st stage , 30 min to 6 hr after ingestion, consists of profuse vomiting and diarrhea (often bloody), abdominal pain, and significant volume losses leading to potential hypovolemic shock. Patients who do not develop GI symptoms within 6 hr of ingestion are unlikely to develop serious toxicity. The 2nd stage , 6-24 hr after ingestion, is often referred to as the “quiescent phase” since the GI symptoms typically have resolved. However, careful clinical examination can reveal subtle signs of hypoperfusion, including tachycardia, pallor, and fatigue. During the 3rd stage , 12-36 hr after ingestion, patients develop multisystem organ failure, shock, hepatic and cardiac dysfunction, acute lung injury, and profound metabolic acidosis. Death usually occurs during the 3rd stage. The 4th stage (hepatic) results in fulminant liver failure and coagulopathy about 2-5 days after ingestion. The 5th stage , 4-6 wk after ingestion, is marked by formation of strictures and signs of GI obstruction.
Symptomatic patients and patients with a large exposure by history should have serum iron levels drawn 4-6 hr after ingestion. Serum iron concentrations of <500 µg/dL 4-8 hr after ingestion suggest a low risk of significant toxicity, whereas concentrations of >500 µg/dL indicate that significant toxicity is likely. Additional laboratory evaluation in the ill patient should include arterial or venous blood gas, CBC, serum glucose level, liver transaminases, and coagulation parameters. Careful attention should be paid to the patient's hemodynamic status. An abdominal radiograph might reveal the presence of iron tablets, although not all formulations of iron are radiopaque.
Treatment.
Close clinical monitoring, combined with aggressive supportive and symptomatic care, is essential to the management of iron poisoning. Activated charcoal does not adsorb iron, and WBI remains the decontamination strategy of choice. Deferoxamine , a specific chelator of iron, is the antidote for moderate to severe iron intoxication (see Table 77.7 ). Indications for deferoxamine treatment include a serum iron concentration >500 µg/dL or moderate to severe symptoms of toxicity (e.g., acidosis), regardless of serum iron concentration. Deferoxamine is preferably given by continuous IV infusion at 15 mg/kg/hr. Hypotension is a common side effect of deferoxamine infusion and is managed by slowing the rate of the infusion and administering fluids and vasopressors as needed. Prolonged deferoxamine infusion (>24 hr) has been associated with pulmonary toxicity (acute respiratory distress syndrome, ARDS) and Yersinia sepsis. The deferoxamine-iron complex can color the urine reddish (“vin rosé”), although the degree of this coloration should not guide therapy. Deferoxamine is typically continued until clinical symptoms and acidosis resolve. Consultation with a PCC or medical toxicologist can yield guidelines for discontinuing deferoxamine.
Oral Hypoglycemics
Oral medications used in the management of type 2 diabetes include sulfonylureas, biguanides (e.g., metformin), thiazolidinediones, and meglitinides. Of these, only the sulfonylureas and meglitinides have the potential to cause profound hypoglycemia in both diabetic and nondiabetic patients. These classes of medications are widely prescribed and thus readily available for both unintentional and intentional exposures. In toddlers, ingestion of a single sulfonylurea tablet can lead to significant toxicity.
Pathophysiology.
Sulfonylureas work primarily by enhancing endogenous insulin secretion. In binding to the sulfonylurea receptor, these drugs induce closure of K+ channels, leading to membrane depolarization, opening of Ca2+ channels, and stimulation of Ca2+ -mediated insulin release. Even in therapeutic use, the duration of hypoglycemic action can last up to 24 hr.
Clinical and Laboratory Manifestations.
Hypoglycemia and symptoms associated with hypoglycemia are the primary clinical manifestations of sulfonylurea toxicity. These signs and symptoms can include diaphoresis, tachycardia, lethargy, irritability, coma, seizures, and even focal neurologic findings. As with other hyperinsulinemic states, sulfonylurea overdoses are associated with a nonketotic hypoglycemia. In the majority of patients, hypoglycemia develops within 6 hr of ingestion but can be delayed up to 16-18 hr after ingestion. Toddlers are particularly susceptible to hypoglycemia during an overnight fast.
Treatment.
Patients with symptomatic hypoglycemia should be promptly treated with dextrose. In patients with mild symptoms, oral dextrose may be sufficient. However, patients with severe symptoms or profound hypoglycemia should be treated with a bolus of IV dextrose. Continuous dextrose infusions and repeated IV dextrose boluses should be avoided if possible, because this can stimulate further insulin release and lead to recurrent and prolonged hypoglycemia. Instead, the preferred antidote for persistent (i.e., requiring ≥2 doses of IV dextrose) sulfonylurea toxicity is octreotide (see Table 77.7 ). Octreotide is a somatostatin analog that inhibits insulin release. Octreotide is given intravenously (IV) or subcutaneously (SC), typically in doses of 1-2 µg/kg (50-100 µg in teens or adults) every 6-8 hr.
Given the potential for significant hypoglycemia, toddlers with witnessed or suspected sulfonylurea ingestions should be admitted to the hospital for serial glucose measurements for at least 12 hr, including an overnight fast. Patients of any age who develop hypoglycemia are also candidates for admission given the prolonged duration of hypoglycemic activity. Prophylactic IV dextrose infusions are not recommended because they can mask the symptoms of toxicity and stimulate further insulin secretion. Patients who require IV dextrose and/or octreotide should be monitored until they can demonstrate euglycemia for at least 8 hr off all therapy.
With the increasing numbers of adolescents with type 2 diabetes, pediatricians should be familiar with the toxic effects of metformin as well. Although metformin does not cause hypoglycemia, its association with lactic acidosis is well documented (metformin-associated lactic acidosis, MALA). This state typically arises after a large overdose in which the agent interferes with the liver's ability to clear lactic acid. Dangerously high serum lactate levels can result, leading to hemodynamic instability. Hemodialysis is usually the best option for patients with severe MALA.
Psychiatric Medications: Antidepressants
Selective serotonin reuptake inhibitors (SSRIs; e.g., fluoxetine, sertraline, paroxetine, citalopram) are the most commonly prescribed class of antidepressants. This trend largely results from their wide therapeutic index and more favorable side effect profile compared with older agents such as tricyclic antidepressants (TCAs; amitriptyline, clomipramine, desipramine, doxepin, nortriptyline, imipramine) and monoamine oxidase inhibitors (MAOIs). Other agents include the serotonin-norepinephrine reuptake inhibitors (SNRIs; e.g., venlafaxine) and atypical antidepressants (e.g., bupropion).
Tricyclic Antidepressants.
Although now prescribed less often for depression, TCAs remain in use for a variety of other conditions, including chronic pain syndromes, enuresis, ADHD, and obsessive-compulsive disorder. TCAs can cause significant toxicity in children, even with ingestion of 1 or 2 pills (10-20 mg/kg).
Pathophysiology.
TCAs achieve their desired antidepressant effects primarily through blockade of norepinephrine and serotonin reuptake. TCAs have complex interactions with other receptor types. Antagonism at muscarinic acetylcholine receptors leads to clinical features of the anticholinergic toxidrome. Antagonism at peripheral α-receptors leads to hypotension and syncope. Key to the toxicity of TCAs is their ability to block fast sodium channels, leading to impaired cardiac conduction and arrhythmias.
Clinical and Laboratory Manifestations.
Cardiovascular and CNS symptoms dominate the clinical presentation of TCA toxicity. Symptoms typically develop within 1-2 hr of ingestion, and serious toxicity usually manifests within 6 hr of ingestion. Patients can have an extremely rapid progression from mild symptoms to life-threatening dysrhythmias. Patients often develop features of the anticholinergic toxidrome , including delirium, mydriasis, dry mucous membranes, tachycardia, hyperthermia, urinary retention, and slow GI motility. CNS toxicity can include lethargy, coma, myoclonic jerks, and seizures. Sinus tachycardia is the most common cardiovascular manifestation of toxicity; however, patients can also develop widening of the QRS complex, premature ventricular contractions, and ventricular dysrhythmias. Refractory hypotension is a poor prognostic indicator and is the most common cause of death in TCA overdose.
An ECG is a readily available bedside test that can help determine the diagnosis and prognosis of the TCA-poisoned patient (Fig. 77.2 ; see Table 77.6 ). A QRS duration >100 msec identifies patients who are at risk for seizures and cardiac arrhythmias. An R wave in lead aVR of ≥3 mm is also an independent predictor of toxicity. Both ECG parameters are superior to measured serum TCA concentrations for identifying patients at risk for serious toxicity, and obtaining levels is rarely helpful in management of the acutely ill patient.
Treatment.
Initial attention should be directed to supporting vital functions, including airway and ventilation as needed. Gastric decontamination can be accomplished with activated charcoal in appropriate patients. Treating clinicians should obtain an ECG as soon as possible and follow serial ECGs to monitor for progression of toxicity. The 4 primary effects described next are seen at the bedside.
1 Altered Mental State.
TCA-poisoned patients can become deeply comatose relatively quickly, so careful and prompt attention to the airway and placement of an endotracheal tube is of paramount importance. The airway should be secured before any GI decontamination efforts.
2 Widened QRS on ECG.
TCAs, as well as with other agents (e.g., diphenhydramine, cocaine), will block the fast Na+ channels on the myocardial cells, slowing the upstroke of the QRS complex. Because the effect on Na+ channels is greatest within the 1st 6 hr, frequent ECGs (i.e., every 20-30 min) during this period are important. As the QRS approaches 160 msec, the risk of the patient developing monomorphic ventricular tachycardia rises to 30%. Sodium, usually in the form of sodium bicarbonate, is the antidote of choice. Indications for sodium bicarbonate include a QRS duration ≥110 msec, ventricular dysrhythmias, and hypotension . Multiple bolus doses of sodium bicarbonate, 1-2 mEq/kg each, may be needed to narrow the QRS to <110 msec. Some prefer then to place the patient on an infusion of sodium bicarbonate, but this may not be necessary if the QRS is carefully monitored after the initial doses and repeat bolus dosing is provided as needed during the 1st 6-12 hr. Hypertonic (3%) saline and/or lipid emulsion therapy may be beneficial in refractory cases.
3 Hypotension.
A direct-acting vasopressor such as norepinephrine or epinephrine is the agent of choice. Boluses of IV crystalloid fluids should be used with caution to prevent fluid overload.
4 Seizures.
Likely a result of the anticholinergic effects of TCAs, seizures are relatively common, typically brief, and should be treated with agents targeting the GABA-receptor complex in the brain. Benzodiazepines are the agent of choice.
Asymptomatic children should receive appropriate decontamination and have continuous cardiac monitoring and serial ECGs for at least 6 hr after exposure. If any manifestations of toxicity develop, the child should be admitted to a monitored setting. Children who remain completely asymptomatic with normal serial ECGs may be candidates for discharge after that monitoring period.
Selective Serotonin Reuptake Inhibitors.
In overdose, SSRIs are considerably less toxic than TCAs. SSRIs are unlikely to cause significant toxicity in exploratory ingestions. Some data suggest that initiating SSRI therapy is associated with an increased risk of suicidal ideation and behavior (see Chapter 40 ).
Pathophysiology.
SSRIs selectively block the reuptake of serotonin in the CNS. In contrast to TCAs and atypical antidepressants, SSRIs do not directly interact with other receptor types.
Clinical and Laboratory Manifestations.
In overdose, the principal manifestations of toxicity are sedation and tachycardia. Cardiac conduction abnormalities (primarily QTc prolongation) and seizures have been described in significant overdoses, especially after ingestions of citalopram. An ECG should be part of the initial assessment after SSRI ingestion. Serum creatine kinase (CK) levels are almost always elevated in a patient with clinically significant serotonin syndrome . Although seen more often after therapeutic use or overdose of several serotonergic agents in combination, the serotonin syndrome has also been described in ingestion of SSRIs alone (Table 77.11 ). Clinically, serotonin syndrome describes a spectrum of altered mental status, autonomic instability, fever, and neuromuscular hyperactivity (hyperreflexia, tremors, clonus in lower extremities > upper extremities). One or all of these signs may be present to varying degrees.
Table 77.11
From Boyer EW, Shannon M: The serotonin syndrome, N Engl J Med 352:1112–1120, 2005.
Treatment.
Initial management includes a careful assessment for signs and symptoms of serotonin syndrome and an ECG. Most patients simply require supportive care and observation until their mental status improves and tachycardia, if present, resolves. Management of serotonin syndrome is directed by the severity of symptoms; possible therapeutic interventions include benzodiazepines in mild cases and intubation, sedation, and paralysis in patients with severe manifestations (e.g., significant hyperthermia). Because agonism at the 5-HT2A serotonin receptor is thought to be primarily responsible for the development of serotonin syndrome, use of the 5-HT2A receptor antagonist cyproheptadine may also be helpful. Cyproheptadine is only available in an oral form.
Atypical Antidepressants.
The atypical antidepressant class includes agents such as venlafaxine and duloxetine (SNRIs), bupropion (dopamine, norepinephrine, and some serotonin reuptake blockade), and trazodone (serotonin reuptake blockade and peripheral α-receptor antagonism). The variable receptor affinities of these agents lead to some distinctions in their clinical manifestations and management.
Clinical and Laboratory Manifestations.
In overdose, venlafaxine and other SNRIs have been associated with cardiac conduction defects, including QRS and QTc prolongation, and seizures. Bupropion warrants special consideration because it is one of the most common etiologies of toxicant-induced seizures in the United States. After ingestion of SR or extended-release (ER) preparations, seizures can occur as late as 18-20 hr after ingestion. In addition, bupropion can cause tachycardia, agitation, and QRS and QTc prolongation. These cardiac effects are thought to result from a reduction in cardiac intracellular coupling caused by inhibition at gap junctions in the heart. Mortality results from not only status epilepticus but also the cardiac conduction disturbances causing ventricular tachycardia. Bupropion is of growing concern with the rising popularity of the drug, especially in the ER formulation. In addition to sedation and signs of serotonin excess, trazodone overdose may be associated with hypotension from blockade of peripheral α-receptors.
Treatment.
Management is directed to clinical signs and symptoms. QRS and QTc interval prolongation after bupropion poisoning is typically resistant to the standard treatments of sodium bicarbonate and magnesium. Seizures are often brief and self-limited but can be treated with benzodiazepines if necessary. A patient poisoned with bupropion who shows unstable hemodynamics with prolonged ECG intervals or persistent seizure activity should receive Intralipid emulsion therapy. Because of the potential for delayed seizures, asymptomatic patients who have ingested an SR preparation of bupropion should be admitted to a monitored setting for at least 20-24 hr. Trazodone-associated hypotension typically responds to fluids, though it can require vasopressors in extreme cases.
Monoamine Oxidase Inhibitors.
Although now rarely used therapeutically, MAOIs remain important agents given their potential for serious and delayed toxicity. Ingestions of only 1 or 2 pills (6 mg/kg) are associated with toxicity in children. Clinical manifestations initially include hypertension, hyperthermia, tachycardia, muscle rigidity, and seizures, followed up to 24 hr later by hemodynamic instability and CV collapse. Any child who ingests a MAOI should be admitted to a monitored setting for at least 24 hr, regardless of symptoms. Management includes blood pressure control, cooling and benzodiazepines for hyperthermia, serial monitoring of CK and renal function, and fluid and vasopressor therapy for hemodynamic instability.
Psychiatric Medications: Antipsychotics
Clinicians are increasingly prescribing antipsychotic medications in the pediatric population. Antipsychotics are usually classified as either typical or atypical. In general, typical agents are associated with more side effects and toxicity than the atypical agents.
Pathophysiology.
Typical or “traditional” antipsychotics (haloperidol, droperidol, thioridazine, chlorpromazine, fluphenazine) are characterized by their antagonism at D2 dopamine receptors. In therapeutic use, these agents are associated with extrapyramidal symptoms, tardive dyskinesia, and development of the neuroleptic malignant syndrome (NMS) . The atypical agents (aripiprazole, clozapine, quetiapine, risperidone, ziprasidone) were developed with relatively less dopamine (D2 -receptor) antagonism in the nigrostriatum in an effort to avoid these side effects and improve their efficacy in managing the “negative” symptoms of schizophrenia. Instead, these agents have complex and varied interactions with multiple receptor types, including α-receptors, serotonin receptors, muscarinic acetylcholine receptors, and histamine receptors.
Clinical and Laboratory Manifestations.
Typical antipsychotic toxicity usually includes sedation, tachycardia, and QTc prolongation. Patients can present with acute dystonia, akathisia, and NMS, although these are seen less frequently in acute overdoses than in therapeutic use. The phenothiazines (e.g., thioridazine) can cause widening of the QRS interval from blockade of fast sodium channels. Clinically, NMS can be difficult to distinguish from serotonin syndrome.
Although the presentation of atypical antipsychotic toxicity can vary based on the receptor affinities of the specific agent, sedation, tachycardia, and QTc prolongation are common. Peripheral α-receptor blockade (e.g., with quetiapine) is associated with hypotension. In therapeutic use, clozapine is associated with agranulocytosis.
Diagnostic testing should include an ECG. Patients with hyperthermia or muscle rigidity should have a serum CK level sent to monitor for possible rhabdomyolysis. Antipsychotic levels are not readily available and are not helpful in managing acute poisoning.
Management.
Initial management involves assessing and supporting vital functions. In some patients, CNS depression may be so profound as to require intubation for airway control. Acute dystonia is treated with diphenhydramine and benztropine. Management of NMS includes conscientious supportive care, IV fluids, cooling, benzodiazepines, and bromocriptine or dantrolene in severe cases. QTc prolongation is managed with repletion of electrolytes (especially calcium, magnesium, and potassium), continuous cardiac monitoring, prevention of bradycardia (overdrive pacing, isoproterenol, atropine), and defibrillation if the patient develops torsades de pointes. Seizures typically are well controlled with benzodiazepines. Hypotension usually responds to boluses of IV fluids, although vasopressor therapy is necessary in some patients.
Household Products
Caustics
Caustics include acids and alkalis as well as a few common oxidizing agents (see Chapter 353 ). Strong acids and alkalis can produce severe injury even in small-volume ingestions.
Pathophysiology.
Alkalis produce a liquefaction necrosis, allowing further tissue penetration of the toxin and setting the stage for possible perforation. Acids produce a coagulative necrosis, which limits further tissue penetration, although perforation can still occur. The severity of the corrosive injury depends on the pH and concentration of the product as well as the length of contact time with the product. Agents with a pH of <2 or >12 are most likely to produce significant injury.
Clinical Manifestations.
Ingestion of caustic materials can produce injury to the oral mucosa, posterior pharynx, vocal cords, esophagus, and stomach. Patients can have significant esophageal injury even in the absence of visible oral burns. Symptoms include pain, drooling, vomiting, abdominal pain, and difficulty swallowing or refusal to swallow. Laryngeal injury can manifest as stridor and respiratory distress, necessitating intubation. In the most severe cases, patients can present in shock after perforation of a hollow viscus. Circumferential burns of the esophagus are likely to cause strictures when they heal, which can require repeated dilation or surgical correction and long-term follow-up for neoplastic changes in adulthood. Caustics on the skin or in the eye can cause significant tissue damage.
Treatment.
Initial treatment of caustic exposures includes thorough removal of the product from the skin or eye by flushing with water. Emesis and lavage are contraindicated . Activated charcoal should not be used because it does not bind these agents and can predispose the patient to vomiting and subsequent aspiration. Stridor or other signs of respiratory distress should alert the provider to the need for a thorough evaluation of the airway for potential intubation or surgical airway management. Endoscopy can be performed within 12-24 hr of ingestion for prognostic and diagnostic purposes in symptomatic patients or those with suspected injury on the basis of history and known characteristics of the ingested product. Endoscopy's role is purely diagnostic. Whether the risks of the procedure are justified is debatable. Expectant management with a period of nothing by mouth (NPO) and proton pump inhibitor therapy is likely appropriate for the majority of patients without airway burns or signs of mediastinitis or peritonitis. Endoscopy is contraindicated in such patients, who instead require immediate surgical consultation. Corticosteroids or prophylactic antibiotics are not beneficial.
Pesticides
Cholinesterase-Inhibiting Insecticides.
The most commonly used insecticides in agriculture are organophosphates and carbamates ; both are inhibitors of cholinesterase enzymes: acetylcholinesterase (AChE), pseudocholinesterase, and erythrocyte AChE. Most pediatric poisonings occur as the result of unintentional exposure to insecticides in and around the home or farm. The chemical warfare weapons known as “nerve agents” are also organophosphate compounds with a similar mechanism of action but much greater potency.
Pathophysiology.
Organophosphates and carbamates produce toxicity by binding to and inhibiting AChE, preventing the degradation of acetylcholine (ACh) and resulting in its accumulation at nerve synapses. If left untreated, organophosphates form an irreversible bond to AChE, permanently inactivating the enzyme. This process, called aging , occurs over a variable time period depending on the characteristics of the specific organophosphate. A period of weeks to months is required to regenerate inactivated enzymes. In contrast, carbamates form a temporary bond to the enzymes, typically allowing reactivation of AChE within 24 hr.
Clinical and Laboratory Manifestations.
Clinical manifestations of organophosphate and carbamate toxicity relate to ACh accumulation at peripheral nicotinic and muscarinic synapses and in the CNS. Symptoms of carbamate toxicity are usually less severe than those seen with organophosphates. A commonly used mnemonic for the symptoms of cholinergic excess at muscarinic receptors is DUMBBELS : diarrhea/defecation, urination, miosis, bronchorrhea/bronchospasm, bradycardia, emesis, lacrimation, and salivation. Nicotinic signs and symptoms include muscle weakness, fasciculation, tremors, hypoventilation (diaphragm weakness), hypertension, tachycardia, and dysrhythmias. Severe manifestations include coma, seizures, shock, arrhythmias, and respiratory failure.
Diagnosis of poisoning is based primarily on history and physical exam findings. Red blood cell cholinesterase and pseudocholinesterase activity levels can be measured in the laboratory. These are only helpful when compared to the patient's known baseline. As such, these assessments are typically limited to farmworkers undergoing ongoing occupational surveillance.
Treatment.
Basic decontamination should be performed, including washing all exposed skin with soap and water and immediately removing all exposed clothing. Activated charcoal is unlikely to be of benefit because these are liquids that are rapidly absorbed. Basic supportive care should be provided, including fluid and electrolyte replacement, intubation, and ventilation if necessary. The use of succinylcholine for rapid sequence intubation should be avoided because the same cholinesterase enzymes that are poisoned metabolize this neuromuscular blocking agent, leading to prolonged paralysis.
Two antidotes are useful in treating cholinesterase inhibitor poisoning: atropine and pralidoxime (see Table 77.7 ). Atropine , which antagonizes the muscarinic ACh receptor, is useful for both organophosphate and carbamate intoxication. Often, large doses of atropine must be administered by intermittent bolus or continuous infusion to control symptoms. Atropine dosing is primarily targeted to drying the respiratory secretions. Pralidoxime breaks the bond between the organophosphate and the enzyme, reactivating AChE. Pralidoxime is only effective if it is used before the bond ages and becomes permanent. Pralidoxime is not necessary for carbamate poisonings because the bond between the insecticide and the enzyme degrades spontaneously.
Without treatment, symptoms of organophosphate poisoning can persist for weeks, requiring continuous supportive care. Even with treatment, some patients develop a delayed polyneuropathy and a range of chronic neuropsychiatric symptoms.
Pyrethrins and Pyrethroids.
Pyrethrins are derived from the chrysanthemum flower and along with pyrethroids, synthetic derivatives, are the most commonly used pesticides in the home. Although >1,000 pyrethrins and pyrethroids exist, <20 are available in the United States, with permethrin being the most common. Exposure to these compounds occurs by inhalation, dermal absorption, or ingestion. Ingestion is the predominant route and typically occurs by eating contaminated foods. Permethrin is also a prescribed medication for the treatment of scabies and lice.
Pathophysiology.
Pyrethrins and pyrethroids prolong the open state of the voltage-gated Na+ channel conduction, which is the main mechanism resulting in its pesticide activity. Pyrethrins have minimal toxicity in mammals because of rapid metabolism, higher affinity for the insect Na+ channel, and decreased activity at higher temperatures seen in warm-blooded animals. Since pyrethroids were specifically manufactured to be more stable in the environment, they have a higher likelihood of toxicity.
Clinical and Laboratory Manifestations.
Pyrethrin exposures can lead to allergic reactions ranging from dermatitis to urticaria to anaphylaxis. Acute exposure can result in headache, nausea, dizziness, tremors, ataxia, choreoathetosis, loss of consciousness, and seizures. The severity of the symptoms depends on the magnitude of the exposure. Reports of acute lung injury have also occurred after pyrethroid exposures, although this is likely from the other components of the insecticide, such as surfactants and solvents. Paresthesias limited to the cutaneous exposure area can also occur following a dermal exposure. Chronic exposures have not been shown to result in any clinical manifestations. Although one can test for urinary pyrethroid metabolites, this is only useful for monitoring occupational exposure and has no role for the acute exposure.
Treatment.
Initial treatment should focus on decontamination, which involves removing all clothing and irrigation of exposed areas. Allergic reactions are treated the same as for antihistamines and corticosteroids. Systemic toxicity should be treated with excellent supportive care, using benzodiazepines for tremors and seizures.
Hydrocarbons
Hydrocarbons include a wide array of chemical substances found in thousands of commercial products. Specific characteristics of each product determine whether exposure will produce systemic toxicity, local toxicity, both, or neither. Nevertheless, aspiration of even small amounts of certain hydrocarbons can lead to serious, potentially life-threatening toxicity.
Pathophysiology.
The most important manifestation of hydrocarbon toxicity is aspiration pneumonitis through inactivation of the type II pneumocytes and resulting in surfactant deficiency (see Chapter 425 ). Aspiration usually occurs during coughing and gagging at the time of ingestion or vomiting after the attempted ingestion of an aliphatic hydrocarbon. The propensity of a hydrocarbon to cause aspiration pneumonitis is inversely proportional to its viscosity, and directly proportional to its volatility. Compounds with low viscosity and high volatility, such as mineral spirits, naphtha, kerosene, gasoline, and lamp oil, spread rapidly across surfaces and cover large areas of the lungs when aspirated. Only small quantities (<1 mL) of such chemicals need be aspirated to produce significant injury. Pneumonitis does not result from dermal absorption of hydrocarbons or from ingestion in the absence of aspiration. Gasoline and kerosene are poorly absorbed, but they often cause considerable irritation of the GI mucosa as they pass through the intestines.
Certain hydrocarbons have unique toxicities and can cause symptoms after ingestion, inhalation, or dermal exposures. Several chlorinated solvents, most notably carbon tetrachloride, can produce hepatic toxicity. Methylene chloride , found in some paint removers, is metabolized to carbon monoxide. Benzene is known to cause cancer, most often acute myelogenous leukemia, after long-term exposure. Nitrobenzene, aniline, and related compounds can produce methemoglobinemia. A number of volatile hydrocarbons, including toluene, propellants, refrigerants, and volatile nitrites, are frequently abused by inhalation. Some of these substances, principally the halogenated hydrocarbons (which contain a chlorine, bromine, or fluorine), can sensitize the myocardium to the effects of endogenous catecholamines. This can result in dysrhythmias and “sudden sniffing death.” Chronic abuse of these agents can lead to cerebral atrophy, neuropsychologic changes, peripheral neuropathy, and kidney disease (see Chapter 140 ).
Clinical and Laboratory Manifestations.
Transient, mild CNS depression is common after hydrocarbon ingestion or inhalation. Aspiration is characterized by coughing, which usually is the 1st clinical finding. Chest radiographs may initially be normal, but they often show abnormalities within 6 hr of exposure in patients who have aspirated. Respiratory symptoms can remain mild or progress rapidly to acute respiratory distress syndrome (ARDS) and respiratory failure. Fever and leukocytosis are common accompanying signs in patients with pneumonitis and do not necessarily imply bacterial superinfection. Chest radiographs can remain abnormal long after the patient is clinically normal. Pneumatoceles can appear on the chest radiograph 2-3 wk after exposure.
After inhalational exposures to halogenated hydrocarbons, patients can present with ventricular dysrhythmias, often refractory to conventional management. Recurrent inhalation of the aromatic hydrocarbon toluene can lead to a type IV renal tubular acidosis.
Treatment.
Emesis and lavage are contraindicated given the risk of aspiration . Activated charcoal is not useful because it does not bind the common hydrocarbons and can also induce vomiting. If hydrocarbon-induced pneumonitis develops, respiratory treatment is supportive (see Chapter 425 ). Neither corticosteroids nor prophylactic antibiotics have shown any clear benefit. Standard mechanical ventilation, high-frequency ventilation, and extracorporeal membrane oxygenation (ECMO) have all been used to manage the respiratory failure and ARDS associated with severe hydrocarbon-induced pneumonitis.
Patients with dysrhythmias in the setting of halogenated hydrocarbon inhalation should be treated with β-blockers (usually esmolol) to block the effects of endogenous catecholamines on the sensitized myocardium.
Toxic Alcohols
Methanol is found in windshield washer fluids, deicers, paint removers, fuel additives, liquid fuel canisters, and industrial solvents. Ethylene glycol is found in antifreeze. Unintentional ingestion is the most common exposure in children, and small-volume ingestions of concentrated products can theoretically cause toxicity. The pathophysiology, acid-base derangements, and treatment of both chemicals are similar, although they differ in their primary end-organ toxicity. In both cases the metabolites of the parent compounds are responsible for the serious clinical effects that can follow exposure.
Isopropyl alcohol (rubbing alcohol), found in hand sanitizers, causes intoxication similar to that associated with ethanol but can also cause a hemorrhagic gastritis and myocardial depression in massive ingestions. Unlike ethylene glycol and methanol, isopropyl alcohol is metabolized to a ketone and does not cause a metabolic acidosis. Management is similar to that of ethanol ingestions (see Chapter 140 ) and is not further discussed here.
Methanol
Pathophysiology.
Methanol is oxidized in the liver by alcohol dehydrogenase to formaldehyde, which is further oxidized to formic acid by aldehyde dehydrogenase. Toxicity is caused primarily by formic acid, which inhibits mitochondrial respiration.
Clinical and Laboratory Manifestations.
Drowsiness, mild inebriation, nausea, and vomiting develop early after methanol ingestion. The onset of serious effects, including profound metabolic acidosis and visual disturbances, is often delayed up to 12-24 hr as the parent methanol undergoes metabolism to its toxic metabolites. This metabolism is further slowed if ethanol has also been ingested, since the liver will preferentially metabolize ethanol. Visual disturbances include blurred or cloudy vision, constricted visual fields, decreased acuity, and the “feeling of being in a snowstorm” and appear only after acidosis is well established. These visual defects may be reversible if treated early, but untreated can lead to permanent blindness. On examination, dilated pupils, retinal edema, and optic disc hyperemia may be noted. Initially, patients have an elevated osmolar gap, then develop an anion gap metabolic acidosis as the parent compound is metabolized to formic acid.
In young children, determining if a significant exposure has occurred is usually difficult based on history. Methanol blood levels are available at some laboratories and should be sent after a concerning exposure. If methanol blood levels are not readily available, estimation of an osmolar gap may be used as a surrogate marker, but a normal osmolar gap does not rule out ingestion of any alcohol. Serum osmolality is measured by the freezing-point depression method and compared with a calculated serum osmolarity.
Treatment.
Treatment is as discussed for ethylene glycol toxicity.
Ethylene Glycol
Pathophysiology.
Ethylene glycol is oxidized by alcohol dehydrogenase in the liver to glycolaldehyde, which is further converted to glycolic acid by aldehyde dehydrogenase. Glycolic acid is responsible for the metabolic acidosis and is further metabolized to glyoxylic and then to oxalic acid. Oxalic acid combines with serum and tissue calcium, forming calcium oxalate crystals that deposit throughout the body, especially in the renal parenchyma, leading to acute tubular necrosis.
Clinical and Laboratory Manifestations.
Early symptoms include nausea, vomiting, CNS depression, and inebriation. Delayed manifestations include an anion gap metabolic acidosis, hypocalcemia, and acute kidney injury. Even later, patients can develop cranial nerve palsies.
Both ethylene glycol and methanol can produce profound, life threatening metabolic acidosis and acidemia, with measured serum bicarbonates that may even be nondetectable. The onset of the acidosis is delayed up to 4-12 hr after ethylene glycol ingestion and may be delayed further with any concomitant ingestion of ethanol. Ethylene glycol blood concentrations are technically difficult to perform and are available only at some larger reference laboratories. In the absence of readily available ethylene glycol concentrations, calculation of the osmolar gap may be helpful as a surrogate marker.
Examination of the urine with a Wood lamp is neither sensitive nor specific for ethylene glycol ingestion. The earliest sign on a urinalysis of ethylene glycol poisoning is usually hematuria. Calcium oxalate crystals can be seen on urine microscopy but might not be evident early after exposure. Electrolytes (including calcium), acid-base status, kidney function, and ECG should be closely monitored in poisoned patients.
Treatment.
Because methanol and ethylene glycol are rapidly absorbed, gastric decontamination is generally not of value. The classic antidote for methanol and ethylene glycol poisoning was ethanol, a preferential substrate for alcohol dehydrogenase, thus preventing the metabolism of parent compounds to toxic metabolites. Fomepizole , a potent competitive inhibitor of alcohol dehydrogenase, has almost entirely replaced ethanol because of its ease of administration, lack of CNS and metabolic effects, and overall excellent patient tolerability profile (see Table 77.7 ). A serum concentration must be interpreted along with the time removed from exposure. A patient with a methanol level of 20 mg/dL 24 hr after exposure had a much larger dose than a patient with the same level only 1 hr after ingestion. Classic indications for fomepizole include ethylene glycol or methanol level >20 mg/dL (assuming no ethanol is present), history of potentially toxic ingestion (e.g., any intentional overdose), or history of ingestion with evidence of acidosis. There are few disadvantages to giving the initial dose of fomepizole to patients with a concerning history of ingestion or lab findings, and given the dosing schedule of fomepizole (every 12 hr), this strategy buys the clinician time to confirm or exclude the diagnosis before giving a 2nd dose. Adjunctive therapy includes folate (methanol toxicity), pyridoxine (ethylene glycol toxicity), and sodium bicarbonate infusion for both (if acidemic). If a child has had an unintentional exposure and the alcohol level cannot be obtained, a reasonable approach is to follow serum chemistries every 4 hr until 12 hr after the exposure. If the bicarbonate level on the chemistry panel does not fall in that period, a toxic exposure is unlikely (assuming no ethanol is present).
Hemodialysis effectively removes ethylene glycol, methanol, and their metabolites (except calcium oxalate) and corrects acid-base and electrolyte disturbances. Fomepizole should be given both before and immediately after dialysis. Indications for dialysis include a methanol level >50 mg/dL, acidosis, severe electrolyte disturbances, and renal failure. However, in the absence of acidosis and kidney failure, even massive ethylene glycol ingestions have been managed without dialysis. Methanol, however, because its elimination in the setting of alcohol dehydrogenase inhibition is prolonged, often warrants dialysis to remove the parent compound. Therapy (fomepizole and/or dialysis) should be continued until ethylene glycol and methanol levels are <20 mg/dL. While the visual effects from methanol poisoning are usually permanent, the kidney injury from ethylene glycol injury is not. Patients requiring hemodialysis after ethylene glycol poisoning will almost always recover complete renal function within 2-6 wk. Consultation with a PCC, medical toxicologist, and nephrologist may be helpful in managing toxic alcohol ingestions.
Plants
Exposure to plants, both inside the home and outside in backyards and fields, is one of the most common causes of unintentional poisoning in children. Fortunately, the majority of ingestions of plant parts (leaves, seeds, flowers) result in either no toxicity or mild, self-limiting effects. However, ingestion of certain plants can lead to serious toxicity (Table 77.12 ).
Table 77.12
AV, Atrioventricular; CNS, central nervous system; CV, cardiovascular; ECG, electrocardiogram; Fab, fragment, antigen binding; GI, gastrointestinal.
The potential toxicity of a particular plant is highly variable, depending on the part of the plant involved (flowers are generally less toxic than the root or seed), the time of year, growing conditions, and the route of exposure. Assessment of the potential severity after an exposure is also complicated by the difficulty in properly identifying the plant. Many plants are known by several common names, which can vary among communities. Poison control centers have access to professionals who can assist in properly identifying plants. They also are well versed in the common poisonous plants in their service area and the seasons when they are more abundant. For these reasons, consultation with the local PCC may be very helpful in the management of these ingestions.
For potentially toxic plant ingestions, consider decontamination with activated charcoal in patients who present within 1-2 hr of ingestion; otherwise, treatment is primarily supportive and based on symptoms. The most common manifestation of toxicity after plant ingestion is GI upset, which can be managed with antiemetics and fluid and electrolyte support. Table 77.12 outlines management strategies for a few specific toxicities.
Toxic Gases
Carbon Monoxide
Although many industrial and naturally occurring gases pose a health risk by inhalation, the most common gas involved in pediatric exposures is carbon monoxide. CO is a colorless, odorless gas produced during the combustion of any carbon-containing fuel; the less efficient the combustion, the greater the amount of CO produced. Wood-burning stoves, kerosene heaters, old furnaces, hot-water heaters, closed-space fires, and automobiles are a few of the potential sources of CO.
Pathophysiology.
CO binds to hemoglobin with an affinity >200 times that of oxygen, forming carboxyhemoglobin (HbCO). In doing so, CO displaces oxygen and creates a conformational change in hemoglobin that impairs the delivery of oxygen to the tissues, leading to tissue hypoxia. HbCO levels are not well correlated with clinical signs of toxicity, likely because CO interacts with multiple proteins in addition to hemoglobin. CO binds to cytochrome oxidase, disrupting cellular respiration. CO displaces nitric oxide (NO) from proteins, allowing NO to bind with free radicals to form the toxic metabolite peroxynitrite, leading to lipid peroxidation and cellular damage. NO is also a potent vasodilator, in part responsible for clinical symptoms such as headache, syncope, and hypotension.
Clinical and Laboratory Manifestations.
Early symptoms are nonspecific and include headache, malaise, nausea, and vomiting. These symptoms are often misdiagnosed as indicating flu or food poisoning. At higher exposure levels, patients can develop mental status changes, confusion, ataxia, syncope, tachycardia, and tachypnea. Severe poisoning is manifested by coma, seizures, myocardial ischemia, acidosis, cardiovascular collapse, and potentially death. Physical examination should focus on the cardiovascular and neurologic systems because these are the most detrimentally effected by CO. Emergency department evaluation should include arterial or venous blood gas analysis with HbCO determined by CO-oximetry, CK level in severely poisoned patients, pregnancy test, and ECG in any patient with cardiac symptoms.
Treatment.
Prevention of CO poisoning is paramount and should involve educational initiatives and the use of home CO detectors. Treatment of CO poisoning focuses on the administration of 100% oxygen to enhance elimination of CO. In ambient air the average half-life of HbCO is 4-6 hr. This is dramatically reduced to 60-90 min by providing 100% oxygen at normal atmospheric pressures by non-rebreather face mask. Severely poisoned patients might benefit from hyperbaric oxygen (HBO) , which decreases the half-life of HbCO to 20-30 min and is thought also to decrease the risk of delayed neurologic sequelae. Although the clinical benefits and referral guidelines for HBO therapy remain controversial, frequently cited indications include syncope, coma, seizure, altered mental status, acute coronary syndrome, HbCO level >25%, abnormal cerebellar examination, and pregnancy. Consultation with a PCC, medical toxicologist, or HBO facility can assist clinicians in determining which patients could benefit from HBO therapy. Sequelae of CO poisoning include persistent and delayed cognitive and cerebellar effects. HBO advocates believe that the risk of such sequelae is minimized through the delivery of 100% oxygen at 3 atm of pressure. Patients typically receive oxygen, by non-rebreather mask or hyperbaric chamber, for 6-24 hr.
Hydrogen Cyanide
Pathophysiology.
Cyanide inhibits cytochrome-c oxidase, part of the electron transport chain, interrupting cellular respiration and leading to profound tissue hypoxia. Patients may be exposed to hydrogen cyanide (HCN) gas in the workplace (manufacturing of synthetic fibers, nitriles, and plastics) or by smoke inhalation in a closed-space fire.
Clinical and Laboratory Manifestations.
Onset of symptoms is rapid after a significant exposure. Clinical manifestations of toxicity include headache, agitation/confusion, sudden loss of consciousness, tachycardia, cardiac dysrhythmias, and metabolic acidosis. Cyanide levels can be measured in whole blood but are not readily available at most institutions. A severe lactic acidosis (lactate >10 mmol/L) in fire victims suggests cyanide toxicity. Impaired oxygen extraction by tissues is implied by elevated mixed-venous oxyhemoglobin saturation, another laboratory finding suggesting cyanide toxicity.
Treatment.
Treatment includes removal from the source of exposure, rapid administration of high concentrations of oxygen, and antidotal therapy. The cyanide antidote kit (no longer manufactured) includes nitrites (amyl nitrite and sodium nitrite) used to produce methemoglobin, which then reacts with cyanide to form cyanomethemoglobin. The 3rd part of the kit is sodium thiosulfate, given to hasten the metabolism of cyanomethemoglobin to hemoglobin and the less toxic thiocyanate. In patients for whom induction of methemoglobinemia could produce more risk than benefit, the sodium thiosulfate component of the kit may be given alone.
The U.S. Food and Drug Administration (FDA) has approved hydroxocobalamin for use in known or suspected cyanide poisoning (see Table 77.7 ). This antidote reacts with cyanide to form the nontoxic cyanocobalamin (vitamin B12 ), which is then excreted in urine. Side effects of hydroxocobalamin include red discoloration of the skin and urine, transient hypertension, and interference with colorimetric lab assays. Hydroxocobalamin has an overall safety profile that appears superior to that of the cyanide antidote kit and thus is the preferred antidote for cyanide poisoning.
Miscellaneous Toxic Agents Found in the Home
Nicotine-Containing Products
Nicotine poisoning has become increasingly common with the recent advent of vaporizer (“vaping”) and e-cigarette devices. Although there are many nicotine-containing products (patches, gums, snuff, chewing tobacco, sprays, lozenges), tobacco cigarettes remain the main source of exposure. Prescription medications (varenicline and cytisine) are available that are partial nicotine receptor agonists. For children, some of the most concerning exposures are from the bottles of liquid nicotine used to refill vaping and e-cigarette devices. These bottles typically do not have childproof caps and contain a large amount of concentrated nicotine.
Pathophysiology.
Nicotine acts on nicotinic ACh receptors in the nervous system, neuromuscular junctions, and adrenal medulla, stimulating neurotransmitter release. Nicotine's effects on the dopaminergic reward pathway play a significant role in its addictive properties. The effects of nicotine are dose dependent; at lower doses it primarily acts on the brain, causing stimulation. At higher doses, nicotine overstimulates receptors, leading to inhibition and resulting in neuromuscular and nervous system blockade.
Clinical and Laboratory Manifestations.
Clinical effects of nicotine also depend on the dose. At low doses typically achieved through smoking, nicotine results in cognitive and mood enhancement, increased energy, and appetite suppression. At higher doses, significant toxicity follows a biphasic pattern, where cholinergic stimulation symptoms predominate and are later followed by inhibition. The first signs of nicotine poisoning are nausea, vomiting, diarrhea, and often muscle fasciculations. Tachycardia and hypertension occur initially, although in severe poisoning these progress to bradycardia, hypotension, coma, and respiratory muscle failure, which typically leads to death if not treated. Serum and urinary levels of nicotine and its metabolite cotinine can be obtained, but these rarely are available in real time and therefore have little effect on diagnosis and management.
Treatment.
Treatment of nicotine poisoning focuses on maximizing symptomatic and supportive care. Aggressive airway management should be the priority, especially in severe poisonings, because death usually occurs from respiratory muscle paralysis. IV fluids with escalation to vasopressors should be used for hypotension. Seizures should be managed with benzodiazepines, barbiturates, or propofol.
Single-Use Detergent Sacs
Commonly known as laundry “pods” for clothing, these products resemble candy to many children. When bitten into, a relatively large dose of concentrated detergent is expelled under pressure onto the posterior pharynx and vocal cords. This can lead to stridor and other signs of respiratory distress. Occasionally, and for unknown reasons, these children may also develop altered mental status. Supportive care with attention to any airway and breathing issues is warranted. Admission to the hospital is often indicated. Importantly, these are not considered caustic ingestions; the pH of these products is in the neutral zone. As such, upper GI endoscopy is rarely indicated. Curiously, laundry detergent drank from a bottle is rarely of significant concern.
Electric Dishwasher Detergent
Especially when in the form of crystals, these products are highly alkaline (pH >13), and exposure by ingestion can cause significant burns to the vocal cords and GI tract. Admission for expectant management or upper GI endoscopy is usually indicated.
Magnets
Most foreign body ingestions pass through the GI tract once known to have passed into the stomach. However, ingestion of ≥2 magnets (unless very weak refrigerator-style magnets) cause concern for bowel obstruction and perforation. Admission for attempted retrieval by endoscopy or clearance by WBI should be considered.
Batteries
Any disk or button-style battery lodged in the esophagus or airway should be considered a true emergency warranting immediate referral to an endoscopist for removal. These batteries can cause necrosis of the tissues in which they are lodged by continued electrical discharge and leaking of their contents (the former is likely the primary method of injury). Mucosal contact for even 2 hr might induce necrosis. Once past the lower esophageal sphincter, button or even larger batteries (e.g., AA, AAA) can usually be allowed to pass through the GI tract with close follow-up.