Genetics of Common Disorders
Bret L. Bostwick, Brendan Lee
Common pediatric diseases are usually multifactorial. The combination of many genes and environmental factors contribute to a complex sequence of events leading to disease. The complexity of the combination of contributing factors increases the challenge of finding genetic variants that cause disease. Genetic tools include the completed human genome sequence, public databases of genetic variants, and the human haplotype map. In addition to public genetic databases, dramatic reduction in the cost of genotyping and DNA sequencing has allowed very large numbers of genetic variants to be efficiently tested in large numbers of patients. Most of these studies focus on common variants (those with frequencies >5%). Technologies for DNA sequencing are allowing whole exome sequencing in many individuals at very low cost. This technology is being used to investigate the role of rare coding sequence variants in common diseases. The incorporation of these tools into large, well-designed population studies is the field of genetic epidemiology . Many new methods for analyzing genetic data have been developed, stimulating a renaissance in applied population genetics.
Major Genetic Approaches to the Study of Common Pediatric Disorders
Bret L. Bostwick, Brendan Lee
Millions of genetic variants are present in every person. Many of these variants have no impact on health, while others have a measureable influence. Sometimes, single-gene mutations consistently cause a disease, as with cystic fibrosis and sickle cell anemia. Other types of genetic variation, however, can contribute much less to the emergence of specific medical conditions, and these are best conceptualized as modifiers of disease risk. Fig. 99.1 demonstrates the relationship between variant frequency and the relative medical impact of the allele. The spectrum of variant impact is logarithmic, ranging widely from a slightly increased risk of illness to predetermined fully expressed disease. Studies aimed at discovering rare variants with outsized health effects only require small sample populations to achieve statistical significance, while those studying common variants require much larger sample sizes because of the small anticipated impact of each variant.

The cumulative risk of many common variants determines genetic susceptibility. For common conditions, the genetic predisposition alone is not sufficient to cause disease. Everyone inherits a different degree of disease vulnerability, which is then augmented by exposure to certain environmental factors. Fig. 99.2 shows a model for the contribution of common genetic variants to individual health. One of the goals in medical genetics is to identify the genes that contribute to initial genetic susceptibility and to help prevent the occurrence of disease, either by avoiding inciting environmental factors or by instituting interventions that reduce risk. For persons who cross the threshold of disease, the goal is to better understand the pathogenesis in the hope that this will suggest better approaches to treatment. Common genetic variation can also influence response to medications and the risk of adverse drug reactions (see Chapter 72 ) and augment the health impacts of environmental toxins.

Complex traits may be inherently difficult to study if the precision of clinical diagnosis (phenotype) is problematic, as often occurs with neurobehavioral traits. A starting point in the genetic analysis of a complex trait is to obtain evidence in support of a genetic contribution and to estimate the relative strength of genetic and environmental factors. Complex traits typically exhibit familial clustering but are not transmitted in a regular pattern as is autosomal dominant or recessive inheritance. Complex traits often show variation among different ethnic or racial groups, possibly reflecting the differences in gene variants among these groups.
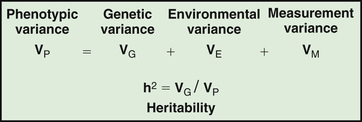
Assessing the potential genetic contribution begins by determining whether the trait is seen among related individuals more often than in the general population. A common measure of familiality is the first-degree relative risk (usually designated by the symbol λs ), which is equal to the ratio of the prevalence rate in siblings and/or parents to the prevalence rate in the general population. The λs for type 1 diabetes is about 15. The relative strength of genetic and nongenetic risk factors can be estimated by variance components analysis. The heritability of a trait is the estimate of the fraction of the total variance contributed by genetic factors (Fig. 99.3 ).
A minority of cases of common diseases such as diabetes may be caused by single-gene mutations (mendelian inheritance), chromosomal disorders, and other genomic disorders. These less common causes of the disease can often provide important insight into the most important molecular pathways involved. Chromosomal regions with genes that might contribute to disease susceptibility could theoretically be located with linkage mapping , which locates regions of DNA that are inherited in families with the specific disease. In practical terms, however, this has become quite difficult for most complex traits either because of a dearth of families or because the effect of individual genetic loci is weak.
Genetic association studies are more powerful in identifying common gene variants (>5% in the population) that confer increased risk of disease, but they fail if the disease-causing gene variants are relatively rare. Detection of the modest effect of each variant and interactions with environmental factors requires well-powered studies that often include thousands of individuals. A number of parallel approaches for analyzing the aggregate effects of rare variants in genes have also been developed. Such rare variant association methods also seem to require large sample sizes because the gene effects have also proved to be relatively weak.
Linkage mapping and association studies require markers along the DNA that can be ascertained, or genotyped , with large-scale, high-throughput laboratory techniques. Markers that are typically used are in the forms of microsatellites and single nucleotide polymorphisms (SNPs ; Fig. 99.4 ). A sample of the same region of genome from 50 people will reveal that approximately 1 in every 200 bases varies from the more common form. Although most SNPs lack any obvious function, a few alter the amino acid sequence of the protein or affect regulation of gene expression. Some of these functional alterations directly affect susceptibility to disease. A complex clinical phenotype can be defined by the presence or absence of a disease as a dichotomous trait , or by selection of a clinically meaningful variable such as serum glucose in type 2 diabetes, which is a continuous or quantitative trait .

Although it might not be possible to define subgroups of patients in advance based on common disease mechanisms, the more uniform the phenotype, the more likely that a genetic study will be successful. Locus heterogeneity refers to the situation in which a trait results from the independent action of more than 1 gene. Allelic heterogeneity indicates that more than 1 variant in a particular gene can contribute to disease risk. The development of a trait or disease from a nongenetic mechanism results in a phenocopy . These 3 factors often contribute to the difficulty in identifying individual disease susceptibility genes, because they reduce the effective size of the study population.
A person bearing any variant or allele (inherited unit, DNA segment, or chromosome) in a gene has a certain probability of being affected with a specific gene variant–associated disease. This is called the penetrance . Some diseases manifest signs only later in life (age-related penetrance), which could lead to misclassifying children who actually have the disease-producing gene as unaffected. Single-gene disorders are typically caused by mutations with relatively high penetrance, but some common variants have very low penetrance because their overall contribution to the disease is small. Many such common variants can contribute to disease risk for a complex trait. Normal human height is influenced by >400 genes.
Ideally, important environmental exposures should be measured and accounted for in a population because there may be a dependent interaction between the environmental factor and specific genetic variant. An example is the likely requirement for a viral infection preceding onset of type 1 diabetes. Although gene-environment interactions are strongly suspected to play an important role in common diseases, it is difficult to identify and measure them. Very large studies with uniform collection of information about environmental exposures are rare. Methods, such as genome-wide analysis of DNA methylation, may show evidence of environmental effects—so-called developmental programming (see Chapter 100 ). This information might be used to discover and validate gene-environment interactions.
Linkage Mapping
Linkage studies were used in the past to isolate genes that cause rare genetic syndromes; modified methods have been used to identify chromosomal regions linked to more common diseases. Linkage studies involve tagging segments of a person's genome with markers that allow identification of segments that have been inherited through the family along with disease. The markers are typically microsatellites or SNPs that define and help to distinguish which type of an allele any person carries. Genotype refers to the combination of alleles at a locus in a diploid organism. Linkage analyses of common diseases have shown inconsistent results. Factors such as heterogeneity, pleiotropy, variable expressivity, and reduced penetrance, in addition to variability in environmental exposures, weaken the power of linkage studies in complex traits.
Genetic Association
For multifactorial common diseases, association analyses may be used to identify causally important genes. There are two types of association study: direct association , in which the causal variant itself is tested to see whether its presence correlates with disease, and indirect association , in which markers that are physically close to the biologically important variant are used as proxies. The correlation of markers with other genetic variants in a small region of the genome is called linkage disequilibrium . Indirect association is enabled by the construction of a detailed genetic map in 3 reference populations (Europeans, Asians, West Africans) through the International HapMap Project. SNPs that tag most of the genome have been identified and can be genotyped at low cost using specially designed microarrays.
Three basic study designs are used for association testing. In a case control design, the frequency of an allele in the affected group is compared with the unaffected group. In a family-based control design, parents or siblings of an affected individual are used as the controls. In a cohort design, large numbers of people are ascertained and then followed for the onset of any number of diseases. The cohort analysis is very expensive, and there are few true cohort studies.
Family-based control study designs are somewhat attractive for pediatric diseases because it is usually possible to enroll parents. These studies solve a major problem in testing for association because the parents are perfectly matched for genetic background. When parents are collected, the statistical test used for these studies is called the transmission disequilibrium test . TDT compares the transmitted genotype with the inferred nontransmitted genotype. The success of all association analysis depends on the design of a well-powered study and an accurately measured trait to avoid phenotypic misclassification. In large, population-based studies, confounding by ethnicity or population stratification could distort results. Some genetic variants are more common in people from a particular ethnic group, which could cause an apparent association of a variant with a disease, when the disease rate happens to be higher in that group. This association would not be a true association between an allele and a disease, because the association would be confounded by genetic background. The family-based tests using the TDT are immune to population stratification. However, TDT and related study designs are inherently less efficient than case control studies. Newer methods for measuring subtle mismatching between cases and controls using many thousands of markers routinely genotyped in genome-wide association studies allow researchers to account for this effect.
Association studies should be a powerful tool to find genetic variation that confers risk to an individual; the effect of any 1 genetic variant will be a very small contribution to the complex disease pathway. Genetic variants have been found that implicate a novel gene in a process, motivating more in-depth research into systems that will affect disease outcome. Associations such as the APOE ε4 variant with an increased risk of Alzheimer disease are noted by many studies. Many published association results are not reproducible; insufficient power and stratification might account for the inconsistencies. As of late 2016, 2,650 studies and 29,954 unique SNP-trait associations have been catalogued (https://www.ebi.ac.uk/gwas/ ).
Low-cost methods for sequencing the complete exomes and genomes of individuals will allow a more comprehensive evaluation of the full range of genetic variants involved in common diseases. Rare genetic variants, including small insertions or deletions, could turn out to be extremely important in explaining the impact of genetic factors in important pediatric diseases such as autism, cardiovascular malformations, and other birth defects. Common traits such as obesity, diabetes, and autoimmune diseases might also be affected by rare variants. In common severe disorders such as intellectual disability and complex heart malformations, de novo mutations (i.e., mutations not present in either parent) are known to play an important role.