Mitochondrial Disease Diagnosis
Marni J. Falk
See also Chapter 105.4 .
Overview of Mitochondrial Disease
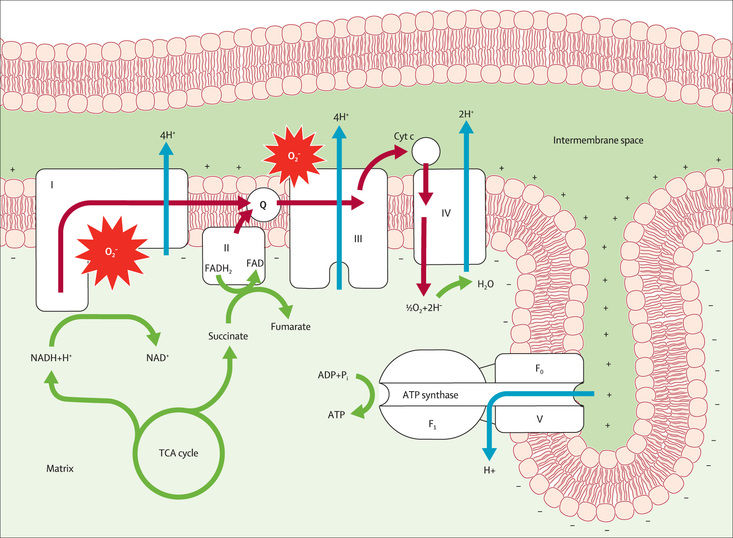
Mitochondrial diseases are multisystemic energy failure states with extensive clinical and genetic heterogeneity. Their common basis is best understood through recognition that mitochondria function as biologic “fuel cells” or “batteries,” producing chemical energy in the form of adenosine triphosphate (ATP) by aerobic metabolism of nutrient-derived reducing equivalents, through the integrated function of the 5-complex mitochondrial respiratory chain (RC) (Fig. 106.1 ). Mitochondria also play other essential roles that can be variably disrupted in disease states, such as regulating calcium homeostasis, diverse aspects of intermediary nutrient metabolism, nucleotide metabolism, and oxidative stress. Primary mitochondrial disease results from deficient RC function, which can be caused by mutations in genes that encode RC subunits, assembly factors or cofactors, components of mitochondrial DNA (mtDNA) metabolism and maintenance, or a host of other basic metabolic processes ongoing within mitochondria. Approximately 1,500 proteins exist within the mitochondrial proteome of different tissues, with variants in more than 350 unique genes across both the nuclear and the mitochondrial genomes already implicated as causal in human mitochondrial disease.
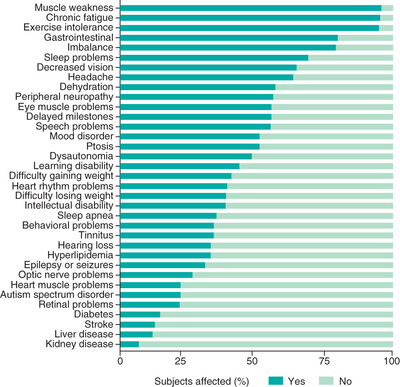
Collectively recognized as the most common group of inherited metabolic diseases, primary (genetic-based) mitochondrial disease has a combined minimal prevalence of 1 in 4,300 individuals across all ages. In addition, secondary mitochondrial dysfunction is broadly implicated in the pathogenesis of a host of complex diseases, ranging from metabolic syndrome to ischemia-reperfusion injury after stroke, to neurodegenerative diseases. Failure of high-energy demand organs in mitochondrial diseases may clinically present as severe neurodevelopmental, cardiac, myopathic, renal, hepatic, endocrine, immune, gastrointestinal, hearing, and vision disabilities, as well as global metabolic instability with lactic acidosis (Fig. 106.2 ) (see Tables 105.2 and 105.3 ). In most mitochondrial disorders, the phenotype may vary depending on the patient's age or the specific gene or genetic variant. Particularly common mitochondrial disease clinical syndromes that present in children include Leigh syndrome (for which there are more than 90 causal genes), mtDNA depletion syndrome (MDS, for which there are dozens of causal genes), mtDNA deletion syndromes (Pearson, Kearns Sayre), primary lactic acidosis, and pyruvate dehydrogenase deficiency. Common clinical features in children present in at least 90 percent of patients include fatigue, exercise intolerance, weakness, gastrointestinal problems, ataxia, and developmental delay. Thus, mitochondrial diseases present to and must be considered by clinicians across every medical specialty.
Patients with suspected mitochondrial disease may frequently experience a diagnostic odyssey , both clinically and genetically. Their extensive phenotypic heterogeneity without a common biomarker (GDF-15 may be one screening test that may be elevated in some mitochondrial myopathies particularly involving mtDNA deletions or depletion, along with lactic acidosis) presents a challenge to the readily available and accurate clinical diagnosis of mitochondrial disorders in many medical settings. Similarly, their extensive genetic heterogeneity involving known etiologies in >300 nuclear genes and all 37 mitochondrial DNA (mtDNA) genes, with likely dozens to hundreds more causative nuclear disease genes awaiting discovery, can make the accurate genetic diagnosis of an individual patient challenging. The diagnostic uncertainty can be further compounded by poor genotype-phenotype correlations and variable clinical presentations of individual gene disorders, high locus heterogeneity (i.e., multiple different causal disease genes) for similar clinical phenotypes, incomplete penetrance for some gene disorders, variable life stressors or environmental exposures that may exacerbate a given child's disease, and the unique biologic aspects of maternal inheritance for the subset of mitochondrial diseases caused by mtDNA gene mutations.
When to Suspect Mitochondrial Disease
Because of failure in the ability to generate cellular energy, mitochondrial diseases can involve any organ system at any age (see Fig. 106.2 ). Mitochondrial disease should be suspected when classic symptoms are present or if unexplained symptoms occur in 3 or more apparently unrelated organs. Individuals may present with a vast array of symptoms, including fatigue, muscle weakness, exercise intolerance, metabolic strokes, seizures, cardiomyopathy, arrhythmias, developmental or cognitive disabilities, autism, diabetes mellitus, other endocrinopathies (adrenal, thyroid), dysautonomia, and autoimmune disorders, as well as impairment of hearing, vision, growth, liver, gastrointestinal (GI), or kidney function. Although individuals may have just one or a few symptoms and a fluctuating disease course in terms of symptom severity, most patients with primary mitochondrial disease tend to develop progressive symptoms over time. A study of patients with mitochondrial diseases showed an average of 16 different clinically significant symptoms per patient, with a range of 7-35. When considering the diagnosis, it is helpful to recognize that most symptoms of mitochondrial disease involve functional, rather than structural, problems.
When mitochondrial disease is considered in the differential diagnosis, it is often helpful to obtain several laboratory screening studies for common biochemical features of mitochondrial disease and overlapping disorders, both at baseline and if unrevealing, during an acute illness or period of decompensation. Blood-based metabolic screening studies include comprehensive chemistry panel, complete blood count with differential, plasma amino acid quantitative analysis, carnitine analysis (total, free, acyl-carnitine profile), ammonia, creatine kinase, and testing for common secondary manifestations of mitochondrial disease (e.g., thyroid screen, lipoprotein profile, hemoglobin A1c ). Urine-based metabolic screening studies include urinalysis, urine organic acid quantitative analysis, and urine amino acid quantitative analysis. Consideration should also be given for screening for congenital disorders of glycosylation or vitamin deficiencies, which may have overlapping clinical features in some cases with mitochondrial disease. Lactic acidemia is neither highly sensitive nor specific for primary mitochondrial disease, but laboratory findings suggestive of primary mitochondrial disease include elevations of blood lactate, pyruvate, lactate:pyruvate ratio, alanine, ratios of alanine to lysine (>3) and alanine to sum of phenylalanine and tyrosine (>4), and anion gap. Biochemical alterations further suggestive of mitochondrial disease may include secondary impairment of fatty acid oxidation with elevation of dicarboxylic acids on acyl-carnitine profile, increased branched-chain amino acids and proline on plasma amino acid analysis, increased tricarboxylic acid cycle intermediates and lactate excretion on urine organic acid analysis, and generalized aminoaciduria on urine amino acid analysis. Growth and differentiation factor 15 (GDF-15 ) may be a useful screening test for mitochondrial depletion based myopathies.
Similarly, when mitochondrial disease is considered in the differential diagnosis, obtaining additional clinical evaluations to carefully phenotype the patient for prevalent or highly morbid and potentially modifiable features of mitochondrial disease is important. Because many individuals with mitochondrial disease develop problems with their vision (reduced visual acuity not correctable with glasses, photophobia or nyctalopia with reduced peripheral vision associated with retinal disease or optic atrophy, ophthalmoplegia, ptosis), hearing (high-frequency sensorineural hearing loss), and heart (arrhythmia, conduction block, cardiomyopathy), carefully evaluating for involvement of these high-energy systems is indicated. Neurologic evaluation is essential because many mitochondrial disease patients experience a range of central (metabolic stroke in cortical or deep gray matter including basal ganglia, midbrain, and/or brainstem, white matter changes, seizures, ataxia, movement disorder, migraine, cognitive changes), peripheral (axonal sensorimotor neuropathy), or autonomic nervous system dysfunction; brain imaging (MRI), spectroscopy (MRS), and on occasion electromyogram or nerve conduction velocity (EMG/NCV) studies can be helpful to support the diagnosis. Formal exercise physiology evaluation can also be useful to quantify and advise patients on their exercise capacity and safety, with some specific features (e.g., reduced V̇O 2 maximal capacity) suggestive of quantifiable mitochondrial dysfunction. Sleep study may be useful for individuals with sleep dysfunction because sleep disorders may mimic mitochondrial disease symptoms, and sleep problems are common and potentially treatable in mitochondrial disease. Gastrointestinal symptoms are common and underrecognized in mitochondrial disease patients, usually involving dysmotility of any portion of the GI tract with reflux, swallowing dysfunction, delayed gastric emptying, feeding and/or growth problems, pseudoobstruction, malabsorption, and constipation. Endocrine abnormalities are also common but underappreciated in many patients, including pituitary, adrenal, thyroid, and pancreatic dysfunction. Such careful phenotyping of patients with suspected mitochondrial disease can thus provide reassurance that the common, and potentially treatable, clinical aspects of mitochondrial disease are not present although they may develop over time, or conversely if identified, increase diagnostic suspicion and direct further diagnostic evaluation.
Mitochondrial Disease Inheritance
Primary mitochondrial disease may result from variants in either nuclear genes or mtDNA genes, which may be inherited from a parent or occur de novo in an affected individual. Thus, all mendelian (autosomal recessive, autosomal dominant, X-linked) or maternal (mtDNA) inheritance patterns can be consistent with mitochondrial diseases (Table 106.1 ). Obtaining a detailed, three-generation pedigree is important to potentially highlight the specific inheritance pattern in a given family. Individuals with inherited mtDNA disorders may report family members related through their maternal lineage (both males and females may be affected, but only affected individuals will be connected through the female germline), with a range of functional problems in different organs, such as migraines, fatigue, exercise intolerance, stroke, diabetes mellitus, thyroid dysfunction, irritable bowel spectrum, mood disorder, or vision and hearing problems. Inherited X-linked disorders typically present with symptoms only, or more severely, in males related through unaffected or minimally affected females. Autosomal recessive disorders are common in pediatric mitochondrial disease, particularly in consanguineous pedigrees, where a rare variant in the general population becomes enriched and passed down through both maternal and paternal lineages to become homozygous in the affected proband and also affect multiple individuals in a given generation without having affected individuals in earlier generations. Autosomal dominant variants may occur de novo or are passed on from either parent to their child, although many disorders may have reduced penetrance, which may make the genetic disorder appear to skip a generation. Identifying a likely inheritance pattern through pedigree analysis can inform accurate interpretation of large-scale genetic diagnostic evaluations, such as multigene sequencing and deletion/duplication analysis panels and exome or genome sequencing. Establishing a correct genetic diagnosis for mitochondrial disease in an affected individual is essential to enable reliable recurrence risk counseling and testing options in a given family, whether in a future pregnancy by chorionic villus sampling (CVS, typically performed at 10-12 weeks’ gestation) or amniocentesis (typically performed at 16-20 weeks’ gestation) or in the in vitro fertilization (IVF) setting with preimplantation genetic diagnosis (PGD) for a specific disease-causing variant.
Table 106.1
Major Molecular Categories of Mitochondrial Genes
| COMPONENT | CAUSAL GENOME | GENE MUTATION EFFECTS | DISEASE EXAMPLES |
|---|---|---|---|
| Electron transport chain enzyme subunits | Nuclear or mtDNA | Decreased functioning of electron transport chain complex |
Complex I deficiency Complex II deficiency |
| Electron transport chain assembly factors | Nuclear | Decreased assembly of electron transport chain enzyme complex |
Complex III deficiency Complex IV deficiency Complex V deficiency |
| Electron transport chain cofactors | Nuclear | Decreased functioning of electron transport chain |
Coenzyme Q10 deficiency Iron sulfur cluster defect Lipoyltransferase deficiency |
| mtDNA translation | Nuclear or mtDNA | Decreased translation of protein-coding mitochondrial DNA genes leading to decreased functioning of electron transport chain enzymes | Combined oxidative phosphorylation complexes deficiency |
| mtDNA maintenance | Nuclear | Increased errors in mitochondrial DNA leading to increased presence of point mutations and deletions, resulting in decreased translation of electron transport chain subunits |
Mitochondrial DNA depletion syndromes Mitochondrial DNA multiple deletion disorders |
| Mitochondrial membrane fission and fusion | Nuclear | Increased mtDNA point mutations and deletions; clumped and fragmented mitochondria |
OPA1 -related conditions MFN2 -related conditions |
From McCormick EM, Muraresku CC, Falk MJ: Mitochondrial genomics: a complex field now coming of age. Curr Genet Med Rep 6:52–61, 2018 (Table 1, p. 57).
Special mention is warranted to consider the unique aspects of maternal inheritance that typify mtDNA disorders. More than 300 disease-causing mtDNA variants have been identified, with extensive variation in disease manifestations and features. Most disease-causing variants are present in only a portion of an individual's mtDNA genomes, a concept known as heteroplasmy . For heteroplasmic mtDNA variants, the precise mutation level (percent) can vary between an individual's different tissues and can change over time, with symptom severity corresponding to different threshold mutation levels that can be difficult to define and that typically vary between organs. An individual's mtDNA genome background set of fixed sequence variants, known as a haplogroup , can also influence the penetrance or severity of a mtDNA disease. When a novel or rare mtDNA variant is identified in a given individual, it may be useful to use highly sensitive sequencing methods to test the levels of that mutation (which may be accurate to detect 1% mutation levels) in their different tissues (blood, urine, buccal, skin cells, muscle), as well as tissues from their mother or maternal relatives, to accurately determine whether it may be causal of disease in that family. Research-based functional testing may also be necessary to characterize fully the effects of a newly recognized mtDNA variant. When it is not known whether an mtDNA variant is maternally inherited or occurs de novo, the recurrence risk to future offspring of their asymptomatic parent is empirically estimated at 1 in 25 (4%), although the empirical recurrence risk rises to 1 in 2 (50%) when the mother is symptomatic.
Diagnostic Testing for Mitochondrial Disease
The diagnosis of mitochondrial disease relies foremost on genetic testing (genomic analysis), with biochemical screens useful in blood or urine and invasive tissue testing often seen as secondary, or sometimes not required at all (Fig. 106.3 ).
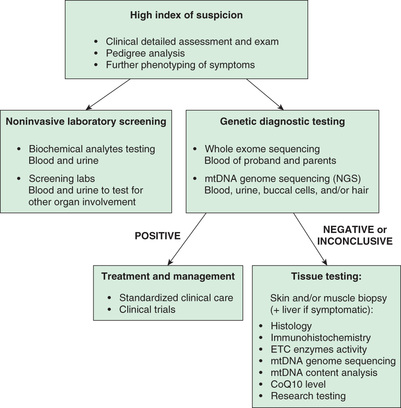
When the clinical evaluation—medical history; detailed review of systems; careful physical, neurologic, and dysmorphic examinations; pedigree-, blood-, and urine-based biochemical screening studies; and additional phenotyping clinical evaluations—is suggestive of mitochondrial disease, a range of clinical diagnostic testing options can be pursued. Absent a known molecular etiology in an affected family member, first-line genetic diagnostic testing may involve a focused panel of hundreds to thousands of known nuclear genes and the mtDNA genome using next-generation sequencing (NGS) methodologies that will detect both single-nucleotide variants and larger-scale gene deletions and duplications. If such testing is unrevealing, clinically based whole exome sequencing (WES) may be pursued. The standard of care is moving to pursue initial diagnostic testing by WES, which is more comprehensive for genes known not only to cause mitochondrial disease, but also to cause all human genetic diseases. The rationale for this evolution in diagnostic testing approach includes the following factors:
- 1. An increasingly similar cost and turnaround-time for panel-based and WES-based massively parallel NGS studies.
- 2. The common genetic diagnostic laboratory practice of generating WES data for all tests ordered, but only evaluating and reporting variants in specific gene subsets when panel-based testing is requested, leaving the remaining genes uninterpreted.
- 3. The mtDNA genome sequence is often included at no extra cost when clinical WES is ordered in blood, but may need to be repeated in a symptomatic tissue (e.g., muscle, liver) to detect heteroplasmic mtDNA variants that may not be present in blood.
- 4. The utility of performing concurrent proband and both parental sample sequencing (trio-based testing), as usually pursued with WES but not panel-based testing, thereby allowing concurrent segregation analysis of a suspected pathogenic variants as well as ready identification of de novo dominant variants in the proband.
- 5. The improved diagnostic yield of exome relative to panel-based testing increasingly being reported by clinical diagnostic laboratories, given the highly heterogeneous nature of mitochondrial disease, rapid rate of change in the recognition of new gene diagnoses making prior established gene panels obsolete, and the extensive phenotypic overlap with non-mitochondrial diseases.
- 6. The ability to utilize WES raw data (either on a research basis or for reanalysis at a later date by the clinical diagnostic laboratory) to highlight and/or identify “novel” gene disorders not previously recognized or associated with human disease.
A mitochondrial disease community resource to centrally curate all mitochondrial disease, gene, and variant knowledge across both genomes is publicly accessible at www.mseqdr.org . Exome sequencing including mtDNA is estimated to identify the definitive genetic etiology for mitochondrial disease in at least 60% of patients in whom it is strongly suspected, reducing the diagnostic odyssey in many patients from decades or years to months.
Tissue-based diagnostic testing has decreased in frequency as a front-line test in all patients with suspected mitochondrial disease, although it still has clinical utility in some cases. These include (1) in the setting of rapidly deteriorating clinical status when genetic testing results may not be available in a timely fashion; (2) when a variant of uncertain significance identified on genomic testing has unclear biochemical consequences; and (3) when uninformative genomic sequencing in blood in an individual with myopathy or muscle symptoms raises concern for other disease processes that may be evident on histology, electron microscopy, immunohistochemistry or enzymatic tissue testing. In addition, some mitochondrial diseases are only evident by tissue-based diagnostic testing. These include mtDNA deletion disorders (typically involving several-thousand nucleotides) not present in blood that cause chronic progressive external ophthalmoplegia (CPEO) or Kearns-Sayre syndrome (KSS) spectrum disorder, as well as different tissue (muscle or liver)-specific mtDNA depletion disorders (e.g., reduced mtDNA tissue content) that confirm a mitochondrial pathophysiology in a given patient and highlight a likely underlying nuclear gene cause for their disease, since mtDNA maintenance requires a host of nuclear-encoded proteins. Muscle analysis for integrated RC oxidative phosphorylation capacity assessment requires analysis of a fresh muscle biopsy only available at a very limited number of sites worldwide, whereas electron transport chain enzyme activity analyses are the accepted gold standard to evaluate for mitochondrial dysfunction in a previously frozen tissue sample, often shipped elsewhere for diagnostic analysis. Skin biopsies are useful to establish fibroblast cell lines in which these same studies of mitochondrial function can be clinically performed. If detected, abnormalities can be revealing of a specific type of mitochondrial disorder, although not all mitochondrial diseases may be expressed or detectable in skin analysis. Thus, if fibroblast testing is unrevealing, more invasive tissue studies may subsequently need to be pursued. Fibroblast cell lines, and occasionally blood-based lymphoblastoid cell lines, also provide a minimally invasive cell source to allow other clinical enzymatic analyses to be performed, as well as novel disease gene validation and research-based therapeutic modeling.
Treatment Principles for Mitochondrial Disease
Effective therapies for both primary and secondary mitochondrial diseases are lacking, because little has been known about the biochemical and physiologic abnormalities that contribute to their diverse clinical manifestations. Clinical complexity and imprecisely defined or understood biochemical phenotypes of different mitochondrial disease subtypes have made it difficult for clinicians to effectively apply or monitor targeted therapies for RC disease. Mitochondrial cocktails of vitamins and supplements variably include vitamins (B1 , B2 , C), antioxidants (CoQ10 , lipoic acid, vitamin E), and metabolic modifiers (creatine, L -carnitine, L -arginine, folinic acid). Although the efficacy, toxicity, and optimal dose of these drugs are not known and have not been objectively assessed in human RC disease patients, they continue to be empirically prescribed in hopes of enhancing residual RC enzymatic function or quenching toxic metabolites theorized to accumulate in RC dysfunction, and because of patient-based reports of improved well-being. However, provision of these therapies has often adopted a one-size-fits-all approach, ignoring the inherent variation in primary mitochondrial disease subtypes, the tissue-specific manifestations, and the major pathogenic factors, such as the predominant downstream metabolic and signaling alterations that occur in different disease subclasses.
Although no cure or U.S. Food and Drug Administration (FDA)–approved therapy yet exists for any mitochondrial disease, improved molecular delineation has enabled selected therapies to advance from the theoretical, empirical, and largely ineffective stage to a promising horizon of rational, personalized, and effective interventions. An increasing number of mitochondrial disease diagnoses have interventions involving the initiation or avoidance of specific medications (corticosteroids, valproic acid, phenytoin, barbiturates, propofol for prolonged duration beyond 30-60 min, certain anesthetics, statins, β-blocking agents, amiodarone, nucleoside reverse transcriptase inhibitors), provision of cofactors or diets, and screening regimens for progressive clinical involvement of modifiable manifestations. General therapies for Leigh syndrome such as L -arginine and citrulline may prevent or reverse neurodevelopmental sequelae from a metabolic stroke. Nutritional therapies in these disorders are tailored to specific disease genes, such as thiamine and biotin for SLC19A3 disease, ubiquinol for PDSS2 (CoQ10 deficiency) disease, and thiamine and the ketogenic diet for PDHA1 (pyruvate dehydrogenase) deficiency. Establishing the precise molecular diagnosis can further be lifesaving by avoiding fasting and mitochondrial-toxic medicines or general anesthetics in specific mitochondrial disease subsets, improving recurrence risk counseling and prevention, enabling targeted screening for reported medical complications, and in some cases providing necessary cofactors or vitamins that may not otherwise have been considered. In addition, reproductive methodologies emerging in some countries for mitochondrial disease prevention, such as mitochondrial replacement technologies (MRTs), are only appropriate to consider in the setting of known pathogenic, inherited mtDNA variants. Finally, the ability to molecularly identify primary mitochondrial disease patients has enabled the design of an increasing number of clinical treatment trials now being planned or underway for a diverse range of symptoms that occur in primary mitochondrial diseases (see www.clinicaltrials.gov ).