Defects in Metabolism of Carbohydrates
Priya S. Kishnani, Yuan-Tsong Chen
Carbohydrate synthesis and degradation provide the energy required for most metabolic processes. The important carbohydrates include 3 monosaccharides—glucose, galactose, and fructose—and a polysaccharide, glycogen. Fig. 105.1 shows the relevant biochemical pathways of these carbohydrates. Glucose is the principal substrate of energy metabolism, continuously available through dietary intake, gluconeogenesis (glucose made de novo from amino acids, primarily alanine), and glycogenolysis (breakdown of glycogen). Metabolism of glucose generates adenosine triphosphate (ATP) via glycolysis (conversion of glucose or glycogen to pyruvate), mitochondrial oxidative phosphorylation (conversion of pyruvate to carbon dioxide and water), or both. Dietary sources of glucose come from polysaccharides, primarily starch, and the disaccharides lactose, maltose, and sucrose. However, oral intake of glucose is intermittent and unreliable. Gluconeogenesis contributes to maintaining euglycemia (normal levels of glucose in the blood), but this process requires time. Hepatic glycogenolysis provides the rapid release of glucose, and is the most significant factor in maintaining euglycemia. Glycogen is also the primary stored energy source in muscle, providing glucose for muscle activity during exercise. Galactose and fructose are monosaccharides that provide fuel for cellular metabolism, though their role is less significant than that of glucose. Galactose is derived from lactose (galactose + glucose), which is found in milk and milk products. Galactose is an important energy source in infants, but it is first metabolized to glucose. Galactose (exogenous or endogenously synthesized from glucose) is also an important component of certain glycolipids, glycoproteins, and glycosaminoglycans. The dietary sources of fructose are sucrose (fructose + glucose, sorbitol) and fructose itself, which is found in fruits, vegetables, and honey.

Defects in glycogen metabolism typically cause an accumulation of glycogen in the tissues, thus the name glycogen storage disease (Table 105.1 ). Defects in gluconeogenesis or the glycolytic pathway, including galactose and fructose metabolism, do not result in an accumulation of glycogen (Table 105.1 ). The defects in pyruvate metabolism in the pathway of the conversion of pyruvate to carbon dioxide and water via mitochondrial oxidative phosphorylation are more often associated with lactic acidosis and some tissue glycogen accumulation.
Table 105.1
Features of the Disorders of Carbohydrate Metabolism
| DISORDERS | BASIC DEFECTS | CLINICAL PRESENTATION | COMMENTS |
|---|---|---|---|
| LIVER GLYCOGENOSES | |||
| Type/Common Name | |||
| Ia/Von Gierke | Glucose-6-phosphatase | Growth retardation, hepatomegaly, hypoglycemia; elevated blood lactate, cholesterol, triglyceride, and uric acid levels |
Common, severe hypoglycemia Adulthood: hepatic adenomas and carcinoma, osteoporosis, pulmonary hypertension, and renal failure |
| Ib | Glucose-6-phosphate translocase | Same as type Ia, with additional findings of neutropenia, periodontal disease, inflammatory bowel disease | 10% of type Ia |
| IIIa/Cori or Forbes | Liver and muscle debrancher deficiency (amylo-1,6-glucosidase) |
Childhood: hepatomegaly, growth retardation, muscle weakness, hypoglycemia, hyperlipidemia, elevated transaminase levels Adult form: muscle atrophy and weakness, peripheral neuropathy, liver cirrhosis and failure, risk for hepatocellular carcinoma |
Common, intermediate severity of hypoglycemia Muscle weakness may progress to need for ambulation assistance such as wheelchair. |
| IIIb | Liver debrancher deficiency; normal muscle enzyme activity | Liver symptoms same as in type IIIa; no muscle symptoms | 15% of type III |
| IV/Andersen | Branching enzyme |
Childhood: failure to thrive, hypotonia, hepatomegaly, splenomegaly, progressive cirrhosis (death usually before 5th yr), elevated transaminase levels; a subset does not have progression of liver disease Adult form: isolated myopathy, central and peripheral nervous system involvement |
Rare neuromuscular variants exist |
| VI/Hers | Liver phosphorylase | Hepatomegaly, typically mild hypoglycemia, hyperlipidemia, and ketosis | Often underdiagnosed, severe presentation also known |
| IX/phosphorylase kinase (PhK) deficiency | Common, X-linked, typically less severe than autosomal forms; clinical variability within and between subtypes; severe cases being recognized across different subtypes | ||
| IX (PHKA2 variant) | Liver PhK | Hypoglycemia, hyperketosis hepatomegaly, chronic liver disease, hyperlipidemia, elevated liver enzymes, growth retardation | X-linked |
| IX (PHKB variant) | Liver and muscle PhK | Hepatomegaly, growth retardation | Autosomal recessive |
| IX (PHKG2 variant) | Liver PhK | More severe than IXa; marked hepatomegaly, recurrent hypoglycemia, liver cirrhosis | Autosomal recessive |
| Glycogen synthase deficiency | Glycogen synthase | Early morning drowsiness and fatigue, fasting hypoglycemia, and ketosis, no hepatomegaly | Decreased liver glycogen store |
| XI/Fanconi-Bickel syndrome | Glucose transporter 2 (GLUT-2) | Failure to thrive, rickets, hepatorenomegaly, proximal renal tubular dysfunction, impaired glucose and galactose utilization | GLUT-2 expressed in liver, kidney, pancreas, and intestine |
| MUSCLE GLYCOGENOSES | |||
| Type/Common Name | |||
| IX (PHKA1 variant) | Muscle PhK | Exercise intolerance, cramps, myalgia, myoglobinuria; no hepatomegaly | X-linked or autosomal recessive |
| II/Pompe infantile | Acid α-glucosidase (acid maltase) | Cardiomegaly, hypotonia, hepatomegaly; onset: birth to 6 mo | Common, cardiorespiratory failure leading to death by age 1-2 yr; minimal to no residual enzyme activity |
| II/Late-onset Pompe (juvenile and adult) | Acid α-glucosidase (acid maltase) | Myopathy, variable cardiomyopathy, respiratory insufficiency; onset: childhood to adulthood | Residual enzyme activity |
| Danon disease | Lysosome-associated membrane protein 2 (LAMP2) | Hypertrophic cardiomyopathy, heart failure | Rare, X-linked |
| PRKAG2 deficiency | Adenosine monophosphate (AMP)–activated protein kinase γ | Hypertrophic cardiomyopathy. Congenital fetal form is rapidly fatal; myopathy, myalgia, seizures | Autosomal dominant |
| V/McArdle | Myophosphorylase | Exercise intolerance, muscle cramps, myoglobinuria, “second wind” phenomenon | Common, male predominance |
| VII/Tarui | Phosphofructokinase | Exercise intolerance, muscle cramps, compensatory hemolytic anemia, myoglobinuria | Prevalent in Japanese and Ashkenazi Jews |
| Late-onset polyglucosan body myopathy | Glycogenin-1 | Adult-onset proximal muscle weakness, nervous system involvement uncommon | Autosomal recessive, rare |
| Phosphoglycerate kinase deficiency | Phosphoglycerate kinase | As with type V | Rare, X-linked |
| Phosphoglycerate mutase deficiency | M subunit of phosphoglycerate mutase | As with type V | Rare, majority of patients are African American |
| Lactate dehydrogenase deficiency | M subunit of lactate dehydrogenase | As with type V | Rare |
| GALACTOSE DISORDERS | |||
| Galactosemia with transferase deficiency | Galactose-1-phosphate uridyltransferase | Vomiting, hepatomegaly, cataracts, aminoaciduria, failure to thrive | Black patients tend to have milder symptoms |
| Galactokinase deficiency | Galactokinase | Cataracts | Benign |
| Generalized uridine diphosphate galactose-4-epimerase deficiency | Uridine diphosphate galactose-4-epimerase | Similar to transferase deficiency with additional findings of hypotonia and nerve deafness | A benign variant also exists |
| FRUCTOSE DISORDERS | |||
| Essential fructosuria | Fructokinase | Urine reducing substance | Benign |
| Fructose-1-phosphate aldolase | Acute: vomiting, sweating, lethargy | ||
| Hereditary fructose intolerance | Chronic: failure to thrive, hepatic failure | Prognosis good with fructose restriction | |
| DISORDERS OF GLUCONEOGENESIS | |||
| Fructose-1,6-diphosphatase deficiency | Fructose-1,6-diphosphatase | Episodic hypoglycemia, apnea, acidosis | Good prognosis, avoid fasting |
| Phosphoenolpyruvate carboxykinase deficiency | Phosphoenolpyruvate carboxykinase | Hypoglycemia, hepatomegaly, hypotonia, failure to thrive | Rare |
| DISORDERS OF PYRUVATE METABOLISM | |||
| Pyruvate dehydrogenase complex defect | Pyruvate dehydrogenase | Severe fatal neonatal to mild late onset, lactic acidosis, psychomotor retardation, failure to thrive | Most commonly caused by E1α subunit, defect X-linked |
| Pyruvate carboxylase deficiency | Pyruvate carboxylase | Same as above | Rare, autosomal recessive |
| Respiratory chain defects (oxidative phosphorylation disease) | Complexes I-V, many mitochondrial DNA mutations | Heterogeneous with multisystem involvement | Mitochondrial inheritance |
| DISORDERS IN PENTOSE METABOLISM | |||
| Pentosuria | L -Xylulose reductase | Urine-reducing substance | Benign |
| Transaldolase deficiency | Transaldolase | Liver cirrhosis and failure, cardiomyopathy | Autosomal recessive |
| Ribose-5-phosphate isomerase deficiency | Ribose-5-phosphate isomerase | Progressive leukoencephalopathy and peripheral neuropathy | |
Glycogen Storage Diseases
Priya S. Kishnani, Yuan-Tsong Chen
The disorders of glycogen metabolism, the glycogen storage diseases (GSDs ), result from deficiencies of various enzymes or transport proteins in the pathways of glycogen metabolism (see Fig. 105.1 ). Glycogen found in these disorders is abnormal in quantity, quality, or both. GSDs are categorized by numerical type in accordance with the chronological order in which these enzymatic defects were identified. This numerical classification is still widely used, at least up to number VII. The GSDs can also be classified by organ involvement into liver and muscle glycogenoses (see Table 105.1 ).
There are more than 12 forms of GSDs. Glucose-6-phosphatase deficiency (type I), lysosomal acid α-glucosidase deficiency (type II), debrancher deficiency (type III), and liver phosphorylase kinase deficiency (type IX) are the most common of those that typically present in early childhood; myophosphorylase deficiency (type V, McArdle disease) is the most common in adolescents and adults. The cumulative frequency of all forms of GSD is approximately 1 in 20,000 live births.
Liver Glycogenoses
The GSDs that principally affect the liver include glucose-6-phosphatase deficiency (type I ), debranching enzyme deficiency (type III ), branching enzyme deficiency (type IV ), liver phosphorylase deficiency (type VI ), phosphorylase kinase deficiency (type IX , formerly GSD VIa), glycogen synthase deficiency (type 0 ), and glucose transporter-2 defect. Because hepatic carbohydrate metabolism is responsible for plasma glucose homeostasis, this group of disorders typically causes fasting hypoglycemia and hepatomegaly. Some (types III, IV, IX) can be associated with liver cirrhosis. Other organs can also be involved and may manifest as renal dysfunction in type I, myopathy (skeletal and/or cardiomyopathy) in types III and IV, as well as in some rare forms of phosphorylase kinase deficiency, and neurologic involvement in types III (peripheral nerves) and IV (diffuse central and peripheral nervous system dysfunction).
Type I Glycogen Storage Disease (Glucose-6-Phosphatase or Translocase Deficiency, Von Gierke Disease)
Type I GSD is caused by the absence or deficiency of glucose-6-phosphatase activity in the liver, kidney, and intestinal mucosa. It has 2 subtypes: type Ia , in which the defective enzyme is glucose-6-phosphatase, and type Ib , in which the defective enzyme is a translocase that transports glucose-6-phosphate across the microsomal membrane. Deficiency of the enzymes in both type Ia and type Ib lead to inadequate hepatic conversion of glucose-6-phosphate to glucose through normal glycogenolysis and gluconeogenesis, resulting in fasting hypoglycemia.
Type I GSD is an autosomal recessive disorder. The gene for glucose-6-phosphatase (G6PC) is located on chromosome 17q21; the gene for translocase (SLC37A4) is on chromosome 11q23. Common pathogenic variants have been identified. Carrier detection and prenatal diagnosis are possible with DNA-based methodologies.
Clinical Manifestations
Patients with type I GSD may present in the neonatal period with hypoglycemia and lactic acidosis but more often present at 3-4 mo of age with hepatomegaly, hypoglycemic seizures, or both. Affected children often have a doll-like face with fat cheeks, relatively thin extremities, short stature, and a protuberant abdomen that is a consequence of massive hepatomegaly. The kidneys are also enlarged, whereas the spleen and heart are not involved.
The biochemical characteristics of type I GSD are hypoglycemia, lactic acidosis, hyperuricemia, and hyperlipidemia. Hypoglycemia and lactic acidosis can develop after a short fast. Hyperuricemia is present in young children; it rarely progresses to symptomatic gout before puberty. Despite marked hepatomegaly, the liver transaminase levels are usually normal or only slightly elevated. Intermittent diarrhea may occur in GSD I. In patients with GSD Ib, the loss of mucosal barrier function as a result of inflammation, which is likely related to the disturbed neutrophil function, seems to be the main cause of diarrhea. Easy bruising and epistaxis are common and are associated with a prolonged bleeding time as a result of impaired platelet aggregation and adhesion.
The plasma may be “milky” in appearance due to strikingly elevated triglyceride levels. Cholesterol and phospholipids are also elevated, but less prominently. The lipid abnormality resembles type IV hyperlipidemia and is characterized by increased levels of very-low-density lipoprotein, low-density lipoprotein, and a unique apolipoprotein profile consisting of increased levels of apolipoproteins B, C, and E, with relatively normal or reduced levels of apolipoproteins A and D. The histologic appearance of the liver is characterized by a universal distention of hepatocytes by glycogen and fat. The lipid vacuoles are particularly large and prominent. There is no associated liver fibrosis.
Although type I GSD affects mainly the liver, multiple organ systems are involved. Delayed puberty is often seen. Females can have ultrasound findings consistent with polycystic ovaries even though other features of polycystic ovary syndrome (acne, hirsutism) are not seen. Nonetheless, fertility appears to be normal, as evidenced in several reports of successful pregnancy in women with GSD I. Increased bleeding during menstrual cycles, including life-threatening menorrhagia, has been reported and could be related to the impaired platelet aggregation. Symptoms of gout usually start around puberty from long-term hyperuricemia. There is an increased risk of pancreatitis, secondary to the lipid abnormalities. The dyslipidemia, together with elevated erythrocyte aggregation, could predispose these patients to atherosclerosis, but premature atherosclerosis has not yet been clearly documented except for rare cases. Impaired platelet aggregation and increased antioxidative defense to prevent lipid peroxidation may function as a protective mechanism to help reduce the risk of atherosclerosis. Frequent fractures and radiographic evidence of osteopenia are common; bone mineral content is reduced, even in prepubertal patients.
By the 2nd or 3rd decade of life, some patients with type I GSD develop hepatic adenomas that can hemorrhage and turn malignant in some cases. Pulmonary hypertension has been seen in some long-term survivors of the disease. Iron-refractory anemia and an increased prevalence of thyroid autoimmunity are also being recognized.
Renal disease is another late complication, and most patients with type I GSD >20 yr of age have proteinuria. Many also have hypertension, renal stones, nephrocalcinosis, and altered creatinine clearance. Glomerular hyperfiltration, increased renal plasma flow, and microalbuminuria are often found in the early stages of renal dysfunction and can occur before the onset of proteinuria. In younger patients, hyperfiltration and hyperperfusion may be the only signs of renal abnormalities. With the advancement of renal disease, focal segmental glomerulosclerosis and interstitial fibrosis become evident. In some patients, renal function has deteriorated and progressed to failure, requiring dialysis and transplantation. Other renal abnormalities include amyloidosis, a Fanconi-like syndrome, hypocitraturia, hypercalciuria, and a distal renal tubular acidification defect.
Patients with GSD Ib can have additional features of recurrent bacterial infections from neutropenia and impaired neutrophil function. Oral involvement including recurrent mucosal ulceration, gingivitis, and rapidly progressive periodontal disease may occur in type Ib. Intestinal mucosa ulceration culminating in GSD enterocolitis is also common. Type 1b is also associated with a chronic inflammatory bowel disease (IBD)–like picture involving the colon that may be associated with neutropenia and/or neutrophil dysfunction; it may resemble ulcerative colitis or Crohn disease.
Diagnosis
The clinical presentation and laboratory findings of hypoglycemia, lactic acidosis, hyperuricemia, and hyperlipidemia lead to a suspected diagnosis of type I GSD. Neutropenia is noted in GSD Ib patients, typically before 1 yr of age. Neutropenia has also been noted in some patients with GSD Ia, especially those with the p.G188A variant. Administration of glucagon or epinephrine leads to a negligible increase, if any, in blood glucose levels, but the lactate level rises significantly. Before the availability of genetic testing, a definitive diagnosis required a liver biopsy. Gene-based variant analysis by single-gene sequencing or gene panels provides a noninvasive way to diagnose most patients with GSD types Ia and Ib.
Treatment
Treatment focuses on maintaining normal blood glucose levels and is achieved by continuous nasogastric (NG) infusion of glucose or oral administration of uncooked cornstarch. In infancy, overnight NG drip feeding may be needed to maintain normoglycemia. NG feedings can consist of an elemental enteral formula or only glucose or a glucose polymer to provide sufficient glucose to maintain euglycemia. During the day, frequent feedings with high-carbohydrate content are typically sufficient.
Uncooked cornstarch acts as a slow-release form of glucose and can be introduced at a dose of 1.6 g/kg every 4 hr for children <2 yr of age. The response of young children is variable. For older children, the cornstarch regimen can be changed to every 6 hr at a dose of 1.6-2.5 g/kg body weight and can be given orally as a liquid. Newer starch products, such as extended-release waxy maize starch, are thought to be longer acting, better tolerated, and more palatable. Medium-chain triglyceride (MCT) supplementation improves metabolic control, leading to improved growth in children. Since fructose and galactose cannot be converted directly to glucose in GSD type I, these sugars should be restricted in the diet. Sucrose (table sugar, cane sugar, other ingredients), fructose (fruit, juice, high-fructose corn syrup), lactose (dairy foods), and sorbitol should be avoided or limited. As a result of these dietary restrictions, vitamins and minerals such as calcium and vitamin D may be deficient, and supplementation is required to prevent nutritional deficiencies.
Dietary therapy improves hyperuricemia, hyperlipidemia, and renal function, slowing the development of renal failure. This therapy fails, however, to normalize blood uric acid and lipid levels completely in some individuals, despite good metabolic control, especially after puberty. The control of hyperuricemia can be further augmented by the use of allopurinol, a xanthine oxidase inhibitor. The hyperlipidemia can be reduced with lipid-lowering drugs such as β-hydroxy-β-methylglutaryl-coenzyme A (HMG-CoA) reductase inhibitors and fibrate (see Chapter 104 ). Microalbuminuria , an early indicator of renal dysfunction in type I disease, is treated with angiotensin-converting enzyme (ACE) inhibitors. Citrate supplements can be beneficial for patients with hypocitraturia by preventing or ameliorating nephrocalcinosis and development of urinary calculi. Thiazide diuretics increase renal reabsorption of filtered calcium and decrease urinary calcium excretion, thereby preventing hypercalciuria and nephrocalcinosis. Growth hormone (GH) should be used with extreme caution and limited to only those with a documented GH deficiency. Even in those patients, there should be close monitoring of metabolic parameters and for the presence of adenomas.
In patients with type Ib GSD, granulocyte and granulocyte-macrophage colony-stimulating factors are successful in correcting the neutropenia, decreasing the number and severity of bacterial infections, and improving the chronic IBD. The minimum effective dose should be used because side effects are noted on these agents, including splenomegaly, hypersplenism, and bone pain. Bone marrow transplantation has been reported to correct the neutropenia of type Ib GSD.
Orthotopic liver transplantation is a potential cure of type I GSD, especially for patients with liver malignancy, multiple liver adenomas, metabolic derangements refractory to medical management, and liver failure. However, this should be considered as a last resort because of the inherent short- and long-term complications. Large adenomas (>2 cm) that are rapidly increasing in size and/or number may necessitate partial hepatic resection. Smaller adenomas (<2 cm) may be treated with percutaneous ethanol injection or transcatheter arterial embolization. Recurrence of liver adenomas is a challenge and may potentiate malignant transformation in these patients, ultimately requiring a liver transplant.
Before any surgical procedure, the bleeding status must be evaluated and good metabolic control established. Prolonged bleeding times can be normalized by the use of intensive intravenous (IV) glucose infusion for 24-48 hr before surgery. DDAVP (1-deamino-8-D -arginine vasopressin) can reduce bleeding complications, but it should be used with caution because of the risk of fluid overload and hyponatremia when administered as an IV infusion. Lactated Ringer solution should be avoided because it contains lactate and no glucose. Glucose levels should be maintained in the normal range throughout surgery with the use of 10% dextrose. Overall, metabolic control is assessed by growth, improvement, and correction of the metabolic abnormalities, such as elevated lactate, glucose, triglyceride, cholesterol, and uric acid levels.
Prognosis
Previously, type I GSD was associated with a high mortality at a young age, and even for those who survived, the prognosis was guarded. Inadequate metabolic control during childhood can lead to long-term complications in adults. Clinical outcomes have improved dramatically with early diagnosis and effective treatment. However, serious complications such as renal disease and formation of hepatic adenomas with potential risk for malignant transformation persist. The ability to identify transformation to hepatocellular carcinoma in the liver adenomas remains a challenge: α-fetoprotein (AFP) and carcinoembryonic antigen (CEA) levels often remain normal in the setting of hepatocellular carcinoma.
Type III Glycogen Storage Disease (Debrancher Deficiency, Limit Dextrinosis)
Type III GSD is caused by deficient activity of the glycogen debranching enzyme . Debranching enzyme, together with phosphorylase, is responsible for complete degradation of glycogen. When debranching enzyme is defective, glycogen breakdown is incomplete, resulting in the accumulation of an abnormal glycogen with short outer-branch chains, which resemble limit dextrin. Symptoms of glycogen debranching enzyme deficiency include hepatomegaly, hypoglycemia, short stature, variable skeletal myopathy, and variable cardiomyopathy. GSD type IIIa usually involves both liver and muscle, whereas in type IIIb , seen in approximately 15% of patients, the disease appears to involve only liver.
Type III GSD is an autosomal recessive disease that has been reported in many different ethnic groups. The frequency is relatively high in Sephardic Jews from North Africa, inhabitants of the Faroe Islands, and in Inuits. The gene for debranching enzyme (AGL) is located on chromosome 1p21. More than 130 different pathogenic variants have been identified; 2 pathogenic variants in exon 3, c.18_19delGA (previously described as c.17_18delAG) and p.Gln6X, are specifically associated with glycogenosis IIIb. Carrier detection and prenatal diagnosis are possible using DNA-based methodologies.
Clinical Manifestations
In infancy and childhood, GSD type III may be indistinguishable from type I GSD because of overlapping features such as hepatomegaly, hypoglycemia, hyperlipidemia, and growth retardation (Fig. 105.2 ). Splenomegaly may be present, but the kidneys are typically not affected. Hepatomegaly in most patients with type III GSD improves with age; however, liver fibrosis, cirrhosis progressing to liver failure, and hepatocellular carcinoma (HCC) are noted in many in late adulthood. Hepatic adenomas occur less often in individuals with GSD III than those with GSD I. The relationship between hepatic adenomas and malignancy in GSD III remains unclear. AFP and CEA levels are not good predictors of the presence of hepatocellular adenomas or malignant transformation. A single case of malignant transformation at the site of adenomas has been noted.
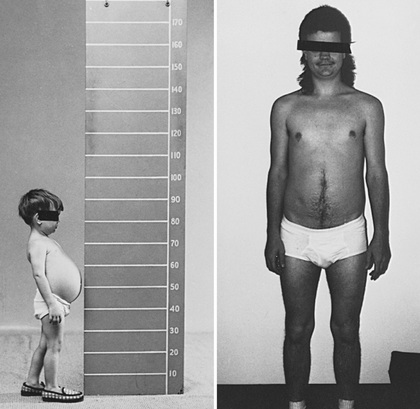
In patients with GSD type IIIa, the muscle weakness is slowly progressive and associated with wasting. The weakness is less remarkable in childhood but can become severe after the 3rd or 4th decade of life. Low bone mineral density in patients with GSD III put them at an increased risk of potential fractures. Myopathy does not follow any particular pattern of involvement; both proximal and distal muscles are involved. Electromyography reveals a widespread myopathy; nerve conduction studies are often abnormal.
Although overt cardiac dysfunction is rare, ventricular hypertrophy is a frequent finding. Cardiac pathology has shown diffuse involvement of various cardiac structures, including vacuolation of myocytes, atrioventricular conduction, and hyperplasia of smooth muscles. Life-threatening arrhythmia and the need for heart transplant have been reported in some GSD III patients. Hepatic symptoms in some patients may be so mild that the diagnosis is not made until adulthood, when the patients show symptoms and signs of neuromuscular disease.
The initial diagnosis has been confused with Charcot-Marie-Tooth disease (see Chapter 631.1 ). Polycystic ovaries are noted; some patients can develop hirsutism, irregular menstrual cycles, and other features of polycystic ovarian syndrome. Fertility does not appear to be affected; successful pregnancies have been reported.
Hypoglycemia and hyperlipidemia are common. In contrast to type I GSD, elevation of liver transaminase levels and fasting ketosis are prominent, but blood lactate and uric acid concentrations are usually normal. Glucagon administration 2 hr after a carbohydrate meal provokes a normal increase in blood glucose; after an overnight fast, however, glucagon may provoke no change in blood glucose level. Serum creatine kinase levels can be useful to identify patients with muscle involvement, although normal levels do not rule out muscle enzyme deficiency.
Diagnosis
The histologic appearance of the liver is characterized by a universal distention of hepatocytes by glycogen and the presence of fibrous septa. The fibrosis and the paucity of fat distinguish type III glycogenosis from type I. The fibrosis, which ranges from minimal periportal fibrosis to micronodular cirrhosis, appears in most cases to be nonprogressive. Overt cirrhosis has been seen in some patients with GSD III.
Patients with myopathy and liver symptoms have a generalized enzyme defect (type IIIa). The deficient enzyme activity can be demonstrated not only in liver and muscle, but also in other tissues such as heart, erythrocytes, and cultured fibroblasts. Patients with hepatic symptoms without clinical or laboratory evidence of myopathy have debranching enzyme deficiency only in the liver, with enzyme activity retained in the muscle (type IIIb). Before the availability of genetic testing, a definitive diagnosis required enzyme assay in liver, muscle, or both. Gene sequencing now allows for diagnosis and subtype assignment in the majority of patients.
Treatment
The mainstay of treatment of GSD III is dietary management, as in GSD I, although it is less demanding. Patients do not need to restrict dietary intake of fructose and galactose, although simple sugars should be avoided to prevent sudden spikes in blood glucose levels. Hypoglycemia is treated with small, frequent meals high in complex carbohydrates, such as cornstarch supplements or nocturnal gastric drip feedings. Additionally, a high-protein diet during the daytime as well as overnight protein enteral infusion is effective in preventing hypoglycemia. The exogenous protein can be used as a substrate for gluconeogenesis which helps to meet energy needs and prevent endogenous protein breakdown. Protein in the diet also reduces the overall starch requirement. Overtreatment with cornstarch should be avoided as it can result in excessive glycogen buildup, which is detrimental and can lead to excessive weight gain. MCT supplementation is being considered as an alternative source of energy. There is no satisfactory treatment for the progressive myopathy other than recommending a high-protein diet and a submaximal exercise program. Close monitoring with abdominal MRI is needed to detect progression of liver fibrosis to cirrhosis and further to HCC. Liver transplantation has been performed in GSD III patients with progressive cirrhosis and/or HCC. There are reports of cardiac transplant in GSD III patients with end stage cardiac disease.
Type IV Glycogen Storage Disease (Branching Enzyme Deficiency, Amylopectinosis, Polyglucosan Disease, or Andersen Disease)
Type IV GSD is caused by the deficiency of branching enzyme activity, which results in the accumulation of an abnormal glycogen with poor solubility. The disease is also known as amylopectinosis because the abnormal glycogen has fewer branch points, more α 1-4 linked glucose units, and longer outer chains, resulting in a structure resembling amylopectin. Accumulation of polyglucosan, which is positive on periodic acid–Schiff (PAS) and partially resistant to diastase digestion, is seen in all tissues of patients, but to different degrees.
Type IV GSD is an autosomal recessive disorder. The glycogen branching enzyme (GBE) gene is located on chromosome 3p21. More than 20 pathogenic variants responsible for type IV GSD have been identified, and their characterization in individual patients can be useful in predicting clinical outcome. The nearly complete absence of GBE activity with null variants has been associated with perinatal death and fatal neonatal hypotonia. Residual GBE enzyme activity >5% and presence of at least 1 missense variant are associated with a nonlethal hepatic cirrhosis phenotype and, in some situations, a lack of progressive liver disease.
Clinical Manifestations
There is a high degree of clinical variability associated with type IV GSD. The most common and classic form is characterized by progressive cirrhosis of the liver and manifests in the 1st 18 mo of life as hepatosplenomegaly and failure to thrive. Cirrhosis may present with portal hypertension, ascites, and esophageal varices and may progress to liver failure, usually leading to death by 5 yr of age. Rare patients survive without progression of liver disease; they have a milder hepatic form and do not require a liver transplant. Extrahepatic involvement in some patients with GSD IV consists of musculoskeletal involvement, particularly cardiac and skeletal muscles, as well as central nervous system (CNS) involvement.
A neuromuscular form of type IV GSD has been reported, with 4 main variants recognized based on age at presentation. The perinatal form is characterized by a fetal akinesia deformation sequence (FADS) and death in the perinatal period. The congenital form presents at birth with severe hypotonia, muscle atrophy, and neuronal involvement, with death in the neonatal period; some patients have cardiomyopathy. The childhood form presents primarily with myopathy or cardiomyopathy. The adult form, adult polyglucosan body disease (APBD), presents as an isolated myopathy or with diffuse CNS and peripheral nervous system dysfunction, accompanied by accumulation of polyglucosan material in the nervous system. Symptoms of neuronal involvement include peripheral neuropathy, neurogenic bladder, and leukodystrophy, as well as mild cognitive decline in some patients. For APBD, a leukocyte or nerve biopsy is needed to establish the diagnosis because branching enzyme deficiency is limited to those tissues.
Diagnosis
Deposition of amylopectin-like materials can be demonstrated in liver, heart, muscle, skin, intestine, brain, spinal cord, and peripheral nerve in type IV GSD. Liver histology shows micronodular cirrhosis and faintly stained basophilic inclusions in the hepatocytes. The inclusions are composed of coarsely clumped, stored material that is PAS positive and partially resistant to diastase digestion. Electron microscopy (EM) shows, in addition to the conventional α and β glycogen particles, accumulation of the fibrillar aggregations that are typical of amylopectin. The distinct staining properties of the cytoplasmic inclusions, as well as EM findings, could be diagnostic. However, polysaccharides with histologic features reminiscent of type IV disease, but without enzymatic correlation, have been observed. The definitive diagnosis rests on the demonstration of the deficient branching enzyme activity in liver, muscle, cultured skin fibroblasts, or leukocytes, or on the identification of pathogenic variants in the GBE gene. Prenatal diagnosis is possible by measuring enzyme activity in cultured amniocytes, chorionic villi, or DNA-based methodologies.
Treatment
There is no specific treatment for type IV GSD. Nervous system involvement, such as gait problems and bladder involvement, requires supportive, symptomatic management. Unlike patients with the other liver GSDs (I, III, VI, IX), those with GSD IV do not have hypoglycemia, which is only seen when there is overt liver cirrhosis. Liver transplantation has been performed for patients with progressive liver disease, but patients must be carefully selected as this is a multisystem disease, and in some patients, extrahepatic involvement may manifest after transplant. The long-term success of liver transplantation is unknown. Individuals with significant diffuse reticuloendothelial involvement may have greater risk for morbidity and mortality, which may impact the success rate for liver transplant.
Type VI Glycogen Storage Disease (Liver Phosphorylase Deficiency, Hers Disease)
Type VI GSD is caused by deficiency of liver glycogen phosphorylase . Relatively few patients are documented, likely because of underreporting of this disease. Patients usually present with hepatomegaly and growth retardation in early childhood. Hypoglycemia, hyperlipidemia, and hyperketosis are of variable severity. Ketotic hypoglycemia may present after overnight or prolonged fasting. Lactic acid and uric acid levels are normal. Type VI GSD presents within a broad spectrum of involvement, some with a more severe clinical presentation. Patients with severe hepatomegaly, recurrent severe hypoglycemia, hyperketosis, and postprandial lactic acidosis have been reported. Focal nodular hyperplasia of liver and hepatocellular adenoma with malignant transformation into carcinoma is reported in some patients. While cardiac muscle was thought to be unaffected, recently mild cardiomyopathy has been reported in a patient with GSD VI.
Treatment is symptomatic and aims to prevent hypoglycemia while ensuring adequate nutrition. A high-carbohydrate, high-protein diet and frequent feeding are effective in preventing hypoglycemia. Blood glucose and ketones should be monitored routinely, especially during periods of increased activity/illness. Long-term follow-up of these patients is needed to expand the understanding of the natural history of this disorder.
GSD VI is an autosomal recessive disease. Diagnosis can be confirmed through molecular testing of the liver phosphorylase gene (PYGL), which is found on chromosome 14q21-22 and has 20 exons. Many pathogenic variants are known in this gene; a splice-site variant in intron 13 has been identified in the Mennonite population. A liver biopsy showing elevated glycogen content and decreased hepatic phosphorylase enzyme activity can also be used to make a diagnosis. However, with the availability of DNA analysis and next-generation sequencing panels, liver biopsies are considered unnecessary.
Type IX Glycogen Storage Disease (Phosphorylase Kinase Deficiency)
Type IX GSD represents a heterogeneous group of glycogenoses. It results from deficiency of the enzyme phosphorylase kinase (PhK), which is involved in the rate-limiting step of glycogenolysis. This enzyme has 4 subunits (α, β, γ, δ), each encoded by different genes on different chromosomes and differentially expressed in various tissues. Pathogenic variants in the PHKA1 gene cause muscle PhK deficiency; pathogenic variants in the PHKA2 and PHKG2 genes cause liver PhK deficiency; pathogenic variants in the PHKB gene cause PhK deficiency in liver and muscle. Pathogenic variants in the PHKG1 gene have not been identified. Defects in subunits α, β, and γ are responsible for liver presentation.
Clinical manifestations of liver PhK deficiency are usually recognizable within the 1st 2 yr of life and include short stature and abdominal distention from moderate to marked hepatomegaly. The clinical severity of liver PhK deficiency varies considerably. Hyperketotic hypoglycemia, if present, can be mild but may be severe in some cases. Ketosis may occur even when glucose levels are normal. Some children may have mild delays in gross motor development and hypotonia. It is becoming increasingly clear that GSD IX is not a benign condition. Severe phenotypes are reported, with liver fibrosis progressing to cirrhosis and HCC, particularly in patients with PHKG2 variants. Progressive splenomegaly and portal hypertension are reported secondary to cirrhosis. Mild cardiomyopathy has been reported in a patient with GSD IX (PHKB variant). Cognitive and speech delays have been reported in a few individuals, but it is not clear whether these delays are caused by PhK deficiency or coincidental. Renal tubular acidosis has been reported in rare cases. Unlike in GSD I, lactic acidosis, bleeding tendency, and loose bowel movements are not characteristic. Although growth is retarded during childhood, normal height and complete sexual development are eventually achieved. As with debrancher deficiency, abdominal distention and hepatomegaly usually decrease with age and may disappear by adolescence. Most adults with liver PhK deficiency are asymptomatic, although further long-term studies are needed to fully assess the impact of this disorder in adults.
Phenotypic variability within each subtype is being uncovered with the availability of molecular testing. The incidence of all subtypes of PhK deficiency is approximately 1 : 100,000 live births.
X-Linked Liver Phosphorylase Kinase Deficiency (From PHKA2 Variants)
X-linked liver PhK deficiency is one of the most common forms of liver glycogenosis in males. In addition to liver, enzyme activity can also be deficient in erythrocytes, leukocytes, and fibroblasts; it is normal in muscle. Typically, a 1-5 yr old boy presents with growth retardation, an incidental finding of hepatomegaly, and a slight delay in motor development. Cholesterol, triglycerides, and liver enzymes are mildly elevated. Ketosis may occur after fasting. Lactate and uric acid levels are normal. Hypoglycemia is typically mild, if present, but can be severe. The response in blood glucose to glucagon is normal. Hepatomegaly and abnormal blood chemistries gradually improve and can normalize with age. Most adults achieve a normal final height and are usually asymptomatic despite a persistent PhK deficiency. It is increasingly being recognized that this disorder is not benign as previously thought, and there are patients with severe disease and long-term hepatic sequelae. In rare cases, liver fibrosis can occur and progress to cirrhosis.
Liver histology shows glycogen-distended hepatocytes, steatosis, and potentially mild periportal fibrosis. The accumulated glycogen (β particles, rosette form) has a frayed or burst appearance and is less compact than the glycogen seen in type I or III GSD. Fibrous septal formation and low-grade inflammatory changes may be seen.
The gene for the common liver isoform of the PhK α subunit, PHKA2, is located on the X chromosome (αL at Xp22.2). Mutations in the PHKA2 gene account for 75% of all PhK cases. X-linked liver PhK deficiency is further subdivided into 2 biochemical subtypes: XLG1, with measurable deficiency of PhK activity in both blood cells and liver, and XLG2, with normal in vitro PhK activity in blood cells and variable activity in liver. It is suspected that XLG2 may be caused by missense variants that affect enzyme regulation, whereas nonsense variants affecting the amount of protein result in XLG1. Female carriers are unaffected.
Autosomal Liver and Muscle Phosphorylase Kinase Deficiency (From PHKB Variants)
PhK deficiency in liver and blood cells with an autosomal recessive mode of inheritance has been reported. Similar to the X-linked form, chief symptoms in early childhood include hepatomegaly and growth retardation. Some patients also exhibit muscle hypotonia. In a few cases where enzyme activity has been measured, reduced PhK activity has been demonstrated in muscle. Mutations are found in PHKB (chromosome 16q12-q13), which encodes the β subunit, and result in liver and muscle PhK deficiency. Several nonsense variants, a single-base insertion, a splice-site mutation, and a large intragenic mutation have been identified. In addition, a missense variant was discovered in an atypical patient with normal blood cell PhK activity.
Autosomal Liver Phosphorylase Kinase Deficiency (From PHKG2 Variants)
This form of PhK deficiency is caused by pathogenic variants in the testis/liver isoform (TL) of the γ subunit gene (PHKG2). In contrast to X-linked PhK deficiency, patients with variants in PHKG2 typically have more severe phenotypes, with recurrent hypoglycemia, prominent hepatomegaly, significant liver fibrosis, and progressive cirrhosis. Liver involvement may present with cholestasis, bile duct proliferation, esophageal varices, and splenomegaly. Other reported presentations include delayed motor milestones, muscle weakness, and renal tubular damage. The spectrum of involvement continues to evolve as more cases are recognized. PHKG2 maps to chromosome 16p12.1-p11.2; many pathogenic variants are known for this gene.
Phosphorylase Kinase Deficiency Limited to Heart
These patients have been reported with cardiomyopathy in infancy and rapidly progress to heart failure and death. Recent studies have shown that this is not a case of cardiac-specific primary PhK deficiency as suspected previously, but rather linked to the γ2 subunit of adenosine monophosphate (AMP)–activated protein kinase (see later). The γ2 subunit is encoded by the PRKAG2 gene.
Diagnosis
PhK deficiency may be diagnosed by demonstration of the enzymatic defect in affected tissues. PhK can be measured in leukocytes and erythrocytes, but because the enzyme has many isozymes, the diagnosis can be easily missed without studies of liver, muscle, or heart. Individuals with liver PhK deficiency also usually have elevated transaminases, mildly elevated triglycerides and cholesterol, normal uric acid and lactic acid concentrations, and normal glucagon responses. Gene sequencing is used for diagnostic confirmation and subtyping of GSD IX.
The PHKA2 gene encoding the α subunit is most frequently involved, followed by the PHKB gene encoding the β subunit. Variants in the PHKG2 gene underlying γ-subunit deficiency are typically associated with severe liver involvement with recurrent hypoglycemia and liver fibrosis.
Treatment and Prognosis
The treatment for liver PhK deficiency is symptomatic. It includes a diet high in complex carbohydrates and proteins and small, frequent feedings to prevent hypoglycemia. Cornstarch can be administered with symptom-dependent dosage and timing (0.6-2.5 g/kg every 6 hr). Oral intake of glucose, if tolerated, should be used to treat hypoglycemia. If not, IV glucose should be given.
Prognosis for the X-linked and certain autosomal forms is typically good; however, long term complications are being recognized. Patients with mutations in the γ subunit typically have a more severe clinical course with progressive liver disease. Liver involvement needs to be monitored in all patients with GSD IX by periodic imaging (abdominal ultrasound or MRI every 6-12 mo) and serial hepatic function tests.
Liver Glycogen Synthase Deficiency
Liver glycogen synthase deficiency type 0 (GSD 0 ) is caused by deficiency of hepatic glycogen synthase (GYS2) activity, leading to a marked decrease of glycogen stored in the liver. The gene GYS2 is located at 12p12.2. Several pathogenic variants have been identified in patients with GSD 0. The disease appears to be rare in humans, and in the true sense, this is not a type of GSD because the deficiency of the enzyme leads to decreased glycogen stores. Patients present in infancy with early-morning (prebreakfast) drowsiness, pallor, emesis, and fatigue and sometimes convulsions associated with hypoglycemia and hyperketonemia. Blood lactate and alanine levels are low, and there is no hyperlipidemia or hepatomegaly. Prolonged hyperglycemia , glycosuria, lactic acidosis, and hyperalaninemia, with normal insulin levels after administration of glucose or a meal, suggest a deficiency of glycogen synthase. Definitive diagnosis may be by a liver biopsy to measure the enzyme activity or identification of pathogenic variants in GYS2 .
Treatment consists of frequent meals, rich in protein and nighttime supplementation with uncooked cornstarch to prevent hypoglycemia and hyperketonemia. Most children with GSD 0 are cognitively and developmentally normal. Short stature and osteopenia are common features. The prognosis seems good for patients who survive to adulthood, including resolution of hypoglycemia, except during pregnancy.
Hepatic Glycogenosis With Renal Fanconi Syndrome (Fanconi-Bickel Syndrome)
Fanconi-Bickel Syndrome is a rare autosomal recessive disorder is caused by defects in the facilitative glucose transporter 2 (GLUT-2), which transports glucose in and out of hepatocytes, pancreatic β cells, and the basolateral membranes of intestinal and renal epithelial cells. The disease is characterized by proximal renal tubular dysfunction, impaired glucose and galactose utilization, and accumulation of glycogen in liver and kidney.
The affected child typically presents in the 1st yr of life with failure to thrive, rickets, and a protuberant abdomen from hepatomegaly and nephromegaly. The disease may be confused with GSD I because a Fanconi-like syndrome can also develop in type I patients. Adults typically present with short stature, dwarfism, and excess fat in the abdomen and shoulders. Patients are more susceptible to fractures because of early-onset generalized osteopenia. In addition, intestinal malabsorption and diarrhea may occur.
Laboratory findings include glucosuria, phosphaturia, generalized aminoaciduria, bicarbonate wasting, hypophosphatemia, increased serum alkaline phosphatase levels, and radiologic findings of rickets. Mild fasting hypoglycemia and hyperlipidemia may be present. Liver transaminase, plasma lactate, and uric acid levels are usually normal. Oral galactose or glucose tolerance tests show intolerance, which could be explained by the functional loss of GLUT-2 preventing liver uptake of these sugars. Tissue biopsy results show marked accumulation of glycogen in hepatocytes and proximal renal tubular cells, presumably from the altered glucose transport out of these organs. Diffuse glomerular mesangial expansion along with glomerular hyperfiltration and microalbuminuria similar to nephropathy in GSD Ia and diabetes have been reported.
This condition is rare, and 70% of patients with Fanconi-Bickel syndrome have consanguineous parents. Most patients have homozygous pathogenic variants; some patients are compound heterozygotes. The majority of variants detected thus far predict a premature termination of translation. The resulting loss of the C-terminal end of the GLUT-2 protein predicts a nonfunctioning glucose transporter with an inward-facing substrate-binding site.
There is no specific treatment. Symptom-dependent treatment with phosphate and bicarbonate can result in growth improvement. Growth may also improve with symptomatic replacement of water, electrolytes, and vitamin D; restriction of galactose intake; and a diet similar to that used for diabetes mellitus, with small, frequent meals and adequate caloric intake.
Muscle Glycogenoses
The role of glycogen in muscle is to provide substrates for the generation of ATP for muscle contraction. The muscle GSDs are broadly divided into 2 groups. The first group is characterized by hypertrophic cardiomyopathy, progressive skeletal muscle weakness and atrophy, or both, and includes deficiencies of acid α-glucosidase , a lysosomal glycogen-degrading enzyme (type II GSD), lysosomal-associated membrane protein 2 (LAMP2 ), and AMP-activated protein kinase γ2 (PRKAG2 ). The 2nd group comprises muscle energy disorders characterized by muscle pain, exercise intolerance, myoglobinuria, and susceptibility to fatigue. This group includes myophosphorylase deficiency (McArdle disease, type V GSD) and deficiencies of phosphofructokinase (type VII ), phosphoglycerate kinase, phosphoglycerate mutase, lactate dehydrogenase, and muscle-specific phosphorylase kinase. Some of these latter enzyme deficiencies can also be associated with compensated hemolysis , suggesting a more generalized defect in glucose metabolism.
Type II Glycogen Storage Disease (Lysosomal Acid α-1,4-Glucosidase Deficiency, Pompe Disease)
Pompe disease, also referred to as GSD type II or acid maltase deficiency , is caused by a deficiency of acid α-1,4-glucosidase (acid maltase), an enzyme responsible for the degradation of glycogen in lysosomes. This enzyme defect results in lysosomal glycogen accumulation in multiple tissues and cell types, predominantly affecting cardiac, skeletal, and smooth muscle cells. In Pompe disease, glycogen typically accumulates within lysosomes, as opposed to its accumulation in cytoplasm in the other glycogenoses. However, as the disease progresses, lysosomal rupture and leakage lead to the presence of cytoplasmic glycogen as well.
Pompe disease is an autosomal recessive disorder. The incidence was thought to be approximately 1 in 40,000 live births in Caucasians and 1 in 18,000 live births in Han Chinese. Newborn screening for Pompe disease in the United States suggests that the prevalence is much higher than previously thought (between 1 in 9,132 and 1 in 24,188). The gene for acid α-glucosidase (GAA) is on chromosome 17q25.2. More than 500 pathogenic variants have been identified that could be helpful in delineating the phenotypes. A splice-site variant (IVS1-13T→G; c.-32-13T>G) is commonly seen in late-onset Caucasian patients.
Clinical Manifestations
Pompe disease is broadly classified into infantile and late-onset forms. Infantile Pompe disease (IPD) is uniformly lethal without enzyme replacement therapy (ERT) with alglucosidase alfa. Affected infants present in the 1st day to weeks of life with hypotonia, generalized muscle weakness with a floppy infant appearance, neuropathic bulbar weakness, feeding difficulties, macroglossia, hepatomegaly, and hypertrophic cardiomyopathy, which if untreated leads to death from cardiorespiratory failure or respiratory infection, usually by 1 yr of age.
Late-onset Pompe disease (LOPD; juvenile-, childhood-, and adult-onset disease) is characterized by proximal limb girdle muscle weakness and early involvement of respiratory muscles, especially the diaphragm. Cardiac involvement ranges from cardiac rhythm disturbances to cardiomyopathy and a less severe, short-term prognosis. Symptoms related to progressive dysfunction of skeletal muscles can start as early as within 1 yr of age to as late as the 6th decade of life. The clinical picture is dominated by slowly progressive proximal muscle weakness with truncal involvement and greater involvement of the lower limbs than the upper limbs. The pelvic girdle, paraspinal muscles, and diaphragm are the muscle groups most seriously affected in patients with LOPD. Other symptoms may include lingual weakness, ptosis, and dilation of blood vessels (e.g., basilar artery, ascending aorta). With disease progression, patients become confined to a wheelchair and require artificial ventilation. The initial symptoms in some patients may be respiratory insufficiency manifested by somnolence, morning headache, orthopnea, and exertional dyspnea, which eventually lead to sleep-disordered breathing and respiratory failure. Respiratory failure is the cause of significant morbidity and mortality in LOPD. Basilar artery aneurysms with rupture also contribute to mortality in some cases. Small-fiber neuropathy presenting as painful paresthesia has been identified in some LOPD patients. Gastrointestinal disturbances such as postprandial bloating, dysphagia, early satiety, diarrhea, chronic constipation, and irritable bowel disease have been reported. Genitourinary tract involvement is not uncommon and may present as bladder and bowel incontinence, weak urine stream or dribbling. If untreated, the age of death varies from early childhood to late adulthood, depending on the rate of disease progression and the extent of respiratory muscle involvement. With the advent of ERT, a new natural history is emerging for both survivors of infantile and LOPD.
Laboratory Findings
These include elevated levels of serum creatine kinase (CK), aspartate transaminase (AST), alanine transaminase (ALT), and lactate dehydrogenase (LDH). Urine glucose tetrasaccharide, a glycogen breakdown metabolite, is a reliable biomarker for disease severity and treatment response. In the infantile form a chest x-ray film showing massive cardiomegaly is frequently the first symptom detected. Electrocardiographic findings include a high-voltage QRS complex, Wolff-Parkinson-White (WPW) syndrome, and a shortened PR interval. Echocardiography reveals thickening of both ventricles and/or the intraventricular septum and/or left ventricular outflow tract obstruction. Muscle biopsy shows the presence of vacuoles that stain positively for glycogen; acid phosphatase is increased, presumably from a compensatory increase of lysosomal enzymes. EM reveals glycogen accumulation within a membranous sac and in the cytoplasm. Electromyography reveals myopathic features with excessive electrical irritability of muscle fibers and pseudomyotonic discharges. Serum CK is not always elevated in adult patients. Depending on the muscle sampled or tested, the muscle histologic appearance and electromyography may not be abnormal.
Some patients with infantile Pompe disease who had peripheral nerve biopsies demonstrated glycogen accumulation in the neurons and Schwann cells.
Diagnosis
Diagnosis of Pompe disease can be made by enzyme assay in dried blood spots, leukocytes, blood mononuclear cells, muscle, or cultured skin fibroblasts demonstrating deficient acid α-glucosidase activity. Gene sequencing showing 2 pathogenic variants in the GAA gene is confirmatory. The enzyme assay should be done in a laboratory with experience using maltose, glycogen, or 4-methylumbelliferyl-α-D -glucopyranoside (4MUG) as a substrate. The infantile form has a more severe enzyme deficiency than the late-onset forms. Detection of percent residual enzyme activity is captured in skin fibroblasts and muscle. Blood-based assays, especially dried blood spots, have the advantage of a rapid turnaround time and are being increasingly used as the first-line tissue to make a diagnosis. A muscle biopsy is often done with suspected muscle disease and a broad differential; it yields faster results and provides additional information about glycogen content and site of glycogen storage within and outside the lysosomes of muscle cells. However, a normal muscle biopsy does not exclude a diagnosis of Pompe disease. Late-onset patients show variability in glycogen accumulation in different muscles and within muscle fibers; muscle histology and glycogen content can vary depending on the site of muscle biopsy. There is also a high risk from anesthesia in infantile patients. An electrocardiogram can be helpful in making the diagnosis in suspected cases of the infantile form and should be done for patients suspected of having Pompe disease before any procedure requiring anesthesia, including muscle biopsy, is performed. Urinary glucose tetrasaccharides can be elevated in the urine of affected patients, and levels are extremely high in infantile patients. Availability of next-generation sequencing panels and whole exome sequencing allows for identification of additional patients with Pompe disease, especially when the diagnosis is ambiguous. Prenatal diagnosis using amniocytes or chorionic villi is available.
Treatment
Enzyme replacement therapy with recombinant human acid α-glucosidase (alglucosidase alfa) is available for treatment of Pompe disease. Recombinant acid α-glucosidase is capable of preventing deterioration or reversing abnormal cardiac and skeletal muscle functions (Fig. 105.3 ). ERT should be initiated as soon as possible across the disease spectrum, especially for babies with the infantile form, because the disease is rapidly progressive. Infants who are negative for cross-reacting immunologic material (CRIM) develop a high-titer antibody against the infused enzyme and respond to the ERT less favorably. Treatment using immunomodulating agents such as methotrexate, rituximab, and intravenous immune globulin (IVIG) have demonstrated efficacy in preventing the development of an immune response to ERT and immune tolerance. Nocturnal ventilatory support, when indicated, should be used; it has been shown to improve the quality of life and is particularly beneficial during a period of respiratory decompensation.
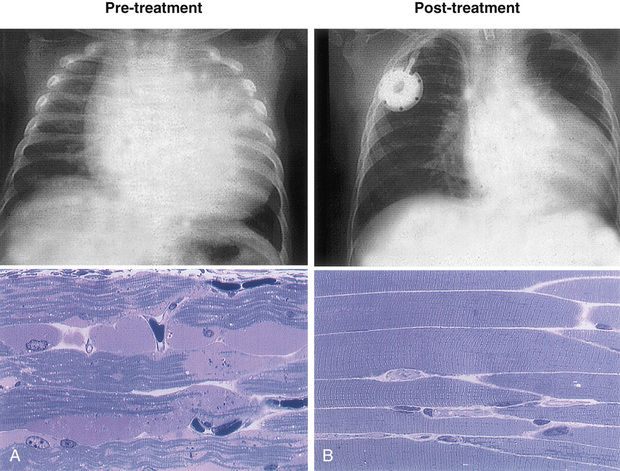
In addition to ERT, other adjunctive therapies have demonstrated benefit in Pompe patients. For patients with the late-onset disease, a high-protein diet may be beneficial. Respiratory muscle strength training has demonstrated improvements in respiratory parameters when combined with ERT. Submaximal exercise regimens are of assistance to improve muscle strength, pain, and fatigue. Other approaches are under clinical development to improve the safety and efficacy of enzyme delivery to affected tissues. These include use of chaperone molecules to enhance rhGAA delivery, and neoGAA, which is a second-generation ERT with a high number of mannose-6-phosphate (M6P) tags that enhances M6P receptor targeting and enzyme uptake. Gene therapy studies to correct the endogenous enzyme production pathways have shown promise.
Early diagnosis and treatment are necessary for optimal outcomes. Newborn screening using blood-based assays in Taiwan has resulted in early identification of Pompe cases and thus improved disease outcomes through the early initiation of ERT.
Glycogen Storage Diseases Mimicking Hypertrophic Cardiomyopathy (Danon Disease)
Danon disease is caused by pathogenic variants in the LAMP2 gene, which leads to a deficiency of lysosomal-associated membrane protein 2 (LAMP2). This leads to accumulation of glycogen in the heart and skeletal muscle, which presents primarily with hypertrophic cardiomyopathy and skeletal muscle weakness. Danon disease can be distinguished from the usual causes of hypertrophic cardiomyopathy (defects in sarcomere-protein genes) by their electrophysiologic abnormalities, particularly ventricular preexcitation and conduction defects. Patients present with cardiac symptoms, including chest pain, palpitations, syncope, and cardiac arrest, usually between ages 8 and 15 yr. Other clinical manifestations in Danon disease include peripheral pigmentary retinopathy, lens changes, and abnormal electroretinograms. This disorder is inherited in an X-linked dominant pattern. Diagnosis can be done by genetic testing for the LAMP2 gene. The prognosis for LAMP2 deficiency is poor, with progressive end-stage heart failure early in adulthood. Treatment is directed toward management of symptoms in affected individuals, including management of cardiomyopathy, correction of arrhythmias, and physical therapy for muscle weakness. Cardiac transplantation has been tried successfully in some patients.
Adenosine Monophosphate–Activated Protein Kinase γ2 Deficiency (PRKAG2 Deficiency)
AMP-activated protein kinase γ2 (PRKAG2) deficiency is caused by pathogenic variants in the PRKAG2 gene mapped to chromosome 7q36. PRKAG2 is required for the synthesis of the enzyme AMP-activated protein kinase (AMPK), which regulates cellular pathways involved in ATP metabolism. Common presentations include hypertrophic cardiomyopathy and electrophysiologic abnormalities such as WPW syndrome, atrial fibrillation, and progressive atrioventricular block. Cardiac involvement is variable and includes supraventricular tachycardia, sinus bradycardia, left ventricular dysfunction, and even sudden cardiac death in some cases. In addition to cardiac involvement, there is a broad spectrum of phenotypic presentations including myalgia, myopathy, and seizures. Cardiomyopathy caused by PRKAG2 variants usually allows for long-term survival, although a rare congenital form presenting in early infancy is associated with a rapidly fatal course. Cardiomyopathy in PRKAG2 syndrome often mimics that in other conditions, especially Pompe disease, and should be considered as a differential diagnosis in infants presenting with severe hypertrophic cardiomyopathy. Treatment is primarily symptomatic, including management of cardiac failure and correction of conduction defects.
Muscle Glycogen Synthase Deficiency
This GSD results from muscle glycogen synthase (glycogen synthase I , GYS1) deficiency. The gene GYS1 has been localized to chromosome 19q13.3. In the true sense, this is not a type of GSD because the deficiency of the enzyme leads to decreased glycogen stores. The disease is extremely rare and has been reported in 3 children of consanguineous parents of Syrian origin. Muscle biopsies showed lack of glycogen, predominantly oxidative fibers, and mitochondrial proliferation. Glucose tolerance was normal. Molecular study revealed a homozygous stop mutation (R462→ter) in the muscle glycogen synthase gene. The phenotype was variable in the 3 siblings and ranged from sudden cardiac arrest, muscle fatigability, hypertrophic cardiomyopathy, an abnormal heart rate, and hypotension while exercising, to mildly impaired cardiac function at rest.
Late-Onset Polyglucosan Body Myopathy (From GYG1 Variants)
Late-onset polyglucosan body myopathy is an autosomal recessive, slowly progressive skeletal myopathy caused by pathogenic variants in the GYG1 gene blocking glycogenin-1 biosynthesis. There is a reduced or complete absence of glyogenin-1, which is a precursor necessary for glycogen formation. Polyglucosan accumulation in skeletal muscles causes adult-onset proximal muscle weakness, prominently affecting hip and shoulder girdles. Cardiac involvement is not seen. Compared with GSD IV–APBD, nervous system involvement is uncommon, although polyglucosan deposition is seen in both disorders. GYG1 is mapped to chromosome 3q24. Muscle biopsies show PAS-positive storage material in 30–40% of muscle fibers. EM reveals the typical polyglucosan structure, consisting of ovoid form composed of partly filamentous material.
Type V Glycogen Storage Disease (Muscle Phosphorylase Deficiency, McArdle Disease)
GSD type V is caused by deficiency of myophosphorylase activity. Lack of this enzyme limits muscle ATP generation by glycogenolysis, resulting in muscle glycogen accumulation, and is the prototype of muscle energy disorders. A deficiency of myophosphorylase impairs the cleavage of glucosyl molecules from the straight chain of glycogen.
Clinical Manifestations
Symptoms usually first develop in late childhood or in the 2nd decade of life. Clinical heterogeneity is uncommon, but cases suggesting otherwise have been documented. Studies have shown that McArdle disease can manifest in individuals as old as 74, as well as in infancy in a fatal, early-onset form characterized by hypotonia, generalized muscle weakness, and respiratory complication. Symptoms are generally characterized by exercise intolerance with muscle cramps and pain. Symptoms are precipitated by 2 types of activity: brief, high-intensity exercise, such as sprinting or carrying heavy loads, and less intense but sustained activity, such as climbing stairs or walking uphill. Most patients can perform moderate exercise, such as walking on level ground, for long periods. Many patients experience a characteristic “second wind” phenomenon, with relief of muscle pain and fatigue after a brief period of rest. As a result of the underlying myopathy, these patients may be at risk for statin-induced myopathy and rhabdomyolysis. While patients typically experience episodic muscle pain and cramping from exercise, 35% of patients with McArdle disease report permanent pain that has a serious impact on sleep and other activities. Studies also suggest that there may also be a link between GSD V and variable cognitive impairment.
Approximately 50% of patients report burgundy-colored urine after exercise as a result of exercise-induced myoglobinuria secondary to rhabdomyolysis . Excessive myoglobinuria after intense exercise may precipitate acute renal failure.
Lab findings show elevated levels of serum CK at rest, which further increases after exercise. Exercise also elevates the levels of blood ammonia, inosine, hypoxanthine, and uric acid, which may be attributed to accelerated recycling of muscle purine nucleotides caused by insufficient ATP production. Type V GSD is an autosomal recessive disorder. The gene for muscle phosphorylase (PYGM) has been mapped to chromosome 11q13.
Diagnosis
The standard diagnosis for GSD V includes a muscle biopsy to measure glycogen content as well as enzyme and sequencing of PYGM . An ischemic exercise test offers a rapid diagnostic screening for patients with a metabolic myopathy. Lack of an increase in blood lactate levels and exaggerated blood ammonia elevations indicate muscle glycogenosis and suggest a defect in the conversion of muscle glycogen or glucose to lactate. The abnormal ischemic exercise response is not limited to type V GSD. Other muscle defects in glycogenolysis or glycolysis produce similar results (deficiencies of muscle phosphofructokinase, phosphoglycerate kinase, phosphoglycerate mutase, or LDH). An ischemic exercise test was once used to be a rapid diagnostic screening for suspected patients but was associated with severe complications and false-positive results. A nonischemic forearm exercise test with high sensitivity that is easy to perform and cost-effective has been determined to be indicative of muscle glycogenosis. However, as with the ischemic test, it cannot differentiate between abnormal exercise responses due to type V disease versus other defects in glycogenolysis or glycolysis or debranching enzyme (noted when the test is done after fasting).
The diagnosis is confirmed by molecular genetic testing of PYGM. A common nonsense variant, p.R49X in exon 1, is found in 90% of Caucasian patients, and a deletion of a single codon in exon 17 is found in 61% of Japanese patients. The p.R49X variant represents 55% of alleles in Spanish patients, whereas the p.W797R variant represents 14% and the p.G204S 9% of pathogenic alleles in the Spanish population. There seems to be an association between clinical severity of GSD V and presence of the D allele of the ACE insertion/deletion polymorphism. This may help explain the spectrum of phenotypic variability manifested in this disorder.
Treatment
Avoidance of strenuous exercise prevents the symptoms; regular and moderate exercise is recommended to improve exercise capacity. Glucose or sucrose given before exercise or injection of glucagon can greatly improve tolerance in these patients. A high-protein diet may increase muscle endurance, and low-dose creatine supplement has been shown to improve muscle function in some patients. The clinical response to creatine is dose dependent; muscle pain may increase on high doses of creatine supplementation. Vitamin B6 supplementation reduces exercise intolerance and muscle cramps. Longevity is not generally affected.
Type VII Glycogen Storage Disease (Muscle Phosphofructokinase Deficiency, Tarui Disease)
Type VII GSD is caused by pathogenic variants in the PFKM gene, located on chromosome 12q13.1, which results in a deficiency of muscle phosphofructokinase enzyme. This enzyme is a key regulatory enzyme of glycolysis and is necessary for the ATP-dependent conversion of fructose-6-phosphate to fructose-1,6-diphosphate. Phosphofructokinase is composed of 3 isoenzyme subunits according to the tissue type and are encoded by different genes: (PFKM [M: muscle], PFKL [L: liver], and PFKP [P: platelet]). Skeletal muscle has only the M subunit, whereas red blood cells (RBCs) express a hybrid of L and M forms. In type VII GSD the M isoenzyme is defective, resulting in complete deficiency of enzyme activity in muscle and a partial deficiency in RBCs.
Type VII GSD is an autosomal recessive disorder with increased prevalence in individuals of Japanese ancestry and Ashkenazi Jews. A splicing defect and a nucleotide deletion in PFKM account for 95% of pathogenic variants in Ashkenazi Jews. Diagnosis based on molecular testing for the common variants is thus possible in this population.
Clinical Manifestations
Although the clinical picture is similar to that of type V GSD, the following features of type VII GSD are distinctive:
- 1. Exercise intolerance, which usually commences in childhood, is more severe than in type V disease and may be associated with nausea, vomiting, and severe muscle pain; vigorous exercise causes severe muscle cramps and myoglobinuria.
- 2. Compensatory hemolysis occurs, as indicated by an increased level of serum bilirubin and an elevated reticulocyte count.
- 3. Hyperuricemia is common and exaggerated by muscle exercise to a greater degree than that observed in type V or III GSD.
- 4. An abnormal polysaccharide is present in muscle fibers; it is PAS positive but resistant to diastase digestion.
- 5. Exercise intolerance is especially worse after carbohydrate-rich meals because the ingested glucose prevents lipolysis, thereby depriving muscle of fatty acid and ketone substrates. This is in contrast to patients with type V disease, who can metabolize blood borne glucose derived from either endogenous liver glycogenolysis or exogenous glucose; indeed, glucose infusion improves exercise tolerance in type V patients.
- 6. The “second wind” phenomenon is absent because of the inability to break down blood glucose.
Several rare type VII variants occur. One variant presents in infancy with hypotonia and limb weakness and proceeds to a rapidly progressive myopathy that leads to death by 4 yr of age. A 2nd variant occurs in infancy and results in congenital myopathy and arthrogryposis with a fatal outcome. A 3rd variant presents in infancy with hypotonia, mild developmental delay, and seizures. An additional presentation is hereditary nonspherocytic hemolytic anemia . Although these patients do not experience muscle symptoms, it remains unclear whether these symptoms will develop later in life. One variant presents in adults and is characterized by a slowly progressive, fixed muscle weakness rather than cramps and myoglobinuria. It may also cause mitral valve thickening from glycogen buildup.
Diagnosis
To establish a diagnosis, a biochemical or histochemical demonstration of the enzymatic defect in the muscle is required. The absence of the M isoenzyme of phosphofructokinase can also be demonstrated in muscle, blood cells, and fibroblasts. Gene sequencing can identify pathogenic variants for the phosphofructokinase gene.
Treatment
There is no specific treatment. Strenuous exercise should be avoided to prevent acute episodes of muscle cramps and myoglobinuria. Consuming simple carbohydrates before strenuous exercise may benefit by improving exercise tolerance. A ketogenic diet has been reported to show clinical improvement in a patient with infantile GSD VII. Drugs such as statins should be avoided. Precautionary measures should be taken to avoid hyperthermia while undergoing anesthesia. Carbohydrate meals and glucose infusions have demonstrated worsening symptoms because of the body's inability to utilize glucose. The administered glucose tends to lower the levels of fatty acids in the blood, a primary source of muscle fuel.
Muscle-Specific Phosphorylase Kinase Deficiency (From PHKA1 Variants)
A few cases of PhK deficiency restricted to muscle are known. Patients, both male and female, present either with muscle cramps and myoglobinuria with exercise or with progressive muscle weakness and atrophy. PhK activity is decreased in muscle but normal in liver and blood cells. There is no hepatomegaly or cardiomegaly. This is inherited in an X-linked or autosomal recessive manner. The gene for the muscle-specific form α subunit (αM) is located at Xq12. Pathogenic variants of the gene have been found in some male patients with this disorder. The gene for muscle γ subunit (γM, PHKG1 ) is on chromosome 7p12. No pathogenic variants in this gene have been reported so far.
Other Muscle Glycogenoses With Muscle Energy Impairment
Six additional defects in enzymes—phosphoglycerate kinase, phosphoglycerate mutase, lactate dehydrogenase, fructose-1,6-bisphosphate aldolase A, muscle pyruvate kinase, and β-enolase in the pathway of the terminal glycolysis—cause symptoms and signs of muscle energy impairment similar to those of types V and VII GSD. The failure of blood lactate to increase in response to exercise is a useful diagnostic test and can be used to differentiate muscle glycogenoses from disorders of lipid metabolism, such as carnitine palmitoyltransferase II deficiency and very-long-chain acyl-CoA dehydrogenase deficiency, which also cause muscle cramps and myoglobinuria. Muscle glycogen levels can be normal in the disorders affecting terminal glycolysis, and assaying the muscle enzyme activity is needed to make a definitive diagnosis. There is no specific treatment (see preceding Treatment section).
Bibliography
Akman HO, Aykit Y, Amuk OC, et al. Late-onset polyglucosan body myopathy in five patients with a homozygous mutation in GYG1 . Neuromuscul Disord . 2016;26(1):16–20.
Byrne BJ, Falk DJ, Pacel CA, et al. Pompe disease gene therapy. Hum Mol Genet . 2011;20:R61–R68.
Case LE, Beckemeyer AA, Kishnani PS. Infantile Pompe disease on ERT—update on clinical presentation, musculoskeletal management, and exercise considerations. Am J Med Genet C Semin Med Genet . 2012;160C:69–79.
Chiang SC, Hwu WL, Lee NC, et al. Algorithm for Pompe disease newborn screening: results from the Taiwan screening program. Mol Genet Metab . 2012;106(3):281–286.
Chien YH, Lee NC, Huang HJ, et al. Later-onset Pompe disease: early detection and early treatment initiation enabled by newborn screening. J Pediatr . 2011;158:1023–1027.
D'Souza R, Levandowski C, Slavov D, et al. Danon disease: clinical features, evaluation, and management. Circ Heart Fail . 2014;7(5):843–849.
DiMauro S, Spiegel R. Progress and problems in muscle glycogenoses. Acta Myol . 2011;30(2):96–102.
Elder ME, Nayak S, Collins SW, et al. B-cell depletion and immunomodulation before initiation of enzyme replacement therapy blocks the immune response to acid alpha-glucosidase in infantile-onset Pompe disease. J Pediatr . 2013;163:847–854.
Hopkins PV, Campbell C1, Klug T, et al. Lysosomal storage disorder screening implementation: findings from the first six months of full population pilot testing in Missouri. J Pediatr . 2015;166(1):172–177.
Kishnani PS, Austin SL, Abdenur JE, et al. Diagnosis and management of glycogen storage disease type I: a practice guideline of the American College of Medical Genetics and Genomics. Genet Med . 2014;16(11):e1.
Kishnani PS, Austin SL, Arn P, et al. Glycogen storage disease type III diagnosis and management guidelines. Genet Med . 2010;12(7):446–463.
Kronn DF, Day-Salvatore D, Hwu WL, et al. Management of confirmed newborn-screened patients with Pompe disease across the disease spectrum. Pediatrics . 2016;140(1):e20160280.
Maga JA, Zhou J, Kambampati R, et al. Glycosylation-independent lysosomal targeting of acid α-glucosidase enhances muscle glycogen clearance in Pompe mice. J Biol Chem . 2013;288(3):1428–1438.
Miteff F, Potter HC, Allen J, et al. Clinical and laboratory features of patients with myophosphorylase deficiency (McArdle disease). J Clin Neurosci . 2011;18(8):1055–1058.
Poelman E, Hoogeveen-Westerveld M, Kross-de Haan MA, et al. High sustained antibody titers in patients with classic infantile Pompe disease following immunomodulation at start of enzyme replacement therapy. J Pediatr . 2018;195:236–243.
Porto A, Brun F, Severini G, et al. Clinical spectrum of PRKAG2 syndrome. Circ Arrhythm Electrophysiol . 2016;9(1):e003121.
Rimoin DL, Connor JM, Pyeritz RE, et al. Emery and Rimoin's principles and practice of medical genetics . Churchill Livingstone Elsevier: New York; 2011.
Roscher A, Patel J, Hewson S, et al. The natural history of glycogen storage disease types VI and IX: long-term outcome from the largest metabolic center in Canada. Mol Genet Metab . 2014;113(3):171–176.
Valayannopoulos V, Bajolle F, Arnoux JB, et al. Successful treatment of severe cardiomyopathy in glycogen storage disease type III with D ,L -3-hydroxybutyrate, ketogenic and high-protein diet. Pediatr Res . 2011;70:638–641.
Valle D, Beaudet AL, Vogelstein B, et al. Online metabolic and molecular bases of inherited disease . [updated 2011] http://www.ommbid.com ; 2006.
Wang DQ, Fiske LM, Carreras CT, et al. Natural history of hepatocellular adenoma formation in glycogen storage disease type 1. J Pediatr . 2011;159:442–446.
Yang CF, Yang CC, Liao HC, et al. Very early treatment for infantile-onset Pompe disease contributes to better outcomes. J Pediatr . 2016;169:174–180.
Defects in Galactose Metabolism
Priya S. Kishnani, Yuan-Tsong Chen
Milk and dairy products contain lactose , the major dietary source of galactose. The metabolism of galactose produces fuel for cellular metabolism through its conversion to glucose-1-phosphate (see Table 105.1 ). Galactose also plays an important role in the formation of galactosides, which include glycoproteins, glycolipids, and glycosaminoglycans. Galactosemia denotes the elevated level of galactose in the blood and is found in 3 distinct inborn errors of galactose metabolism in 1 of the following enzymes: galactose-1-phosphate uridyl transferase, galactokinase, and uridine diphosphate galactose-4-epimerase. The term galactosemia, although adequate for the deficiencies in any of these disorders, generally designates the transferase deficiency.
Galactose-1-Phosphate Uridyl Transferase Deficiency Galactosemia
Two forms of the deficiency exist: infants with complete or near-complete deficiency of the enzyme (classic galactosemia) and those with partial transferase deficiency. Classic galactosemia is a serious disease with onset of symptoms typically by the 2nd half of the 1st wk of life. The incidence is predicted to be 1 in 60,000 live births. The newborn infant receives high amounts of lactose (up to 40% in breast milk and certain formulas), which consists of equal parts of glucose and galactose. Without the transferase enzyme, the infant is unable to metabolize galactose-1-phosphate, the accumulation of which results in injury to kidney, liver, and brain. This injury may begin prenatally in the affected fetus by transplacental galactose derived from the diet of the heterozygous mother or by endogenous production of galactose in the fetus.
Clinical Manifestations
The diagnosis of uridyl transferase deficiency should be considered in newborn or young infants with any of the following features within a few days or weeks after birth: jaundice, hepatomegaly, vomiting, hypoglycemia, seizures, lethargy, irritability, feeding difficulties, poor weight gain or failure to regain birthweight, and aminoaciduria. Untreated children may show nuclear cataracts, vitreous hemorrhage, hepatic failure, cirrhosis, ascites, splenomegaly, or intellectual disability. Patients with galactosemia are at increased risk for Escherichia coli neonatal sepsis; the onset of sepsis often precedes the diagnosis of galactosemia. Pseudotumor cerebri can occur and cause a bulging fontanel. Complete withdrawal of lactose from the diet results in improvement of the acute symptoms. If untreated, death from liver and kidney failure and sepsis may follow within days. When the diagnosis is not made at birth, damage to the liver (cirrhosis) and brain (intellectual disability) becomes increasingly severe and irreversible.
Partial transferase deficiency is generally asymptomatic. It is more common than classic galactosemia and is diagnosed in newborn screening because of moderately elevated blood galactose and/or low transferase activity. Galactosemia should be considered for the newborn or young infant who is not thriving or who has any of the preceding findings. Light and electron microscopy of hepatic tissue reveals fatty infiltration, the formation of pseudoacini, and eventual macronodular cirrhosis. These changes are consistent with a metabolic disease but do not indicate the precise enzymatic defect.
Diagnosis
The initial diagnosis of galactosemia is done by demonstration of a reducing substance in several urine specimens collected while the patient is on a diet containing human milk, cow's milk, or any other formula containing lactose. The reducing substance detected in urine by Clinitest (e.g., glucose, galactose) can be identified by chromatography or an enzymatic test specific for galactose. Galactose can be detected in urine, provided the milk feeding was within the last few hours and the child is not vomiting excessively. Clinistix urine test results are usually negative because the test relies on the action of glucose oxidase, which is specific for glucose but is nonreactive with galactose. Amino acids may be detected in urine since they are excreted together with glucose because of a proximal renal tubular syndrome. Since galactose is injurious to persons with galactosemia, diagnostic challenge tests dependent on administering galactose orally or intravenously should not be used. Direct enzyme assay using erythrocytes establishes the diagnosis. The clinician needs to confirm that the patient did not receive a blood transfusion before the collection of the blood sample, because a diagnosis could be missed. A novel method utilizes nonradioactive ultraviolet (UV) light and high-performance liquid chromatography (HPLC) to accurately detect levels of galactose-1-phosphate uridyl transferase in erythrocytes.
Genetics
Transferase deficiency is an autosomal recessive disorder. Based on newborn screening in the United States, the frequency of the disease is approximately 1 in 47,000 live births. There are several enzymatic variants of galactosemia. The Duarte variant, a single– amino acid substitution (p.N314D), has diminished RBC enzyme activity (50% of normal), but usually is of no clinical significance. This variant is the most common, with a carrier frequency of 12% in the general population. Those who are heterozygous for the Duarte variant of galactosemia typically have 25% of normal galactose activity, few symptoms, elevated metabolites, and no need for intervention. Other similar variants expressing little enzyme activity typically require no intervention. Some black patients have milder symptoms despite the absence of measurable transferase activity in erythrocytes; these patients retain 10% enzyme activity in liver and intestinal mucosa, whereas most white patients have no detectable activity in any of these tissues. More than 230 identifiable pathogenic variants have been associated with transferase deficiency. In blacks, 62% of alleles are represented by the p.S135L variant, a variant that is responsible for a milder disease course. In the white population, 70% of alleles are represented by the p.Q188R and p.K285N missense variants and are associated with severe disease. Carrier testing and prenatal diagnosis can be performed by direct enzyme analysis of amniocytes or chorionic villi; testing can also be DNA based.
Treatment and Prognosis
With the availability of newborn screening for galactosemia, it is possible to identify and treat patients earlier than before. All galactose-containing foods should be removed from the diet on initial suspicion of galactosemia. Various non–lactose-containing milk substitutes are available (casein hydrolysates, soybean-based formula). Elimination of galactose from the diet along with adequate calcium supplementation reverses growth failure and renal and hepatic dysfunction. Cataracts regress, and most patients have no impairment of vision. Early diagnosis and treatment have improved the prognosis of galactosemia. On long-term follow-up, however, patients still manifest ovarian failure with primary or secondary amenorrhea, decreased bone mineral density, developmental delay, and learning disabilities that increase in severity with age. Hypergonadotropic hypogonadism is reported in 80% to >90% of female patients with classic galactosemia. Although most women with classic galactosemia are infertile when they reach childbearing age, a small number have given birth. Most patients manifest speech disorders, whereas a smaller number demonstrate poor growth and impaired motor function and balance (with or without overt ataxia). The relative control of galactose-1-phosphate levels does not always correlate with long-term outcome, leading to the belief that other factors, such as elevated galactitol, decreased uridine diphosphate galactose (a donor for galactolipids and proteins), and endogenous galactose production may be responsible.
Galactokinase Deficiency
The deficient enzyme is galactokinase , which normally catalyzes the phosphorylation of galactose. The principal metabolites accumulated are galactose and galactitol. Two genes are reported to encode galactokinase: GK1 on chromosome 17q24 and GK2 on chromosome 15. Cataracts are usually the sole manifestation of galactokinase deficiency; pseudotumor cerebri is a rare complication. The affected infant is otherwise asymptomatic. Heterozygous carriers may be at risk for presenile cataracts. Lab findings show an increased concentration of blood galactose levels, provided the infant has been fed a lactose-containing formula. The diagnosis is made by demonstrating an absence of galactokinase activity in erythrocytes or fibroblasts. Transferase activity is normal. Treatment is dietary restriction of galactose.
Uridine Diphosphate Galactose-4-Epimerase Deficiency
There are 2 distinct forms of epimerase deficiency. The first is a benign form that is diagnosed incidentally through newborn screening programs. Affected individuals are asymptomatic because the enzyme deficiency is limited to leukocytes and erythrocytes. This form does not require treatment. The second variety is severe because the epimerase deficiency is more generalized. Clinical manifestations resemble transferase deficiency, with the additional symptoms of hypotonia and nerve deafness. Clinical symptoms improve with restriction of galactose in diet. Although the severe form of galactosemia is rare, it must be considered in a symptomatic patient with measurable galactose-1-phosphate who has normal transferase activity. The abnormally accumulated metabolites are similar to those in transferase deficiency; however, there is also an increase in cellular uridine diphosphate (UDP) galactose. Diagnosis is confirmed by the assay of epimerase in erythrocytes.
Patients with the severe form of epimerase deficiency cannot synthesize UDP galactose from UDP glucose and are galactose dependent. Because galactose is an essential component of many nervous system structural proteins, patients are placed on a galactose-restricted diet rather than a galactose-free diet.
Infants with the mild form of epimerase deficiency have not required treatment. It is advisable to follow urine specimens for reducing substances and exclude aminoaciduria within a few weeks of diagnosis while the infant is still on lactose-containing formula.
The gene for UDP galactose-4-epimerase (GALE) is located on chromosome 1 at 1p36. Carrier detection is possible by measurement of epimerase activity in the erythrocytes. Prenatal diagnosis for the severe form of epimerase deficiency can be done using an enzyme assay of cultured amniotic fluid cells.
Bibliography
Fridovich-Keil JL, Walter JH. Galactosemia. The online metabolic & molecular bases of inherited disease . McGraw-Hill: New York; 2009 http://www.ommbid.com .
Potter NL, Nievergelt Y, Shriberg LD. Motor and speech disorders in classic galactosemia. JIMD Rep . 2013;11:31–41.
Rimoin DL, Connor JM, Pyeritz RE, et al. Emery and Rimoin's principles and practice of medical genetics . Churchill Livingstone Elsevier: New York; 2011.
Waisbren SE, Potter NL, Gordon CM, et al. The adult galactosemic phenotype. J Inherit Metab Dis . 2012;35(2):279–286.
Defects in Fructose Metabolism
Priya S. Kishnani, Yuan-Tsong Chen
Two inborn errors are known in the specialized pathway of fructose metabolism: benign or essential fructosuria and hereditary fructose intolerance. Fructose-1,6-bisphosphatase deficiency, although strictly speaking not a defect of the specialized fructose pathway, is discussed in Chapter 105.4 .
Deficiency of Fructokinase (Essential or Benign Fructosuria)
Deficiency of fructokinase is not associated with any clinical manifestations. Fructosuria is an accidental finding usually made because the asymptomatic patient's urine contains a reducing substance. No treatment is necessary, and the prognosis is excellent. Inheritance is autosomal recessive with an incidence of 1 in 120,000 live births. The gene encoding fructokinase (KHK) is located on chromosome 2p23.3.
Fructokinase catalyzes the first step of metabolism of dietary fructose: conversion of fructose to fructose-1-phosphate (see Fig. 105.1 ). Without this enzyme, ingested fructose is not metabolized; its level is increased in the blood, and it is excreted in urine because practically no renal threshold exists for fructose. Clinitest results reveal the urinary reducing substance, which can be identified as fructose by chromatography.
Deficiency of Fructose-1,6-Bisphosphate Aldolase (Aldolase B, Hereditary Fructose Intolerance)
Deficiency of fructose-1,6-bisphosphate aldolase (aldolase-B ) is a severe condition of infants caused by a deficiency of aldolase B activity in the liver, kidney, and intestine. This enzyme catalyzes the hydrolysis of fructose-1,6-bisphosphate into triose phosphate and glyceraldehyde phosphate. The same enzyme also hydrolyzes fructose-1-phosphate. In the absence of enzyme activity, there is a rapid accumulation of fructose-1-phosphate, which presents with severe symptoms when fructose-containing food is ingested.
Epidemiology and Genetics
The exact incidence of hereditary fructose intolerance (HFI) is unknown but is estimated to be as high as 1 in every 26,000 live births. HFI is inherited in an autosomal recessive manner. The ALDOB gene is mapped to chromosome 9q22.3. At least 40 pathogenic variants causing HFI are known. The most common pathogenic variant identified in northern Europeans is a single missense variant, a G→C transversion in exon 5 resulting in the normal alanine at position 149 being replaced by proline. This variant, along with 2 other missense variants (p.A174D and p.N334K), account for 80–85% of HFI in Europe and the United States. Diagnosis of HFI can be made by direct DNA analysis for the common variants and phosphorus magnetic resonance spectroscopy.
Clinical Manifestations
Affected individuals remain asymptomatic until fructose or sucrose (table sugar) is introduced in diet (usually from fruit, fruit juice, or sweetened cereal). Signs and symptoms typically manifest in infancy when foods or formulas containing these sugars are introduced. Certain patients are very sensitive to fructose, whereas others can tolerate moderate intakes (up to 250 mg/kg/day). The average intake of fructose in Western societies is 1-2 g/kg/day. Early clinical manifestations resemble galactosemia and include jaundice, hepatomegaly, vomiting, lethargy, irritability, and convulsions. There may also be a higher incidence of celiac disease in HFI patients (>10%) than in the general population (1–3%). As they grow older, patients usually develop an aversion to fructose-containing foods due to associated symptoms of nausea, vomiting, and abdominal pain.
Characteristic lab findings include lactic acidosis, hypophosphatemia, hyperuricemia, and hypermagnesemia. A prolonged clotting time, hypoalbuminemia, elevation of bilirubin and transaminase levels, and proximal tubular dysfunction are also seen. Acute fructose ingestion produces symptomatic hypoglycemia; the higher the intake, the more severe the clinical picture. Chronic ingestion results in failure to thrive and hepatic disease. If the intake of fructose persists, hypoglycemic episodes recur, leading to progressive renal and hepatic failure and eventually death.
Diagnosis
The presence of a reducing substance in urine during an acute episode raises the possibility of HFI. Oral fructose challenge is no longer considered a diagnostic approach because of high risk to the patient, who can become acutely ill after the test. Definitive diagnosis is made by demonstration of 2 pathogenic variants in ALDOB on molecular genetic testing. A common pathogenic variant (substitution of Pro for Ala at position 149) accounts for 53% of HFI alleles worldwide. An alternative is to show deficient hepatic fructose 1-phosphate aldolase (aldolase B) activity on liver biopsy.
Treatment
Acute episodes are managed symptomatically by correcting hypoglycemia with IV glucose (dextrose) administration, providing supportive treatment of hepatic insufficiency, and correcting metabolic acidosis. Complete elimination of fructose usually rapidly reverses symptoms and results in normalization of related metabolic disturbances. The cornerstone of long-term treatment is the complete restriction of all sources of sucrose, fructose, and sorbitol from the diet. It may be difficult because these sugars are widely used additives, found even in most medicinal preparations. With treatment, liver and kidney dysfunction improves, and catch-up in growth is common. Intellectual development is usually unimpaired. As the patient matures, symptoms become milder even after fructose ingestion; the long-term prognosis is good. Because of voluntary dietary avoidance of sucrose, affected patients have few dental caries. Care should be taken to avoid fructose-containing IV fluids during hospitalizations.
Bibliography
Bouteldja N, Timson DJ. The biochemical basis of hereditary fructose intolerance. J Inherit Metab Dis . 2010;33(2):105–112.
Rimoin DL, Connor JM, Pyeritz RE, et al. Emery and Rimoin's principles and practice of medical genetics . Churchill Livingstone Elsevier: New York; 2011.
Steinmann B, Gitzemann R, van den Berghe G. Disorders of fructose metabolism. The online metabolic and molecular bases of inherited disease–OMMBID . McGraw-Hill: New York; 2009 http://www.ommbid.com .
Defects in Intermediary Carbohydrate Metabolism Associated With Lactic Acidosis
Priya S. Kishnani, Yuan-Tsong Chen
Lactic acidosis (type B3 ) occurs with defects of carbohydrate metabolism that interfere with the conversion of pyruvate to glucose via the pathway of gluconeogenesis or to carbon dioxide and water via the mitochondrial enzymes of the Krebs cycle. Fig. 105.4 depicts the relevant metabolic pathways. Type I GSD, fructose-1,6-diphosphatase deficiency, and phosphoenolpyruvate carboxylase deficiency are disorders of gluconeogenesis associated with lactic acidosis. Pyruvate dehydrogenase complex deficiency, respiratory chain defects, and pyruvate carboxylase deficiency are disorders in the pathway of pyruvate metabolism causing lactic acidosis. Lactic acidosis (type B3) can also occur in defects of fatty acid oxidation, organic acidurias (see Chapters 103.6 , 103.10 , and 104.1 ), or biotin utilization diseases (type B3) (Table 105.2 ). These disorders are easily distinguishable by the presence of abnormal acyl carnitine profiles, amino acids in the blood, and unusual organic acids in the urine. Blood lactate, pyruvate, and acyl carnitine profiles, and the presence of these unusual urine organic acids should be determined in infants and children with unexplained acidosis, especially if there is an increase of anion gap.
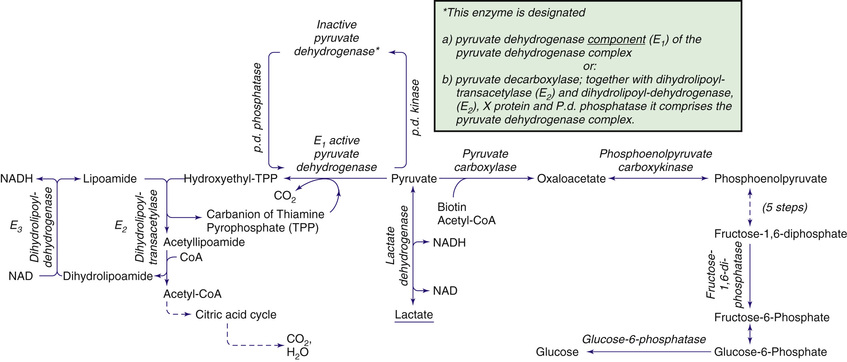
Lactic acidosis unrelated to an enzymatic defect occurs in hypoxemia (type A lactic acidosis). In this case, as well as in defects in the respiratory chain, the serum pyruvate concentration may remain normal (<1.0 mg/dL, with increased lactate:pyruvate ratio), whereas pyruvate is usually increased when lactic acidosis results from an enzymatic defect in gluconeogenesis or pyruvate dehydrogenase complex (both lactate and pyruvate are increased, and the ratio is normal). Lactate and pyruvate should be measured in the same blood specimen and on multiple blood specimens obtained when the patient is symptomatic because lactic acidosis can be intermittent. Fig. 105.5 is an algorithm for the differential diagnosis of lactic acidosis. Lactic acidosis is also noted with various underlying diseases (type B1 ) and drugs or toxins (type B2 ) (Table 105.2 ).

Disorders of Gluconeogenesis
Deficiency of Glucose-6-Phosphatase (Type I Glycogen Storage Disease)
Type I GSD is the only glycogenosis associated with significant lactic acidosis . The chronic metabolic acidosis predisposes these patients to osteopenia; after prolonged fasting, the acidosis associated with hypoglycemia is a life-threatening condition (see Chapter 105.1 ).
Fructose-1,6-Diphosphatase Deficiency
Fructose-1,6-diphosphatase deficiency impairs the formation of glucose from all gluconeogenic precursors, including dietary fructose. Hypoglycemia occurs when glycogen reserves are limited or exhausted. The clinical manifestations are characterized by life-threatening episodes of acidosis, hypoglycemia, hyperventilation, convulsions, and coma. In about half the cases, the deficiency presents in the 1st wk of life. In infants and small children, episodes are triggered by febrile infections and gastroenteritis if oral food intake decreases. The frequency of the attacks decreases with age. Laboratory findings include low blood glucose, high lactate and uric acid levels, and metabolic acidosis. In contrast to HFI, there is usually no aversion to sweets; renal tubular and liver function is normal.
The diagnosis is established by demonstrating an enzyme deficiency in either liver or intestinal biopsy. The enzyme defect can also be demonstrated in leukocytes in some cases. The gene coding for fructose-1,6-diphosphatase (FBP1) is located on chromosome 9q22; pathogenic variants are characterized, making carrier detection and prenatal diagnosis possible. Treatment of acute attacks consists of correction of hypoglycemia and acidosis by IV glucose infusion; the response is usually rapid. Avoidance of fasting, aggressive management of infections, and restriction of fructose and sucrose from the diet can prevent further episodes. For long-term prevention of hypoglycemia, a slowly released carbohydrate such as cornstarch is useful. Patients who survive childhood develop normally.
Phosphoenolpyruvate Carboxykinase Deficiency
Phosphoenolpyruvate carboxykinase (PEPCK ) is a key enzyme in gluconeogenesis. It catalyzes the conversion of oxaloacetate to phosphoenolpyruvate (see Fig. 105.4 ). PEPCK deficiency is both a mitochondrial enzyme deficiency and a cytosolic enzyme deficiency, encoded by 2 distinct genes.
PEPCK deficiency has been reported in only a few cases. The clinical features are heterogeneous, with hypoglycemia, lactic acidemia, hepatomegaly, hypotonia, developmental delay, and failure to thrive as the major manifestations. There may be multisystem involvement, with neuromuscular deficits, hepatocellular damage, renal dysfunction, and cardiomyopathy. The diagnosis is based on the reduced activity of PEPCK in liver, fibroblasts, or lymphocytes. Fibroblasts and lymphocytes are not suitable for diagnosing the cytosolic form of PEPCK deficiency because these tissues possess only mitochondrial PEPCK. To avoid hypoglycemia, patients should receive treatment with slow-release carbohydrates such as cornstarch, and fasting should be avoided.
Disorders of Pyruvate Metabolism
Pyruvate is formed from glucose and other monosaccharides, from lactate, and from alanine. It is metabolized through 4 main enzyme systems: lactate dehydrogenase, alanine transaminase, pyruvate carboxylase, and pyruvate dehydrogenase complex. Deficiency of the M subunit of LDH causes exercise intolerance and myoglobinuria (see Chapter 105.1 ).
Pyruvate Dehydrogenase Complex Deficiency
After entering the mitochondria, pyruvate is converted into acetyl-CoA by the pyruvate dehydrogenase complex (PDHC ), which catalyzes the oxidation of pyruvate to acetyl-CoA, which then enters the tricarboxylic acid cycle for ATP production. The complex comprises 5 components: E1 , an α-ketoacid decarboxylase; E2 , a dihydrolipoyl transacylase; E3 , a dihydrolipoyl dehydrogenase; protein X , an extra lipoate-containing protein; and pyruvate dehydrogenase phosphatase. The most common is a defect in the E1 (see Fig. 105.4 ).
Deficiency of the PDHC is the most common of the disorders leading to lactic acidemia and CNS dysfunction. The CNS dysfunction occurs because the brain obtains its energy primarily from oxidation of glucose. Brain acetyl-CoA is synthesized almost exclusively from pyruvate.
The E1 defects are caused by pathogenic variants in the gene coding for E1 α subunit, which is X-linked dominant. Although X-linked, its deficiency is a problem in both male and female patients, despite only one E1 α allele in females carrying a variant.
Clinical Manifestations
PDHC deficiency has a wide spectrum of presentations, from the most severe neonatal presentation to a mild late-onset form. The neonatal onset is associated with lethal lactic acidosis, white matter cystic lesions, agenesis of the corpus callosum, and the most severe enzyme deficiency. Infantile onset can be lethal or associated with psychomotor delay and chronic lactic acidosis, cystic lesions in the brainstem and basal ganglia, and pathologic features resembling Leigh disease (see later and Chapter 616.2 ). Neurologic symptoms in PDHC can be categorized into 2 groups: abnormal brain development, seen in both males and females, and brain lesions and epilepsy, seen in male patients only. Older children, usually boys, may have less acidosis, have greater enzyme activity, and manifest ataxia with high-carbohydrate diets. Intelligence may be normal. Patients of all ages may have facial dysmorphology, features similar to those of fetal alcohol syndrome.
The E2 and protein X–lipoate defects are rare and result in severe psychomotor retardation. The E3 lipoamide dehydrogenase defect leads to deficient activity not only in the PDHC, but also in the α-ketoglutarate and branched-chain ketoacid dehydrogenase complexes. This deficiency is more common in the Ashkenazi Jewish population. The reactive oxygen species generated by the pathogenic variants responsible for lipoamide dehydrogenase deficiency may in fact explain certain disease characteristics and suggest the utility of antioxidant therapy. Pyruvate dehydrogenase phosphatase deficiency has also been reported. These other PDHC defects have clinical manifestations within the variable spectrum associated with PDHC deficiency caused by E1 deficiency.
Treatment
The general prognosis is poor, except in rare patients in whom variants are associated with altered affinity for thiamine pyrophosphate, who may respond to thiamine supplementation. Because carbohydrates can aggravate lactic acidosis, a ketogenic diet is recommended. The diet has been found to lower the blood lactate level; the long-term benefit to patient outcome is unclear. A potential treatment strategy is to maintain any residual PDHC in its active form by oral administration of dichloroacetate , an inhibitor of E1 kinase. Beneficial effects of controlling postprandial lactic acidosis have been shown in some patients. Young children with congenital acidosis generally tolerate dichloroacetate well, but continued exposure is associated with peripheral neuropathy, a condition that could be attributable to the drug or the disease.
Deficiency of Pyruvate Carboxylase
Pyruvate carboxylase is a mitochondrial, biotin-containing enzyme essential in the process of gluconeogenesis; it catalyzes the conversion of pyruvate to oxaloacetate. The enzyme is also essential for Krebs cycle function as a provider of oxaloacetate and is involved in lipogenesis and formation of nonessential amino acids. Clinical manifestations of this deficiency have varied from neonatal severe lactic acidosis accompanied by hyperammonemia, citrullinemia, and hyperlysinemia (type B ) to late-onset mild to moderate lactic acidosis and developmental delay (type A ). In both types, patients who survived usually had severe psychomotor retardation with seizures, spasticity, and microcephaly. Some patients have pathologic changes in the brainstem and basal ganglia that resemble Leigh disease . The clinical severity appears to correlate with the level of the residual enzyme activity. A “benign” form of pyruvate carboxylase deficiency has also been described, characterized by recurrent attacks of lactic acidosis and mild neurologic deficits (type C ). Laboratory findings are characterized by elevated levels of blood lactate, pyruvate, alanine, and ketonuria. In the case of type B, blood ammonia, citrulline, and lysine levels are also elevated, which might suggest a primary defect of the urea cycle. The mechanism is likely caused by depletion of oxaloacetate, which leads to reduced levels of aspartate, a substrate for argininosuccinate synthase in the urea cycle (see Chapter 103.12 ). The gene for pyruvate carboxylase (PC) is located on chromosome 11q13.4-q13.5, and about 15 pathogenic variants have been identified.
Treatment consists of avoidance of fasting and eating a carbohydrate meal before bedtime. During acute episodes of lactic acidosis, patients should receive continuous IV glucose. Aspartate and citrate supplements restore the metabolic abnormalities; whether this treatment can prevent the neurologic deficits is not known. Liver transplantation has been attempted; its benefit remains unknown. Diagnosis of pyruvate carboxylase deficiency is made by the measurement of enzyme activity in liver or cultured skin fibroblasts and must be differentiated from holocarboxylase synthase or biotinidase deficiency.
Deficiency of Pyruvate Carboxylase Secondary to Deficiency of Holocarboxylase Synthase or Biotinidase
Deficiency of either holocarboxylase synthase (HCS ) or biotinidase, which are enzymes of biotin metabolism, result in multiple-carboxylase deficiency (pyruvate carboxylase and other biotin-requiring carboxylases and metabolic reactions) and in clinical manifestations associated with the respective deficiencies, as well as rash, lactic acidosis, and alopecia (see Chapter 103.6 ). The course of HCS or biotinidase deficiency can be protracted, with intermittent exacerbation of chronic lactic acidosis, failure to thrive, seizures, and hypotonia leading to spasticity, lethargy, coma, and death. Auditory and optic nerve dysfunction can lead to deafness and blindness, respectively. Late-onset milder forms have also been reported. Laboratory findings include metabolic acidosis and abnormal organic acids in the urine. In HCS deficiency, biotin concentrations in plasma and urine are normal. Diagnosis can be made in skin fibroblasts or lymphocytes by assay for HCS activity, and in the case of biotinidase, in the serum by a screening blood spot.
Treatment consists of biotin supplementation, 5-20 mg/day, and is generally effective if treatment is started before the development of brain damage. Patients identified through newborn screening and treated with biotin have remained asymptomatic.
Both enzyme deficiencies are autosomal recessive disorders. The incidence of HCS deficiency is approximately 1 in 87,000 live births. HCS and biotinidase (BTD) are located on chromosome 21q22 and 3p25, respectively. Ethnic-specific pathogenic variants in the HCS gene have been identified. Two common pathogenic variants (del7/ins3 and p.R538C) in the BTD account for 52% of all pathogenic alleles in symptomatic patients with biotinidase deficiency.
Mitochondrial Respiratory Chain Defects (Oxidative Phosphorylation Disease)
The mitochondrial respiratory chain catalyzes the oxidation of fuel molecules and transfers the electrons to molecular oxygen with concomitant energy transduction into adenosine triphosphate (oxidative phosphorylation ) (see Chapter 106 ). The respiratory chain produces ATP from adenosine diphosphate and inorganic phosphate utilizing the energy from electrons transferred from nicotinamide adenine dinucleotide (NADH) or flavin adenine dinucleotide and includes 5 specific complexes (I: NADH–coenzyme Q reductase; II: succinate–coenzyme Q reductase; III: coenzyme QH2 cytochrome-c reductase; IV: cytochrome-c oxidase; V: ATP synthase). Each complex is composed of 4-35 individual proteins and, with the exception of complex II (which is encoded solely by nuclear genes), is encoded by nuclear or mitochondrial DNA (inherited only from the mother by mitochondrial inheritance). Defects in any of these complexes or assembly systems produce chronic lactic acidosis, presumably because of a change of the reduction-oxidation state with increased concentrations of NADH (see Table 105.3 ).
In contrast to PDHC or pyruvate carboxylase deficiency, skeletal muscle and heart are usually involved in the respiratory chain disorders. On muscle biopsy, ragged red fibers indicating mitochondrial proliferation are very suggestive when present (see Fig. 105.5 ). Because of the ubiquitous nature of oxidative phosphorylation, a defect of the mitochondrial respiratory chain accounts for a vast array of clinical manifestations and should be considered in patients in all age-groups presenting with multisystem involvement. Some deficiencies resemble Leigh disease , whereas others cause infantile myopathies such as MELAS (mitochondrial encephalopathy, lactic acidosis, and stroke-like episodes), MERRF (myoclonic epilepsy and ragged red fibers), and Kearns-Sayre syndrome (external ophthalmoplegia, acidosis, retinal degeneration, heart block, myopathy, and high cerebrospinal fluid protein) (Table 105.3 ) (see Chapters 616.2 and 629.4 ). There is a higher incidence of psychiatric disorders in adults with a primary oxidative phosphorylation disease than in the general population. Elevated serum growth and differentiation factor (GDF)-15 levels help screen for mitochondrial disorders.
Table 105.3
Clinical and Genetic Heterogeneity of Disorders Related to Mutations in Mitochondrial DNA*
| SYMPTOMS, SIGNS, AND FINDINGS | LARGE DELETIONS IN MITOCHONDRIAL DNA | MUTATION IN TRANSFER RNA | MUTATION IN RIBOSOMAL RNA | MUTATION IN MESSENGER RNA | |||||
|---|---|---|---|---|---|---|---|---|---|
| KSS | PEO | PS | MERRF | MELAS | AID | NARP | MILS | LHON | |
| CENTRAL NERVOUS SYSTEM | |||||||||
| Seizures | − | − | − | +++ | + | − | − | + | − |
| Ataxia | + | − | − | + | + | + | ± | − | |
| Myoclonus | − | − | − | + | ± | − | − | − | − |
| Psychomotor retardation | − | − | − | − | − | − | − | + | − |
| Psychomotor regression | + | − | − | ± | + | − | − | − | − |
| Hemiparesis and hemianopia | − | − | − | − | +++ | − | − | − | − |
| Cortical blindness | − | − | − | − | + | − | − | − | − |
| Migraine-like headaches | − | − | − | − | + | − | − | − | − |
| Dystonia | − | − | − | − | + | − | − | + | ± |
| PERIPHERAL NERVOUS SYSTEM | |||||||||
| Peripheral neuropathy | ± | − | − | ± | ± | − | + | − | − |
| MUSCLE | |||||||||
| Weakness and exercise intolerance | + | +++ | − | + | + | − | + | + | − |
| Ophthalmoplegia | + | + | ± | − | − | − | − | − | − |
| Ptosis | + | − | − | − | − | − | − | − | |
| EYE | |||||||||
| Pigmentary retinopathy | + | − | − | − | − | − | + | ± | − |
| Optic atrophy | − | − | − | − | − | − | ± | ± | |
| BLOOD | |||||||||
| Sideroblastic anemia | ± | − | + | − | − | − | − | − | − |
| ENDOCRINE SYSTEM | |||||||||
| Diabetes mellitus | ± | − | − | − | ± | − | − | − | − |
| Short stature | + | − | − | + | + | − | − | − | − |
| Hypoparathyroidism | ± | − | − | − | − | − | − | − | − |
| HEART | |||||||||
| Conduction disorder | + | − | − | − | ± | − | − | − | ± |
| Cardiomyopathy | ± | − | − | − | ± | + | − | ± | − |
| GASTROINTESTINAL SYSTEM | |||||||||
| Exocrine pancreatic dysfunction | ± | − | + | − | − | − | − | − | − |
| Intestinal pseudoobstruction | − | − | − | − | + | − | − | − | − |
| EAR, NOSE, AND THROAT | |||||||||
| Sensorineural hearing loss | ± | − | − | + | + | + | ± | − | − |
| KIDNEY | |||||||||
| Fanconi syndrome | − | ± | − | ± | − | − | − | − | |
| LABORATORY FINDINGS | |||||||||
| Lactic acidosis | + | ± | + | + | + | ± | ± | ± | − |
| Ragged-red fibers on muscle biopsy | + | + | ± | + | + | − | − | − | − |
| MODE OF INHERITANCE | |||||||||
| Maternal | − | − | − | + | + | − | + | + | + |
| Sporadic | + | + | + | − | − | − | − | − | − |
* Characteristic constellations of symptoms and signs are in bold .
+, Presence of a symptom, sign, or finding; −, absence of a symptom, sign, or finding; ±, possible presence of a symptom, sign, or finding; AID, aminoglycoside-induced deafness; KSS, Kearns-Sayre syndrome; LHON, Leber hereditary optic neuropathy; MELAS, mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes; MERRF, myoclonic epilepsy with ragged-red fibers; MILS, maternally inherited Leigh syndrome; NARP, neuropathy, ataxia, and retinitis pigmentosa; PEO, progressive external ophthalmoplegia; PS, Pearson syndrome.
From DiMauro S, Schon EA: Mitochondrial respiratory-chain diseases, N Engl J Med 348:2656–2668, 2003. Copyright 2003 Massachusetts Medical Society. All rights reserved.
Diagnosis requires demonstration of abnormalities of oxidative phosphorylation enzyme complex activities in tissues or of mitochondrial DNA or a nuclear gene coding for mitochondrial functions, or both (Fig. 105.6 ). Muscle histology, including EM, can detect ragged red fibers and other abnormalities typical of mitochondrial myopathies. Analysis of oxidative phosphorylation complexes I-IV from intact mitochondria isolated from fresh skeletal muscle is the most sensitive assay for mitochondrial disorders; however, electron transport chain testing of flash-frozen muscle provides an alternative approach when fresh muscle testing is not available. Next-generation sequencing of mitochondrial DNA and panels of nuclear genes provides a noninvasive alternative to diagnosis. Specific criteria may assist in making a diagnosis (Table 105.4 ). Table 105.5 lists clues to the diagnosis of mitochondrial diseases.
Table 105.4
Mitochondrial Disease Criteria (Simplified Version for Bedside Use)*
| I. CLINICAL SIGNS AND SYMPTOMS, 1 POINT/SYMPTOM (max. 4 points) | ||||
|---|---|---|---|---|
| A. Muscular Presentation (max. 2 points) | B. CNS Presentation (max. 2 points) | C. Multisystem Disease (max. 3 points) | II. Metabolic/Imaging Studies (max. 4 points) | III. Morphology (max. 4 points) |
| Ophthalmoplegia † | Developmental delay | Hematology | Elevated lactate † | Ragged red/blue fibers ‡ |
| Facies myopathica | Loss of skills | GI tract | Elevated L/P ratio | COX-negative fibers ‡ |
| Exercise intolerance | Stroke-like episode | Endocrine/growth | Elevated alanine † | Reduced COX staining ‡ |
| Muscle weakness | Migraine | Heart | Elevated CSF lactate † | Reduced SDH staining |
| Rhabdomyolysis | Seizures | Kidney | Elevated CSF protein | SDH positive blood vessels † |
| Abnormal EMG | Myoclonus | Vision | Elevated CSF alanine † | Abnormal mitochondria/EM † |
| Cortical blindness | Hearing | Urinary TA excretion † | ||
| Pyramidal signs | Neuropathy | Ethylmalonic aciduria | ||
| Extrapyramidal signs | Recurrent/familial | Stroke-like picture/MRI | ||
| Brainstem involvement | Leigh syndrome/MRI † | |||
| Elevated lactate/MRS | ||||
* Score 1: mitochondrial disorder unlikely; score 2 to 4: possible mitochondrial disorder; score 5 to 7: probable mitochondrial disorder; score 8 to 12: definite mitochondrial disorder.
† This specific symptom scores 2 points.
‡ This symptom in a higher percentage scores 4 points.
GI, gastrointestinal; L/P, lactate/pyruvate; COX, cytochrome C oxidase; SDH, succinate dehydrogenase; EM, electron microscopy; EMG, electromyography; TA, tricarbon acid.
From Morava E, van den Heuvel L, Hol F, et al: Mitochondrial disease criteria – diagnostic applications in children. Neurology 67:1823-1826, 2006, p 1824.
The majority of mitochondrial disorders are caused by nuclear genes involved in mitochondrial function, and >300 genes have been included in nuclear gene panels for mitochondrial disorder diagnosis. However, pathogenic variants can be identified in 50% or fewer of patients diagnosed clinically with a mitochondrial disorder. An important consideration is that many genetic and multifactorial conditions have been associated with defects in 1 or more of the 4 complexes assayed in mitochondrial oxidative phosphorylation testing. These latter conditions feature so-called secondary mitochondrial dysfunction, because the conditions are not considered to be mitochondrial disorders per se.
Treatment remains largely symptomatic and does not significantly alter the outcome of disease. Some patients appear to respond to cofactor supplements, typically coenzyme Q10 ± L -carnitine at pharmacologic doses. The addition of creatine monohydrate and α-lipoic acid supplementation may add a significant benefit. EPI-743 is a parobenzoquinone like agent that has protective activity against oxidative injury; it is a promising agent in the treatment of mitochondrial disorders, including Leigh syndrome.
Leigh Disease (Subacute Necrotizing Encephalomyelopathy)
Leigh disease is a heterogeneous neurologic disease characterized by demyelination, gliosis, necrosis, relative neuronal sparing, and capillary proliferation in specific brain regions (see Chapter 616.2 ). Patients with Leigh disease frequently present with feeding and swallowing problems, failure to thrive, and developmental delay. The presentation is highly variable and may include seizures, altered consciousness, pericardial effusion, and dilated cardiomyopathy. Diagnosis is usually confirmed by radiologic or pathologic evidence of symmetric lesions affecting the basal ganglia, brainstem, and subthalamic nuclei. Patients with Leigh disease have defects in several enzyme complexes. Dysfunction in cytochrome-c oxidase (complex IV) is the most commonly reported defect, followed by NADH–coenzyme Q reductase (complex I), PDHC, and pyruvate carboxylase (see Chapter 106 ). Pathogenic variants in the nuclear SURF1 gene, which encodes a factor involved in the biogenesis of cytochrome-c oxidase and mitochondrial DNA variants in the adenosine triphosphatase 6 coding region, have been reported in patients with Leigh disease in association with complex IV deficiency. The most common mitochondrial DNA variant in Leigh disease is the T8993G variant in MT-ATP6 . The prognosis for Leigh syndrome is poor. In a study of 14 cases, there were 7 fatalities before age 1.5 yr.
Lactic acidosis, hypoglycemia, and encephalopathy have also been reported in patients with thiamine transporter deficiency and with pyridoxine-dependent epilepsy. Both disorders should improve by the provision of thiamine and pyridoxine, respectively.
Bibliography
Barnerias C, Saudubray JM, Touti G, et al. Pyruvate dehydrogenase complex deficiency: four neurological phenotypes with differing pathogenesis. Dev Med Child Neurol . 2010;52(2):1–9.
Davis RL, Liang C, Sue CM. A comparison of current serum biomarkers as diagnostic indicators of mitochondrial diseases. Neurology . 2016;86:2010–2015.
Fassone E, Rahman S. Complex I deficiency: clinical features, biochemistry, and molecular genetics. J Med Genet . 2012;49(9):578–590.
Ghaddhab C, Morin C, Brunel-Guitton C, et al. Premature ovarian failure in French Canadian Leigh syndrome. J Pediatr . 2017;184:227–229.
Koopman WJH, Distelmaier F, Smeitink JA, et al. OXPHOS mutations and neurodegeneration. EMBO J . 2013;32(1):9–29.
Koopman WJH, Willema PHGM, Smeitink JAM. Monogenic mitochondrial disorders. N Engl J Med . 2012;366:1132–1140.
Mercimek-Mahmutoglu S, Horvath GA, Coulter-Mackie M, et al. Profound neonatal hypoglycemia and lactic acidosis caused by pyridoxine-dependent epilepsy. Pediatrics . 2012;129:e1368–e1372.
Montero R, Yubero D, Villarroya J, et al. GDF-15 is elevated in children with mitochondrial diseases and is induced by mitochondrial dysfunction. PLoS ONE . 2016;11(2):e0148709.
Pèrez-Dueñas B, Serrano M, Rebollo M, et al. Reversible lactic acidosis in a newborn with thiamine transporter-2 deficiency. Pediatrics . 2013;131:e1670–e1675.
Rimoin DL, Connor JM, Pyeritz RE, et al. Emery and Rimoin's principles and practice of medical genetics . Churchill Livingstone Elsevier: New York; 2011.
Steinmann B, Gitzemann R, van den Berghe G. Disorders of fructose metabolism. The online metabolic and molecular bases of inherited disease–OMMBID . McGraw-Hill: New York; 2009 http://www.ommbid.com .
Vernon C, LeTourneau JL. Lactic acidosis: recognition, kinetics, and associated prognosis. Crit Care Clin . 2010;26:255–283.
Vernon HJ. Inborn errors of metabolism: advances in diagnosis and therapy. JAMA Pediatr . 2015;169(8):778–782.
Wallace DC. Mitochondrial DNA mutations in disease and aging. Environ Mol Mutagen . 2010;51(5):440–450.
Defects in Pentose Metabolism
Priya S. Kishnani, Yuan-Tsong Chen
Approximately 90% of glucose metabolism in the body is via the glycolytic pathway, with the remaining 10% via the hexose monophosphate pathway. The hexose monophosphate shunt leads to formation of pentoses, as well as providing NADH. One of the metabolites is ribose-5-phosphate, which is used in the biosynthesis of ribonucleotides and deoxyribonucleotides. Through the transketolase and transaldolase reactions, the pentose phosphates can be converted back to fructose-6-phosphate and glucose-6-phosphate.
Essential Pentosuria
Essential pentosuria is a benign disorder encountered principally in Ashkenazi Jews and is an autosomal recessive trait. The urine contains L -xylulose , which is excreted in increased amounts because of a block in the conversion of L -xylulose to xylitol as a result of xylitol dehydrogenase deficiency . The condition is usually discovered accidentally in a urine test for reducing substances. No treatment is required.
Transaldolase Deficiency
Few patients have reported symptoms that include liver cirrhosis, hepatosplenomegaly, severe neonatal hepatopathy, and cardiomyopathy. Biochemical abnormalities revealed elevated levels of arabitol, ribitol, and erythritol in the urine. Erythronic acid has been identified by urine nuclear magnetic resonance spectroscopy as another hallmark metabolite. Enzyme assay in the lymphoblasts and fibroblasts demonstrated low transaldolase activity, which was confirmed by pathogenic variants in the transaldolase gene. In addition, measurement of transaldolase activity in fibroblasts, lymphoblasts, or liver tissue, as well as assessing urinary concentrations of polyols, also can be used to confirm the diagnosis.
Ribose-5-Phosphate Isomerase Deficiency
Only one case of this disorder has been reported. The affected male had psychomotor delay from early in life and developed epilepsy at 4 yr of age. Thereafter, a slow neurologic regression developed, with prominent cerebellar ataxia, some spasticity, optic atrophy, and a mild sensorimotor neuropathy. MRI of the brain at ages 11 and 14 yr showed extensive abnormalities of the cerebral white matter. Proton magnetic resonance spectroscopy (MRS) of the brain revealed elevated levels of ribitol and D -arabitol. These pentitols were also increased in urine and plasma similar to the patient found in transaldolase deficiency. Enzyme assays in cultured fibroblasts showed deficient ribose-5-phosphate isomerase activity, which was confirmed by a molecular study. These results, combined with a study of ribose-5-phosphate isomerase–deficient mice, demonstrated that the specific genetic pairing of a null allele with an allele coding for a form of the enzyme that is only partly active, allowing for cell type–dependent expression deficits, is a contributing factor to the rarity of the disease. Ribose-5-phosphate isomerase deficiency may represent an example of a single-gene disease that appears seldom because of its complex molecular etiology.
Bibliography
Copeland WC. Defects of mitochondrial DNA replication. J Child Neurol . 2014;29(9):1216–1224.
Pierce SB, Spurrell CH, Mandell JB, et al. Garrod's fourth inborn error of metabolism solved by the identification of mutations causing pentosuria. Proc Natl Acad Sci USA . 2011;108(45):18313–18317.
Rimoin DL, Connor JM, Pyeritz RE, et al. Emery and Rimoin's principles and practice of medical genetics . Churchill Livingstone Elsevier: New York; 2011.
Valle D, Beaudet AL, Vogelstein B, et al. Online metabolic and molecular bases of inherited disease . [updated 2011] http://www.ommbid.com ; 2006.
Wamelink MMC, Struys EA, Jakobs C. The biochemistry, metabolism and inherited defects of the pentose phosphate pathway: a review. J Inherit Metab Dis . 2008;31:703–717.
Disorders of Glycoprotein Degradation and Structure
Margaret M. McGovern, Robert J. Desnick
The disorders of glycoprotein degradation and structure include several lysosomal storage diseases that result from defects in glycoprotein degradation, and the congenital disorders of glycosylation (see Chapter 105.7 ). Glycoproteins are macromolecules composed of oligosaccharide chains linked to a peptide backbone. They are synthesized by 2 pathways: the glycosyltransferase pathway, which synthesizes oligosaccharides linked O -glycosidically to serine or threonine residues; and the dolichol, lipid-linked pathway, which synthesizes oligosaccharides linked N -glycosidically to asparagine.
The glycoprotein lysosomal storage diseases result from the deficiency of the enzymes that normally participate in the degradation of oligosaccharides and include sialidosis, galactosialidosis, aspartylglucosaminuria, and α-mannosidosis. In some instances the underlying abnormality that leads to glycoprotein accumulation also results in abnormal degradation of other classes of macromolecules that contain similar oligosaccharide linkages, such as certain glycolipids and proteoglycans. In these cases the underlying enzymatic deficiency results in the accumulation of both glycoproteins and glycolipids. The classification of these types of disorders as lipidoses or glycoproteinoses depends on the nature of the predominantly stored substance. In general, the glycoprotein disorders are characterized by autosomal recessive inheritance and a progressive disease course with clinical features that resemble those seen in the mucopolysaccharidoses.
Sialidosis and Galactosialidosis
Sialidosis is an autosomal recessive disorder that results from the primary deficiency of neuraminidase because of mutations in the gene (NEU1 ) that encodes this protein, located on chromosome 6p21.33. In contrast, galactosialidosis is caused by the deficiency of 2 lysosomal enzymes—neuraminidase and β-galactosidase. The loss of these enzymatic activities results from mutations in a single gene, CTSA, located on chromosome 20q13.12, that encodes the protective protein cathepsin A, which functions to stabilize these enzymatic activities. Neuraminidase normally cleaves terminal sialyl linkages of several oligosaccharides and glycoproteins. Its deficiency results in the accumulation of oligosaccharides, and the urinary excretion of sialic acid terminal oligosaccharides and sialylglycopeptides. Examination of tissues from affected individuals reveals pathologic storage of substrate in many tissues, including liver, bone marrow, and brain.
The clinical phenotype associated with neuraminidase deficiency is variable and includes type I sialidosis, which usually presents in the 2nd decade of life with myoclonus and cherry-red spots in the macula. These patients typically present secondary to gait disturbances, myoclonus, or visual complaints. In contrast, type II sialidosis occurs at several ages of onset (congenital, infantile, and juvenile), depending on the severity of the gene mutation. The congenital and infantile forms result from isolated neuraminidase deficiency, whereas the juvenile form results from both neuraminidase and β-galactosidase deficiency. The congenital type II disease is characterized by hydrops fetalis, neonatal ascites, hepatosplenomegaly, stippling of the epiphyses, periosteal cloaking, and stillbirth or death in infancy. The type II infantile form presents in the 1st yr of life with dysostosis multiplex, moderate global developmental delays, visceromegaly, corneal clouding, cherry-red maculae, and seizures. The juvenile type II form of sialidosis, which is sometimes designated galactosialidosis, has a variable age of onset ranging from infancy to adulthood. In infancy, the phenotype is similar to that of GM1 gangliosidosis, with edema, ascites, skeletal dysplasia, and cherry-red spots. Patients with later-onset disease have dysostosis multiplex, visceromegaly, intellectual disability, dysmorphism, corneal clouding, progressive neurologic deterioration, and cherry-red spots.
No specific therapy exists for any form of the disease, although studies in animal models have demonstrated improvement in the phenotype after bone marrow transplantation. The diagnosis of sialidosis and galactosialidosis is achieved by the demonstration of the specific enzymatic deficiency or by mutations in the responsible gene. Prenatal diagnosis using cultured amniotic cells or chorionic villi is available by demonstrating the enzyme defect and/or specific gene mutations.
Aspartylglucosaminuria
This is a rare autosomal recessive lysosomal storage disorder, except in Finland, where the carrier frequency is estimated at 1 in 36 adults, the high frequency due to a founder gene. The disorder results from the deficient activity of aspartylglycosaminidase and the subsequent accumulation of aspartylglycosamine, particularly in the liver, spleen, and thyroid. The gene for the enzyme (AGA) has been localized to chromosome 4q32-33, and the DNA and gene have been isolated and sequenced. In the Finnish population, a single AGA mutation encoding p.C163S accounts for most mutant alleles, whereas outside of Finland, a large number of private mutations have been described.
Affected individuals with aspartylglucosaminuria typically present in the 1st yr of life with recurrent infections, diarrhea, and umbilical hernias. Coarsening of the facies and short stature usually develop later. Other features include joint laxity, macroglossia, hoarse voice, crystal-like lens opacities, hypotonia, and spasticity. Psychomotor development is usually near normal until age 5 yr, when a decline is noted. Behavioral abnormalities are typically seen, and IQ values in affected adults are usually<40 (severe intellectual disability). Survival to adulthood is common, with most early deaths attributable to pneumonia or other pulmonary causes. Definitive diagnosis requires demonstration of markedly deficient aspartylglucosaminidase in peripheral blood leukocytes, and/or the specific AGA mutation(s). Several patients have undergone allogeneic bone marrow transplants, but this approach has not proved effective, and no specific treatment is available. Prenatal diagnosis is available by the determination of aspartylglucosaminidase deficiency and/or the specific AGA mutations in cultured amniocytes or chorionic villi.
α-Mannosidosis
This autosomal recessive disorder results from the deficient activity of α-mannosidase and the accumulation of mannose-rich compounds. The gene MAN2B1 encoding the enzyme has been localized to chromosome 19p13.2-q12, and the cDNA and gene sequence have been determined. To date >140 gene mutations have been reported. Affected patients display clinical heterogeneity. There is a severe infantile form, or type I disease, and a milder juvenile variant, type II disease. All patients have psychomotor retardation, facial coarsening, and dysostosis multiplex. The infantile form of the disorder, however, is characterized by more rapid cognitive deterioration, with death occurring between ages 3 and 10 yr. Patients with the infantile form also have more severe skeletal involvement and hepatosplenomegaly. The juvenile disorder is characterized by onset of symptoms in early childhood or adolescence, with milder somatic features and survival to adulthood. Hearing loss, destructive synovitis, pancytopenia, and spastic paraplegia have been reported in type II patients. The diagnosis is made by the demonstration of the marked deficiency of α-mannosidase activity in white blood cells or cultured fibroblasts. Clinical trials of ERT with recombinant human α-mannosidase are underway. Prenatal diagnosis can be made by demonstrating the enzyme defect and/or the specific gene mutations in cultured amniocytes or chorionic villi.
Bibliography
Arvio M, Mononen I. Aspartylglycosaminuria: a review. Orphanet J Rare Dis . 2016;11:162–172.
Borgwardt L, Stensland HM, Olsen KJ, et al. Alpha-mannosidosis: correlation between phenotype, genotype and mutant MAN2B1 subcellular localization. Orphanet J Rare Dis . 2015;10:70–86.
Freeze HH, Eklund EA, Ng BG, et al. Neurological aspects of human glycosylation disorders. Annu Rev Neurosci . 2015;38:105–125.
Stroobants S, Damme M, Van der Jeugd A, et al. Long-term enzyme replacement therapy improves neurocognitive functioning and hippocampal synaptic plasticity in immune-tolerant alpha-mannosidosis mice. Neurobiol Dis . 2017;106:255–268.
Tegtmeyer LC, Rust S, van Scherpenzeel M, et al. Multiple phenotypes in phosphoglucomutase 1 deficiency. N Engl J Med . 2014;370:533–542.
Congenital Disorders of Glycosylation
Eva Morava, Peter Witters
Keywords
- glycosylation
- deglycosylation
- glycans
- transferrin isoelectric focusing
- TIEF
- cerebro-ocular dysplasia–muscular dystrophy
- phosphomannomutase-2 deficiency
- PMM2-CDG
- mannosephosphoisomerase deficiency
- MPI-CDG
- mannose therapy
- glucosyltransferase-1 deficiency
- ALG6-CDG
- UDP-GlcNAc:Dol-P-GlcNAc-P transferase deficiency
- DPAGT1-CDG
- muscle-eye-brain disease
- hyperphosphatasia
- PIGA-CDG
- steroid 5α-reductase deficiency
- SRD5A3-CDG
- cutis laxa
- Golgi-α1-2 mannosidase-1 deficiency
- MAN1B1-CDG
- phosphoglucomutase-1 deficiency
- PGM1-CDG
- D -galactose
- Golgi homeostasis
- manganese transporter defect
- SLC39A8-CDG
- N -glycanase-1 deficiency
- NGLY1
Glycosylation is the complex multistep metabolic process of adding (oligo)saccharides to proteins and lipids. The classification of disorders of hypoglycosylation is based on biochemical structures: (1) defects in protein N -linked glycosylation, (2) defects in protein O -linked glycosylation, (3) defects in glycosphingolipid and in glycosylphosphatidylinositol-anchor glycosylation, and (4) defects in multiple glycosylation pathways and in other pathways (Fig. 105.7 ). No disorders are known to result from abnormal C -linked glycosylation. Congenital disorders of glycosylation are labeled based on their genetic defect (CDG) .
Protein glycosylation is an essential pathway. Most functional proteins are glycosylated, including serum proteins (e.g., transferrin, ceruloplasmin, TBG), hormones (e.g., TSH, FSH, FH, ACTH, IGFBP3), and clotting and anticoagulation factors (e.g., factors IX and XI, antithrombin). Membrane proteins are also highly glycosylated. Important intracellular glycoproteins include enzymes such as glycosyltransferases or lysosomal enzymes.
N -glycans are linked to the amide group of asparagine. They are synthetized in a complicated process throughout the cytoplasm, endoplasmic reticulum (ER), and Golgi complex, starting with sugar activation and nucleotide sugar synthesis, then oligosaccharide assembly, and finally glycan processing (Fig. 105.8 ). The majority of the pediatric disorders are N -glycosylation disorders. O -glycans are linked to the hydroxyl group of serine or threonine. These diverse glycoproteins are mostly formed in the Golgi complex; their defects can involve xylosylation, fucosylation, mannosylation, or other modifications. An important focus is O -mannosylation defects because of their relevance for dystroglycanopathies.
Lipid glycosylation is an essential process for the synthesis of ceramide and ganglioside synthesis. Glycosylphosphatidylinositols (GPIs) are very special glycolipids that link various proteins to the plasma membrane, as complex lipid-sugar anchors (GPI anchors, see Fig. 105.7 ).
Congenital disorders of glycosylation (CDG) are predominantly multisystem diseases, caused by >140 different genetic defects in glycoprotein and glycolipid glycan synthesis. This rapidly growing group is one of the newest and largest metabolic disorder groups. Most patients described with CDG have N -glycosylation defects, followed by the fastest-growing group of CDGs, involving multiple glycosylation pathways and dolicholphosphate synthesis. Smaller groups are O -glycosylation disorders and disorders of glycosylphosphatidylinositol. The “oldest” CDG is PMM2-CDG, in which the genetic defect leads to the loss of phosphomannomutase 2 (PMM2), the enzyme that catalyzes the conversion of mannose-6-phosphate into mannose-1-phosphate. The majority of CDGs have an autosomal recessive inheritance. Only 2 N -linked CDGs are autosomal dominant, GANAB-CDG and PRKCSH-CDG. The dominantly inherited O -linked CDGs include EXT1/EXT2-CDG, POFUT1-CDG, and POGLUT1-CDG. X-linked CDGs include ALG13-CDG, SSR4-CDG, PIGA-CDG, SLC35A2-CDG, ATP6AP2-CDG and ATP6AP1-CDG.
Some CDGs are lethal; 20% of PMM2-CDG patients die in the 1st 2 yr of life. Some patients, however, stabilize throughout young adulthood. Almost any clinical phenotype can be present in a patient with CDG. It can affect any organ or organ system and most often includes the central nervous system (CNS). The most common clinical features include developmental and speech delay, seizures, ataxia, spasticity, peripheral neuropathy, hypotonia, strabismus, abnormal fat distribution, visual loss, cardiomyopathy, feeding difficulties, liver dysfunction, endocrine abnormalities, bleeding diathesis, and thrombosis (Fig. 105.9 and Table 105.6 ). Single-organ presentations are rare in CDGs (e.g., TUSC3-CDG and ST3GAL3-CDG: brain; DHDDS-CDG: retina; ALG14-CDG: neuromuscular junction; POFUT1-CDG and POGLUT1-CDG: skin; SEC23B-CDG: red cell lineage; EXT1/EXT2-CDG: cartilage; TMEM199-CDG: liver). Many CDGs are recognizable syndromes. CDG should be considered in any patient with a developmental disability or an unexplained clinical condition, especially in multisystem disease with neurologic involvement.
Table 105.6
Clinical and Laboratory Features in Common Congenital Disorders of Glycosylation (CDGs), with Clinically Recognizable Phenotype and Abnormal Glycosylation, Detectable by Serum Transferrin Isoform Analysis (TIEF)
| DEFECTIVE GENE | MOST FREQUENT CLINICAL FEATURES | SUGGESTIVE FEATURES | LABORATORY ABNORMALITIES | OTHER BIOCHEMICAL ANOMALIES |
|---|---|---|---|---|
| PMM2 | Strabismus, nystagmus, smooth philtrum, large ears, vomiting, diarrhea, FTT, axial hypotonia, cerebellar vermis hypoplasia, ataxia, psychomotor disability, seizures, spasticity, neuropathy, pigmentary retinitis | Inverted nipples and/or abnormal fat pads, stroke-like episodes | Elevated serum transaminases, hypoalbuminemia, decreased factor IX, XI and AT activity, low serum ceruloplasmin and TBG levels | Type 1 serum TIEF, decreased PMM activity in leukocytes and fibroblasts |
| PMI | Cholestasis, hepatomegaly, feeding difficulties, recurrent vomiting, chronic diarrhea, ascites, recurrent thrombosis, gastrointestinal bleeding |
Hyperinsulinism, protein losing enteropathy Normal intelligence and absence of neurologic features |
Elevated transaminases, hypoalbuminemia, hypoglycemia, decreased factor IX, XI, and AT-III activity | Type 1 serum TIEF, decreased PMI activity in leukocytes and fibroblasts |
| ALG6 | Hypotonia, muscle weakness, seizures, ataxia, intellectual disability, behavioral abnormalities | (Distal limb malformations) | Elevated serum transaminases; hypoalbuminemia; decreased factor IX, XI, and AT activity; low serum IgG level | Type 1 serum TIEF, abnormal LLO results in fibroblasts |
| DPAGT1 | Microcephaly, brain malformations, hypotonia, severe psychomotor disability, seizures, spasticity, proximal weakness, failure to thrive, joint contractures |
Congenital myasthenia phenotype In multisystem phenotype: cataract |
Decreased AT, protein C, and protein S activity; increased creatine kinase; hypoalbuminemia; normal creatine kinase in myasthenia | Type 1 serum TIEF |
| SRD5A3 | Developmental delay, hypotonia, ataxia, cerebellar vermis hypoplasia, intellectual disability, speech delay, visual loss | Congenital cataract, retinal and iridic coloboma, glaucoma, optic nerve dysplasia, ichthyosis | Low anticoagulation factors (AT, protein C, and protein S activity), increased serum transaminases | Type 1 serum TIEF but reported false-negative TIEF |
| ATP6V0A2 | Generalized cutis laxa, hypotonia, strabismus, characteristic facial features, joint laxity, seizures, motor and language developmental delay, spontaneous improvement of cutis laxa by aging | Cobblestone-like brain dysgenesis | Mild coagulation abnormalities, increased serum transaminase levels |
Type 2 serum TIEF but reported false-negative TIEF Abnormal apoC-III IEF, characteristic MALDI TOF profile (Note abnormal skin histology) |
| ATP6V1A and ATP6V1E1 | Cardiovascular anomalies | Mild coagulation abnormalities and increased serum transaminase levels, hypercholesterolemia | ||
| PGM1 | Pierre Robin sequence, cholestasis, short stature, dilated cardiomyopathy, | Cleft palate, hyperinsulinism, normal intelligence | Hypoglycemia, increased serum transaminase levels, decreased AT | Mixed type 1/ 2 serum TIEF, decreased fibroblast PGM1 activity |
| MAN1B1 | Developmental delay, speech delay, intellectual disability, muscle weakness | Obesity, autistic features, inverted nipples, characteristic face | Increased serum transaminase levels, low AT | Type 2 serum TIEF, abnormal apoC-III IEF, diagnostic MALDI TOF profile |
| TMEM199 | Cholestasis, hepatomegaly, liver steatosis, liver fibrosis, liver failure, spontaneous bleedings, motor developmental delay | Normal intelligence | Decreased serum ceruloplasmin, increased serum transaminase levels, hypercholesterolemia, high AP | Type 2 serum TIEF, abnormal apoC-III IEF, characteristic MALDI TOF profile |
| CCDC115 | Hepatomegaly | |||
| ATP6AP1 and ATP6AP2 | Immune deficiency | |||
| SLC39A8 | Seizures, hypsarrhythmia, hypotonia, developmental and speech delay, FTT | Dwarfism, craniosynostosis, rhizomelia, Leigh disease | Decreased serum manganese, high serum transaminases, abnormal coagulation | Type 2 serum TIEF, abnormal apoC-III, characteristic MALDI TOF profile |
AP, Alkaline phosphatase; AT, antithrombin; apoC-III: apolipoprotein C-III; FTT, failure to thrive; LLO, lipid-linked oligosaccharides; MALDI-TOF, matrix-assisted laser desorption/ionization time of flight; TBG, thyroxine-binding globulin; TIEF, transferrin isoelectric focusing.
There are also congenital disorders of deglycosylation , including known lysosomal disorders and a severe neurologic condition caused by defective N -glycanase function (NGLY1 defect).
Laboratory evaluations in most N -linked CDGs rely on a primary screening method called serum transferrin isoelectric focusing (TIEF) . Transferrin isoforms, which are hyposialylated (missing terminal sialic acid residues), show different cathodal shifts depending on either missing glycan chains or truncated glycans. A type 1 pattern suggests an early metabolic defect in the cytosolic-ER–related glycan synthesis and assembly. A type 2 pattern suggests Golgi-related glycan-processing defects (Fig. 105.10 ).
Isoelectric focusing of apolipoprotein C-III (IEF apoC-III) , a serum mucine type O -glycosylated protein, can detect some O -glycosylation disorders (combined N - and O -linked glycosylation defects). Mass spectrometry in serum for type 1 defects is highly sensitive for mild glycosylation abnormalities. Glycomics by matrix-assisted laser desorption/ionization time of flight (MALDI-TOF ) can be diagnostic in specific types of CDG (mostly Golgi related with a type 2 pattern). Dolichol-linked glycan or lipid-linked oligosaccharide (LLO) analysis is a complicated but sensitive method to detect ER-related N -glycan assembly (CDG type 1) defects in patient fibroblasts. GPI-anchor defects can be suspected based on recurrent elevation of alkaline phosphatase levels in blood.
Dystroglycanopathies can be confirmed based on abnormal immunohistochemistry in muscle biopsy. Fluorescence-activated cell sorting (FACS ) analysis of the membrane-anchored markers CD16 and CD24 in leukocytes is highly suggestive for a GPI-anchor abnormality, especially when alkaline phosphatase in blood is significantly elevated. Enzyme analysis in blood is only available for a few, more common CDGs (PMM2-CDG, MPI-CDG, PGM1-CDG); it is more reliable in fibroblasts.
With an abnormal TIEF pattern result or clinical suspicion of any type of CDG, most metabolic centers use a direct CDG gene panel analysis or next-generation sequencing (NGS; whole exome sequencing) (see Fig. 105.10 ).
Congenital Disorders of Protein N -Glycosylation
Phosphomannomutase-2 Deficiency (PMM2-CDG)
Clinical Manifestations
PMM2-CDG is the most common and easily recognizable CDG. Most patients have alternating strabismus, characteristic facial features (short nose, long philtrum, large ears) (Fig. 105.9E ), inverted nipples and/or abnormal fat pads (Fig. 105.9A -C ), feeding difficulties, axial hypotonia, and decreased reflexes, already in the 1st few mo of life. Nystagmus (caused by pontocerebellar and vermis hypoplasia; Fig. 105.9F ) is also common. Psychomotor disability is present in most patients, but normal intellectual development has been described in a few patients. Most patients develop a multisystem disease, and <25% show an isolated neurologic phenotype without other organ involvement, normal endocrine regulation, and no coagulopathy. The neurologic involvement is quite diverse, with ataxia, seizures, spasticity, and peripheral neuropathy (Fig. 105.9D ) the most common features. Dystonia, stroke-like episodes, and proximal myopathy can also occur. PMM2-CDG is not a progressive disease, but certain features, when present, typically appear at different ages during the disease. From birth, pericardial fluid collection, cardiomyopathy, or chronic vomiting/diarrhea can occur; after 7 yr, retinitis pigmentosa and cataract; and after puberty, scoliosis, neuropathy, and recurrent thrombotic events. Liver function anomalies are mild, and only a few patients develop cholestasis or liver fibrosis. Most patients have a hypergonadotropic hypogonadism; no successful pregnancies have been reported. Intellectual disability can be mild to severe; speech development is frequently delayed and can even be absent. Autistic behavior is common, although patients usually have a cheerful personality.
Pathophysiology
Phosphomannomutase 2 catalyzes the conversion of mannose-6-phosphate to mannose-1-phosphate, essential for the formation of activated mannose units used in the synthesis of the growing glycan chain in the ER. Hypoglycosylation leads to abnormal function affecting many essential glycoproteins, such as coagulation and anticoagulation factors, endocrine regulation, transport proteins, liver function, and immune, membrane, and receptor proteins.
Diagnosis
The primary screening method for PMM2-CDG is serum transferrin glycoform analysis, which is most frequently performed by TIEF. Intact transferrin has 4 negatively charged sialic acid residues (tetrasialotransferrin). Transferrin glycoforms, missing terminal sialic acid residues, show different cathodal shifts, less abundant tetrasialotransferrin, increased disialotransferrin, and some a-sialotransferrin (see Fig. 105.10 ). This is the so-called type 1 pattern, suggestive of a defect in glycan assembly in the cytosol-ER. Transferrin isoforms are also detectable by mass spectrometry. Certain other disorders can cause a false-positive transferrin isoform pattern, including galactosemia, hereditary fructose intolerance, and excessive alcohol use. PMM enzyme analysis is available in leukocytes and fibroblasts.
The presence of elevated serum transaminases, hypoalbuminemia, decreased factor IX and XI and antithrombin activity, or low ceruloplasmin or thyroxine-binding globulin (TBG) level is highly suggestive of CDG, including the most common type, PMM2-CDG.
PMM2-CDG is autosomal recessive. Genetic testing is mostly performed by direct sequencing. The most frequent pathogenic variant (c.422G>A; R141H) is present in 75% of patients of Caucasian origin. The exact incidence of PMM2-CDG is not known, but it is estimated to be as high as 1 in 40,000-80,000 in Europe. Prenatal diagnosis is only reliable by genetic testing.
Treatment
The therapy in PMM2-CDG relies on supportive treatment. Even with the best treatment, mortality is about 20% in the 1st 2 yr of life, mostly from cardiac or kidney involvement and severe infections. Current recommended therapy includes adequate nutrition, diet or tube feeding if needed, cardiac support, hormone supplements, physical and occupational therapy, speech therapy, seizure management, and strabismus surgery. Therapeutic developments include targeted mannose-phosphate treatment, and chaperone therapy; these are only in preclinical trial phases.
Mannosephosphoisomerase Deficiency (MPI-CDG)
Clinical Manifestations
MPI deficiency is a recognizable and treatable CDG. Most patients show early symptoms of liver disease (cholestasis, elevated transaminases) and feeding difficulties, with recurrent vomiting and chronic diarrhea, most frequently with protein-losing enteropathy. Life-threatening episodes might appear as early as the 1st few mo of life with recurrent thrombosis and severe gastrointestinal bleeding because of severe coagulation abnormalities. Hypoglycemia is usually caused by hyperinsulinism. Hypoalbuminemia can be severe; patients might develop visible abdominal distention from a combination of ascites and hepatomegaly. Patients with MPI-CDG have no other organ involvement, and the CNS is not affected. There are no dysmorphic features. The liver disease frequently progresses to fibrosis or cirrhosis.
Pathophysiology
Mannosephosphoisomerase (MPI) catalyzes the conversion of fructose-6-phosphate to mannose-6-phosphate, 1 step before PMM2, therefore blocking the formation of activated mannose units (GDP mannose) for oligosaccharide synthesis. Hypoglycosylation leads to abnormal glycoprotein function the same as in PMM2-CDG, especially coagulation and anticoagulation factors, liver function, and hormone receptors.
Diagnosis
The primary screening method in a suspected MPI-CDG patient is serum transferrin isoform analysis by TIEF (see Fig. 105.10 ) or MS analysis. MPI deficiency leads to a type 1 pattern, as seen in PMM2 deficiency. MPI enzyme analysis is available in leukocytes and fibroblasts. The presence of elevated serum transaminases, hypoalbuminemia, decreased factor IX and XI and antithrombin activity, hyperinsulinism, and nonketotic hypoglycemia are highly suggestive for MPI-CDG.
MPI-CDG is autosomal recessive. Genetic testing is mostly performed by direct sequencing. The exact incidence of MPI-CDG is not known, but it is estimated at 1 : 800 000 in Europe. Prenatal diagnosis is only reliable by genetic testing. Although this is a rare CDG, early diagnosis is imperative because it is treatable.
Treatment
MPI-CDG is the first CDG type treatable by dietary therapy. Mannose therapy is clinically effective by both IV and oral supplementation of 1 g/kg/day divided into 3-4 doses. A known side effect is hemolysis. The treatment uses an alternative pathway: mannose can be phosphorylated by hexokinases to mannose 6-phosphate, bypassing the MPI defect. The clinical symptoms improve rapidly, but liver function might further deteriorate. Liver fibrosis and cirrhosis might necessitate liver transplantation, which will resolve the metabolic disease. The oldest patient known with MPI-CDG has survived into her late 30s.
Glucosyltransferase-1 Deficiency (ALG6-CDG)
Clinical Manifestations
ALG6-CDG is the 2nd most common CDG. Most patients have hypotonia, muscle weakness, seizures, and ataxia. To date, no patient with ALG6-CDG has normal intelligence. Speech delay and nystagmus are common neurologic signs. Brachydactyly, skeletal abnormalities, and transverse limb defects have been observed. Strabismus and characteristic facial dysmorphism are rare (hypertelorism, oval face, short nose). Inverted nipples and/or abnormal fat pads are exceptional in ALG6-CDG.
The most severe ALG6-CDG patients show a multisystem phenotype in the 1st few mo of life, including severe infections, protein-losing enteropathy, hypoalbuminemia, anemia, and failure to thrive. Autistic behavior and mood changes have been observed in several patients. The oldest patient to date is almost 45 yr.
Pathophysiology
The metabolic problem is caused by defective binding of the 1st of 3 glucoses to the lipid-linked oligosaccharide in the ER. This glucose binding is essential for attachment of the oligosaccharyltransferase enzyme complex to the newly built oligosaccharide chain and the ability to transfer it to the protein. This leads to protein hypoglycosylation and abnormal glycoprotein function similar to PMM2-CDG and MPI-CDG. Laboratory abnormalities are also similar, including abnormalities in coagulation and anticoagulation factors, liver function, thyroid hormones, and immunoglobulins (IgG).
Diagnosis
The primary screening method in a suspected ALG6-CDG patient is serum transferrin glycoform analysis by TIEF or MS analysis. ALG6 deficiency leads to a type 1 pattern (see Fig. 105.10 ), as seen in PMM2 and MPI deficiency. There is no available enzyme analysis, although lipid-linked oligosaccharides could be evaluated in patient fibroblasts.
ALG6-CDG is autosomal recessive. Genetic testing is mostly performed by direct sequencing. The most common mutations are p.A333V and p.I299Del. Prenatal diagnosis is only reliable by genetic testing. The exact incidence of ALG6-CDG is not known.
Treatment
The current therapy in ALG6-CDG relies on supportive treatment. Mortality is about 10% in the 1st years of life, mostly from protein-losing enteropathy and severe infections.
UDP-GlcNAc:Dol-P-GlcNAc-P Transferase Deficiency (DPAGT1-CDG)
Clinical Manifestations
DPAGT1 deficiency is a recognizable and potentially treatable CDG. About one third of patients show the congenital myasthenia phenotype, indistinguishable from other genetic congenital myasthenias. Creatine kinase (CK) levels are normal. These patients have a relatively good prognosis, especially with early myasthenia therapy. The other patients show a multisystem phenotype with microcephaly, brain malformations, hypotonia, severe psychomotor disability, seizures, spasticity, failure to thrive, joint contractures, and cataracts.
Pathophysiology
DPAGT1 defect leads to very early arrest of glycan synthesis outside the ER membrane, by slowing down the addition of the 2nd GlcNAc sugar to the phosphorylated dolichol arm. Abnormal receptor glycosylation in the neuromuscular junction leads to myasthenia. Hypoglycosylation in the multisystem type leads to abnormal glycoprotein function similar to that in PMM2-CDG, especially involving the anticoagulation factors, and interestingly leading to high serum CK (in contrast to the congenital myasthenia phenotype) and hypoalbuminemia.
Diagnosis
The primary screening method is serum transferrin glycoform analysis or MS analysis. Most patients show a type 1 pattern (see Fig. 105.10 ), but patients with the congenital myasthenia phenotype can show normal screening. There is no clinically available enzyme analysis.
DPAGT1-CDG is autosomal recessive. Genetic testing is mostly performed by direct sequencing. The exact incidence is not known. Prenatal diagnosis is only reliable by genetic testing. Because of the false-negative TIEF results in several patients with the myasthenic phenotype, congenital myasthenia panel testing is suggested in suspected cases, especially for determining the potential therapy.
Treatment
The congenital myasthenia phenotype is frequently treatable by high-dose pyridostigmine, eventually enhanced with salbutamol. In the multisystem phenotype of DPAGT1-CDG, treatment is supportive.
Congenital Disorders of Protein O -Glycosylation
Cerebro-Ocular Dysplasia–Muscular Dystrophy and Muscle-Eye-Brain Disease Spectrum (POMT1-CDG, POMT2-CDG, POMGNT1-CDG)
From isolated muscular dystrophy to Walker Warburg syndrome , this group of O -linked glycosylation disorders presents with severe muscle weakness, congenital eye malformations, and neuronal migration defects. Pachygyria, cobblestone dysgenesis, hydrocephalus, polymicrogyria, heterotopias, and corpus callosum agenesis are variably present. Eye malformations include anophthalmia, microphthalmia, congenital cataract, or colobomas. Congenital muscular dystrophy is associated with significant CK level elevations. There is severe psychomotor disability.
The underlying metabolic defect is the abnormal synthesis of the O- mannosylglycan core, which is essential for the proper glycosylation of α-dystroglycan. The α-dystroglycan is heavily O -glycosylated with mannose residues and is expressed in both muscle and brain. Defective mannosylation of α-dystroglycan leads to muscle degeneration and migration defects. Muscle biopsy shows abnormal α-dystroglycan staining on immunohistochemistry.
Transferrin isoelectric focusing is normal in patients with isolated O -mannosylation defects. There is also no clinically available enzyme analysis. Diagnosis is based on histology (muscle biopsy) and genetic analysis.
POMT1-CDG, POMT2-CDG, POMGNT1-CDG are the most common autosomal recessive α-dystroglycanopathies. Additional gene defects occur in the pathway; POMK, FKTN, FKRP, LARGE, B4GAT1, TMEM5, and ISPD have been described in association with human disease. The exact incidence of α-dystroglycanopathies is not known.
In α-dystroglycanopathies the treatment is supportive.
Defects in Lipid Glycosylation and in Glycosylphosphatidylinositol Anchor Biosynthesis
Hyperphosphatasia–Intellectual Disability Syndromes: PIGA Deficiency (PIGA-CDG)
This clinically recognizable syndrome is an epilepsy syndrome with intellectual disability, hypotonia, dysmorphic facial features, skin anomalies, congenital brain malformations, and behavioral abnormalities, including autism. Other organ malformations, including cardiac and renal defects, have also been reported. (Note that somatic mutations with PIGA defect can also lead to paroxysmal nocturnal hemoglobinuria.)
N -acetylglucosamine (GlcNAc) cannot be efficiently transferred to phosphatidylinositol for glycophosphatidylinositol synthesis. Abnormal anchoring of alkaline phosphatase leads to hyperphosphatasemia in blood and loss of specific surface antigens on blood cells.
Transferrin isoform analysis is normal in GPI-anchor defects. FACS analysis of the membrane-anchored markers CD16 and CD24 in leukocytes is highly suggestive for a GPI-anchor abnormality, especially in association with increased levels of serum alkaline phosphatase. Mutation analysis confirms the defect.
PIGA-CDG is X-linked. The exact incidence is not known. A similar phenotype has been described in PIGO, PIGV, PIGY, PIG, PGAP2, and PGAP3 defects.
In PIGA-CDG the treatment is supportive.
Defects in Multiple Glycosylation Pathways and in Other Pathways, Including Dolicholphosphate Biosynthesis Defects
Steroid 5α-Reductase Deficiency (SRD5A3-CDG)
Clinical Manifestations
SRD5A3 deficiency is a clinically recognizable CDG, originally described as a multiple–congenital malformation syndrome. About 20 patients have been diagnosed at different ages, including one at 45 yr. Patients have hypotonia, ataxia, and eye abnormalities, including congenital cataract, retinal and iridic colobomas, glaucoma, optic nerve dysplasia, and visual loss. Cerebellar vermis hypoplasia can be variable. Intellectual disability has been described in all affected patients thus far. About one third of patients have severe congenital ichthyosis . Hypertrichosis and dysmorphic facial features are common, including squared face, high forehead, large ears, and coarsening. Some children with SRD5A3-CDG have a severe autism spectrum disorder. Skeletal abnormalities (scoliosis) and cardiac malformations are less common.
Pathophysiology
SRD5A3 deficiency leads to abnormal dolichol synthesis affecting early glycan synthesis outside the ER membrane and affects O -mannosylation and GPI-anchor synthesis. Hypoglycosylation affects anticoagulation factors and leads to increased serum transaminases.
Diagnosis
The primary screening method in a suspected SRD5A3-CDG patient is serum transferrin glycoform analysis or MS analysis. Most patients show a type 1 pattern (see Fig. 105.10 ), but several false-negative cases have been described. There is no clinically available enzyme analysis.
SRD5A3-CDG is autosomal recessive. Genetic testing is mostly performed by direct sequencing. The exact incidence is not known.
Treatment
In SRD5A3-CDG the treatment is supportive.
Autosomal Recessive Cutis Laxa Type 2 (ARCL-2A or ATP6V0A2-CDG, ATP6V1A-CDG and ATP6V1E1-CDG)
Clinical Manifestations
ATP6V02-CDG is a multiple-malformation syndrome originally described as cutis laxa syndrome and recently discovered to be a combined N - and O -linked glycosylation disorder. Patients show generalized cutis laxa with inelastic, sagging skin at birth, hypotonia, strabismus, myopia, characteristic facial features, and joint laxity. The facial features include hypertelorism, short nose, long philtrum, down-slanting palpebral fissures with sagging eyelids, and sagging cheeks. Cardiovascular involvement is rare, and there is variable CNS involvement. Seizures and motor and language developmental disability are common, but normal intelligence has been described as well. Sensorineural hearing loss is sometimes observed. Some patients have vermis hypoplasia, and several children have been described with cobblestone like dysgenesis and partial pachygyria on brain MRI. Skeletal abnormalities and short stature are common, as well as late-closing fontanels, and/or brachydactyly and scoliosis. There is frequently enamel dysplasia. The skin features spontaneously improve with age. ATP6V1A-CDG and ATP6V1E1-CDG show a highly overlapping phenotype with associated cardiovascular symptoms and hypercholesterolemia.
Pathophysiology
ATP6V0A2 is a membrane subunit of the proton pump of the vesicular adenosine triphosphatase (V-ATPase) complex. Abnormal function of the V-ATPase complex alters the pH gradient in the secretory pathway and affects the maturation and transport of several glycosyltransferases and elastic fibers (e.g., elastin). ATP6V1A and ATP6V1E1 are other complex subunits affecting ATP6V0A2 function and cause secondary ATPase deficiency. Both N - and O -linked glycosylation are affected. There are mild coagulation abnormalities and high serum transaminase levels in some patients.
Diagnosis
The primary screening method in a suspected ATP6V0A2-CDG patient is serum transferrin glycoform analysis or MS analysis. Most patients show a type 2 pattern (see Fig. 105.10 ), but false-negative cases have been described before age 6 wk. Apolipoprotein III-C (apoC-III) is a mucin-type secretory glycoprotein that is only O -glycosylated. ApoC-III IEF shows a hypoglycosylation pattern in patients, even when the TIEF is falsely negative. Skin biopsy in patients show classic histologic changes of cutis laxa with diminished, short, abnormal, and fuzzy elastic fibers.
ATP6V0A2-CDG is autosomal recessive. Genetic testing is mostly performed by direct sequencing. The exact incidence is not known. ATP6V1A and ATP6V1E1 defects have been recently described.
Treatment
In autosomal recessive cutis laxa type 2, the treatment is supportive. There is continuous and spontaneous improvement of skin symptoms throughout the disease course, especially in ATP6V0A2-CDG.
Golgi-α1-2 Mannosidase-1 Deficiency (MAN1B1-CDG)
Clinical Manifestations
MAN1B1 defect was originally described as an intellectual disability syndrome in association with dysmorphic features. Additional patients were recognized with psychomotor disability, muscle hypotonia, and inverted nipples in association with truncal obesity. The degree of intellectual disability is quite variable. Autistic behaviors, eating disorders, and aggressive behavior are frequent features. More than 30 patients have been reported.
Pathophysiology
MAN1B1 codes for a Golgi mannosidase, which is essential for the final “trimming” of mannose units during the glycan processing in the Golgi. Hypermannosylation leads to abnormal, truncated glycans and CDG-II. The glycosylation abnormality in serum is relatively mild. Increased serum transaminases and abnormal coagulation are uncommon.
Diagnosis
Most patients show a mild type 2 pattern by TIEF, but false-negative cases have been described. MALDI-TOF analysis shows characteristic, hybrid glycans in serum. In suspected cases, direct sequence analysis is recommended, even if the TIEF is normal.
MAN1B1-CDG is autosomal recessive. The exact incidence is unknown; several adult patients are known.
Treatment
Only supportive treatment is available.
Phosphoglucomutase-1 Deficiency (PGM1-CDG)
Clinical Manifestations
PGM1-CDG is a disorder presenting with midline malformations (cleft palate, Pierre Robin sequence, bifid uvula), liver dysfunction, hypoglycemia, and short stature in almost all patients. Hypoglycemia is usually caused by hyperinsulinism in the 1st years of life. It can resolve with aging; ketotic hypoglycemia has also been observed. Cholestasis, liver fibrosis, and even cirrhosis have been described in a few patients. About one third of patients also show proximal muscle weakness and dilated cardiomyopathy; the latter led to mortality in at least 7 reported cases. Other malformations, including cardiac and skeletal anomalies, have also been described. Wound healing is frequently abnormal, and there is a very high risk for bleeding during surgery. Intelligence is normal.
Pathophysiology
Phosphoglucomutase 1 (PGM1) is an essential enzyme for glycogenolysis and glycolysis. It also provides substrates for nucleotide sugars needed for normal glycosylation. PGM1 regulates the bidirectional conversion of glucose-1-phosphate and glucose-6-phosphate. During fasting it leads to a glycogenosis-like phenotype (also called GSD XIV, MIM 614921). PGM1-CDG affects both the ER- and Golgi-related glycosylation and causes a mixed type 1/type 2 hypoglycosylation pattern. Abnormal serum proteins include coagulation and anticoagulation factors, insulin-like growth factor–binding protein 3 (IGFBP3), TBG, and thyroid-stimulating hormone (TSH), in addition to serum transaminases, hypoglycemia, and elevated CK.
Diagnosis
The primary screening method in a suspected PGM1-CDG is serum transferrin glycoform analysis or MS analysis. Patients show a mixed type 1/type 2 pattern.
PGM1-CDG is autosomal recessive. It is among the relatively common CDGs; >40 patients have been described. Enzyme testing is possible in blood, but is more reliable in fibroblasts. Direct sequencing is available for testing.
Treatment
PGM1-CDG seems to be the 2nd treatable CDG besides MPI-CDG. D -Galactose is hypothesized to restore the balance in the availability of different nucleotide sugars. Adding 1 g/kg/day D -galactose for a few weeks to the diet improves glycosylation significantly, although the TIEF pattern does not fully normalize. This treatment improves liver transaminases and antithrombin levels and in some patients the hormonal status. The effect of D -galactose on hypoglycemic episodes and the myopathy is not yet clear. Larger, long-term dietary trials are ongoing.
Disorders of Golgi Homeostasis: TMEM199-, CCDC115-, ATP6AP2-CDG, and ATP6AP1-CDG
Clinical Manifestations
These 4 disorders are clinically and biochemically indistinguishable. They have been described with liver function anomalies, cholestasis, fibrosis, and cirrhosis with liver failure, necessitating liver transplantation in a few patients. The phenotype resembles Wilson disease , especially because of low serum ceruloplasmin and copper levels, but there is no Kayser-Fleischer ring. In CCDC115-CDG there are frequently also neurologic features. The intellectual outcome is variable. Additional abnormalities include hypercholesterolemia and elevated alkaline phosphatase. In ATP6AP1-CDG there is also immunologic involvement.
Pathophysiology
TMEM199-, CCDC115-, ATP6AP1-CDG, and ATP6AP2-CDG are important for Golgi homeostasis. The exact pathologic mechanism is not yet known, but it is hypothesized that the secondary Golgi dysfunction affects and delays the normal glycosylation process.
Diagnosis
The primary screening method in a patient with suspected PGM1-CDG is serum transferrin glycoform analysis or MS analysis. Patients show a type 2 pattern (see Fig. 105.10 ). ApoC-III IEF is abnormal. Glycomics results by MALDI-TOF analysis are characteristic but cannot discriminate between the 3 defects. Final diagnosis requires mutation analysis.
TMEM199-CDG and CCDC115-CDG are autosomal recessive, whereas ATP6AP1-CDG, and ATP6AP2-CDG are X-linked.
Treatment
Treatment is supportive; 2 patients successfully underwent liver transplantation.
Manganese Transporter Defect: SLC39A8-CDG
Clinical Manifestations
This intriguing disorder was originally described as a neurologic disease with hypotonia, seizures (hypsarrhythmia), and developmental disability. Some of the later-described patients had severe skeletal dysplasia with rhizomelic chondrodysplasia, craniosynostosis, and dwarfism. Mitochondrial dysfunction (Leigh disease, cerebral lactic acidemia, dystonia) may also be present.
Pathophysiology
SLC39A8 is a membrane transporter, responsible for the manganese (Mn) transmembrane transport. SLC39A8 deficiency affects all Mn-dependent enzymes and therefore different parts of the metabolism. Since several glycosyltransferases (e.g. β-1,4-galactosyltransferase) are Mn dependent, a secondary Golgi glycosylation occurs with a type 2 glycosylation defect. Low serum Mn levels are suggestive but not always present in patients.
Diagnosis
The primary screening method in a suspected SLC39A8-CDG is serum transferrin glycoform analysis or MS analysis. Patients show a type 2 pattern (see Fig. 105.10 ). MALDI-TOF analysis is suggestive, but not discriminative. Final diagnosis requires mutation analysis.
SLC39A8-CDG is an autosomal recessive disease. Its incidence is unknown.
Treatment
Besides supportive treatment, a few patients showed biochemical and clinical improvement (better seizure control) with oral D -galactose (1-3 g/kg/day) therapy.
Congenital Disorders of Deglycosylation
N-Glycanase 1 Deficiency (NGLY1 Defect)
Clinical Manifestations
Patients with NGLY1 deficiency do have a glycosylation disorder, but not from the deficient synthesis; rather, it is caused by deficient breakdown of glycoproteins. The phenotype comprises severe CNS involvement, microcephaly, intellectual disability, seizures, neuropathy, movement disorders, and hypotonia. The presence of alacrimia , hypolacrimia, or chalazion is highly suggestive for the diagnosis, but not all patients have problems with tearing. Other features include failure to thrive, intrauterine growth restriction, and liver involvement. Some patients have a recognizable oval face with a short nose, flat profile, and hypertelorism. Masklike face also occurs, imitating the phenotype of mitochondrial disorders, especially when serum lactic acid levels are also elevated.
Pathophysiology
N -glycanase is responsible for the deglycosylation of misfolded N -linked glycoproteins. The enzyme is essential for cutting off the glycans before the proteins are degraded in the ER. The increased abundance of misfolded N -glycans increases ER stress, which has been suggested as a possible reason for lactate elevation in several patients. Serum transaminase and α-fetoprotein levels are also frequently increased.
Diagnosis
Serum transferrin isoform analysis shows a normal pattern. Final diagnosis requires mutation analysis.
NGLY1-CDG is an autosomal recessive disease. The most common mutation is c.1201A>T/p.R401X. The exact incidence of the condition is unknown, but >20 patients have been reported in a few years since the discovery of the disease.
Treatment
Only supportive treatment is available for the patient with NGLY1 deficiency.
Therapeutic Summary
Most CDGs are only treatable with supportive therapy. The initially discovered oral mannose treatment in MPI-CDG (1 g/kg/day) has proved to be efficient for coagulation problems and protein-losing enteropathy but cannot prevent liver fibrosis in all patients. Liver transplantation in MPI-CDG has been successful in a few patients. Oral D-galactose in PGM1-CDG (1g/kg/day) improves serum transaminases and coagulation, and has a positive effect on endocrine function, but cannot restore glycosylation fully. Seizure frequency improved in patients with SLC39A8-CDG receiving oral D -galactose treatment (1 g/kg/day) and oral Mn intake. The congenital myasthenic syndrome in DPAGT1-CDG, GFPT1-CDG and GMPPB-CDG has been successfully treated with high dose of cholinesterase inhibitors. Several CDG have been positively controlled by transplantation; including DOLK-CDG (DK1-CDG; heart transplantation) PGM3-CDG (hematopoietic stem cell transplantation), CCDC155-CDG (liver transplantation).
Additional CDG treatment options are available for disorders not described in this chapter. Patients with CAD-CDG show significant clinical improvement on receiving oral uridine therapy, especially with seizure control. Two children with SLC35C1-CDG–defective immune function improved on oral fucose therapy. GNE-CDG patients showed significant improvement in muscle strength on N -acetylmannoseamin therapy. Several dietary trials are currently ongoing in different CDG.
Bibliography
De Lonlay P, Seta N. The clinical spectrum of phosphomannose isomerase deficiency, with an evaluation of mannose treatment for CDG-Ib. Biochim Biophys Acta . 2009;1792(9):841–843.
Freeze HH, Chong JX, Bamshad MJ, Ng BG. Solving glycosylation disorders: fundamental approaches reveal complicated pathways. Am J Hum Genet . 2014;94:161–175.
Freeze HH, Eklund EA, Ng BG, Patterson MC. Neurology of inherited glycosylation disorders. Lancet Neurol . 2012;11:453–466.
Funke S, Gardeitchik T, Kouwenberg D, et al. Perinatal and early infantile symptoms in congenital disorders of glycosylation. Am J Med Genet . 2013;161A:578–584.
Jaeken J, Hennet T, Matthijs G, Freeze HH. CDG nomenclature: time for a change!. Biochim Biophys Acta . 2009;1792:825–826.
Jaeken J, et al. Sialic acid-deficient serum and cerebrospinal fluid transferrin in a newly recognized genetic syndrome. Clin Chim Acta . 1984;144:245–247.
Lam C, Ferreira C, Krasnewich D, et al. Prospective phenotyping of NGLY1-CDDG, the first congenital disorder of deglycosylation. Genet Med . 2017;19(2):160–168.
Lefeber DJ, Morava E, Jaeken J. How to find and diagnose a CDG due to defective N -glycosylation. J Inherit Metab Dis . 2011;34(4):849–852.
Mohamed M, Kouwenberg D, Gardeitchik T, et al. Metabolic cutis laxa syndromes. J Inherit Metab Dis . 2011;34:907–916.
Morava E. Galactose supplementation in 1 phosphoglucomutase-1 deficiency: review and outlook for a novel treatable CDG. Mol Genet Metab . 2014;112:275–279.
Morava E, Tiemes V, Thiel C, et al. ALG6-CDG: a recognizable phenotype with epilepsy, proximal muscle weakness, ataxia and behavioral and limb anomalies. J Inherit Metab Dis . 2016;39(5):713–723.
Morava E, Wevers RA, Cantagrel V, et al. A novel cerebello-ocular syndrome with abnormal glycosylation due to abnormalities in dolichol metabolism. Brain . 2010;133:3210–3220.
Pérez-Cerdá C, Girás ML, Serrano M, et al. A population-based study on congenital disorders of protein N - and combined with O -glycosylation: experience in clinical and genetic diagnosis. J Pediatr . 2017;183:170–177.
Rymen D, Winter J, van Hasselt PM, et al. Key features and clinical variability of COG6-CDG. Mol Genet Metab . 2015;116(3):163–170.
Scott K, Gadomski T, Kozicz T, Morava E. Congenital disorders of glycosylation: new defects and still counting. J Inherit Metab Dis . 2014;37:609–617.
Wong SY, Beamer LJ, Gadomski T, et al. Defining the phenotype and assessing severity in phosphoglucomutase-1 deficiency. J Pediatr . 2016;175:130–136.e8.