Miscellaneous Conditions Associated With Arthritis
Angela Byun Robinson, C. Egla Rabinovich
Relapsing Polychondritis
Relapsing polychondritis (RP) is a rare condition characterized by episodic chondritis causing cartilage destruction and deformation of the ears (sparing the earlobes), nose, larynx, and tracheobronchial tree. Antibodies to matrillin-1 and collagen (type II, IX and XI) are present in approximately 60% of patients with RP, suggesting an autoimmune pathogenesis. Patients may experience arthritis, uveitis, and hearing loss resulting from inflammation near the auditory and vestibular nerves. Children may initially relate episodes of intense erythema over the outer ears. Other dermatologic manifestations may include erythema nodosum, maculopapular rash, and purpura. Cardiac involvement, including conduction defects and coronary vasculitis, has been reported. Severe, progressive, and potentially fatal disease resulting from destruction of the tracheobronchial tree and airway obstruction is unusual in childhood. Diagnostic criteria established for adults are useful guidelines for evaluating children with suggestive symptoms (Table 194.1 ). The clinical course of RP is variable; flares of disease are often associated with elevations of acute-phase reactants and may remit spontaneously. Although seen more often in the adult population, RP may coexist with other rheumatic disease (e.g., systemic lupus erythematosus, Sjögren syndrome, Henoch-Schönlein purpura) in up to 30% of patients. The differential diagnosis includes ANCA-associated vasculitis (granulomatosis with polyangiitis) (see Chapter 192.4 ) and Cogan syndrome , which is characterized by auditory nerve inflammation and keratitis but not chondritis. Many children respond to nonsteroidal antiinflammatory drugs, but some require corticosteroids or other immunosuppressive agents (azathioprine, methotrexate, hydroxychloroquine, colchicine, cyclophosphamide, cyclosporine, and anti–tumor necrosis factor [TNF] agents), as reported in small series and case reports.
Table 194.1
| MAJOR |
|
Typical inflammatory episodes of ear cartilage Typical inflammatory episodes of nose cartilage Typical inflammatory episodes of laryngotracheal cartilage |
| MINOR |
|
Eye inflammation (conjunctivitis, keratitis, episcleritis, uveitis) Hearing loss Vestibular dysfunction Seronegative inflammatory arthritis |
* The diagnosis is established by the presence of 2 major or 1 major and 2 minor criteria. Histologic examination of affected cartilage is required when the presentation is atypical.
Data from Michet CJ Jr, McKenna CH, Luthra HS, et al: Relapsing polychondritis: survival and predictive role of early disease manifestations, Ann Intern Med 104:74-78, 1986.
Mucha-Habermann Disease/Pityriasis Lichenoides Et Varioliformis Acuta
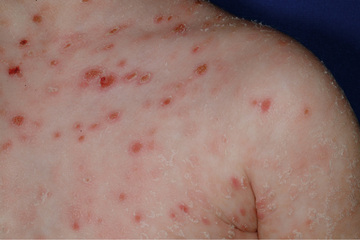
Pityriasis lichenoides et varioliformis acuta (PLEVA ) is a benign, self-limited cutaneous vasculitis characterized by episodes of macules, papules, and papulovesicular lesions that can develop central ulceration, necrosis, and crusting (Fig. 194.1 ). Different stages of development are usually seen at once. PLEVA fulminans or febrile ulceronecrotic Mucha-Habermann disease (FUMHD ) is the severe, life threatening form of PLEVA. Large, coalescing, ulceronecrotic lesions are seen, accompanied by high fever and elevated erythrocyte sedimentation rate (ESR). Systemic manifestations can include interstitial pneumonitis, abdominal pain, malabsorption, arthritis, and neurologic manifestations. PLEVA has a male predominance and occurs more frequently in childhood. The diagnosis is confirmed by biopsy of skin lesions, which reveals perivascular and intramural lymphocytic inflammation affecting capillaries and venules in the upper dermis that may lead to keratinocyte necrosis. When disease is severe, corticosteroids have been used with questionable effect, and methotrexate has been reported to induce rapid remission in resistant cases. Cyclosporine and anti-TNF agents have also been efficacious in case reports.
Sweet Syndrome
Sweet syndrome, or acute febrile neutrophilic dermatosis , is a rare entity in children. It is characterized by fever, elevated neutrophil count, and raised, tender erythematous plaques and nodules over the face, extremities, and trunk. Skin biopsy reveals neutrophilic perivascular infiltration in the upper dermis. Female predominance is seen in the adult population, whereas gender distribution is equal in children. Established criteria are useful for diagnosis (Table 194.2 ). Children can also have arthritis, sterile osteomyelitis, myositis, and other extracutaneous manifestations. Sweet syndrome may be idiopathic or secondary to malignancy (particularly acute myelogenous leukemia), drugs (granulocyte colony-stimulating factor, tretinoin or trimethoprim-sulfamethoxazole), or rheumatic diseases (Behçet disease, antiphospholipid antibody syndrome, systemic lupus erythematosus). The condition usually responds to treatment with corticosteroids, treatment of underlying disease, or removal of associated medication.
Hypertrophic Osteoarthropathy
Children with chronic disease, especially pulmonary or cardiac disease, can demonstrate clubbing of the terminal phalanges and have associated periosteal reaction and arthritis. These findings characterize the classic presentation of hypertrophic osteoarthropathy. HOA can be primary (idiopathic) or secondary. Although rare, secondary HOA is more common in children and is seen in those with chronic pulmonary disease (cystic fibrosis), congenital heart disease, gastrointestinal disease (malabsorption syndromes, biliary atresia, inflammatory bowel disease), and malignancy (nasopharyngeal sarcoma, osteosarcoma, Hodgkin disease). It may precede diagnosis of cardiopulmonary disease or malignancy. The pathogenesis of secondary HOA is unknown; symptoms often improve if the underlying condition is treated successfully. HOA-related pain can be disabling; in adults, management with bisphosphonates has been reported. Evaluation of children presenting with HOA should include a chest radiograph to evaluate for pulmonary disease or intrathoracic mass. Autosomal recessive mutations in prostaglandin pathway genes have recently been described in primary HOA, also described as pachydermoperiostosis .
Plant Thorn Synovitis
A diagnosis of plant thorn synovitis should be considered in children with monoarticular arthritis nonresponsive to antiinflammatory therapy. Acute or chronic arthritis can occur after a plant thorn or other foreign object penetrates a joint. Unlike septic arthritis, children with plant thorn synovitis are usually afebrile. The most common organism seen with plant thorn synovitis is Pantoea agglomerans , although cultures are often negative. The initial injury may be unknown or forgotten, making diagnosis difficult. Ultrasound or MRI can be useful in identifying the foreign body. Removal of the foreign body using arthroscopy, followed by an antibiotic course, is the accepted therapy.
Pigmented Villonodular Synovitis
Proliferation of synovial tissue is seen in pigmented villonodular synovitis (PVNS). This proliferation is localized or diffuse and can affect the joint, tendon sheath, or bursa. Macrophages and multinucleated giant cells with brownish hemosiderin are present histologically. It is unclear if the etiology of PVNS is inflammatory or neoplastic in nature. Although findings are not pathognomonic, MRI with contrast is a useful diagnostic tool by which PVNS can be seen as a mass or bone erosion. Brown or bloody synovial fluid is seen with arthrocentesis, but the diagnosis is made by tissue biopsy. Surgical removal of the affected tissue is the therapeutic modality, and with diffuse disease, a total synovectomy is recommended.