Vasculitis Syndromes
Vidya Sivaraman, Edward C. Fels, Stacy P. Ardoin
Childhood vasculitis encompasses a broad spectrum of diseases that share inflammation of the blood vessels as the central pathophysiology. The pathogenesis of the vasculitides is generally idiopathic. Some forms of vasculitis are associated with infectious agents and medications, whereas others may occur in the setting of preexisting autoimmune disease. The pattern of vessel injury provides insight into the form of vasculitis and serves as a framework to delineate the different vasculitic syndromes. The distribution of vascular injury includes small vessels (capillaries, arterioles, and postcapillary venules), medium vessels (renal arteries, mesenteric vasculature, and coronary arteries), and large vessels (the aorta and its proximal branches) (Fig. 192.1 ). Additionally, some forms of small vessel vasculitis are characterized by the presence of antineutrophil cytoplasmic antibodies (ANCAs) , whereas others are associated with immune complex deposition in affected tissues. A combination of clinical features, histologic appearance of involved vessels, and laboratory data is used to classify vasculitis (Tables 192.1 to 192.3 ). A nomenclature system from the 2012 International Chapel Hill Consensus Conference has proposed using the pathologic diagnosis rather than eponyms for vasculitis nomenclature. For example, Henoch-Schönlein purpura would be referred to as IgA vasculitis. Additionally, the classification criteria endorsed by the European League Against Rheumatism, Pediatric Rheumatology International Trial Organization, and Pediatric Rheumatology European Society (EULAR/ PRINTO/PRES) have been validated in childhood vasculitis. (Table 192.1 ).
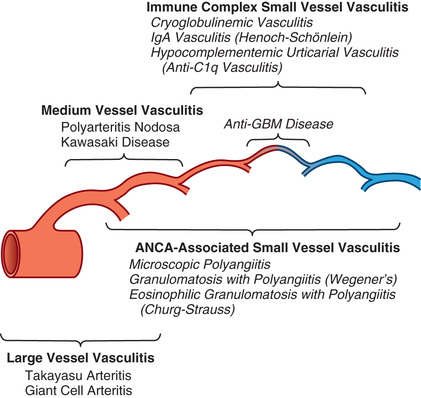
Table 192.3
Clinicopathologic Characteristics of Vasculitides in Childhood
| SYNDROME | FREQUENCY | VESSELS AFFECTED | CHARACTERISTIC PATHOLOGY |
|---|---|---|---|
| Polyarteritis | |||
| Polyarteritis nodosa | Rare | Medium-size and small muscular arteries and sometimes arterioles | Focal segmental (often near bifurcations); fibrinoid necrosis; gastrointestinal, renal microaneurysms; lesions at various stages of evolution |
| Kawasaki disease | Common | Coronary and other muscular arteries | Thrombosis, fibrosis, aneurysms, especially of coronary vessels |
| Leukocytoclastic Vasculitis | |||
| Henoch-Schönlein purpura (IgA vasculitis) | Common | Arterioles and venules, often small arteries and veins | Leukocytoclasis; mixed cells, eosinophils, IgA deposits in affected vessels |
| Hypersensitivity angitis | Rare | Arterioles and venules | Leukocytoclastic or lymphocytic, varying eosinophils, occasionally granulomatous; widespread lesions at same stage of evolution |
| Granulomatous Vasculitis | |||
| Granulomatosis with polyangiitis (Wegener granulomatosis) | Rare | Small arteries and veins, occasionally larger vessels | Upper and lower respiratory tract, necrotizing granulomata glomerulonephritis |
| Eosinophilic granulomatosis with polyangiitis (Churg-Strauss syndrome) | Rare | Small arteries and veins, often arterioles and venules | Necrotizing extravascular granulomata; lung involvement; eosinophilia |
| Giant Cell Arteritis | |||
| Takayasu arteries | Uncommon | Large arteries | Granulomatous inflammation, giant cells; aneurysms, dissection |
| Temporal arteritis | Rare | Medium-size and large arteries | Granulomatous inflammation, giant cell arteries |
Adapted from Cassidy JT, Petty RE: Textbook of pediatric rheumatology, ed 6, Philadelphia, 2011, Elsevier Saunders.
Childhood vasculitis varies from a relatively benign and self-limited disease such as Henoch-Schönlein purpura to catastrophic disease with end-organ damage, as seen in granulomatosis with polyangiitis (formerly Wegener granulomatosis). Vasculitis generally manifests as a heterogeneous multisystem disease. Although some features, such as purpura, are easily identifiable, others, such as hypertension secondary to renal artery occlusion or glomerulonephritis, can be subtler. Ultimately, the key to recognizing vasculitis relies heavily on pattern recognition. Demonstration of vessel injury and inflammation on biopsy or vascular imaging is required to confirm a diagnosis of vasculitis.
Henoch-Schönlein Purpura
Vidya Sivaraman, Edward C. Fels, Stacy P. Ardoin
Henoch-Schönlein purpura (HSP ) is the most common vasculitis of childhood and is characterized by leukocytoclastic vasculitis and immunoglobulin A deposition in the small vessels in the skin, joints, gastrointestinal tract, and kidney. According to the 2012 International Chapel Hill Consensus Conference nomenclature, HSP is also referred to as IgA vasculitis , based on the presence of vasculitis with predominance of IgA deposits affecting small vessels.
Epidemiology
HSP occurs worldwide and affects all ethnic groups but is more common in white and Asian populations. The incidence of HSP is estimated at 14-20 per 100,000 children per year and affects males more than females, with a 1.2-1.8 : 1 male/female ratio. Approximately 90% of HSP cases occur in children, usually between ages 3 and 10 yr. HSP is distinctly less common in adults, who often have severe and chronic complications. HSP is more common in the winter and spring and is unusual in summer months. Many cases of HSP follow a documented upper respiratory infection.
Pathology
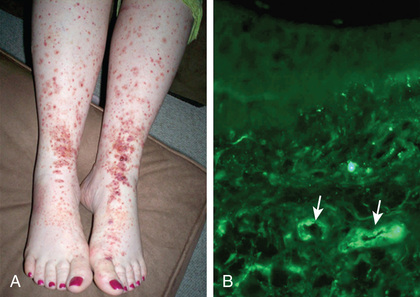
Skin biopsies demonstrate leukocytoclastic vasculitis of the dermal capillaries and postcapillary venules. The inflammatory infiltrate includes neutrophils and monocytes. Renal histopathology typically shows endocapillary proliferative glomerulonephritis, ranging from a focal segmental process to extensive crescentic involvement. In all tissues, immunofluorescence identifies IgA deposition in walls of small vessels (Fig. 192.2 ), accompanied to a lesser extent by deposition of C3, fibrin, and IgM.
Pathogenesis
The exact pathogenesis of HSP remains unknown. Given the seasonality of HSP and the frequency of preceding upper respiratory infections, infectious triggers such as group A β-hemolytic streptococcus, Staphylococcus aureus, mycoplasma, and adenovirus have been suspected. The common finding of deposition of IgA, specifically IgA1 , suggests that HSP is a disease mediated by IgA and IgA immune complexes. HSP occasionally clusters in families, suggesting a genetic component. HLA-B34 and HLA-DRB1*01 alleles have been linked to HSP nephritis. Patients with familial Mediterranean fever, hereditary periodic fever syndromes, and complement deficiencies are at increased risk for developing HSP, suggesting that genetically determined immune dysregulation may contribute.
Clinical Manifestations
The hallmark of HSP is its rash : palpable purpura starting as pink macules or wheals and developing into petechiae, raised purpura, or larger ecchymoses. Occasionally, bullae and ulcerations develop. The skin lesions are usually symmetric and occur in gravity-dependent areas (lower extremities), extensor aspect of the upper extremities or on pressure points (buttocks) (Figs. 192.2 and 192.3 ). The skin lesions often evolve in groups, typically lasting 3-10 days, and may recur up to 4 mo after initial presentation. Subcutaneous edema localized to the dorsa of hands and feet, periorbital area, lips, scrotum, or scalp is also common.
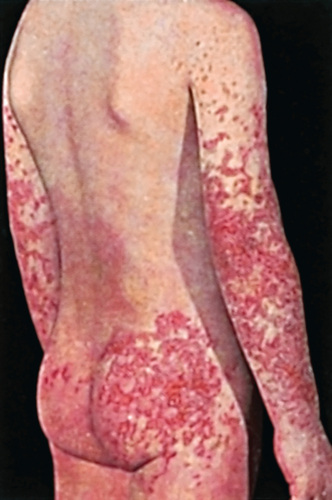
Musculoskeletal involvement, including arthritis and arthralgias, is common, occurring in up to 75% of children with HSP. The arthritis tends to be self-limited and oligoarticular, with a predilection for large joints such as the knees and ankles, and does not lead to deformities. Periarticular swelling and tenderness without erythema or effusions are common. The arthritis usually resolves within 2 wk but can recur.
Gastrointestinal (GI) manifestations occur in up to 80% of children with HSP and include abdominal pain, vomiting, diarrhea, paralytic ileus, and melena. Intussusception, mesenteric ischemia, and intestinal perforation are rare but serious complications. Endoscopic evaluation is usually not needed but may identify vasculitis of the intestinal tract.
Renal involvement occurs in up to 30% of children with HSP, manifesting as microscopic hematuria, proteinuria, hypertension, frank nephritis, nephrotic syndrome, and acute or chronic renal failure. However, progression to end-stage renal disease (ESRD) is uncommon in children (1–2%) (see Chapter 538.3 ). Renal manifestations can be delayed for several months after the initial illness, so close follow-up with serial urinalyses and blood pressure monitoring is necessary.
Neurologic manifestations of HSP, caused by hypertension (posterior reversible encephalopathy syndrome) or central nervous system (CNS) vasculitis, may also occur, including intracerebral hemorrhage, seizures, headaches, depressed level of consciousness, cranial or peripheral neuropathies, and behavior changes. Other, less common potential manifestations of HSP are inflammatory eye disease, carditis, pulmonary hemorrhage, orchitis, and testicular torsion.
Diagnosis
The diagnosis of HSP is clinical and often straightforward when the typical rash is present. However, in at least 25% of cases, the rash appears after other manifestations, making early diagnosis challenging. Table 192.4 summarizes the EULAR/PRES classification criteria for HSP. Most patients are afebrile.
The differential diagnosis for HSP depends on specific organ involvement but usually includes other small vessel vasculitides, infections, acute poststreptococcal glomerulonephritis, hemolytic-uremic syndrome, coagulopathies, and other acute intraabdominal processes. Additional disorders in the differential include papular-purpuric glove and sock syndrome, systemic lupus erythematosus (SLE), other vasculitides (urticarial, hypersensitivity), and thrombocytopenia.
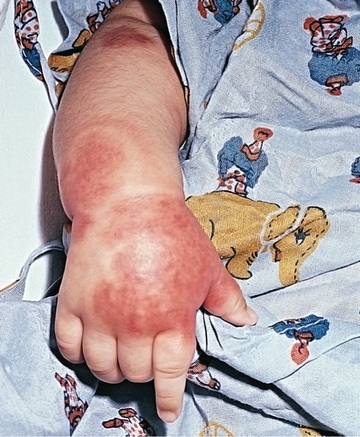
Infantile acute hemorrhagic edema (AHE) , an isolated cutaneous leukocytoclastic vasculitis that affects infants <2 yr of age, resembles HSP clinically. AHE manifests as fever; tender edema of the face, scrotum, hands, and feet; and ecchymosis (usually larger than the purpura of HSP) on the face and extremities (Fig. 192.4 ). The trunk is spared, but petechiae may be seen in mucous membranes. The patient usually appears well except for the rash. The platelet count is normal or elevated, and the urinalysis results are normal. The younger age, the nature of the lesions, absence of other organ involvement, and a biopsy may help distinguish infantile AHE from HSP.
Laboratory Findings
No laboratory finding is diagnostic of HSP. Common but nonspecific findings include leukocytosis, thrombocytosis, mild anemia, and elevations of erythrocyte sedimentation rate (ESR) and C-reactive protein (CRP). The platelet count is normal in HSP . Occult blood is frequently found in stool specimens. Serum albumin levels may be low because of renal or intestinal protein loss. Autoantibody testing such as antinuclear antibody (ANA) is not useful diagnostically except to exclude other diseases. Serum IgA values are often elevated but are not routinely measured. Assessment of renal involvement with blood pressure, urinalysis, and serum creatinine is necessary.
Ultrasound is often used in the setting of GI complaints to look for bowel wall edema or the rare occurrence of an associated intussusception. Barium enema can also be used to both diagnose and treat intussusception. Although often unnecessary in typical HSP, biopsies of skin and kidney can provide important diagnostic information, particularly in atypical or severe cases, and characteristically show leukocytoclastic vasculitis with IgA deposition in affected tissues.
Treatment
Treatment for mild and self-limited HSP is supportive, with an emphasis on ensuring adequate hydration, nutrition, and analgesia. Corticosteroids are most often used to treat significant GI involvement or other life-threatening manifestations. Glucocorticoids such as oral prednisone (1-2 mg/kg/day), or in severe cases, intravenous (IV) methylprednisolone for 1-2 wk, followed by taper, reduce abdominal and joint pain but do not alter overall prognosis. Corticosteroids are not routinely recommended for prevention of complications such as nephritis. Rapid tapering of corticosteroids may lead to a flare of HSP symptoms. Although few data are available to demonstrate efficacy, intravenous immune globulin (IVIG) and plasma exchange are sometimes used for severe disease. In some patients, chronic HSP renal disease is managed with a variety of immunosuppressants, including azathioprine, cyclophosphamide, cyclosporine, and mycophenolate mofetil. ESRD develops in <5% of children with HSP nephritis.
Complications
Acutely, serious GI involvement, including intussusception and intestinal perforation, imparts significant morbidity and mortality. Renal disease is the major long-term complication, occurring in 1–2% of children with HSP. Renal disease can develop up to 6 mo after diagnosis but rarely does so if the initial urinalysis findings are normal. Therefore, it is recommended that children with HSP undergo serial monitoring of blood pressure and urinalysis for at least 6 mo after diagnosis to monitor for development of nephritis.
Prognosis
Overall, the prognosis for childhood HSP is excellent, and most children experience an acute, self-limited course lasting on average 4 wk. However, 15–60% of children with HSP experience 1 or more recurrences, typically within 4-6 mo of diagnosis. With each relapse, symptoms are usually milder than at presentation. Children with a more severe initial course are at higher risk for relapse. The long-term prognosis usually depends on the severity and duration of GI or renal involvement. Chronic renal disease develops in 1–2% of children with HSP, and <5% of those with HSP nephritis go on to have ESRD. The risk of HSP recurrence and graft loss following renal transplantation is estimated at 7.5% after 10 yr.
Bibliography
Chartapisak W, Opastiraku S, Willis NS, et al. Prevention and treatment of renal disease in Henoch-Schönlein purpura: a systematic review. Arch Dis Child . 2009;94:132–137.
Coulombe J, Jean SE, Hatami A, et al. Pigmented purpuric dermatosis: clinicopathologic characterization in a pediatric series. Pediatr Dermatol . 2015;32(3):358–362.
Dayanir YO, Akdilli A, Karaman CZ, et al. Epididymoorchitis mimicking testicular torsion in Henoch-Schönlein purpura. Eur Radiol . 2011;11:2267–2269.
Dudley JS. Randomised, double-blind, placebo-controlled trial to determine whether steroids reduce the incidence and severity of nephropathy in Henoch-Schönlein purpura (HSP). Arch Dis Child . 2013;98(10):756–763.
He X, Yu C, Zhao P, et al. The genetics of Henoch-Schönlein purpura: a systematic review and meta-analysis. Rheumatol Int . 2013;33:1387–1395.
Jauhola O, Ronkainen J, Koskimies O, et al. Clinical course of extrarenal symptoms in HSP: a 6-month prospective study. Arch Dis Child . 2010;95:871–876.
Jauhola O, Ronkainen J, Koskimies O, et al. Renal manifestations of HSP in a 6-month prospective study of 223 children. Arch Dis Child . 2010;95:877–882.
Jauhola O, Ronkainen J, Koskimies O, et al. Outcome of Henoch-Schönlein purpura 8 yr after treatment with a placebo or prednisone at disease onset. Pediatr Nephrol . 2012;28:933–939.
Kanaan N, Mourad G, Thervet E. Recurrence and graft loss after kidney transplantation for Henoch-Schönlein purpura nephritis: a multicenter analysis. Clin J Am Soc Nephrol . 2011;6:1768–1772.
Jennette JC, Falk RJ. Revised International Chapel Hill Consensus Conference nomenclature of vasculitides. Arthritis Rheum . 2013;65(1):1–11.
Mir S, Yavascan O, Mutlubas F, et al. Clinical outcome in children with Henoch-Schönlein nephritis. Pediatr Nephrol . 2007;22:64–70.
Ozen S, Pistorio A, Iusan SM, et al. EULAR/PRINTO/PRES criteria for Henoch-Schönlein purpura, childhood polyarteritis nodosa, childhood Wegener granulomatosis and childhood Takayasu arteritis: Ankara 2008. Part II. Final classification criteria. Ann Rheum Dis . 2010;69:798–806.
Ronkainen J, Koskimies O, Ala-Houhala M, et al. Early prednisone therapy in Henoch-Schönlein purpura: a randomized, double-blind, placebo-controlled trial. J Pediatr . 2006;149:241–247.
Weiss PF, Feinstein JA, Luan X, et al. Effects of corticosteroid on Henoch-Schönlein purpura: a systematic review. Pediatrics . 2007;120:1079–1087.
Weiss PF, Klink AJ, Hexem K, et al. Variation in inpatient therapy and diagnostic evaluation of children with Henoch-Schönlein purpura. J Pediatr . 2009;155:812–818.
Weiss PF, Klink AJ, Localio R, et al. Corticosteroids may improve clinical outcomes during hospitalization for Henoch-Schönlein purpura. Pediatrics . 2010;126:674–6781.
Yang YS, Yu HH, Chiang BL. The diagnosis and classification of Henoch-Schönlein purpura: an updated review. Autoimmun Rev . 2014;13:355–358.
Takayasu Arteritis
Vidya Sivaraman, Edward C. Fels, Stacy P. Ardoin
Takayasu arteritis (TA ), also known as pulseless disease , is a chronic large vessel vasculitis of unknown etiology that predominantly involves the aorta and its major branches.
Epidemiology
Although TA occurs worldwide and can affect all ethnic groups, the disease is most common in Asians. Age of onset is typically between 10 and 40 yr. Most children are diagnosed as adolescents, on average at age 13 yr. Up to 20% of individuals with TA are diagnosed before 19 yr. Younger children may be affected, but diagnosis in infancy is rare. TA preferentially affects females, with a reported 2-4 : 1 female/male ratio in children and adolescents and a 9 : 1 ratio among adults. Occlusive complications are more common in the United States, Western Europe, and Japan, whereas aneurysms predominate in Southeast Asia and Africa.
Pathology
TA is characterized by inflammation of the vessel wall, starting in the vasa vasorum. Involved vessels are infiltrated by T cells, natural killer cells, plasma cells, and macrophages. Giant cells and granulomatous inflammation develop in the media. Persistent inflammation damages the elastic lamina and muscular media, leading to blood vessel dilation and the formation of aneurysms. Progressive scarring and intimal proliferation can result in stenotic or occluded vessels. The subclavian, renal, and carotid arteries are the most commonly involved aortic branches; pulmonary, coronary, and vertebral arteries may also be affected.
Pathogenesis
The etiology of TA remains unknown. The presence of abundant T cells with a restricted repertoire of T-cell receptors in TA vascular lesions points to the importance of cellular immunity and suggests the existence of a specific but unknown aortic tissue antigen. Expression of interleukin (IL)-1, IL-6, and tumor necrosis factor (TNF)-α is reported to be higher in patients with active TA than in patients with inactive TA and in healthy controls. In some patient populations, IL-1 genetic polymorphisms are linked to TA. Some individuals with TA have elevated serum values of antiendothelial antibodies. The increased prevalence of TA in certain ethnic populations and its occasional occurrence in monozygotic twins and families suggest a genetic predisposition to the disease.
Clinical Manifestations
The diagnosis of TA is challenging, because early disease manifestations are often nonspecific. As a result, diagnosis can be delayed for several months, and the time to diagnosis is usually longer in children than in adults. Fever, malaise, weight loss, headache, hypertension, myalgias, arthralgias, dizziness, and abdominal pain are common early complaints in the pre-pulseless phase of the disease. Among children, hypertension and headache are particularly common presenting manifestations and should prompt consideration of TA when present without alternative explanation. Some individuals with TA report no systemic symptoms and instead present with vascular complications. It is only after substantial vascular injury that evidence of hypoperfusion becomes clinically evident. Later manifestations of disease include diminished pulse, asymmetric blood pressure, claudication, Raynaud phenomenon, renal failure, and symptoms of pulmonary or cardiac ischemia. Inflammation can extend to the aortic valve, resulting in valvular insufficiency. Other findings may include pericardial effusion, pericarditis, pleuritis, splenomegaly, and arthritis.
Supradiaphragmatic (aortic arch) disease often manifests with CNS (stroke, transient ischemic attack) and cardiac (heart failure, palpitations) symptoms. Infradiaphragmatic (mid-aortic syndrome) disease may produce hypertension, abdominal bruits, and pain. Most patients have involvement in both areas.
Diagnosis
Specific pediatric criteria for TA have been proposed (Table 192.5 ). Radiographic demonstration of large vessel vasculitis is necessary . A thorough physical examination is required to detect an aortic murmur, diminished or asymmetric pulses, and vascular bruits. Four extremity blood pressures should be measured; >10 mm Hg asymmetry in systolic pressure is indicative of disease.
Differential Diagnosis
In the early phase of TA, when nonspecific symptoms predominate, the differential diagnosis includes a wide array of systemic infections, autoimmune conditions, and malignancies. Although giant cell arteritis , also known as temporal arteritis, is a common large vessel vasculitis in older adults, this entity is rare in childhood. Noninflammatory conditions that can cause large vessel compromise include fibromuscular dysplasia, Marfan syndrome, and Ehlers-Danlos syndrome.
Laboratory Findings
The laboratory findings in TA are nonspecific, and there is no specific diagnostic laboratory test. ESR and CRP value are typically elevated, and other nonspecific markers of chronic inflammation may include leukocytosis, thrombocytosis, anemia of chronic inflammation, and hypergammaglobulinemia. Autoantibodies, including ANA and ANCA, are not useful in diagnosing TA except to help exclude other autoimmune diseases.
Radiographic assessment is essential to establish large vessel arterial involvement. Conventional arteriography of the aorta and major branches, including carotid, subclavian, pulmonary, renal, and mesenteric branches, can identify luminal defects, including dilation, aneurysms, and stenoses, even in smaller vessels such as the mesenteric arteries. Fig. 192.5 shows a conventional arteriogram in a child with TA. Although not yet thoroughly validated in TA, magnetic resonance angiography (MRA) and computed tomography angiography (CTA) also provide important information about vessel wall thickness and enhancement, although they may not image smaller vessels as well as conventional angiography. Positron emission tomography (PET) may detect vessel wall inflammation but has not been studied extensively. Ultrasound with duplex color flow Doppler imaging may identify vessel wall thickening and assesses arterial flow. Echocardiography is recommended to assess for aortic valvular involvement. Serial vascular imaging is usually necessary to assess response to treatment and to detect progressive vascular damage.
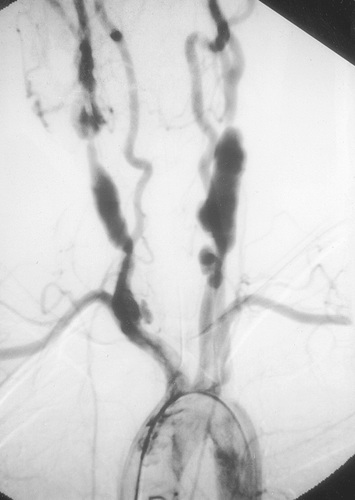
Treatment
Glucocorticoids are the mainstay of therapy, typically starting with high doses (1-2 mg/kg/day of prednisone or methylprednisolone IV) followed by gradual dosage tapering. When TA progresses or recurs, steroid-sparing therapy is often required, usually involving methotrexate or azathioprine. Cyclophosphamide is reserved for severe or refractory disease. Results of small case series also suggest that mycophenolate mofetil or anti–TNF-α therapy may be beneficial in select patients. Anti–IL-6 therapy with tocilizumab has shown promising results in a small case series of children with TA. Antihypertensive medications are often necessary to control blood pressure caused by renovascular disease.
Complications
Progressive vascular damage can result in arterial stenoses, aneurysms, and occlusions, which produce ischemic symptoms and can be organ or life threatening. Potential ischemic complications include stroke, renal impairment or failure, myocardial infarction, mesenteric ischemia, and limb-threatening arterial disease. When these complications occur or are imminent, intervention with surgical vascular grafting or catheter-based angioplasty and stent placement may be necessary to restore adequate blood flow. A high rate of recurrent stenosis has been reported after angioplasty and stent placement. Aortic valve replacement may be required if significant aortic insufficiency develops.
Prognosis
Although up to 20% of individuals with TA have a monophasic course and achieve sustained remission, most suffer relapses. Survival for individuals with TA has improved considerably over the decades, although higher mortality rates are reported in children and adolescents. The overall estimated survival for individuals with TA is 93% at 5 yr and 87% at 10 yr. However, morbidity from vascular complications remains high, particularly when there is evidence of ongoing active inflammation as detected by elevated CRP or ESR. Given the chronic endothelial insult and inflammation, children and adolescents with TA are probably at high risk for accelerated atherosclerosis. Early detection and treatment are critical to optimizing outcome in TA.
Bibliography
Alibaz-Oner F, Aydin SZ, Direskeneli H. Advances in the diagnosis, assessment and outcome of Takayasu's arteritis. Clin Rheumatol . 2013;32:541–546.
Brunner J, Feldman BM, Tyrrell PN, et al. Takayasu arteritis in children and adolescents. Rheumatology . 2010;1806–1814.
Batu ED, Sönmez HE, Hazrolan T, et al. Tocilizumab treatment in childhood Takayasu arteritis: case series of four patients and systematic review of the literature. Semin Arthritis Rheum . 2017;46(4):529–535.
Cakar N, Yalcinkaya F, Duzova A, et al. Takayasu arteritis in children. J Rheumatol . 2008;35:913–919.
Filocamo G, Buoncompagni A, Viola S, et al. Treatment of Takayasu's arteritis with tumor necrosis factor antagonists. J Pediatr . 2008;153:432–434.
Forsey J, Dhandayuthapani G, Hamilton MCK, et al. Takayasu arteritis: key clinical factors for early diagnosis. Arch Dis Child Educ Pract Ed . 2011;96:176–182.
Kalangos A, Christenson JT, Cikirikcioglu M, et al. Long-term outcome after surgical intervention and interventional procedures for the management of Takayasu's arteritis in children. J Thorac Cardiovasc Surg . 2006;132:656–664.
Liang P, Tan-Ong M, Hoffman GS. Takayasu's arteritis: vascular interventions and outcomes. J Rheumatol . 2004;31:102–106.
Maksimowicz-McKinnon K, Clark TM, Hoffman GS. Limitations of therapy and a guarded prognosis in an American cohort of Takayasu arteritis patients. Arthritis Rheum . 2007;56:1000–1009.
Ozen S, Duzova A, Bakkaloglu A, et al. Takayasu arteritis in children: preliminary experience with cyclophosphamide induction and corticosteroids followed by methotrexate. J Pediatr . 2007;150:72–76.
Park MC, Lee SW, Park YB, et al. Clinical characteristics and outcomes of Takayasu's arteritis: analysis of 108 patients using standardized criteria for diagnosis, activity assessment, and angiographic classification. Scand J Rheumatol . 2005;34:284–292.
Park MC, Lee SW, Park YB, et al. Serum cytokine profiles and their correlations with disease activity in Takayasu's arteritis. Rheumatology (Oxford) . 2006;45:545–548.
Saadoun D, Lambert M, Mirault T, et al. Retrospective analysis of surgery versus endovascular intervention in Takayasu arteritis: a multicenter experience. Circulation . 2012;125:813–819.
Polyarteritis Nodosa and Cutaneous Polyarteritis Nodosa
Vidya Sivaraman, Edward C. Fels, Stacy P. Ardoin
Polyarteritis nodosa (PAN ) is a systemic necrotizing vasculitis affecting small and medium-size arteries. Aneurysms and stenoses form at irregular intervals throughout affected arteries. Cutaneous PAN is limited to the skin.
Epidemiology
PAN is rare in childhood. Boys and girls are equally affected, and the mean age at presentation is 9 yr. The cause is unknown, but the development of PAN following infections, including group A streptococcus and chronic hepatitis B, suggests that PAN may represent a postinfectious autoimmune response. Infections with other organisms, including Epstein-Barr virus, Mycobacterium tuberculosis , cytomegalovirus, parvovirus B19, and hepatitis C virus, have also been associated with PAN. There is a possible association between PAN and familial Mediterranean fever.
Pathology
Biopsies show necrotizing vasculitis with granulocytes and monocytes infiltrating the walls of small and medium-size arteries (Fig. 192.6 ). Involvement is usually segmental and tends to occur at vessel bifurcations. Granulomatous inflammation is not present, and deposition of complement and immune complexes is rarely observed. Different stages of inflammation are found, ranging from mild inflammatory changes to panmural fibrinoid necrosis associated with aneurysm formation, thrombosis, and vascular occlusion.
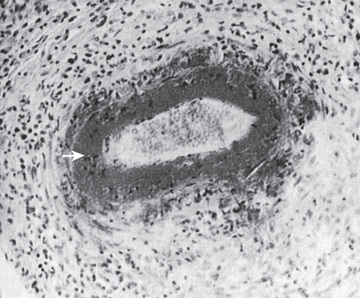
Pathogenesis
Immune complexes are believed to be pathogenic, but the mechanism is poorly understood. It is not known why PAN has a predilection for small and medium-size blood vessels. The inflamed vessel wall becomes thickened and narrowed, impeding blood flow and contributing to end-organ damage characteristic of this disease. Although there is no clear genetic association with PAN, PAN-like vasculitis is a component of 3 recently described monogenic autoinflammatory conditions.
Deficiency in adenosine deaminase 2 (DADA2) , caused by mutations in the CECR1 gene, causes a familial form of vasculitis in Georgian Jewish patients with an autosomal recessive inheritance (see Chapter 188 ).
Clinical Manifestations
The clinical presentation of PAN is variable but generally reflects the distribution of inflamed vessels. Constitutional symptoms are present in most children at disease onset. Weight loss and severe abdominal pain suggest mesenteric arterial inflammation and ischemia. Renovascular arteritis can cause hypertension, hematuria, or proteinuria, although glomerulonephritis is not typical. Cutaneous manifestations include purpura, livedo reticularis, ulcerations, digital ischemia, and painful nodules. Arteritis affecting the nervous system can result in cerebrovascular accidents, transient ischemic attacks, psychosis, and ischemic motor or sensory peripheral neuropathy (mononeuritis multiplex ). Myocarditis or coronary arteritis can lead to heart failure and myocardial ischemia; pericarditis and arrhythmias have also been reported. Arthralgias, arthritis, or myalgias are frequently present. Less common symptoms include testicular pain that mimics testicular torsion, bone pain, and vision loss as a result of retinal arteritis. The pulmonary vasculature is usually spared in PAN.
Diagnosis
The diagnosis of PAN requires demonstration of vessel involvement on biopsy or angiography (Table 192.6 ). Biopsy of cutaneous lesions shows small or medium vessel vasculitis (see Fig. 192.6 ). Kidney biopsy in patients with renal manifestations may show necrotizing arteritis. Electromyography in children with peripheral neuropathy identifies affected nerves, and sural nerve biopsy may reveal vasculitis. Conventional arteriography is the gold standard diagnostic imaging study for PAN and reveals areas of aneurysmal dilation and segmental stenosis, the classic “beads on a string” appearance (Fig. 192.7 ). MRA and CTA, less invasive imaging alternatives, are gaining acceptance, but may not be as effective in identifying small vessel disease or in younger children.
Table 192.6
| CRITERION | FINDINGS |
|---|---|
| Histopathology | Necrotizing vasculitis in medium or small arteries |
| Angiographic abnormalities | Angiography showing aneurysm, stenosis, or occlusion of medium or small artery not from noninflammatory cause |
| Cutaneous findings | Livedo reticularis, tender subcutaneous nodules, superficial skin ulcers, deep skin ulcers, digital necrosis, nail bed infarctions, or splinter hemorrhages |
| Muscle involvement | Myalgia or muscle tenderness |
| Hypertension | Systolic or diastolic blood pressure >95th percentile for height |
| Peripheral neuropathy | Sensory peripheral neuropathy, motor mononeuritis multiplex |
| Renal involvement | Proteinuria (>300 mg/24 hr equivalent), hematuria or red blood cell casts, impaired renal function (glomerular filtration rate <50% normal) |
* The presence of 5 criteria provides 89.6% sensitivity and 99.6% specificity for the diagnosis of childhood-onset polyarteritis nodosa.
Adapted from Ozen S, Pistorio A, Iusan SM, et al: EULAR/PRINTO/PRES criteria for Henoch-Schönlein purpura, childhood polyarteritis nodosa, childhood Wegener granulomatosis and childhood Takayasu arteritis: Ankara 2008. Part II. Final classification criteria, Ann Rheum Dis 69:798–806; 2010.
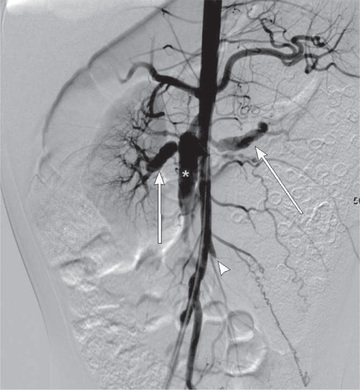
Differential Diagnosis
Early skin lesions may resemble those of HSP, although the finding of nodular lesions and presence of systemic features help distinguish PAN. Because pulmonary vascular involvement is very rare in PAN, pulmonary lesions suggest ANCA-associated vasculitis or Goodpasture disease. Other rheumatic diseases, including SLE, have characteristic target-organ involvement and associated autoantibodies distinguishing them from PAN. Prolonged fever and weight loss should also prompt consideration of inflammatory bowel disease or malignancy.
Laboratory Findings
Nonspecific laboratory findings include elevations of ESR and CRP, anemia, leukocytosis, and hypergammaglobulinemia. Abnormal urine sediment, proteinuria, and hematuria indicate renal disease. Laboratory findings may be normal in cutaneous PAN or similar to those of systemic PAN. Elevated hepatic enzyme values may suggest hepatitis B or C infection. Serologic tests for hepatitis (hepatitis B surface antigen and hepatitis C antibody) should be performed in all patients.
Treatment
Oral prednisone (1-2 mg/kg/day) or IV pulse methylprednisolone (30 mg/kg/day) are the mainstay of therapy. Oral or IV cyclophosphamide are often used as adjunctive therapy, and plasma exchange may be warranted for life-threatening disease. If hepatitis B is identified, appropriate antiviral therapy should be initiated (see Chapter 385 ). Most cases of cutaneous PAN can be treated with less intense therapy such as corticosteroids alone, nonsteroidal antiinflammatory drugs (NSAIDs), and methotrexate. Azathioprine, mycophenolate mofetil, IVIG, thalidomide, cyclosporine, and anti-TNF agents such as infliximab have all been reported as successful in treatment of refractory cutaneous or systemic PAN, although clinical trials are lacking. If an infectious trigger for PAN is identified, antibiotic prophylaxis can be considered.
Complications
Cutaneous nodules may ulcerate and become infected. Hypertension and chronic renal disease may develop from renovascular involvement in PAN. Cardiac involvement may lead to decreased cardiac function or coronary artery disease. Mesenteric vasculitis can predispose to bowel infarction, rupture, and malabsorption. Stroke and rupture of hepatic arterial aneurysm are uncommon complications of this disorder.
Prognosis
The course of PAN varies from mild disease with few complications to a severe, multiorgan disease with high morbidity and mortality. Poor prognostic factors in PAN include elevated serum creatinine, proteinuria, severe GI involvement, cardiomyopathy, and CNS involvement. Early and aggressive immunosuppressive therapy increases the likelihood of clinical remission. Compared with disease in adults, childhood PAN is associated with less mortality. Cutaneous PAN is unlikely to transition to systemic disease. Early recognition and treatment of the disease are important to minimizing potential long-term vascular complications.
Bibliography
Dedeoglu F, Sundel RP. Vasculitis in children. Rheum Dis Clin North Am . 2007;33:555–583.
Eleftheriou D, Dillon M, Tullus K, et al. Systemic polyarteritis nodosa in the young: a single center experience over 32 years. Arthritis Rheum . 2013;65(9):2476–2485.
Elkan PN, Pierce SB, Segel R, et al. Mutant adenosine deaminase 2 in a polyarteritis nodosa vasculopathy. N Engl J Med . 2014;370:921–930.
Guillevin L, Lhote F, Gayraud M, et al. Prognostic factors in polyarteritis nodosa and Churg-Strauss syndrome: a prospective study in 342 patients. Medicine (Baltimore) . 1996;75:17–29.
Liu Y, Ramot Y, Torrelo A, et al. Mutations in proteasome subunit β type 8 cause chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature with evidence of genetic and phenotypic heterogeneity. Arthritis Rheum . 2012;64(3):895–907.
Liu Y, Jesus AA, Marrero B, et al. Activated STING in a vascular and pulmonary syndrome. N Engl J Med . 2014;371(6):507–518.
Nakamura T, Kanazawa N, Ikeda T, et al. Cutaneous polyarteritis nodosa: revisiting its definition and diagnostic criteria. Arch Dermatol Res . 2009;301:117–121.
Ozen S, Pistorio A, Iusan SM, et al. EULAR/PRINTO/PRES criteria for Henoch-Schönlein purpura, childhood polyarteritis nodosa, childhood Wegener granulomatosis and childhood Takayasu arteritis: Ankara 2008. Part II. Final classification criteria. Ann Rheum Dis . 2010;69:798–806.
Reddy VB, Schloemer N. Polyarteritis nodosa in a 9-year-old boy. J Pediatr . 2013;162:216.
Villiger PM, Guillevin L. Microscopic polyangiitis: clinical presentation. Autoimmun Rev . 2010;9:812–819.
Yalcinkaya F, Ozcakar B, Kasapcoupur O, et al. Prevalence of the MEFV gene mutations in childhood polyarteritis nodosa. J Pediatr . 2007;151:675–678.
Antineutrophilic Cytoplasmic Antibody–Associated Vasculitis
Vidya Sivaraman, Edward C. Fels, Stacy P. Ardoin
The ANCA-associated vasculitides are characterized by small vessel involvement, circulating ANCAs, and paucity of immune complex deposition in affected tissues, thus the term pauci-immune vasculitis . ANCA-associated vasculitis is categorized into 3 distinct forms: granulomatosis with polyangiitis (GPA) , formerly Wegener granulomatosis; microscopic polyangiitis (MPA) ; and eosinophilic granulomatosis with polyangiitis , formerly Churg-Strauss syndrome (CSS ) (see Table 192.1 ).
Epidemiology
GPA is a necrotizing granulomatous small and medium vessel vasculitis that occurs at all ages and targets the upper and lower respiratory tracts and the kidneys. Although most cases of GPA occur in adults, the disease also occurs in children with a mean age at diagnosis of 14 yr. There is a female predominance of 3-4 : 1, and pediatric GPA is most prevalent in Caucasians.
MPA is a small vessel necrotizing vasculitis with clinical features similar to those of GPA, but without granulomas and upper airway involvement. CSS is a small vessel necrotizing granulomatous (allergic granulomatosis) vasculitis associated with a history of refractory asthma and peripheral eosinophilia. MPA and CSS are rare in children, and there does not appear to be a gender predilection in either disease.
Pathology
Necrotizing vasculitis is the cardinal histologic feature in both GPA and MPA. Kidney biopsies typically demonstrate crescentic glomerulonephritis with little or no immune complex deposition (“pauci-immune”), in contrast to biopsies from patients with SLE. Although granulomatous inflammation is common in GPA and CSS, it is typically not present in MPA. Biopsies showing perivascular eosinophilic infiltrates distinguish CSS syndrome from both MPA and GPA (Table 192.7 ).
Table 192.7
Differential Diagnostic Features of Small Vessel Vasculitis
| FEATURE | HENOCH-SCHÖNLEIN PURPURA | GRANULOMATOSIS WITH POLYANGIITIS | CHURG-STRAUSS SYNDROME* | MICROSCOPIC POLYANGIITIS |
|---|---|---|---|---|
| Signs and symptoms of small vessel vasculitis † | + | + | + | + |
| IgA-dominant immune deposits | + | − | − | − |
| Circulating antineutrophil cytoplasmic antibodies | − | + (PR3) | + (MPO > PR3) | + (MPO) |
| Necrotizing vasculitis | − | + | + | + |
| Granulomatous inflammation | − | + | + | − |
| Asthma and eosinophilia | − | − | + | − |
* Eosinophilic granulomatosis with polyangiitis.
† Signs and symptoms of small vessel vasculitis include purpura, other rash, arthralgias, arthritis, and constitutional symptoms.
MPO, Myeloperoxidase-reactive antibodies; PR3, proteinase-3–reactive antibodies; +, present; −, absent.
Adapted from Jeannett JC, Falk RJ: Small-vessel vasculitis, N Engl J Med 337:1512–1523, 1997.
Pathogenesis
The etiology of ANCA-associated vasculitis remains unknown, although neutrophils, monocytes, and endothelial cells are involved in disease pathogenesis. Neutrophils and monocytes are activated by ANCAs, specifically by the ANCA-associated antigens proteinase-3 (PR3) and myeloperoxidase (MPO), and release proinflammatory cytokines such as TNF-α and IL-8. Localization of these inflammatory cells to the endothelium results in vascular damage characteristic of the ANCA vasculitides. Why the respiratory tract and kidneys are preferential targets in GPA and MPA is unknown.
Clinical Manifestations
Early disease course is characterized by nonspecific constitutional symptoms, including fever, malaise, weight loss, myalgias, and arthralgias. In GPA, upper airway involvement can manifest as sinusitis, nasal ulceration, epistaxis, otitis media, and hearing loss. Lower respiratory tract symptoms in GPA include cough, wheezing, dyspnea, and hemoptysis. Pulmonary hemorrhage can cause rapid respiratory failure. Compared with adults, childhood GPA is more frequently complicated by subglottic stenosis (Fig. 192.8 ). Inflammation-induced damage to the nasal cartilage can produce a saddle nose deformity (Fig. 192.8 ). Ophthalmic involvement includes conjunctivitis, scleritis, uveitis, optic neuritis, and invasive orbital pseudotumor (causing proptosis). Perineural vasculitis or direct compression on nerves by granulomatous lesions can cause cranial and peripheral neuropathies. Hematuria, proteinuria, and hypertension in GPA signal renal disease. Cutaneous lesions include palpable purpura and ulcers. Venous thromboembolism is a rare but potentially fatal complication of GPA. The frequencies of organ system involvement throughout the disease course in GPA follow: respiratory tract, 74%; kidneys, 83%; joints, 65%; eyes, 43%; skin, 47%; sinuses, 70%; and nervous system, 20%. Table 192.8 lists the classification criteria for pediatric-onset GPA.
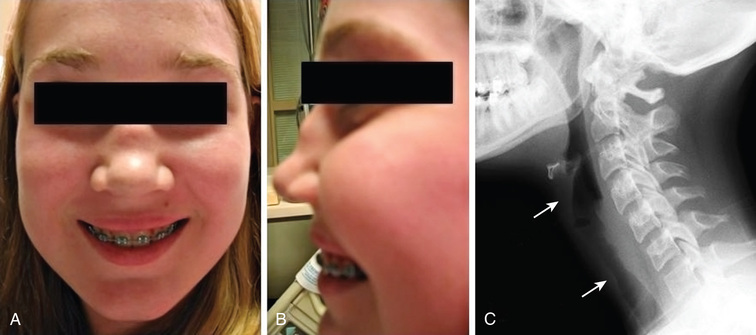
The clinical presentation of MPA closely resembles that of GPA, although sinus disease is less common; systemic features of fever, malaise, weight loss, myalgias, and arthralgias may be dominant. MPA predominantly affects the kidney and lungs; other organ systems include skin, CNS, muscle, heart, and eyes.
CSS frequently causes inflammation of the upper and lower respiratory tracts, but cartilage destruction is rare. CSS may initially demonstrate chronic or recurrent rhinitis/sinusitis, nasal polyposis, nonfixed pulmonary lesions, and difficult-to-treat asthma. Eosinophilia (>10% of leukocytes) with pulmonary infiltrates may precede a vasculitic phase. Other organ involvement includes skin, cardiac, peripheral neuropathy, GI tract, and muscle. Renal involvement in CSS is uncommon.
Diagnosis
GPA should be considered in children who have recalcitrant sinusitis, pulmonary infiltrates, and evidence of nephritis. Chest radiography often fails to detect pulmonary lesions, and chest CT may show nodules, ground-glass opacities, mediastinal lymphadenopathy, and cavitary lesions (Fig. 192.9 ). The diagnosis is confirmed by the presence of c-ANCA with anti-PR3 specificity (PR3-ANCAs) and the finding of necrotizing granulomatous vasculitis on pulmonary, sinus, or renal biopsy. The ANCA test result is positive in approximately 90% of children with GPA, and the presence of anti-PR3 increases the specificity of the test.
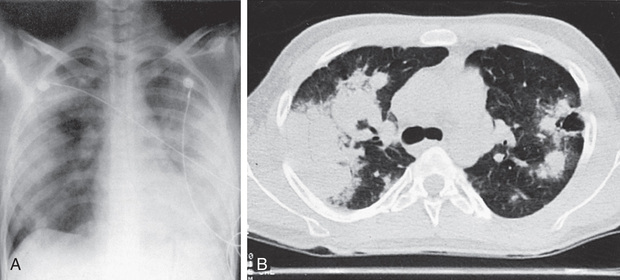
In MPA, ANCAs are also frequently present (70% of patients) but are usually p-ANCA with reactivity to MPO (MPO-ANCAs). MPA can be distinguished from PAN by the presence of ANCAs and the tendency for small vessel involvement. The ANCA test result is positive in 50–70% of cases of CSS, and MPO-ANCAs are more common than PR3-ANCAs. In addition, the presence of chronic asthma and peripheral eosinophilia suggests the diagnosis of CSS.
Differential Diagnosis
ANCAs are absent in other granulomatous diseases, such as sarcoidosis and tuberculosis. Goodpasture syndrome is characterized by antibodies to glomerular basement membrane. Medications such as propylthiouracil, hydralazine, and minocycline are associated with drug-induced ANCA (usually perinuclear ANCA) vasculitis. SLE and HSP can manifest as pulmonary hemorrhage and nephritis.
Laboratory Findings
Nonspecific laboratory abnormalities include elevated ESR and CRP values, leukocytosis, and thrombocytosis, which are present in most patients with an ANCA-associated vasculitis but are nonspecific. Anemia may be caused by chronic inflammation or pulmonary hemorrhage. ANCA antibodies show 2 distinct immunofluorescence patterns: perinuclear (p-ANCA) and cytoplasmic (c-ANCA). In addition, ANCAs can also be defined by their specificity for PR3 or MPO antigen. GPA is strongly associated with c-ANCAs/anti-PR3 antibodies, whereas 75% of patients with MPA have a positive p-ANCA (see Table 192.7 ). There is no clear correlation between ANCA titers and disease activity or relapse.
Treatment
When the lower respiratory tract or kidneys are significantly involved, initial induction therapy usually consists of prednisone (oral 2 mg/kg/day oral or IV methylprednisolone 30 mg/kg/day × 3 days) in conjunction with daily oral or monthly IV cyclophosphamide. Rituximab , a monoclonal antibody to CD20 on activated B cells, is an option for induction therapy in ANCA-positive vasculitides, although it has been studied primarily in adults. Plasmapheresis in conjunction with methylprednisolone has a role in the therapy of patients with severe disease manifestations such as pulmonary hemorrhage or ESRD, with the potential for reducing dialysis dependency. Patients are transitioned to a less toxic maintenance medication (usually methotrexate, azathioprine, or mycophenolate mofetil) within 3-6 mo once remission is achieved. Trimethoprim-sulfamethoxazole (one 180 mg/800 mg tablet 3 days/wk) is often prescribed both for prophylaxis against Pneumocystis jiroveci infection and to reduce upper respiratory bacterial colonization with S. aureus, which may trigger disease activity. If disease is limited to the upper respiratory tract, corticosteroids (1-2 mg/kg/day) and methotrexate (0.5-1.0 mg/kg/wk) may be first-line treatment.
Mepolizumab , an anti–IL-5 monoclonal antibody, may have a role in the treatment of eosinophilic granulomatosis with polyangiitis (CSS).
Complications
Upper respiratory tract lesions can invade the orbit and threaten the optic nerve, and lesions in the ear can cause permanent hearing loss. Respiratory complications include potentially life-threatening pulmonary hemorrhage and upper airway obstruction caused by subglottic stenosis. Chronic lung disease secondary to granulomatous inflammation, cavitary lesions, and scarring can predispose to infectious complications. Chronic glomerulonephritis may progress to ESRD in a subset of patients with advanced or undertreated disease.
Prognosis
The course is variable, but disease relapse occurs in up to 60% of patients. Mortality has been reduced with the introduction of cyclophosphamide and other immunosuppressive agents. Compared with adults, children are more likely to develop multiorgan involvement, renal involvement, and subglottic stenosis.
Bibliography
Akikusa JD, Schneider R, Harvey EA, et al. Clinical features and outcome of pediatric Wegener's granulomatosis. Arthritis Rheum . 2007;57:837–844.
Berden A, Goceroglu A, Jayne D, et al. Diagnosis and management of ANCA-associated vasculitis. BMJ . 2012;344:40–44.
Bosch X, Guilabert A, Espinosa G, et al. Treatment of antineutrophil cytoplasmic antibody–associated vasculitis. JAMA . 2007;298:655–668.
Cabral DA, Canter DL, Muscal E, et al. Comparing presenting clinical features in 48 children with microscopic polyangiitis to 183 children who have granulomatosis with polyangiitis (Wegener's): an ARChiVe cohort study. Arthritis Rheumatol . 2016;68(10):2514–2526.
Cabral DA, Uribe AG, Benseler S, et al. Classification, presentation, and initial treatment of Wegener's granulomatosis in childhood. Arthritis Rheum . 2009;60(11):3413–3424.
Falk RJ, Gross WL, Guillevin L, et al. Granulomatosis with polyangiitis (Wegener's): an alternative name for Wegener's granulomatosis. Arthritis Rheum . 2011;63(4):863–864.
Finkielman JD, Merkel PA, Schroeder D, et al. Antiproteinase 3 antineutrophil cytoplasmic antibodies and disease activity in Wegener granulomatosis. Ann Intern Med . 2007;147:611–619.
Gendelman S, Zeft A, Spaulding SJ. Childhood-onset eosinophillic granulomatosis with polyangiitis (formerly Churg-Strauss syndrome): a contemporary single-center cohort. J Rheumatol . 2013;40:929–935.
Guillevin L, Pagnoux C, Karras A, et al. Rituximab versus azathioprine for maintenance in ANCA-associated vasculitis. N Engl J Med . 2014;371:1771–1780.
Jayne DR, Gaskin G, Rasmussen N, et al. Randomized trial of plasma exchange or high-dosage methylprednisolone as adjunctive therapy for severe renal vasculitis. J Am Soc Nephrol . 2007;18(7):2180–2188.
Kallenberg CG. Pathogenesis of PR3-ANCA associated vasculitis. J Autoimmun . 2008;30:29–36.
Levine D, Akikusa J, Manson D, et al. Chest CT findings in pediatric Wegener's granulomatosis. Pediatr Radiol . 2007;37:57–62.
Lyons PA, Rayner TF, Trivedi S, et al. Genetically distinct subsets within ANCA-associated vasculitis. N Engl J Med . 2012;367(3):214–222.
Mahr A, Moosig F, Neumann T, et al. Eosinophilic granulomatosis with polyantiitis (Churg-Strauss): evolutions in classification, etiopathogenesis, assessment and management. Curr Opin Rheumatol . 2014;26(1):16–23.
Pagnoux C, Mahr A, Hamidou MA, et al. Azathioprine or methotrexate maintenance for ANCA-associated vasculitis. N Engl J Med . 2008;359:2790–2802.
Simmon E, Tramma D, Bowen C, et al. ANCA-associated glomerulonephritis/systemic vasculitis in childhood: clinical features—outcome. Pediatr Nephrol . 2012;27:1911–1920.
Sinico RA, Di Toma L, Radice A. Renal involvement in anti-neutrophil cytoplasmic autoantibody–associated vasculitis. Autoimmun Rev . 2013;12:477–482.
Siomou E, Tramma D, Bowen C, et al. ANCA-associated glomerulonephritis/systemic vasculitis in childhood: clinical features–outcome. Pediatr Nephrol . 2012;10:1911–1920.
Specks U, Merkel PA, Seo P, et al. Efficacy of remission-induction regimens for ANCA-associated vasculitis. N Engl J Med . 2013;369:417–426.
Stone JH, Merkel PA, Spiera R, et al. Rituximab versus cyclophosphamide for ANCA-associated vasculitis. N Engl J Med . 2010;363:221–232.
Wechsler ME, Akuthota P, Jayne D, et al. Mepolizumab or placebo for eosinophilic granulomatosis with polyangiitis. N Engl J Med . 2017;376(20):1921–1932.
Other Vasculitis Syndromes
Vidya Sivaraman, Edward C. Fels, Stacy P. Ardoin
Other vasculitic conditions can occur in childhood; the most common is Kawasaki disease (see Chapter 191 ). Behçet disease is a rare form of vasculitis seen in children of Turkish and Mediterranean descent, characterized by the triad of recurrent aphthous stomatitis, genital ulcers, and uveitis (see Chapter 186 ).
Hypersensitivity vasculitis is a cutaneous vasculitis triggered by medication or toxin exposure. The rash consists of palpable purpura or other, nonspecific rash. Skin biopsies reveal characteristic changes of leukocytoclastic vasculitis (small vessels with neutrophilic perivascular or extravascular neutrophilic infiltration) (Table 192.9 ). Hypocomplementemic urticarial vasculitis involves small vessels and manifests as recurrent urticaria that resolves over several days but leaves residual hyperpigmentation. This condition is associated with low levels of complement component C1q and systemic findings that include fever, GI symptoms, arthritis, and glomerulonephritis. Some patients with urticarial vasculitis have normal complement levels. Cryoglobulinemic vasculitis can complicate mixed essential cryoglobulinemia and is a small vessel vasculitis affecting skin, joints, kidneys, and lungs.
Table 192.9
| CRITERION | DEFINITION |
|---|---|
| Age at onset >16 yr | Development of symptoms after 16 yr of age |
| Medication at disease onset | Medication that may have been a precipitating factor was taken at the onset of symptoms |
| Palpable purpura | Slightly elevated purpuric rash over 1 or more areas; does not blanch with pressure and is not related to thrombocytopenia |
| Maculopapular rash | Flat and raised lesions of various sizes over 1 or more areas of the skin |
| Biopsy, including arteriole and venule | Histologic changes showing granulocytes in a perivascular or extravascular location |
* For purposes of classification, a patient is said to have hypersensitivity vasculitis if at least 3 of these criteria are present. The presence of ≥3 criteria has a diagnostic sensitivity of 71.0% and specificity of 83.9%. The age criterion is not applicable for children.
Adapted from Calabrese LH, Michel BA, Bloch DA, et al: The American College of Rheumatology 1990 criteria for the classification of hypersensitivity vasculitis, Arthritis Rheum 33:1108–1113, 1990 (Table 2, p 1110); and Textbook of pediatric rheumatology, ed 7, Philadelphia, 2016, Elsevier (Table 38.2, p 511).
Primary angiitis of the central nervous system represents vasculitis confined to the CNS and requires exclusion of other systemic vasculitides. Large vessel disease (angiography positive) may be progressive or nonprogressive and may manifest with focal deficits similar to an occlusive stroke, with hemiparesis, focal gross or fine motor deficits, language disorders, or cranial nerve deficits. Diffuse cognitive, memory, and concentration deficits as well as behavioral disorders are seen in 30–40% of patients. Small vessel disease (angiography negative, biopsy positive) more often results in language problems and diffuse deficits, such as cognitive, memory, behavior, and concentration problems, as well as focal seizures. In both types of cerebral angiitis, patients may have an elevated ESR or CRP and abnormal CSF findings (increased protein, pleocytosis), although these are not consistent findings in all patients. Diagnosis remains a challenge, and brain biopsy is often indicated to confirm the diagnosis and exclude vasculitis mimics such as infections that could worsen with immunosuppressive therapy (Table 192.10 ).
Nonprogressive angiography-positive CNS vasculitis, also known as transient CNS angiopathy, represents a more benign variant and can be seen after varicella infection. Cogan syndrome is rare in children; its potential clinical manifestations include constitutional symptoms; inflammatory eye disease such as uveitis, episcleritis, or interstitial keratitis; vestibuloauditory dysfunction (vertigo, hearing loss, tinnitus); arthritis; and large vessel vasculitis or aortitis. Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL ) is caused by mutations in the NOTCH3 gene and manifests with stroke, mood changes, cognitive decline, and migraines; it is a vasculitis mimic and demonstrates osmophilic granules in cerebral arteries. CARASIL (cerebral autosomal recessive arteriopathy with subcortical infarcts and leukoencephalopathy) is another mimic of angiitis caused by mutations in the HTRA1 gene. It manifests with early-onset hair loss, spasticity, stroke, memory loss, and personality changes.
Identification of these vasculitis syndromes requires a comprehensive history and physical examination. Table 192.11 outlines other diagnostic considerations. Although tailored to disease severity, treatment generally includes prednisone (up to 2 mg/kg/day). Potent immunosuppressive medications, such as cyclophosphamide, are often indicated, particularly in primary angiitis of the CNS to prevent rapid neurologic decline. For hypersensitivity vasculitis, withdrawal of the triggering medication or toxin is indicated if possible.
Table 192.11
CNS, Central nervous system; CT, computed tomography; MRA, magnetic resonance angiography.
Bibliography
Abril A. Churg-Strauss syndrome: an update. Curr Rheumatol Rep . 2011;13:489–495.
Benseler SM. Central nervous system vasculitis in children. Curr Rheumatol Rep . 2006;8:442–449.
Cassidy JT, Petty RE. Systemic vasculitis. Textbook of pediatric rheumatology . ed 5. Elsevier Saunders: Philadelphia; 2005.
Dedeoglu F, Sundel RP. Vasculitis in children. Rheum Dis Clin North Am . 2007;33:555–583.
Gray PEA, Bock A, Ziegler DS, et al. Neonatal Sweet syndrome: a potential marker of serious systemic illness. Pediatrics . 2012;129(5):e1333–e1359.
Gowdie P, Twitt M, Benseler SM. Primary and secondary central nervous system vasculitis. J Child Neurol . 2012;27(11):1448–1459.
Van Mater H. Pediatric inflammatory brain diseases: a diagnostic approach. Curr Opin Rheumatol . 2014;26(5):553–561.