Hereditary Periodic Fever Syndromes and Other Systemic Autoinflammatory Diseases
James W. Verbsky
The hereditary periodic fever syndromes are a group of monogenic diseases that present with recurrent bouts of fever and associated pleural and/or peritoneal inflammation, arthritis, and various types of skin rash. A number of identifiable disorders present with recurrent episodes of inflammation, although fevers may not be common feature. Therefore the term systemic autoinflammatory diseases is used to include all diseases that present with seemingly unprovoked episodes of inflammation, without the high-titer autoantibodies or antigen-specific T cells typically seen in autoimmune diseases. Whereas the autoimmune diseases are disorders of the adaptive immune system, driven by B and T lymphocyte effector cells, autoinflammatory diseases largely represent disorders of the phylogenetically more primitive innate immune system, mediated by myeloid effector cells and germline-encoded receptors. Autoinflammatory diseases exhibit episodic or persistent inflammation characterized by an acute-phase response with elevation of the erythrocyte sedimentation rate (ESR), C-reactive protein (CRP), and serum amyloid A (AA). In some patients, untreated autoinflammatory disorders over time will lead to AA amyloidosis (see Chapter 189 ).
It is important to note that autoinflammatory disorders are rare, whereas fever in childhood caused by innocuous illness is very common. The approach to a child with fevers should include a detailed history, physical examination, and limited laboratory investigations to rule out other conditions that lead to fevers, including autoimmune disorders and malignancies (Table 188.1 ). If there is evidence of recurrent infections with fevers, an immune deficiency could be considered and evaluated. If the workup is reassuring, the inflammatory episodes resolve, and the child is otherwise well without unusual physical findings, observance is often warranted because these episodes are likely to resolve as the child's immune system matures.
Classification of Autoinflammatory Disorders
Because of the rapidly expanding number of autoinflammatory disorders and their varied clinical presentation, it can be difficult to group these disorders in a meaningful manner. Some autoinflammatory disorders present with prominent fevers and are known as hereditary periodic fever syndromes . These include 2 disorders with an autosomal recessive mode of inheritance, familial Mediterranean fever (FMF ; MIM249100) and the hyperimmunoglobulinemia D (hyper-IgD) with periodic fever syndrome (HIDS ; MIM260920). Hereditary periodic fever syndromes with an autosomal dominant mode of inheritance include the tumor necrosis factor (TNF) receptor–associated periodic syndrome (TRAPS ; MIM191190) and a spectrum of disorders known as the cryopyrin-associated periodic syndromes (CAPS ), or cryopyrinopathies. From mildest to most severe, CAPS include the familial cold autoinflammatory syndrome (FCAS1 ; MIM120100), Muckle-Wells syndrome (MWS ; MIM191100), and neonatal-onset multisystem inflammatory disease (NOMID ; MIM607115) (also known as chronic infantile neurologic cutaneous and articular syndrome, CINCA ) (Table 188.2 ).
Table 188.2
| DISEASE | GENETIC DEFECT/PRESUMED PATHOGENESIS | INHERITANCE | AFFECTED CELLS | FUNCTIONAL DEFECTS | ASSOCIATED FEATURES |
|---|---|---|---|---|---|
| Familial Mediterranean fever | Mutations of MEFV (lead to gain of pyrin function, resulting in inappropriate IL-1β release) | AR | Mature granulocytes, cytokine-activated monocytes | Decreased production of pyrin permits ASC-induced IL-1 processing and inflammation following subclinical serosal injury; macrophage apoptosis decreased | Recurrent fever, serositis, and inflammation responsive to colchicine. Predisposes to vasculitis and inflammatory bowel disease |
| Mevalonate kinase deficiency (hyper IgD syndrome) | Mutations of MVK (lead to a block in the mevalonate pathway). Interleukin-1β mediates the inflammatory phenotype | AR | Affecting cholesterol synthesis; pathogenesis of disease is unclear | Periodic fever and leukocytosis with high IgD levels | |
| Muckle–Wells syndrome | Mutations of NLRP3 (also called PYPAF1 or NALP3 ) lead to constitutive activation of the NLRP3 inflammasome | AD | PMNs, monocytes | Defect in cryopyrin, involved in leukocyte apoptosis and NF-κB signaling and IL-1 processing | Urticaria, SNHL, amyloidosis |
| Familial cold autoinflammatory syndrome |
Mutations of NLRP3 (see above) Mutations of NLRP12 |
AD | PMNs, monocytes | Same as above | Nonpruritic urticaria, arthritis, chills, fever, and leukocytosis after cold exposure |
| Neonatal-onset multisystem inflammatory disease (NOMID) or chronic infantile neurologic cutaneous and articular syndrome (CINCA) | Mutations of NLRP3 (see above) | PMNs, chondrocytes | Same as above | Neonatal-onset rash, chronic meningitis, and arthropathy with fever and inflammation | |
| TNF receptor–associated periodic syndrome (TRAPS) | Mutations of TNFRSF1A (resulting in increased TNF inflammatory signaling) | AD | PMNs, monocytes | Mutations of 55-kDa TNF receptor leading to intracellular receptor retention or diminished soluble cytokine receptor available to bind TNF | Recurrent fever, serositis, rash, and ocular or joint inflammation |
| Pyogenic sterile arthritis, pyoderma gangrenosum, acne (PAPA) syndrome | Mutations of PSTPIP1 (also called C2BP1 ) (affects both pyrin and protein tyrosine phosphatase to regulate innate and adaptive immune responses) | AD | Hematopoietic tissues, upregulated in activated T cells | Disordered actin reorganization leading to compromised physiologic signaling during inflammatory response | Destructive arthritis, inflammatory skin rash, myositis |
| Blau syndrome | Mutations of NOD2 (also called CARD15 ) (involved in various inflammatory processes) | AD | Monocytes | Mutations in nucleotide binding site of CARD15, possibly disrupting interactions with lipopolysaccharides and NF-κB signaling | Uveitis, granulomatous synovitis, campodactyly, rash, and cranial neuropathies, 30% develop Crohn disease |
| Chronic recurrent multifocal osteomyelitis and congenital dyserythropoietic anemia (Majeed syndrome) | Mutations of LPIN2 (increased expression of the proinflammatory genes) | AR | Neutrophils, bone marrow cells | Undefined | Chronic recurrent multifocal osteomyelitis, transfusion-dependent anemia, cutaneous inflammatory disorders |
| Early-onset inflammatory bowel disease | Mutations in IL-10 (results in increase of many proinflammatory cytokines) | AR | Monocyte/macrophage, activated T cells | IL-10 deficiency leads to increase of TNFγ and other proinflammatory cytokines | Enterocolitis, enteric fistulas, perianal abscesses, chronic folliculitis |
| Early-onset inflammatory bowel disease | Mutations in IL-10RA (see above) | AR | Monocyte/macrophage, activated T cells | Mutation in IL-10 receptor alpha leads to increase of TNFγ and other proinflammatory cytokines | Enterocolitis, enteric fistulas, perianal abscesses, chronic folliculitis |
| Early-onset inflammatory bowel disease | Mutations in IL-10RB (see above) | AR | Monocyte/macrophage, activated T cells | Mutation in IL-10 receptor beta leads to increase of TNFγ and other proinflammatory cytokines | Enterocolitis, enteric fistulas, perianal abscesses, chronic folliculitis |
AD, autosomal dominant; AR, autosomal recessive; Ig, immunoglobulin; IL, interleukin; NF-κB, nuclear factor-κB; PMN, polymorphonuclear neutrophil; SNHL, sensorineural hearing loss; TNF, tumor necrosis factor.
From Verbsky JW, Routes JR: Recurrent fever, infections, immune disorders, and autoinflammatory diseases. In Kliegman RM, Lyse PS, Bordini BJ, et al, editors: Nelson pediatric symptom-based diagnosis. Philadelphia, 2018, Elsevier, Table 41-5.
A variety of mendelian autoinflammatory disorders may or may not exhibit prominent fevers and are not considered periodic fever syndromes, but do have continuous or repeated episodes of spontaneous inflammation with unique clinical characteristics. These include the syndrome of pyogenic arthritis with pyoderma gangrenosum and acne (PAPA ; MIM604416), deficiency of the interleukin-1 (IL-1) receptor antagonist (DIRA ; MIM612852), Blau syndrome caused by mutations in NOD2 (also known as early-onset sarcoidosis ; MIM186580), autoinflammation with phospholipase Cγ2 -associated antibody deficiency and immune dysregulation (APLAID ; MIM614878), and deficiency of adenosine deaminase-2 (DADA2 ). Other disorders include congenital sideroblastic anemia with B-cell immunodeficiency, periodic fevers, and developmental delay (SIFD ) caused by biallelic mutations of the TRNT1 gene (MIM616084), autoinflammation with infantile enterocolitis caused by mutations in NLRC4 (AIFEC ; MIM616060), familial cold autoinflammatory syndrome type 2 caused by mutations in NLRP12 (FCAS2 ; MIM611762), CARD14 (MIM607211), and deficiency in IL-36 receptor antagonist (DITRA ; 614204).
In addition to the previous autoinflammatory disorders, a variety of disorders are characterized by inappropriate interferon expression , the interferonopathies . Type 1 interferons (e.g., IFN-α, IFN-β) are cytokines expressed by many cells in response to viral infections. Disorders that result in spontaneous interferon production and inflammatory manifestations include STING-associated vasculopathy of infancy (SAVI ; MIM615934) and chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature (CANDLE ; MIM256040).
There are also a number of autoinflammatory disorders with a complex mode of inheritance. These include the syndrome of periodic fever with aphthous stomatitis, pharyngitis, and adenitis (PFAPA ) and chronic recurrent multifocal osteomyelitis (CRMO ; MIM259680). Other genetically complex disorders that are sometimes considered autoinflammatory include systemic-onset juvenile idiopathic arthritis (see Chapter 180 ), Behçet disease (see Chapter 186 ), and Crohn disease (see Chapter 362.2 ).
Distinguishing autoinflammatory disorders from one another can be difficult because their presentations can vary, and many display similarities. Some disorders have characteristic fever patterns (Fig. 188.1 ), whereas others have characteristic skin findings that can aid in a diagnosis (Table 188.3 ). Others can have characteristic physical features or organ involvement. Some of these disorders have bone involvement (Table 188.4 ). Other clinical features can also be helpful, such as ethnicity, age of onset, triggers, laboratory testing, and response to therapies (Table 188.5 ). Genetic panels are increasingly being used to screen for most if not all of these defects in a single test, rather than individual genetic assessment based on clinical findings.

Table 188.4
Autoinflammatory Bone Disorders
| CRMO | MAJEED SYNDROME | DIRA | CHERUBISM | CMO AND LUPO MICE | |
|---|---|---|---|---|---|
| Ethnicity | Worldwide, but mostly European | Arabic | European, Puerto Rican, Arabic | Worldwide | Occurs in various backgrounds |
| Fever | Uncommon | Common | Uncommon | No | Not assessed |
| Sites of osseous involvement | Metaphyses of long bones > vertebrae, clavicle, sternum, pelvis, others | Similar to CRMO | Anterior rib ends, metaphyses of long bones, vertebrae, others |
Mandible > maxilla Rarely ribs |
Vertebrae hind > forefeet |
| Extraosseous manifestations | PPP, psoriasis, IBD, others | Dyserythropoietic anemia, Sweet syndrome, HSM, growth failure | Generalized pustulosis, nail changes, lung disease, vasculitis | Cervical lymphadenopathy | Dermatitis, extramedullary hematopoiesis, splenomegaly |
| Family history of inflammatory disorders | Psoriasis, PPP, arthritis, IBD, others | Psoriasis in some obligate carriers | No known associations | No known associations | Heterozygotes normal |
| Inheritance | Not clear | Autosomal recessive | Autosomal recessive | Autosomal dominant; incomplete penetrance | Autosomal recessive |
| Gene defect | Unknown | LPIN2 | IL1RN | SH3BP2 >> PTPN11 | Pstpip2 |
| Protein name | ? | Lipin2 | IL-1Ra | SH3BP2 | PSTPIP2 (MAYP) |
| Protein function | ? | Fat metabolism: (PAP enzyme activity), ↑ message to oxidative stress, ? role in mitosis | Antagonist of IL-1 receptor | ↑ Myeloid cell response to M-CSF and RANKL, ↑ TNF-α expression in macrophages | Macrophage proliferation, macrophage recruitment to sites of inflammation, cytoskeletal function |
| Cytokine abnormalities | ↑ serum TNF-α | Not tested | ↑ IL-1α, IL-1β, MIP-1α, TNF-α, IL-8, IL-6 ex vivo monocyte assay; skin reveals ↑ IL-17 staining | ↑ serum TNF-α in mouse model |
cmo: ↑ serum IL-6, MIP-1α, TNF-α, CSF-1, IP-10 Lupo: ↑ serum MIP-1α, IL-4, RANTES, TGF-β |
CRMO, Chronic recurrent multifocal osteomyelitis; CSF, colony-stimulating factor; DIRA, deficiency of interleukin-1 receptor antagonist; HSM, hepatosplenomegaly; IBD, inflammatory bowel disease; IL, interleukin; IL-1Ra, interleukin-1 receptor antagonist; IP-10, interferon-inducible protein-10; M-CSF, macrophage colony-stimulating factor; MIP-1α, macrophage inflammatory protein-1α; PAP, phosphatidate phosphatase; PPP, palmar-plantar pustulosis; PSTPIP2, proline-serine-threonine phosphatase interacting protein; RANKL, receptor activator of nuclear factor-κB ligand; RANTES, regulated on activation, normal T cell expressed and secreted; SH3BP2, SH3 binding protein 2; TGF, transforming growth factor; TNF-α, tumor necrosis factor alpha.
From Ferguson PJ, Laxer RM: Autoinflammatory bone disorders. In Cassidy JT, Petty RE, Laxer RM, et al, editors: Textbook of pediatric rheumatology, ed 6, Philadelphia, 2010, Saunders (Table 44-2).
Table 188.5
Clues That May Assist in Diagnosis of Autoinflammatory Syndromes
| AGE OF ONSET | |
| At birth | NOMID, DIRA, MWS |
| Infancy and 1st yr of life | HIDS, FCAS, NLRP12 |
| Toddler | PFAPA |
| Late childhood | PAPA |
| Most common of autoinflammatory syndromes to have onset in adulthood | TRAPS, DITRA |
| Variable (mostly in childhood) | All others |
| ETHNICITY AND GEOGRAPHY | |
| Armenians, Turks, Italian, Sephardic Jews | FMF |
| Arabs | FMF, DITRA (Arab Tunisian) |
| Dutch, French, German, Western Europe | HIDS, MWS, NLRP12 |
| United States | FCAS |
| Can occur in blacks (West Africa origin) | TRAPS |
| Eastern Canada, Puerto Rico | DIRA |
| Worldwide | All others |
| TRIGGERS | |
| Vaccines | HIDS |
| Cold exposure | FCAS, NLRP12 |
| Stress, menses | FMF, TRAPS, MWS, PAPA, DITRA |
| Minor trauma | PAPA, MWS, TRAPS, HIDS |
| Exercise | FMF, TRAPS |
| Pregnancy | DITRA |
| Infections | All, especially DITRA |
| ATTACK DURATION | |
| <24 hr | FCAS, FMF |
| 1-3 days | FMF, MWS, DITRA (fever) |
| 3-7 days | HIDS, PFAPA |
| >7 days | TRAPS, PAPA |
| Almost always “in attack” | NOMID, DIRA |
| INTERVAL BETWEEN ATTACKS | |
| 3-6 wk | PFAPA, HIDS |
| >6 wk | TRAPS |
| Mostly unpredictable | All others |
| Truly periodic | PFAPA, cyclic neutropenia |
| USEFUL LABORATORY TESTS | |
| Acute-phase reactants must be normal between attacks | PFAPA |
| Urine mevalonic acid in attack | HIDS |
| IgD > 100 mg/dL | HIDS |
| Proteinuria (amyloidosis) | FMF, TRAPS, MWS, NOMID |
| RESPONSE TO THERAPY | |
| Corticosteroid dramatic | PFAPA |
| Corticosteroid partial | TRAPS, FCAS, MWS, NOMID, PAPA* |
| Colchicine | FMF, PFAPA (30% effective) |
| Cimetidine | PFAPA (30% effective) |
| Etanercept | TRAPS, FMF arthritis |
| Anti–IL-1 dramatic | DIRA (anakinra), FCAS, MWS, NOMID, PFAPA |
| Anti–IL-1 mostly | TRAPS, FMF |
| Anti–IL-1 partial | HIDS, PAPA |
* For intraarticular corticosteroids.
DIRA, Deficiency of IL-1 receptor antagonist; DITRA, deficiency of IL-36 receptor antagonist (generalized pustular psoriasis); FCAS, familial cold autoinflammatory syndrome; FMF, familial Mediterranean fever; HIDS, hyper-IgD syndrome; IL, interleukin; MWS, Muckle-Wells syndrome; NLRP, nucleotide oligomerization domain–like receptor family, pyrin domain; NOMID, neonatal-onset multisystem inflammatory disorder; PAPA, pyogenic sterile arthritis, pyoderma gangrenosum, acne syndrome; PFAPA, periodic fever, aphthous stomatitis, pharyngitis, adenitis; TRAPS, tumor necrosis factor receptor–associated periodic syndrome.
From Hashkes PJ, Toker O: Autoinflammatory syndromes, Pediatr Clin North Am 59:447–470, 2012 (Table 2).
Autoinflammatory Diseases With Periodic or Prominent Fevers
The first descriptions of autoinflammatory disorders focused on genetic diseases that presented with prominent fevers, the periodic fever syndromes. As new autoinflammatory diseases were discovered, it was clear that a variety of inflammatory disorders can occur in the absence of fever.
Familial Mediterranean Fever
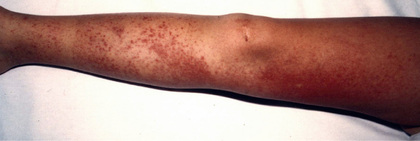
FMF is a recessively inherited autoinflammatory disease usually characterized by recurrent, short-lived (1-3 days), self-limited episodes of fever, serositis, mono- or pauciarticular arthritis, or an erysipeloid rash, sometimes complicated by AA amyloidosis. Most patients with FMF present with symptoms in childhood, with 90% presenting before age 20. Clinical features of FMF may include fever, serositis presenting as pleuritic chest pain or severe abdominal pain, arthritis, and rash. The pleural pain is typically unilateral, whereas the abdominal pain (sterile peritonitis) can be generalized or localized to 1 quadrant, similar to other forms of peritonitis. FMF-associated arthritis occurs primarily in the large joints, may be accompanied by large, neutrophil-rich effusions, and is usually nonerosive and nondestructive. The hallmark cutaneous finding is an erysipeloid erythematous rash that overlies the ankle or dorsum of the foot (Fig. 188.2 ). Other clinical findings include scrotal pain caused by inflammation of the tunica vaginalis testis, febrile myalgia, exercise-induced myalgia (particularly common in children), and an association with various forms of vasculitis, including Henoch-Schönlein purpura, in as many as 5% of pediatric patients. FMF episodes may be triggered by stress, menses, or infections. Between flares, patients are generally symptom free but may have persistent elevation of their inflammatory markers. The attack frequency can vary from weekly to 1-2 flares per year. Table 188.6 lists diagnostic criteria for FMF.
FMF is caused by autosomal recessive mutations in MEFV , a gene encoding a 781 amino acid protein denoted pyrin (Greek for “fever”). Pyrin is expressed in granulocytes, monocytes, and dendritic cells (DCs) and in peritoneal, synovial, and dermal fibroblasts. The N-terminal approximately 90 amino acids of pyrin are the prototype for a motif (the PYRIN domain) that mediates protein-protein interactions and is found in >20 different human proteins that regulate inflammation and apoptosis. Many of the FMF-associated mutations in pyrin are found at the C-terminal B30.2 domain of pyrin, encoded by exon 10 of MEFV. More than 50 such FMF mutations are listed in an online database (http://fmf.igh.cnrs.fr/ISSAID/infevers/ ), almost all of which are missense substitutions. Homozygosity for the M694V mutation may be associated with an earlier age of onset, arthritis, and an increased risk of amyloidosis. The substitution of glutamine for glutamic acid at residue 148 (E148Q) is considered either a mild mutation or a functional polymorphism in the pyrin protein. The carrier frequency of FMF mutations among several Mediterranean populations is very high, suggesting the possibility of a heterozygote advantage.
FMF occurs primarily among ethnic groups of Mediterranean ancestry, most frequently Jews, Turks, Armenians, Arabs, and Italians. Because of a higher frequency of the M694V mutation, FMF is more severe and more readily recognized in the Sephardic (North African) than the Ashkenazi (East European) Jewish population. With the advent of genetic testing, mutation-positive FMF has been documented worldwide, although at lower frequency than in the Mediterranean basin and Middle East.
Through PYRIN-domain interactions, pyrin can activate caspase-1 , the enzyme that converts the 31 kDa pro–IL-1β molecule into the biologically active 17 kDa IL-1β, which is a major mediator of fever and inflammation. FMF mutations lead to a gain-of-function activation of caspase-1 and IL-1β–dependent inflammation, with a gene-dosage effect. These results may explain why as many as 30% of heterozygous carriers of FMF mutations have biochemical evidence of inflammation.
Prophylactic daily oral colchicine decreases the frequency, duration, and intensity of FMF flares. This regimen also prevents the development of systemic AA amyloidosis. Colchicine is generally well tolerated and safe in children, with the most common side effects being diarrhea and other gastrointestinal (GI) complaints. Some patients develop lactose intolerance while taking colchicine. GI side effects can be minimized by initiating therapy at a low dose (for young children, 0.3 mg/day) and slowly titrating upward. A dose-related transaminitis may also be observed; bone marrow suppression is rarely seen at the dosages prescribed for FMF. Pediatric patients may require doses of colchicine similar to those needed in adults (1-2 mg/day), reflecting that children metabolize the drug more rapidly than adults. It is not always possible to find a tolerated dose of colchicine at which all symptoms are suppressed, but approximately 90% of patients have a marked improvement in disease-related symptoms. A small percentage of FMF patients are either unresponsive to or intolerant of therapeutic doses of colchicine. Based on the role of pyrin in IL-1β activation, a trial demonstrated the safety and effectiveness of rilonacept, an IL-1 inhibitor, in FMF; there are case reports of the effectiveness of anakinra, a recombinant interleukin-1 receptor (IL-1R) antagonist.
Amyloidosis is the most serious complication of FMF, and in its absence FMF patients may live a normal life span. Amyloidosis may develop when serum AA, an acute-phase reactant found at extremely high levels in the blood during FMF attacks, is cleaved to produce a 76–amino acid fragment that misfolds and deposits ectopically, usually in the kidneys, GI tract, spleen, lungs, testes, thyroid, and adrenals. Rarely, cardiac amyloidosis may develop; macroglossia and amyloid neuropathy are generally not seen with the amyloidosis of FMF. The most common presenting sign of AA amyloidosis is proteinuria. The diagnosis is then usually confirmed by rectal or renal biopsy. In a small number of case reports, mostly from the Middle East, amyloidosis may actually precede overt FMF attacks, presumably because of subclinical inflammation. Risk factors for the development of amyloidosis in FMF include homozygosity for the M694V MEFV mutation, polymorphisms of the serum AA gene (encoding AA), noncompliance with colchicine treatment, male gender, and a positive family history of AA amyloid. For unclear reasons, country of origin is also a major risk factor for amyloidosis in FMF, with patients raised in the Middle East having a much higher risk than genotypically identical patients raised in the West. Aggressive lifelong suppression of the acute-phase reactants should be the goal in patients with FMF amyloidosis, and documented cases show this may result in resorption of amyloid deposits. The natural history of untreated amyloidosis in FMF is the inexorable progression to renal failure, often within 3-5 yr.
Hyperimmunoglobulinemia D With Periodic Fever Syndrome
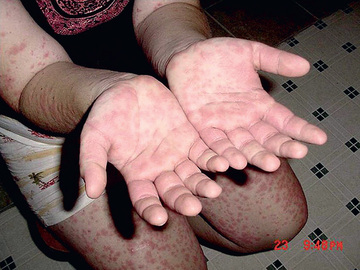
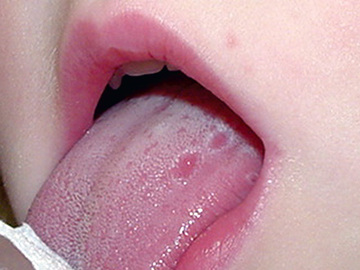
HIDS, also known as mevalonate kinase deficiency , was initially described in a cohort of Dutch patients and occurs primarily in patients of Northern European descent. HIDS is recessively inherited and caused by mutations of MVK , a gene that encodes mevalonate kinase (MK). The clinical features of HIDS generally appear within the 1st 6 mo of life. Febrile attacks last 3-7 days, with abdominal pain often accompanied by diarrhea, nausea, and vomiting. Other clinical manifestations include cervical lymphadenopathy, diffuse macular rash, aphthous ulcers, headaches, and occasional splenomegaly (Figs. 188.3 to 188.5 ). Arthritis or arthralgia can be present in an oligoarticular or polyarticular pattern. Inflammatory disease–like illness and Kawasaki disease–like presentation have also been reported. Attacks are often precipitated by intercurrent illness, immunizations, and surgery. Families frequently recount flares around the time of birthdays, holidays, and family vacations. The symptoms of HIDS may persist for years but tend to become less prominent in adulthood. Patients with HIDS usually have a normal life span. Unlike FMF and TRAPS, the incidence of AA amyloidosis is quite low. Complete MK deficiency results in mevalonic aciduria that presents with severe mental retardation, ataxia, myopathy, cataracts, and failure to thrive (see Chapter 103 ).
MK is expressed in multiple tissues and catalyzes the conversion of mevalonic acid to 5-phosphomevalonic acid in the biosynthesis of cholesterol and nonsterol isoprenoids. Patients with HIDS-associated mutations have greatly reduced, but not absent, MK enzymatic activity. HIDS patients usually have low-normal serum cholesterol levels, but the deficiency of isoprenoids may cause increased IL-1β production by aberrant activation of the small guanosine triphosphatase Rac1. Temperature elevation may further exacerbate this process by more complete inhibition of MK activity, leading to a possible positive feedback loop.
The diagnosis of HIDS may be confirmed either by 2 mutations in MVK (approximately 10% of patients with seemingly typical disease have only a single identifiable mutation) or by elevated levels of mevalonate in the urine during acute attacks. HIDS-associated mutations are distributed throughout the MK protein, but the 2 most common mutations are the substitution of isoleucine for valine at residue 377 (V377I), a variant that is quite common in the Dutch population, and the substitution of threonine for isoleucine at residue 268 (I268T). The eponymous elevation in serum IgD levels is not universally present, especially in young children; IgA levels can also be elevated. Conversely, serum IgD levels may be increased in other autoinflammatory disorders as well as in some chronic infections. During attacks, leukocytosis and increased serum levels of acute-phase reactants and proinflammatory cytokines are frequently present. Table 188.7 lists diagnostic criteria for HIDS.
Standards for the treatment of HIDS are evolving. Very few patients respond to colchicine, and milder disease courses may respond to nonsteroidal antiinflammatory drugs (NSAIDs). Corticosteroids are of limited utility. Small trials of both etanercept and either intermittent or daily anakinra in HIDS are promising.
Tumor Necrosis Factor Receptor–Associated Periodic Syndrome
TRAPS is characterized by recurrent fevers and localized inflammation and is inherited in an autosomal dominant manner. TRAPS has a number of distinguishing clinical and immunologic features. TRAPS was first recognized in patients of Irish descent and denoted familial Hibernian fever to draw a contrast with FMF, but the current nomenclature was proposed when mutations in TNFRSF1A were discovered not only in the original Irish family, but in families from a number of other ethnic backgrounds. TNFRSF1A encodes the 55 kDa receptor (denoted p55, TNFR1, or CD120a) for TNF-α that is widely expressed on a number of cell types. A 2nd 75 kDa receptor is largely restricted to leukocytes.
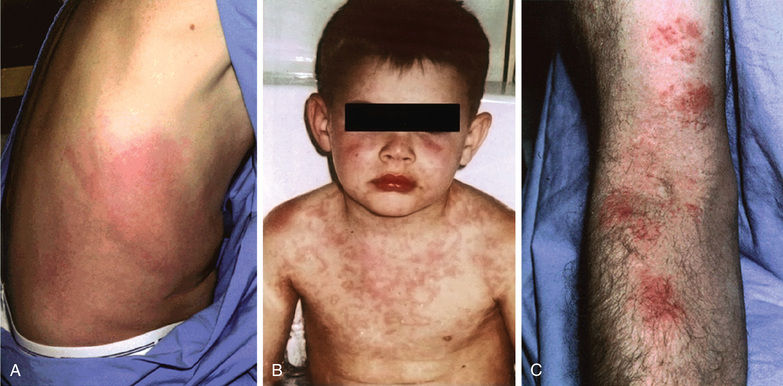
Patients with TRAPS typically present within the 1st decade of life with flares that occur with variable frequency but of often substantially longer duration than FMF or HIDS flares. The febrile episodes of TRAPS last at least 3 days and can persist for weeks. There may be pleural and peritoneal involvement. At times, patients present with signs of an acute abdomen; on exploration such patients have sterile peritonitis , sometimes with adhesions from previous episodes. Patients may also have nausea and frequently report constipation at the onset of flares that progresses to diarrhea by the conclusion. Ocular signs include periorbital edema and conjunctivitis. TRAPS patients may also experience severe myalgia and on imaging, the muscle groups may have focal areas of edema. Many rashes can be seen in TRAPS patients, but the most common is an erythematous macular rash that on biopsy contains superficial and deep perivascular infiltrates of mononuclear cells. Patients often report that the rash migrates distally on a limb during its course with an underlying myalgia and can resemble cellulitis. Other rashes include erythematous annular patches as well as a serpiginous rash (Fig. 188.6 ). Approximately 10–15% of patients with TRAPS may develop AA amyloidosis; the presence of cysteine mutations and a positive family history are risk factors for this complication. If amyloidosis does not develop, TRAPS patients have a normal life expectancy. Table 188.8 lists diagnostic criteria.
Almost all the TRAPS-associated mutations are in the extracellular domain of the TNFR1 protein, with about one-third involving the substitution of another amino acid for a highly conserved cysteine residue, thus disrupting disulfide bonds and leading to protein misfolding. A number of other missense mutations not involving cysteine residues have been shown to have a similar effect on TNFR1 protein folding. Misfolded TNFR1 aggregates intracellularly and leads to constitutive signaling through mitogen-activated protein kinases or nuclear factor (NF)-κB, resulting in the release of proinflammatory cytokines such as IL-6, IL-1β and TNF-α. The substitution of glutamine for arginine at residue 92 (R92Q) and the substitution of leucine for proline at residue 46 (P46L) are seen in >1% of the white and black population, respectively. These variants do not lead to the same biochemical or signaling abnormalities seen with more-severe TRAPS mutations, and as with E148Q in FMF, debate surrounds whether they are mild mutations or functional polymorphisms.
Colchicine is generally not effective in TRAPS. For relatively mild disease, NSAIDs may suffice. For more severe disease with infrequent attacks, corticosteroids at the time of an attack may be effective, but it is not unusual for steroid requirements to increase over time. Etanercept is often effective in reducing the severity and frequency of flares, but longitudinal follow-up of TRAPS patients treated with etanercept indicates waning efficacy with time. Of note, treatment of TRAPS with anti-TNF-α monoclonal antibodies has sometimes led to a paradoxical worsening of disease. Clinical responses to anakinra, canakinumab, a monoclonal anti–IL-1β antibody, and tocilizumab, a monoclonal anti-IL6 antibody, has been favorable in TRAPS patients.
Cryopyrin-Associated Periodic Fever Syndromes
CAPS represent a spectrum of clinical disorders, including familial cold autoinflammatory syndrome (FCAS), Muckle-Wells syndrome (MWS), and neonatal-onset multisystem inflammatory disorder (NOMID). Although 3 separate clinical diagnoses have been defined, it should be emphasized that the cryopyrinopathies are really a continuum of disease severity. This spectrum of illness is caused by mutations in NLRP3 (formerly known as CIAS1 ), which encodes a protein called cryopyrin ; >100 disease-associated NLRP3 mutations have been enumerated on the Infevers online database. Advances in next-generation sequencing have also permitted the identification of symptomatic individuals with somatic NLRP3 mosaicism.
NLRP3 is a PYRIN domain-containing protein that is strongly expressed in myeloid cells and to a lesser degree in other tissues. It is a part of a macromolecular complex termed the NLRP3 inflammasome that activates pro–IL-1β to its mature form in response to a variety of endogenous danger-associated molecular patterns and pathogen-associated molecular patterns. Patients with cryopyrinopathies have gain-of-function mutations in NLRP3 that result in constitutive or easily-triggered activation of the NLRP3 inflammasome.
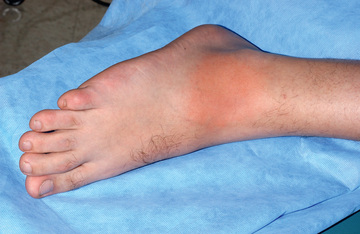
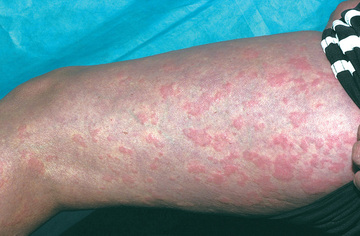
The cryopyrinopathies are characterized by recurrent fevers and an urticaria-like rash that develops early in infancy (Fig. 188.7 ). Histopathologic examination reveals a perivascular neutrophilic infiltrate without the mast cells or mast cell degranulation seen with true urticaria. In patients with FCAS, febrile attacks generally begin 1-3 hr after generalized cold exposure. FCAS patients also experience polyarthralgia of the hands, knees, and ankles, and conjunctivitis may also develop during attacks. FCAS episodes are self-limited and generally resolve within 24 hr. AA amyloidosis rarely occurs in FCAS. Table 188.9 lists diagnostic criteria for FCAS.
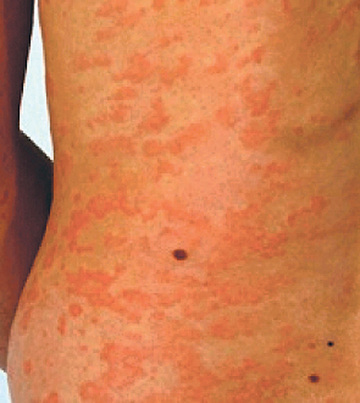
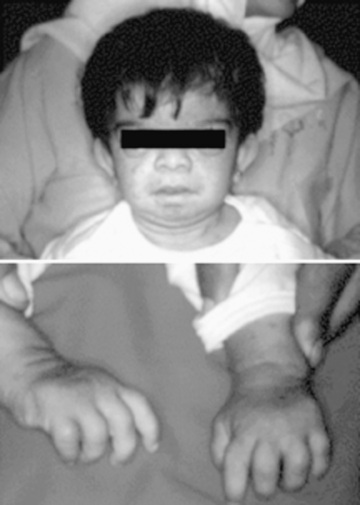
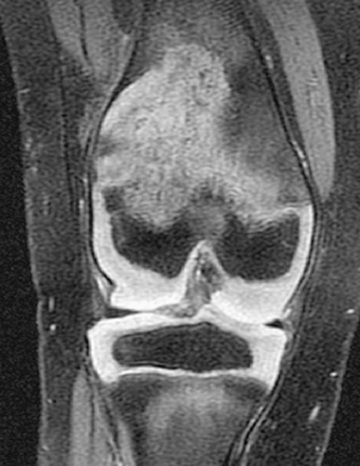
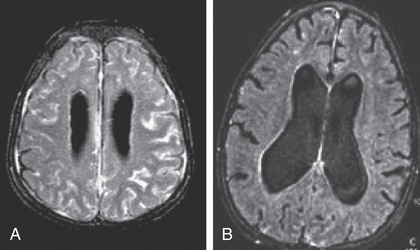
In contrast to FCAS, the febrile episodes of MWS are not cold induced but are characterized by the same urticarial-like rash seen in FCAS (Fig. 188.8 ). Many MWS patients also develop progressive sensorineural hearing loss, and untreated, approximately 30% of MWS patients develop AA amyloidosis. NOMID patients present in the neonatal period with a diffuse, urticarial rash, daily fevers, and dysmorphic features (Fig. 188.9 ). Significant joint deformities, particularly of the knees, may develop because of bony overgrowth of the epiphyses of the long bones (Fig. 188.10 ). NOMID patients also develop chronic aseptic meningitis, leading to increased intracranial pressure, optic disc edema, visual impairment, progressive sensorineural hearing loss, and intellectual disability (Fig. 188.11 ).
Targeted therapy with anakinra (recombinant IL-1R antagonist) has been life changing for NOMID patients , not only controlling fever and rash, but also preventing end-organ damage. Anakinra, rilonocept, and canakinumab are all effective in both FCAS and MWS; they are approved by the U.S. Food and Drug Administration (FDA) for both conditions. Aggressive IL-1 blockade has resulted in attenuation of amyloidosis in the cryopyrinopathies.
Other Mendelian Autoinflammatory Diseases
Syndrome of Pyogenic Arthritis With Pyoderma Gangrenosum and Acne
PAPA syndrome is a rare autosomal dominant disorder caused by mutations in PSTPIP1, a gene that encodes the cytoskeletal proline serine threonine phosphatase–interacting protein-1 (PSTPIP). The PSTPIP1 protein interacts with a number of immunologically important molecules, including CD2, the Wiskott-Aldrich syndrome protein (WASP), and pyrin. PAPA-associated PSTPIP1 mutations greatly increase its affinity to pyrin and cause increased IL-1β production.
Clinical manifestations of PAPA syndrome begin in early childhood with recurrent episodes of sterile, pyogenic arthritis that leads to erosions and joint destruction, and appears to develop spontaneously or after minor trauma. Fever is not a dominant feature. Cutaneous manifestations tend to develop in adolescence, at which time patients are prone to developing severe cystic acne. Additionally, PAPA patients commonly develop ulcerating pyoderma gangrenosum lesions (Fig. 188.12 ), and some develop pathergy reactions.

The treatment of PAPA syndrome may involve the use of corticosteroids, IL-1 antagonists, and TNF-α inhibitors, sometimes in combination. The joint manifestations of PAPA appear to respond to IL-1 blockade, whereas the cutaneous manifestations seem to respond more favorably to TNF-α blockade. Local measures, such as joint aspiration and drainage and intensive wound care, are also important in the care of PAPA patients, as is pain management for cutaneous disease. Caution should be taken when prescribing sulfonamides because some PAPA patients develop pancytopenia.
Deficiency of Interleukin-1 Receptor Antagonist
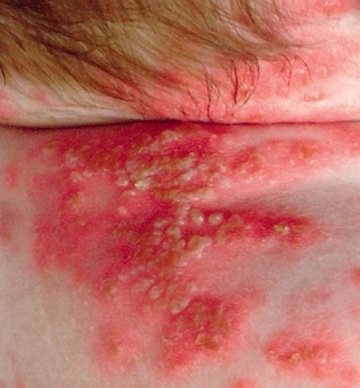
DIRA is an autosomal recessive autoinflammatory disease that is distinct from the cryopyrinopathies. DIRA typically presents in the neonatal period with systemic inflammation and a neutrophilic pustulosis, sterile multifocal osteomyelitis, widening of the anterior ends of the ribs, periostitis, and osteopenia (Figs. 188.13 and 188.14 ). Although fever is not a prominent clinical feature, patients do have greatly elevated acute-phase reactants. Multiorgan failure and pulmonary interstitial fibrosis can occur and can be fatal.
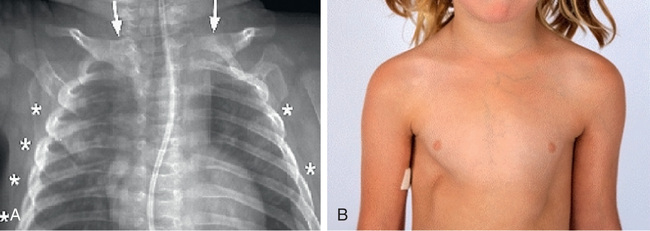
DIRA is caused by loss-of-function mutations in IL1RN , encoding the IL-1R antagonist. Because of the lack of antagonistic activity, the cells are hyperresponsive to IL-1β stimulation. Numerous treatments for DIRA have been tried, including NSAIDs, glucocorticoids, intravenous immune globulin (IVIG), methotrexate, cyclosporine, and etanercept. However, anakinra is the treatment of choice, essentially replacing the lost protein and resulting in a rapid clinical response. Anakinra is dosed daily, with the dose titrated to achieve a normal CRP. There are now longer-acting anti-IL-1 agents, canakinumab and rilonacept, which are effective and require less frequent dosing than anakinra.
Blau Syndrome
Blau syndrome is a rare autosomal dominant disorder that manifests as early-onset (<5 yr of age) granulomatous arthritis, uveitis, and rash. The arthritis may affect the ankles and wrists and may lead to flexion contractures of the fingers and toes (camptodactyly). Early-onset sarcoidosis presents with a similar clinical picture, sometimes with visceral involvement, and both conditions are caused by mutations in the caspase recruitment domain protein 15 (CARD15), also known as nucleotide-binding oligomerization domain-2 protein (NOD2). NOD2 is an intracellular sensor of bacterial products in DCs, myelomonocytic cells, and Paneth cells. Mutations in the NACHT oligomerization domain of this protein cause Blau syndrome/early-onset sarcoidosis, whereas variants primarily in the leucine-rich repeat domain are associated with susceptibility to Crohn disease . Corticosteroids have been the mainstay of therapy for Blau syndrome. There are a number of case reports of the beneficial effects of TNF-α inhibitors in Blau syndrome.
Autoinflammation With Phospholipase Cγ2 –Associated Antibody Deficiency and Immune Dysregulation
APLAID is a dominantly inherited disorder characterized by recurrent blistering skin lesions, bronchiolitis, arthralgia, ocular inflammation, enterocolitis, absence of autoantibodies, and mild immunodeficiency. Rash is the first manifestation of APLAID, which is described as a full-body epidermolysis bullosa–like eruption. Over time, this rash changes to recurrent plaques and vesiculopustular lesions that are triggered by heat and sunlight. Colitis also presents in childhood before age 5 yr. Ocular manifestations begin before age 1 yr and include corneal ulcerations and erosions as well as cataracts. Immune manifestations include markedly decreased class-switched memory B cells, resulting in low IgM and IgA.
Patients with APLAID show a gain-of-function missense mutation in the autoinhibitory region of phospholipase Cγ2 (PLCγ2 ), leading to increased activity of downstream mediators and stimulation of lymphocytes. Despite the enhanced signaling, the resulting populations of immune cells have poor function. Interestingly, a different mutation in the PLCγ2 complex leads to a syndrome known as PLCγ2 -associated antibody deficiency and immune dysregulation (PLAID) , characterized by cold-induced urticaria, hypogammaglobulinemia with resulting susceptibility to infection, and autoimmunity.
Because of the low number of affected patients described, there are no agreed treatment regimens for APLAID. Patients have been treated with NSAIDs, and corticosteroids can be effective, but side effects limit their long term use. TNF-α inhibitors and IL-1 inhibitors have been used with some success.
Deficiency of Adenosine Deaminase 2
DADA2 is an autoinflammatory disorder caused by loss-of-function mutations in CECR1 , encoding adenosine deaminase 2. DADA2 presents with recurrent fevers and a spectrum of vascular manifestations that includes livedo racemosa, early-onset ischemic lacunar strokes, and systemic vasculitis of medium side vessels similar to polyarteritis nodosa . The lacunar strokes, typically affecting the deep brain nuclei and the brainstem, transpire before age 5 yr and typically occur during inflammatory episodes. The livedoid rash is also a prominent feature during inflammatory episodes, and biopsies demonstrate a predominance of neutrophils and macrophages as well as vasculitis in medium-sized vessels. Acute-phase reactants are typically elevated. Other features include ophthalmologic involvement, various degrees of lymphopenia, hypogammaglobulinemia (usually IgM), hepatosplenomegaly, portal hypertension, and neutropenia. Patients may meet criteria for polyarteritis nodosa and can exhibit digit necrosis and Raynaud phenomenon.
ADA2 is produced primarily by monocytes and macrophages, is found in plasma, and appears to act as a growth and differentiation factor for a subset of inflammatory macrophages. Numerous antiinflammatories have been tried in patients with DAD2, including glucocorticoids and cyclophosphamide. TNF-α inhibitors (etanercept or adalimumab) are the mainstay of treatment, and anecdotal reports have shown a benefit of anakinra. Macrophages and monocytes are the main sources of ADA2, raising the possibility of bone marrow transplant to achieve a permanent cure.
Sideroblastic Anemia With Immunodeficiency, Fevers, and Development Delay
SIFD is a syndrome characterized by systemic inflammation, fevers, enteritis, and sideroblastic anemia and caused by biallelic mutations in TRNT1 . SIFD presents in infancy with fever, elevated inflammatory markers, gastroenteritis, and anemia. Bone marrow biopsies demonstrate ringed sideroblasts. Other features include hypogammaglobulinemia, B-cell lymphopenia, developmental delay, and variable neurodevelopmental degeneration, seizures, and sensorineural hearing loss. Brain imaging was notable for cerebellar atrophy, delayed white matter myelination, and decreased perfusion. Other, isolated clinical features include nephrocalcinosis, aminoaciduria, ichthyotic skin, cardiomyopathy, and retinitis pigmentosa. TRNT1 is an RNA polymerase that is necessary for maturation of cytosolic and mitochondrial transfer RNAs, by the addition of 2 cytosines and 1 adenosine to the tRNA ends.
Symptomatic treatment with regular blood transfusions and immunoglobulin replacement therapy is the mainstay of SIFD therapy. Iron overload from the transfusion often requires chelation therapy. Anakinra relieved the febrile episodes in one patient but did not alter the other clinical manifestations. Patients with SIFD have a high mortality rate. One patient underwent hematopoietic bone marrow transplantation at 9 mo of age that resulted in correction of the hematologic and immunologic abnormalities.
Deficiency of Interleukin-36 Receptor Antagonist (DITRA)
DITRA is characterized by episodes of diffuse erythematous pustular rash (generalized pustular psoriasis), fevers, general malaise, and systemic inflammation. Attacks can be triggered by events such as infections, pregnancy, or menstruation or can occur randomly. The underlying genetic etiology has been determined to be autosomal recessive mutations in the IL36RN gene, which encodes an IL-36R antagonist. IL-36 is related to and acts similarly to IL1R antagonist, preventing production of inflammatory cytokines such as IL-8. Interestingly, the rash of DITRA is similar to the rash of DIRA (IL-1R deficiency; see earlier), but DITRA is largely skin limited. DITRA has been treated with various modalities, including vitamin A analogs, cyclosporine, methotrexate, and TNF-α inhibitors. The use of anakinra has been described in case reports and results in alleviation of the symptoms.
Familial Cold Autoinflammatory Syndrome Type 2
Mutations in NLRP12 lead to a periodic fever syndrome characterized by fevers >40°C, arthralgias, and myalgias lasting from 2-10 days. This disorder is named FCAS2 because these episodes can be precipitated by cold. Clinical findings may include an urticarial-like rash, abdominal pain and vomiting, aphthous ulcers, and lymphadenopathy. As with Muckle-Wells syndrome, sensorineural hearing loss and optic neuritis have been described. NALP12 is a member of the CATERPILLAR family of proteins, which are important in innate immunity. Similar to Toll-like receptors (TLRs) that act to recognize pathogen-associated molecular patterns (PAMPs), NLRP12 also senses PAMPs and can lead to the activation of the inflammasome and generation of IL-1β. Treatment of NALP12 mutations was difficult until the advent of anti–IL-1 agents (e.g., anakinra), which are the preferred treatment for FCAS2 and result in remarkable resolution of symptoms. Colchicine can be partially effective, and systemic glucocorticoids can reduce the duration of the attacks.
Autoinflammation With Enterocolitis
A disorder caused by mutations in NLRC4 was described with neonatal-onset enterocolitis, fever, and autoinflammatory episodes. Inflammatory markers are typically elevated, including CRP and ferritin. Macrophage activation syndrome , characterized by pancytopenia, hypertriglyceridemia, and coagulopathies, is common during acute flares, which can be precipitated by emotional and physical stress. Recurrent myalgias with febrile episodes often occur as well. This disorder is caused by gain-of-function missense mutations in NOD-like receptor C4 (NLRC4), which normally aids in the activation of the inflammasome. The resulting protein leads to constitutive production of IL-1. The mainstay of treatment is anti-IL-1 agents such as anakinra, canakinumab, and rilonacept. Before their diagnosis, patients with NLRC4 mutations had been treated with colchicine and oral glucocorticoids, with varying success.
Majeed Syndrome
Majeed syndrome is an autosomal recessive disorder caused by mutations in the LPIN2 gene (see Table 188.4 ). The clinical manifestation of Majeed syndrome begin in childhood with recurrent fevers, sterile osteomyelitis, congenital dyserythropoietic anemia (CDA), neutrophilic dermatosis, failure to thrive, and hepatomegaly. Treatment of Majeed syndrome has included NSAIDs, corticosteroids, and IL-1R antagonist. How mutations in LPIN2 lead to an autoinflammatory disorder is not known.
Interferonopathies
Type 1 interferons (IFN-α and IFN-β) are the first line of defense against viral infections and are produced by a variety of cell types. During viral infections, a variety of products are made by the virus, including ssRNA, dsRNA, and CpG-containing DNA, and are recognized by intracellular sensors. These sensors then induce type 1 IFN production that activates IFN receptors and activates IFN-responsive genes to help control the spread of the virus until the adaptive immune system can be activated to clear the virus. Inappropriate activation of these pathways leads to IFN production and interferonopathies.
Chronic Atypical Neutrophilic Dermatosis With Lipodystrophy and Elevated Temperature
CANDLE syndrome, also known as proteasome-associated autoinflammatory syndrome (PRAAS) or joint contractures, muscular atrophy, panniculitis-induced lipodystrophy (JMP) syndrome , is an autosomal recessive disease. Patients present early in life with recurrent fevers and systemic inflammation; skin involvement, including annular erythema, erythema nodosum–like panniculitis, or neutrophilic dermatosis; small joint contractures; lipodystrophy; muscle atrophy or myositis; violaceous eyelid swelling; and anemia. Conjunctivitis, aseptic meningitis, and organomegaly are common. Acute-phase reactants and platelet counts are elevated. Autoimmunity can occur, including Coombs-positive hemolytic anemia and hypothyroidism. Intelligence and development are typically spared, although mild developmental delays have been reported. CANDLE is caused by loss-of-function mutations in PSMB8, the gene that encodes the β5i subunit of the proteasome. Proteosomes are important in the degradation of ubiquinated proteins to ensure proper protein homeostasis, and defects in proteasomes result in cellular stress and inflammatory cytokine release, including type 1 interferons.
There is no established treatment for CANDLE, although multiple treatment modalities have been attempted, including colchicine, dapsone, cyclosporine, infliximab, and etanercept, all with minimal success. Glucocorticoids and methotrexate have provided slight improvement in symptoms. Anakinra has not proved successful, whereas IL-6–blocking agents have shown some benefit. Since interferon receptors use the JAK/STAT pathway to signal, JAK inhibitors (tofacitinib, ruxolitinib, and baricitinib) show promise.
STING-Associated Vasculopathy With Onset in Infancy
SAVI is a rare disorder that presents in infancy. It is caused by mutations in the TMEM173 gene, which encodes for the stimulator of interferon genes (STING). Systemic inflammation is an early manifestation, with fever and elevated inflammatory markers. Skin involvement includes a neutrophilic rash as well as violaceous lesions of fingers, toes, nose, cheeks, and ears. These lesions worsen over time and can become necrotic with vascular occlusion. Histology of the lesions reveals dermal inflammation with leukocytoclastic vasculitis and microthrombotic angiopathy. Since STING is also expressed in pulmonary epithelium, SAVI patients also developed pulmonary complications, including paratracheal adenopathy, interstitial lung disease, and fibrosis.
STING is an adapter protein of the intracellular DNA sensing machinery and mediates the production of interferon-β (IFN-β). The IFN-β then signals through the IFN receptor by activating the JAK/STAT signaling pathway and downstream IFN-responsive genes, including IL-6 and TNF-α. Mutations in STING that cause SAVI are de novo gain-of-function mutations that activate spontaneous IFN-β production.
Treatment options for patients with SAVI are limited at this time, although recent data with JAK inhibitors (tofacitinib, ruxolitinib, and baricitinib) have shown promise in blocking IFN-β receptor signaling and the activation of IFN-response genes.
Genetically Complex Autoinflammatory Diseases
Periodic Fever, Aphthous Stomatitis, Pharyngitis, and Adenitis
PFAPA is the most common recurrent fever syndrome in children. It usually presents between ages 2 and 5 yr with recurring episodes of fever, malaise, exudative-appearing tonsillitis with negative throat cultures, cervical lymphadenopathy, oral aphthae, and less often headache, abdominal pain, and arthralgia. The episodes last 4-6 days, regardless of antipyretic or antibiotic treatment, and often occur with clock-like regularity on 3-6 wk cycles. Findings during the episodes may include mild hepatosplenomegaly, mild leukocytosis, and elevated acute-phase reactants. Both the frequency and the intensity of the episodes diminish with increasing age. The etiology and pathogenesis of PFAPA remain unknown.
Most patients show dramatic response to a single oral dose of prednisone (0.6-2.0 mg/kg), although this approach does not prevent recurrence and may actually shorten the interval between flares. Cimetidine at 20-40 mg/kg/day is effective at preventing recurrences in approximately one third of cases. Small series have shown that anakinra may be effective during a flare, but because corticosteroids are effective, this may not be a cost-effective approach. Colchicine may extend the time between flares. Complete resolution has been reported after tonsillectomy, although medical management should be the first approach.
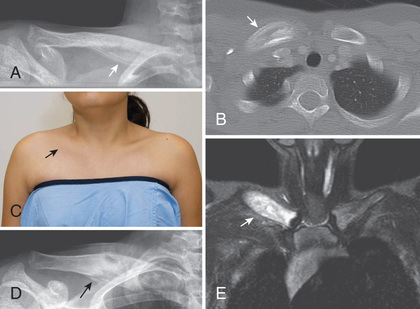
Chronic Recurrent Multifocal Osteomyelitis
CRMO is a form of inflammatory bone disease most frequently seen in children (see Table 188.4 ). Histologically and radiologically, CRMO is virtually indistinguishable from infectious osteomyelitis (Fig. 188.15 ). Patients typically present with bone pain and may also have fever, soft tissue swelling, and elevated acute-phase reactants. Cultures are sterile. Typically involved bones include the distal femur, proximal tibia or fibula, spine, and pelvis. Both metaphyseal and epiphyseal lesions may occur; premature physeal closure may develop. Less frequently involved bones include the clavicle and mandible. The differential diagnosis includes infectious osteomyelitis, histiocytosis, and malignancy (neuroblastoma, lymphoma, leukemia, Ewing sarcoma). SAPHO (synovitis, acne, pustulosis, hyperostosis, and osteitis) may be an adult equivalent to CRMO. The etiology of sporadic CRMO is unknown. CRMO is seen in Majeed syndrome (see earlier), in association with inflammatory bowel disease, and inflammatory skin disease such as palmoplantar pustulosis. Initial therapy includes NSAIDs. Second-line treatments include corticosteroids, TNF inhibitors, and bisphosphonates.