Disorders of the Exocrine Pancreas
Steven L. Werlin, Michael Wilschanski
Disorders Associated With Pancreatic Insufficiency
Other than cystic fibrosis (CF), conditions that cause pancreatic insufficiency are very rare in children. They include Shwachman-Diamond syndrome (SDS), Johanson-Blizzard syndrome, Ivemark syndrome, Pearson syndrome, isolated enzyme deficiencies, enterokinase deficiency (see Chapter 364 ), chronic pancreatitis, protein-calorie malnutrition (see Chapters 57 and 364 ), and IMNEPD (infantile onset multisystem neurologic, endocrine, and pancreatic disease).
Cystic Fibrosis (see Chapter 432 )
By the end of the 1st yr of life, 85–90% of children with CF have pancreatic insufficiency, which, if untreated, will lead to malnutrition. Treatment of the associated pancreatic insufficiency leads to improvement in absorption, better growth, and more normal stools. Pancreatic function can be monitored in children with CF with serial measurements of fecal elastase. Ten to 15% of children present with a neonatal intestinal obstruction called meconium ileus; in later life a common intestinal complication is distal intestinal obstruction syndrome which is unique to CF. Ten percent of CF patients develop severe liver disease. Ten to 15% of CF patients are pancreatic sufficient and their presentation tends to be later in life, including recurrent pancreatitis, male infertility, and chronic bronchiectasis. CF is part of the newborn screen in every state in the United States and in most countries in the Western world.
Shwachman-Diamond Syndrome (see Chapter 157 )
SDS is an autosomal recessive syndrome (1/20,000 births) caused by a mutation of the Shwachman-Bodian-Diamond syndrome (SBDS) gene on chromosome 7, which causes ribosomal dysfunction in 90–95% of patients. Signs and symptoms of SDS include pancreatic insufficiency; neutropenia, which may be cyclic; neutrophil chemotaxis defects; metaphyseal dysostosis; failure to thrive; and short stature. Some patients with SDS have liver or kidney involvement, dental disease, or learning difficulty. SDS is a common cause of congenital neutropenia.
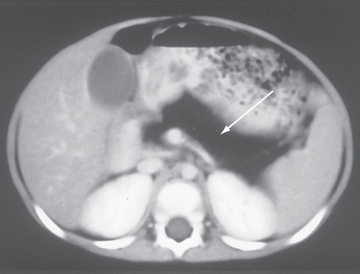
Patients typically present in infancy with poor growth and steatorrhea. More varied phenotypes have been described including absence of pancreatic lipomatosis on imaging, normal fecal elastase levels, and normal skeletal survey. These children can be readily differentiated from those with CF by their normal sweat chloride levels, lack of mutations in the CF gene, characteristic metaphyseal lesions, and fatty pancreas characterized by a hypodense appearance on CT and MRI scans (Fig. 376.1 ).
Despite adequate pancreatic replacement therapy and correction of malabsorption, poor growth commonly continues. Pancreatic insufficiency is often transient, and steatorrhea frequently spontaneously improves with age. Recurrent pyogenic infections (otitis media, pneumonia, osteomyelitis, dermatitis, sepsis) are frequent and are a common cause of death. Thrombocytopenia is found in 70% of patients and anemia in 50%. Development of aplastic anemia or a myelodysplastic syndrome can occur, with transformation to acute myeloid leukemia in 24%. The pancreatic acini are replaced by fat with little fibrosis. Islet cells and ducts are normal. Bone marrow transplant is the treatment of choice in patients who develop acute myeloid leukemia.
Pearson Syndrome
Pearson (marrow-pancreas) syndrome is caused by a contiguous mitochondrial gene depletion involving several mitochondrial genes affecting oxidative phosphorylation, that manifests in infants with severe macrocytic anemia and variable thrombocytopenia . The bone marrow demonstrates vacuoles in erythroid and myeloid precursors as well as ringed sideroblasts. In addition to its role in severe bone marrow failure, pancreatic insufficiency contributes to growth failure. Mitochondrial DNA mutations are transmitted through maternal inheritance to both sexes or are sporadic.
Johanson-Blizzard Syndrome
The features of Johanson-Blizzard syndrome include exocrine pancreatic deficiency, aplasia or hypoplasia of the alae nasi, congenital deafness, hypothyroidism, developmental delay, short stature, ectodermal scalp defects, absence of permanent teeth, urogenital malformations, and imperforate anus. This syndrome is caused by a mutation in the UBR1 gene found on chromosome 15. The UBR1 protein acts as a ubiquitin ligase.
Isolated Enzyme Deficiencies
Isolated deficiencies of trypsinogen, enterokinase, lipase, and colipase have been reported. Although enterokinase is a brush-border enzyme, deficiency causes pancreatic insufficiency because enterokinase is required to activate trypsinogen to trypsin in the duodenum. Deficiencies of trypsinogen or enterokinase manifest with failure to thrive, hypoproteinemia, and edema. Isolated amylase deficiency is typically developmental and resolves by age 2-3 yr.
Other Syndromes Associated With Pancreatic Insufficiency
Pancreatic agenesis, congenital pancreatic hypoplasia, and congenital rubella are rare causes of pancreatic insufficiency. Pancreatic insufficiency has also been reported in duodenal atresia and stenosis and may also be seen in infants with familial or nonfamilial hyperinsulinemic hypoglycemia, who require 95–100% pancreatectomy to control hypoglycemia. Mutations in at least 6 genes have been described. Pancreatic insufficiency, which may be found in children with celiac disease and undernutrition, recovers with nutritional rehabilitation.
IMNEPD is a rare disease due to mutations in the PTRH2 gene. Neurologic features dominate the phenotype (microcephaly, intellectual disability, cerebellar atrophy, deafness, and neuropathy), but pancreatic insufficiency is seen in most patients.