Cystic Fibrosis
Marie E. Egan, Michael S. Schechter, Judith A. Voynow
Cystic fibrosis (CF) is an inherited multisystem disorder of children and adults; it is the most common life-limiting recessive genetic trait among whites. Dysfunction of the cystic fibrosis transmembrane conductance regulator (CFTR) protein, the primary defect, leads to a wide and variable array of presenting manifestations and complications.
CF is responsible for most cases of exocrine pancreatic insufficiency in early life and is the major cause of severe chronic lung disease in children. It is also responsible for many cases of hyponatremic salt depletion, nasal polyposis, pansinusitis, rectal prolapse, pancreatitis, cholelithiasis, and nonautoimmune insulin-dependent hyperglycemia. Because CF may manifest as failure to thrive and hepatic dysfunction, including cirrhosis, this disorder enters into the differential diagnosis of many pediatric conditions (Table 432.1 ).
Table 432.1
Complications of Cystic Fibrosis
Adapted from Silverman FN, Kuhn JP: Essentials of Caffey's pediatric x-ray diagnosis, Chicago, 1990, Year Book, p. 649.
Genetics
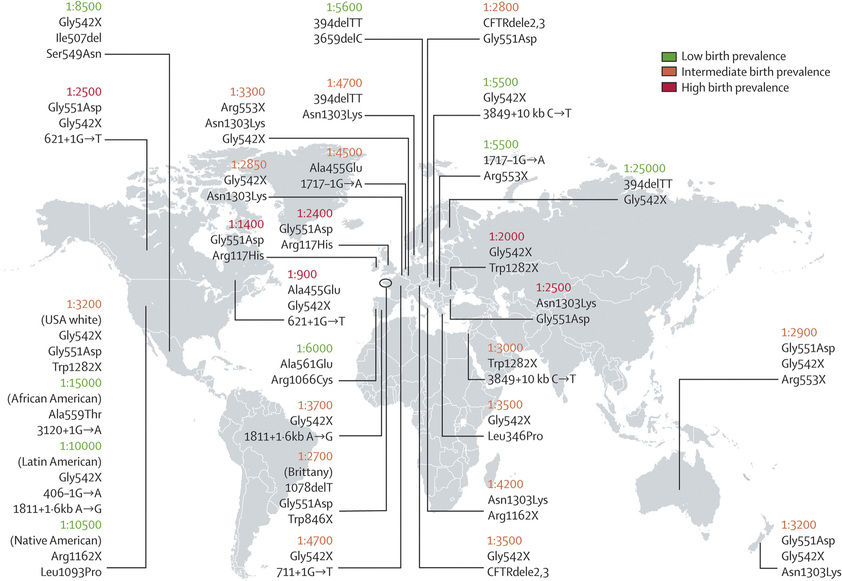
CF occurs most frequently in white populations of northern Europe, North America, and Australia/New Zealand. The prevalence in these populations varies but approximates 1 in 3,500 live births (1 in 9,200 individuals of Hispanic descent and 1 in 15,000 African Americans). Although less frequent in African, Hispanic, Middle Eastern, South Asian, and eastern Asian populations, the disorder does exist in these populations as well (Fig. 432.1 ).
CF is inherited as an autosomal recessive trait. The CF gene codes for the CFTR protein, which is 1,480 amino acids. CFTR is expressed largely in epithelial cells of airways, the gastrointestinal tract (including the pancreas and biliary system), the sweat glands, and the genitourinary system. CFTR is a member of the adenosine triphosphate–binding cassette superfamily of proteins. It functions as a chloride channel and has other regulatory functions that are perturbed variably by the different mutations. More than 1,900 CFTR polymorphisms have been described, many of which are not clearly of clinical significance. Those that are associated with clinical manifestations may be grouped into 6 main classes based upon how they impact upon protein structure and function (Table 432.2 ; Fig. 432.2 ). Mutation class I-III are generally considered to be severe mutations in that they lead to a complete or nearly complete absence of CFTR function, whereas class IV-VI mutations are associated with some residual functional protein. The most prevalent mutation of CFTR is the deletion of a single phenylalanine residue at amino acid 508 (F508del). This mutation is responsible for the high incidence of CF in northern European populations and is considerably less frequent in other populations, such as those of southern Europe and Israel. Nearly 50% of individuals with CF in the United States Cystic Fibrosis Foundation (CFF) Patient Registry are homozygous for F508del, and approximately 87% carry at least 1 F508del gene. Remaining patients have an extensive array of mutations, none of which has a prevalence of more than several percentage points, except in certain populations; for example, the W1282X mutation occurs in 60% of Ashkenazi Jews with CF. Through the use of probes for 40 of the most common mutations, the genotype of 80–90% of Americans with CF can be ascertained. Genotyping using a discreet panel of mutation probes is quick and less costly than more comprehensive sequencing and is the approach typically used in state newborn screening programs. In remaining patients, sequencing the entire CFTR gene and looking for deletions and duplications are necessary to establish the genotype. As sequencing technologies evolve and costs decrease, sequencing the entire CFTR gene may become mainstream for all patients.
Table 432.2
One Proposed Classification of Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) Mutations
| CLASS | EFFECT ON CFTR | FUNCTIONAL CFTR PRESENT? | SAMPLE MUTATIONS |
|---|---|---|---|
| I | Lack of protein production | No | Stop codons (designation in X; e.g., Trp1282X, Gly542X); splicing defects with no protein production (e.g., 711+1G→T, 1717-1G→A) |
| II | Defect in protein trafficking with ubiquitination and degradation in endoplasmic reticulum/Golgi body | No/substantially reduced | Phe508del, Asn1303Lys, Gly85Gly, leu1065Pro, Asp507, Ser549Arg |
| III | Defective regulation; CFTR not activated by adenosine triphosphate or cyclic adenosine monophosphate | No (nonfunction CFTR present in apical membrane) | Gly551Asp, Ser492Phe, Val520Phe, Arg553Gly, Arg560Thr, Arg560Ser |
| IV | Reduced chloride transport through CFTR at the apical membrane | Yes | Ala455Glu, Arg117Cys, Asp1152His, Leu227Arg, Arg334Trp, Arg117His* |
| V | Splicing defect with reduced production of CFTR | Yes | 3849+10kbC→T, 1811+16kbA→G, IVS8-5T, 2789+5G→A |
* Function of Arg117His depends on the length of the polythymidine track on the same chromosome in intron 8 (IVS8): 5T, 7T, or 9T. There is more normal CFTR function with a longer polythymidine track.
From O'Sullivan BP, Freedman SD: Cystic fibrosis, Lancet 373:1891–1902, 2009.

The relationship between CFTR genotype and clinical phenotype is highly complex. CFTR mutation class is strongly associated with pancreatic dysfunction and will usually predict this manifestation in any given patient. Respiratory complications and lung function decline are also correlated with mutation class severity but with greater variation due to the influence of non-CFTR modifier gene polymorphisms and environmental influences on the manifestations of lung disease in any one individual. Studies have identified specific non-CFTR modifier genes of importance; genome-wide association studies identified a polymorphism on chromosome 11 in the intergenic region between EHF (an epithelial transcription factor) and APIP (an inhibitor of apoptosis) that is associated with lung disease severity and may influence the expression of EHF and APIP, as well as other genes in the region, including PDHX , CD44 , and ELF5 . A region on chromosome 20 may also be found to relate to lung disease severity. This region encompasses several genes (MC3R, CASS4, AURKA) that may play a role in lung host defense involving neutrophil function, apoptosis, and phagocytosis. Genome-wide association studies analysis also identified genetic regions that predispose to risk for liver disease, CF-related diabetes, and meconium ileus.
The high-frequency of CFTR mutations has been ascribed to resistance to the morbidity and mortality associated with infectious dysenteries through the ages. Cultured CF intestinal epithelial cells homozygous for the F508del mutation are unresponsive to the secretory effects of cholera toxin. CFTR heterozygous mice experience less mortality when treated with cholera toxin than their unaffected wild-type littermates.
Pathogenesis
A number of long-standing observations of CF are of fundamental pathophysiologic importance; they include failure to clear mucous secretions, a paucity of water in mucous secretions, an elevated salt content of sweat and other serous secretions, and chronic infection limited to the respiratory tract. In addition, there is a greater negative potential difference across the respiratory epithelia of patients with CF than across the respiratory epithelia of control subjects. Aberrant electrical properties are also demonstrated for CF sweat gland duct and rectal epithelia. The membranes of CF epithelial cells are unable to secrete chloride or bicarbonate in response to cyclic adenosine monophosphate–mediated signals, and at least in the respiratory epithelial cells, excessive amounts of sodium are absorbed through these membranes. These defects can be traced to a dysfunction of CFTR. CFTR function is highly regulated and energy dependent; it requires both cyclic adenosine monophosphate–stimulated protein kinase A phosphorylation of the regulatory domain and ATP binding and hydrolysis at the nucleotide binding domains. CFTR also interacts with other ion channels, signal transduction proteins, and the cytoskeleton (Fig. 432.3 and see Fig. 432.2 ).
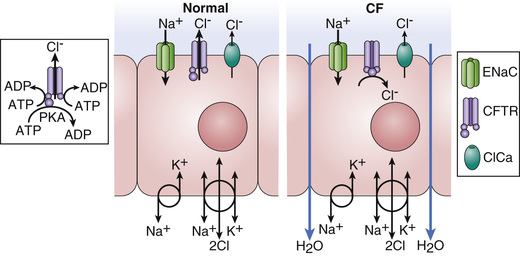
Many hypotheses have been postulated to explain how CFTR dysfunction results in the clinical phenotype (Fig. 432.4 ). It is likely that no one hypothesis explains the full spectrum of disease. One model is that airway hydration homeostasis requires both CFTR and P2Y2 -regulated calcium-activated chloride secretion. When extracellular ATP is depleted such as after viral infections, calcium-activated chloride secretion is not activated and the failure of mutant CFTR chloride secretion results in dehydrated airway secretions, increased concentration of mucin solids, and more viscoelastic mucus that is not cleared by normal mucociliary transport. Another mechanism that is supported by both primary human airway studies and investigations in the CF pig is that mutant CFTR causes failure of HCO3 − secretion and a more acidic airway surface liquid, which increases mucous viscoelasticity resulting in poor mucociliary clearance. Mucous secretions are tethered to submucosal gland ducts and are retained and obstruct airways, starting with those of the smallest caliber, the bronchioles. Airflow obstruction at the level of small airways is the earliest observable physiologic abnormality of the respiratory system. CFTR dysfunction in airway smooth muscle has been implicated in tracheal and airway abnormalities in humans and in animal models of the disease (pig and mice). These data suggest that CFTR expression in this nonepithelial tissue contributes to airway constriction.
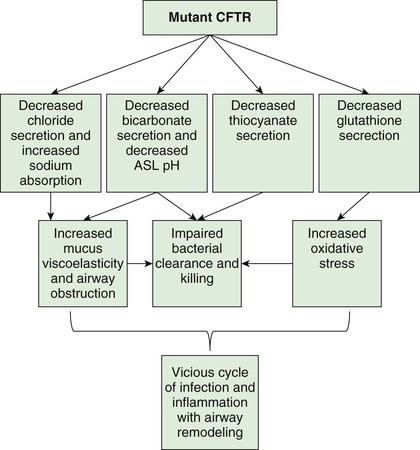
It is plausible that similar pathophysiologic events take place in the pancreatic and biliary ducts (and in the vas deferens), leading to desiccation of proteinaceous secretions and obstruction. Because the function of sweat gland duct cells is to absorb rather than secrete chloride, salt is not retrieved from the isotonic primary sweat as it is transported to the skin surface; chloride and sodium levels are consequently elevated.
Chronic infection in CF is limited to the airways. One explanation for infection is a sequence of events starting with failure to clear inhaled bacteria promptly and then proceeding to persistent infection and an inflammatory response in airway walls. Another explanation for early infection is the failure of innate immune proteins to kill bacteria in an abnormally acidic airway milieu. In addition, it has been proposed that abnormal CFTR creates a proinflammatory state or amplifies the inflammatory response to initial infections (viral or bacterial). Some investigators have identified primary differences in CF-affected immune cells (including macrophage, neutrophils, lymphocytes, and dendritic cells) and have suggested that these alterations contribute to this proinflammatory state as well as a dysregulated immune response. It appears that inflammatory events occur first in small airways, perhaps because it is more difficult to clear altered secretions and microorganisms from these regions. The agents of airway injury include neutrophil products, such as oxidative radicals and proteases, and immune reaction products. These inflammatory products further aggravate airway obstruction by increasing mucin secretion and altering mucin structure to promote both intramolecular and intermolecular interactions. Excessive inflammatory cell polymers in CF sputum, including DNA, filamentous actin, and glycosaminoglycans, further contribute to abnormal mucous viscoelastic properties and airway obstruction. Chronic bronchiolitis and bronchitis are the initial lung manifestations (see Chapter 418 ), but after months to years, structural changes in airway walls produce bronchiolectasis and bronchiectasis . With advanced lung disease, infection may extend to peribronchial lung parenchyma.
A central feature of lung disease in patients with CF is the high prevalence of airway infection with Staphylococcus aureus (see Chapter 208.1 ), Pseudomonas aeruginosa (see Chapter 232.1 ), and Burkholderia cepacia complex (see Chapter 232.2 ), organisms that rarely infect the lungs of other individuals. It has been postulated that the CF airway epithelial cells or surface liquids may provide a favorable environment for harboring these organisms. CF airway epithelium may be compromised in its innate defenses against these organisms, through either acquired or genetic alterations. Antimicrobial activity is diminished in CF secretions; this diminution may be related to hyperacidic surface liquids or other effects on innate immunity. Another puzzle is the propensity for P. aeruginosa to undergo mucoid transformation in the CF airways. The complex polysaccharide produced by these organisms generates a biofilm that provides a hypoxic environment and thereby protects Pseudomonas against antimicrobial agents.
Altered lipid homeostasis has been implicated as a predisposing factor for respiratory tract infection and inflammation. Concentrations of lipoxins—molecules that suppress neutrophilic inflammation—are suppressed in CF airways. There is an imbalance of lipids with increased arachidonic acid and decreased docosahexaenoic acid, which promotes inflammation. There is also an imbalance of ceramide in the CF airway that is proinflammatory. Supporting the idea that altered lipid uptake affects infection and inflammation is the observation that the 10–15% of individuals with CF who retain substantial exocrine pancreatic function have delayed acquisition of P. aeruginosa and slower deterioration of lung function. However, it appears that nutritional factors are contributory only because preservation of pancreatic function does not preclude development of typical lung disease.
The variation in progression of lung disease seen in patients with CF is largely influenced by social and physical environment factors, whose impact matches that of CFTR genotype. Exposure to environmental tobacco smoke and outdoor air pollutants, and early acquisition of respiratory virus infections, as well as pathogenic organisms like P. aeruginosa and methicillin-resistant S. aureus, have been implicated as causes of worsening disease. Sex/gender disparities also seem to exist, with females having a poorer prognosis. Although studies have suggested that estrogen may influence disease exacerbations, the gap seems to be narrowing in the past decade.
Although most CF care is delivered at specialty centers and is broadly influenced by current clinical guidelines, there is enough variability in treatment approaches to cause large variation in respiratory and nutritional outcomes across the care networks in both North America and Europe. Social determinants of health are associated with significant disparities in outcome; socioeconomic status has been shown to be a strong predictor of mortality, as well as both nutritional status and lung function on both sides of the Atlantic. The specific mechanism of effect is unclear, but evidence suggests a role for socioeconomic status–related differences in health behaviors and disease self-management practices, stress and mental health issues, and environmental tobacco smoke exposure. Differential access to specialty care and medications is not a major factor in North American children (lack of insurance in some adults is a problem); however, differences in disease outcomes across European countries of varying wealth are quite clear.
Pathology
The earliest pathologic lesion in the lung is that of bronchiolitis (mucous plugging and an inflammatory response in the walls of the small airways); with time, mucous accumulation and inflammation extend to the larger airways (bronchitis ) (see Chapter 418.2 ). Goblet cell hyperplasia and submucosal gland hypertrophy become prominent pathologic findings, which is most likely a response to chronic airway infection. Organisms appear to be confined to the endobronchial space; invasive bacterial infection is not characteristic. With long-standing disease, evidence of airway destruction such as bronchiolar obliteration, bronchiolectasis, and bronchiectasis (see Chapter 430 ) becomes prominent. Imaging modalities demonstrate both increased airway wall thickness and luminal cross-sectional area relatively early in lung disease evaluation. Bronchiectatic cysts and emphysematous bullae or subpleural blebs are frequent with advanced lung disease, the upper lobes being most commonly involved. These enlarged air spaces may rupture and cause pneumothorax. Interstitial disease is not a prominent feature, although areas of fibrosis appear eventually. Bronchial arteries are enlarged and tortuous, contributing to a propensity for hemoptysis in bronchiectatic airways. Small pulmonary arteries eventually display medial hypertrophy, which would be expected in secondary pulmonary hypertension.
The paranasal sinuses are uniformly filled with secretions containing inflammatory products, and the epithelial lining displays hyperplastic and hypertrophied secretory elements (see Chapter 408 ). Polypoid lesions within the sinuses and erosion of bone have been reported. The nasal mucosa may form large or multiple polyps , usually from a base surrounding the ostia of the maxillary and ethmoidal sinuses.
The pancreas is usually small, occasionally cystic, and often difficult to find at postmortem examination. The extent of involvement varies at birth. In infants, the acini and ducts are often distended and filled with eosinophilic material. In 85–90% of patients, the lesion progresses to complete or almost complete disruption of acini and replacement with fibrous tissue and fat. Infrequently, foci of calcification may be seen on radiographs of the abdomen. The islets of Langerhans contain normal-appearing β cells, although they may begin to show architectural disruption by fibrous tissue in the 2nd decade of life.
The intestinal tract shows only minimal changes. Esophageal and duodenal glands are often distended with mucous secretions. Concretions may form in the appendiceal lumen or cecum. Crypts of the appendix and rectum may be dilated and filled with secretions.
Focal biliary cirrhosis secondary to blockage of intrahepatic bile ducts is uncommon in early life, although it is responsible for occasional cases of prolonged neonatal jaundice. This lesion becomes much more prevalent and extensive with age and is found in 70% of patients at postmortem examination. This process can proceed to symptomatic multilobular biliary cirrhosis that has a distinctive pattern of large irregular parenchymal nodules and interspersed bands of fibrous tissue. Approximately 30–70% of patients have fatty infiltration of the liver, in some cases despite apparently adequate nutrition. At autopsy, hepatic congestion secondary to cor pulmonale is frequently observed. The gallbladder may be hypoplastic and filled with mucoid material and often contains stones. The epithelial lining often displays extensive mucous metaplasia. Atresia of the cystic duct and stenosis of the distal common bile duct have been observed.
Glands of the uterine cervix are distended with mucus, copious amounts of which collect in the cervical canal. In >95% of males, the body and tail of the epididymis, the vas deferens, and the seminal vesicles are obliterated or atretic, resulting in male infertility.
Clinical Manifestations
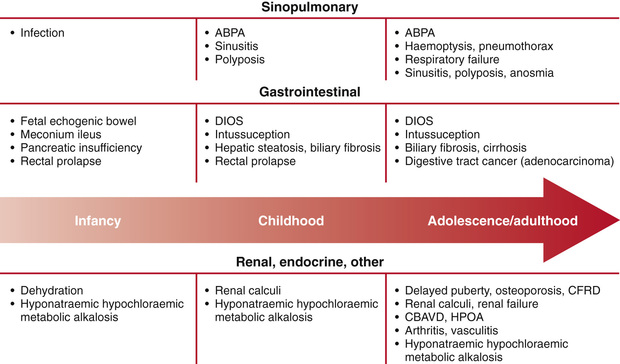
Since the universal adoption of CF newborn screening in the United States and overseas, as well as the evolution of aggressive and proactive treatment approaches, the clinical face of CF is very different from what it was in earlier decades. Diagnosis is typically accomplished before 1 mo of age, prior to any obvious clinical symptoms or signs, and treatment is targeted on immediately correcting nutritional deficiencies and delaying the respiratory complications of the disease. The interaction of mutational heterogeneity and environmental factors leads to highly variable involvement of the lungs, pancreas, and other organs. A summary of the time course of potential development of clinical manifestations is shown in Fig. 432.5 .
Respiratory Tract
Infants diagnosed by CF newborn screening are generally asymptomatic from a respiratory standpoint. Nonetheless, the majority are infected with S. aureus
, Haemophilus influenza
, or even P. aeruginosa
within the 1st mo of life, and chest CT scans show characteristic heterogeneous air trapping in 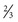 of infants by their first birthday, and bronchiectasis is found in more than 10% of 1 yr olds and ∼60% of 5 yr olds. The earliest symptom is usually cough that may begin with a viral respiratory tract infection but then persists unless treated with antibiotics. With treatment, the generally realized goal is for patients to remain asymptomatic throughout childhood, except for the periodic development of cough, chest congestion, sputum production, and/or wheezing that define a pulmonary exacerbation
.
of infants by their first birthday, and bronchiectasis is found in more than 10% of 1 yr olds and ∼60% of 5 yr olds. The earliest symptom is usually cough that may begin with a viral respiratory tract infection but then persists unless treated with antibiotics. With treatment, the generally realized goal is for patients to remain asymptomatic throughout childhood, except for the periodic development of cough, chest congestion, sputum production, and/or wheezing that define a pulmonary exacerbation
.
The rate of progression of lung disease is the chief determinant of morbidity and mortality. As lung disease slowly progresses, chronic cough, sputum production, exercise intolerance, shortness of breath, and failure to thrive are noted. Cor pulmonale, respiratory failure, and death eventually supervene unless lung transplantation is accomplished; this has become increasingly uncommon in childhood. Infection with certain strains of B. cepacia and other multidrug-resistant organisms may be associated with particularly rapid pulmonary deterioration and death.
Eventual physical findings include increased anteroposterior diameter of the chest, generalized hyperresonance, scattered or localized coarse crackles, and digital clubbing. Expiratory wheezes may be heard, a manifestation of airway inflammation and edema that may or may not be associated with bronchodilator responsiveness. Cyanosis is a late sign. Common pulmonary complications include atelectasis, hemoptysis, pneumothorax, and cor pulmonale; these usually appear in late adolescence or beyond.
Even though the paranasal sinuses are virtually always opacified radiographically, acute sinusitis is infrequent. Nasal obstruction and rhinorrhea are common, caused by inflamed, swollen mucous membranes or, in some cases, nasal polyposis. Nasal polyps are most troublesome between 5 and 20 yr of age.
Intestinal Tract
In 15–20% of newborn infants with CF, the ileum is completely obstructed by meconium (meconium ileus ). The frequency is greater among siblings born subsequent to a child with meconium ileus and is particularly striking in monozygotic twins, reflecting a genetic contribution from one or more unknown modifying genes. Abdominal distention, emesis, and failure to pass meconium appear in the first 24-48 hr of life (see Chapters 123.1 and 356.2 ) and often requires surgical intervention. Abdominal radiographs (Fig. 432.6 ) show dilated loops of bowel with air-fluid levels and, frequently, a collection of granular, “ground-glass” material in the lower central abdomen. Rarely, meconium peritonitis results from intrauterine rupture of the bowel wall and can be detected radiographically as the presence of peritoneal or scrotal calcifications.
Ileal obstruction with fecal material (distal intestinal obstruction syndrome [DIOS] ) occurs in older children, causing cramping abdominal pain, abdominal distention, and obstruction that can be treated with medical approaches to bowel evacuation.
More than 85% of children with CF have exocrine pancreatic insufficiency, causing protein and fat malabsorption. Symptoms, if untreated, include frequent, bulky, greasy stools and failure to gain weight even when food intake appears to be large. Weight gain can be challenging, but attainment of normal growth and development is an expectation of treatment. A protuberant abdomen, decreased muscle mass, poor growth, and delayed maturation are classic and rarely seen physical signs. Excessive flatus may be a problem. Supplementation with fat-soluble vitamin preparations has made deficiencies of vitamin A, E, and K unusual, but vitamin D deficiency continues to be prevalent and, although rickets is rare, osteoporosis is common, especially in older patients and those with more severe lung disease. Class IV-VI mutations are associated with pancreatic sufficiency, but patients with these mutations are prone to pancreatitis when they reach adolescence.
Historically a relatively common event, rectal prolapse occurs much less frequently as the result of earlier diagnosis and initiation of pancreatic enzyme replacement therapy.
Biliary Tract
Infants may occasionally present with neonatal jaundice suggestive of biliary obstruction. Evidence for liver dysfunction is most often detected in the first 15 yr of life and can be found in up to 30% of individuals. Biliary cirrhosis becomes symptomatic in only 5–7% of patients. Manifestations can include icterus, ascites, hematemesis from esophageal varices, and evidence of hypersplenism. Biliary colic secondary to cholelithiasis may occur in the 2nd decade or later. Liver disease occurs independent of genotype but is associated with meconium ileus and pancreatic insufficiency.
Cystic Fibrosis–Related Diabetes and Pancreatitis
Endocrine pancreatic insufficiency tends to develop in the 2nd decade and beyond and is more common in patients with a family history of type II diabetes mellitus. It most commonly begins with postprandial hyperglycemia and may or may not be accompanied by weight loss or flattening weight gain. Fasting hyperglycemia and elevated hemoglobin A1c are later manifestations. Ketoacidosis usually does not occur, but eye, kidney, and other vascular complications have been noted in patients living ≥10 yr after the onset of hyperglycemia. Recurrent, acute pancreatitis occurs occasionally in individuals who have residual exocrine pancreatic function and may be the sole manifestation of homozygotic CFTR mutations.
Genitourinary Tract
Virtually all males are azoospermic because of failure of development of wolffian duct structures, but sexual function is generally unimpaired. The female fertility rate is diminished, especially in women who have poor nutrition or advanced lung disease. Pregnancy is generally tolerated well by women with good pulmonary function but may accelerate pulmonary progression in those with advanced lung problems and may lead to glucose intolerance. Urinary incontinence associated with cough occurs in 18–47% of female children and adolescents.
Sweat Glands
Excessive loss of salt in the sweat predisposes young children to salt depletion episodes, especially during episodes of gastroenteritis and during warm weather. These children may present with hypochloremic alkalosis. Hyponatremia is a risk particularly in warm climates. Frequently, parents notice salt frosting of the skin or a salty taste when they kiss the child. A few genotypes are associated with normal sweat chloride values.
Diagnosis and Assessment
The diagnosis of CF has been based on a positive quantitative sweat test (Cl− ≥ 60 mEq/L) in conjunction with one or more of the following features: identification of 2 CFTR mutations, typical chronic obstructive pulmonary disease, documented exocrine pancreatic insufficiency, and a positive family history. With newborn screening, diagnosis is often made prior to obvious clinical manifestations such as failure to thrive and chronic cough. Diagnostic criteria have been recommended to include additional testing procedures (Table 432.3 ).
Table 432.3
Sweat Testing
The sweat test, which involves using pilocarpine iontophoresis to collect sweat and performing chemical analysis of its chloride content, is the standard approach to diagnosis of CF. The procedure requires care and accuracy. An electric current is used to carry pilocarpine into the skin of the forearm and locally stimulate the sweat glands. If an adequate amount of sweat is collected, the specimens are analyzed for chloride concentration. Infants with a positive newborn screen for CF should have the sweat chloride testing performed after 36-wk corrected gestational age and at a weight greater than 2 kg and at age greater than 10 days to increase the likelihood of sufficient sweat collection for an accurate study. Positive results should be confirmed; for a negative result, the test should be repeated if suspicion of the diagnosis remains.
More than 60 mmol/L of chloride in sweat is diagnostic of CF when one or more other criteria are present. In individuals with a positive newborn screen, a sweat chloride level less than 30 mmol/L indicates that CF is unlikely. Borderline (or intermediate) values of 30-59 mmol/L have been reported in patients of all ages who have CF with atypical involvement and require further testing. Table 432.4 lists the conditions associated with false-negative and false-positive sweat test results.
DNA Testing
Several commercial laboratories test for 30-96 of the most common CFTR mutations. This testing identifies ≥90% of individuals who carry 2 CF mutations. Some children with typical CF manifestations are found to have 1 or no detectable mutations by this methodology. Some laboratories perform comprehensive mutation analysis screening for all the >1,900 identified mutations.
Other Diagnostic Tests
The finding of increased potential differences across nasal epithelium (nasal potential difference) that is the increased voltage response to topical amiloride application, followed by the absence of a voltage response to a β-adrenergic agonist, has been used to confirm the diagnosis of CF in patients with equivocal or frankly normal sweat chloride values. This testing is primarily used in research applications and has never undergone extensive validation as a clinical tool.
Pancreatic Function
The diagnosis of pancreatic malabsorption can be made by the quantification of elastase-1 activity in a fresh stool sample by an enzyme-linked immunosorbent assay specific for human elastase. The quantification of fat malabsorption with a 72-hr stool collection is rarely necessary in the clinical setting. CF-related diabetes affects approximately 20% of adolescents and 40–50% of adults, and clinical guidelines recommend yearly oral glucose tolerance testing (OGTT) after age 10. OGTT may sometimes be clinically indicated at an earlier age. Spot testing of blood and urine glucose levels and glycosylated hemoglobin levels are not sufficiently sensitive.
Radiology
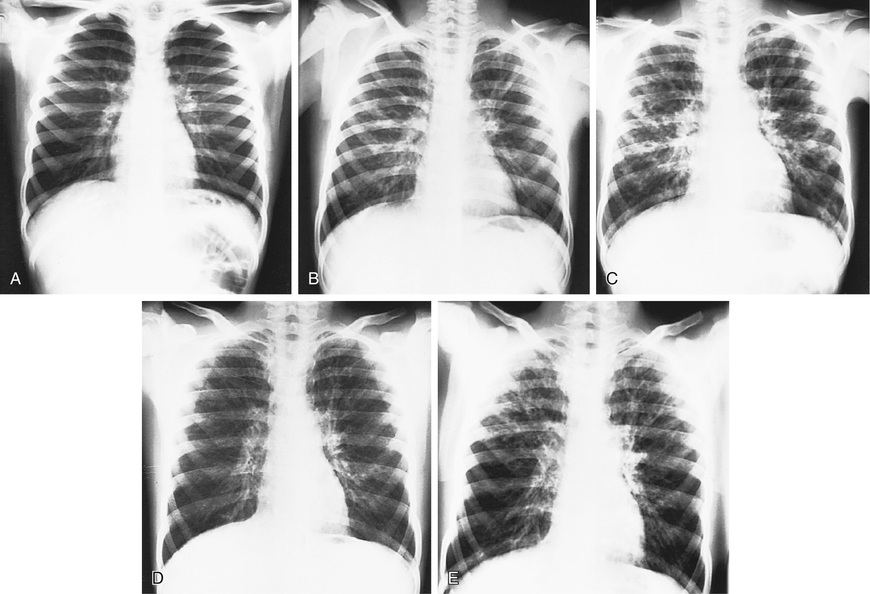
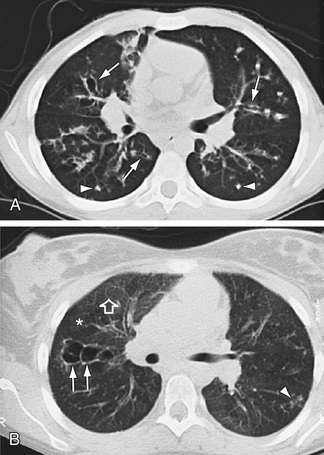
Hyperinflation of lungs occurs early and is often accompanied by nonspecific peribronchial thickening (Fig. 432.7 ). Bronchial thickening and plugging and ring shadows suggesting bronchiectasis usually appear first in the upper lobes. Nodular densities, patchy atelectasis, and confluent infiltrate follow. Hilar lymph nodes may be prominent. With advanced disease, impressive hyperinflation with markedly depressed diaphragms, anterior bowing of the sternum, and a narrow cardiac shadow are noted. Cyst formation, extensive bronchiectasis, dilated pulmonary artery segments, and segmental or lobar atelectasis is often apparent with advanced disease. Most CF centers obtain chest radiographs (posteroanterior [PA] and lateral) at least annually. Standardized scoring of radiologic changes has been used to follow progression of lung disease. CT of the chest can detect heterogeneous hyperinflation and localized thickening of bronchial airway walls, mucous plugging, focal hyperinflation, and early bronchiectasis (Fig. 432.8 ). CT abnormalities are commonly seen at a young age, even in asymptomatic children with normal lung function.
Radiographs of paranasal sinuses reveal panopacification and, often, failure of frontal sinus development. CT provides better resolution of sinus changes if this information is required clinically. Fetal ultrasonography may show pancreatic changes indicative of CF and suggest ileal obstruction with meconium early in the second trimester, but this finding is not predictive of meconium ileus at birth.
Pulmonary Function
Infant pulmonary function testing is done routinely for clinical evaluation at a few CF centers but, given its complexity and the need for sedation, for the most part it is reserved for research protocols. Lung clearance index (LCI) measured by multiple breath washout can be done in infants and young children and is a sensitive measure of ventilation inhomogeneity caused by small airways disease. Currently it is primarily used for research, but given its ease and applicability it may be adopted as a standard monitoring tool in the future as CF care centers become more accustomed to its use.
Standard pulmonary function studies are usually obtained starting at about 4 yr of age and are routinely done by age 6. Forced expiratory volume in 1 sec (FEV1 ) is the measurement that has been shown to correlate most closely with mortality and shows a gradual decline averaging 2–3% per year throughout childhood. Although a small number of children may already show evidence of airway obstruction by age 6, trends over the past several decades, as reported by the CFF patient registry, show a steady improvement in average FEV1 of the CF population, and as of 2015 ∼75% had normal or near-normal lung function at age 18 yr. Residual volume and functional residual capacity are increased early in the course of lung disease and are the cause of decreasing forced vital capacity (FVC) measurement. Restrictive changes, characterized by declining total lung capacity and vital capacity, correlate with extensive lung injury and fibrosis and are a late finding. Testing at each clinic visit is recommended to evaluate the course of the pulmonary involvement and allow for early intervention when clinically significant decrements are documented—this is probably the most sensitive indicator of a pulmonary exacerbation that should be treated with systemic antibiotics.
Microbiologic Studies
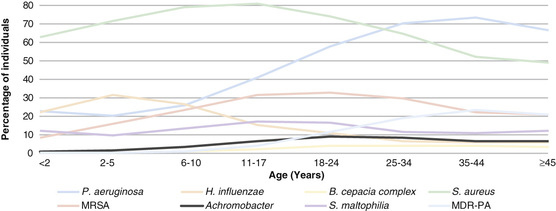
H. influenza and S. aureus are the most common organisms recovered in young children (Fig. 432.9 ). Pseudomonas may be acquired early and is eventually an organism of key significance. P. aeruginosa appears to have a special propensity for the CF airway and over time characteristically develops a biofilm associated with a mucoid appearance in the microbiology lab and which correlates with more rapid progression of lung disease. Once P. aeruginosa develops a mucoid phenotype, it is extremely difficult to eradicate from the airway. A wide range of other organisms are frequently recovered, particularly in advanced lung disease; they include a variety of Gram-negative rods including the Burkholderia cepacia complex, which may be associated with a fulminant downhill course (the cepacia syndrome); Stenotrophomonas maltophilia , and Achromobacter xylosoxidans; assorted fungi, especially Aspergillus fumigatus , which is most important due to the relatively common development of allergic bronchopulmonary aspergillosis ; and nontuberculous mycobacterial species, especially Mycobacterium avium complex and Mycobacterium abscessus . Airway cultures are obtained regularly, most typically using oropharyngeal swabs in young children, and then sputum (which may be induced) in older children capable of expectoration. Oropharyngeal swabs typically give a good indication of the lower airway flora, but fiberoptic bronchoscopy may be used to gather lower respiratory tract secretions of infants and young children who do not expectorate if there is a concern for false-negative cultures, especially regarding the presence of P. aeruginosa .
The CF airway microbiome consists of a large number of additional organisms, especially anaerobes that are identified through antigen detection but not culture methods. The significance of this finding and its therapeutic implications remain somewhat unclear, but it has long been appreciated that response to antibiotic treatment of pulmonary exacerbations is not always predictable based upon culture and sensitivity of airway cultures.
Newborn Screening
Newborn screening for CF is mandated in all 50 states and is the most common way that CF is diagnosed. A variety of newborn screening algorithms are in place to identify infants with CF. Most algorithms use a combination of immunoreactive trypsinogen (IRT) results and limited DNA testing on blood spots; because not all mutations can be found using this approach, babies with an elevated IRT and a single detected mutation are considered a positive screen, and all positive screens are followed by a confirmatory sweat analysis. Depending upon race and ethnicity, about 10–15% of infants with a positive screen based on the finding of only 1 CF mutation will be found to have CF. This screening test is ≈95% sensitive and should result in a median age at diagnosis of less than 1 mo. Newborn diagnoses can prevent early nutritional deficiencies and improve long-term growth and may improve cognitive function. Importantly, good nutritional status (50 percentile weight for length or 50 percentile body mass index) is associated with better lung function at 6 yr of age.
An occasional patient may be missed by newborn screening, and those caring for adolescents and adults need to be aware that most of those older patients were not screened at birth and may present at later ages, into late adulthood. Prior to the advent of newborn screening, infants and children commonly presented with malabsorption and failure to thrive, in addition to respiratory symptoms. Most older patients whose diagnosis was missed early in life will have unusual class IV, V, or VI mutations and therefore normal pancreatic function. They will more typically present with chronic productive cough due to either bronchitis or chronic sinusitis and may have nasal polyps or allergic bronchopulmonary aspergillosis or unexplained bronchiectasis. The most common nonrespiratory manifestations will be congenital bilateral absence of the vas deferens (CBAVD) (in males) or recurrent pancreatitis. It is important to recognize that sweat testing at an adept lab (typically limited to CF Foundation accredited care centers) is the most accurate way to diagnose CF in this group. CFTR mutation testing with standard panels is never as sensitive as sweat testing and will frequently miss the unusual mutations that are seen more commonly in people who present late in this manner.
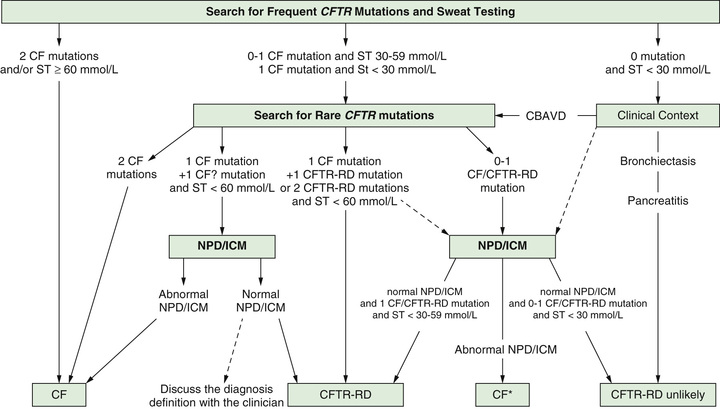
There is a subset of infants with a positive newborn screen for CF who have a nondiagnostic sweat chloride (30-59 mmol/L) and/or 1 or 2 CFTR mutations that is not clearly disease causing. These infants have CFTR -related metabolic syndrome (CRMS) (also called CFTR-related disease) and should be followed in a CF center closely through the 1st yr and then annually to evaluate them for the development of CF symptoms. Indeed, in some (∼10%) patients, the sweat test becomes clearly abnormal over time and they can be diagnosed as having CF. Because CRMS is a condition defined by asymptomatic detection in the context of newborn screening and CF newborn screening has been commonly performed only in the past decade or so, it is not clear whether some children in this group will eventually develop manifestations of CFTR-related disorder, such as CBAVD, chronic sinusitis, recurrent pancreatitis, or even bronchiectasis. An approach to the evaluation of patients with CRMS is seen in Fig. 432.10 .
Treatment
General Approach to Care
Initial efforts after diagnosis should be intensive and should include baseline assessment, initiation of treatment to prevent pulmonary involvement in young infants or reverse it in those diagnosed later, nutritional maintenance or remediation, and education of the patient and parents. Follow-up evaluations are scheduled every 1-3 mo, depending on the age at diagnosis, because many aspects of the condition require careful monitoring. An interval history and physical examination should be obtained at each visit. A sputum sample or, if that is not available, a lower pharyngeal swab taken during or after a forced cough is obtained for culture and antibiotic susceptibility studies. Because irreversible loss of pulmonary function from low-grade infection can occur gradually and without acute symptoms, emphasis is placed on a thorough pulmonary history and physical exam and routine pulmonary function testing. Table 432.5 lists symptoms and signs that suggest the need for more intensive antibiotic and physical therapy (PT). Protection against exposure to methicillin-resistant S. aureus, P. aeruginosa, B. cepacia, and other resistant Gram-negative organisms is essential, including contact isolation procedures and careful attention to cleaning of inhalation therapy equipment. A nurse, physical therapist, respiratory therapist, social worker, and dietitian, as members of the multidisciplinary care team, should evaluate children regularly and contribute to the development of a comprehensive daily care plan. Considerable education and programs to empower families and older children to take responsibility for care are likely to result in the best adherence to daily care programs. Screening patients and caregivers for anxiety and depression annually is expected to identify issues that can interfere with adherence to daily care. Standardization of practice, on the part of both caregivers and families, as well as close monitoring and early intervention for new or increasing symptoms appears to result in the best long-term outcomes.
Table 432.5
Symptoms and Signs Associated With Exacerbation of Pulmonary Infection in Patients With Cystic Fibrosis
|
SYMPTOMS SIGNS |
From Ramsey B: Management of pulmonary disease in patients with cystic fibrosis, N Engl J Med 335:179, 1996.
Because secretions of CF patients are not adequately hydrated, attention in early childhood to oral hydration, especially during warm weather or with acute gastroenteritis, may minimize complications associated with impaired mucous clearance. Intravenous therapy for dehydration should be initiated early.
The goal of therapy is to maintain a stable condition for prolonged periods. This can be accomplished for most patients by interval evaluation and adjustments of the home treatment program. Some children have episodic acute or low-grade chronic lung infection that progresses. For these patients, intensive inhalation and airway clearance and intravenous antibiotics are indicated. Improvement is most reliably accomplished in a hospital setting; selected patients have demonstrated successful outcomes while completing these treatments at home. Intravenous antibiotics may be required infrequently or as often as every 2-3 mo. The goal of treatment is to return patients to their previous pulmonary and functional status.
The basic daily care program varies according to the age of the child, the degree of pulmonary involvement, other system involvement, and the time available for therapy. The major components of this care are pulmonary and nutritional therapies. Because therapy is medication intensive, iatrogenic problems frequently arise. Monitoring for complications is also an important part of management
Pulmonary Therapy
The object of pulmonary therapy is to clear secretions from airways and to control infection. When a child is not doing well, every potentially useful aspect of therapy should be reconsidered.
Inhalation Therapy
Human recombinant DNase (2.5 mg) enzymatically dissolves extracellular DNA released by neutrophils, a major contributor to the characteristically sticky and viscous CF airway secretions. It is usually given as a single daily aerosol dose, improves pulmonary function, decreases the number of pulmonary exacerbations, and promotes a sense of well-being. Benefit for those with mild, moderate, and severe lung disease has been documented. Improvement is sustained for 12 mo or longer with continuous therapy.
Nebulized hypertonic saline, acting as a hyperosmolar agent, is believed to draw water into the airway and rehydrate mucus and the periciliary fluid layer, resulting in improved mucociliary clearance. Seven percent hypertonic saline nebulized 2-4 times daily increases mucous clearance and reduces pulmonary exacerbation, with only a slight short-term improvement in pulmonary function.
Airway Clearance Therapy
Airway clearance treatment begins in infancy with chest percussion (with or without postural drainage) and derives its rationale from the idea that cough clears mucus from large airways, but chest vibrations are required to shear secretions for the airway wall and move secretions from small airways, where expiratory flow rates are low. Chest PT can be particularly useful for patients with CF because they accumulate secretions in small airways first, even before the onset of symptoms. Cessation of chest PT in children with mild to moderate airflow limitation results in deterioration of lung function within 3 wk, and prompt improvement of function occurs when therapy is resumed, but it is less clear which available modality is best. Airway clearance therapy is recommended 2-4 times a day, depending on the severity of lung dysfunction, and usually increased during acute exacerbations. Cough, huffing, or forced expirations are encouraged intermittently throughout the session. Vest-type mechanical percussors (high-frequency chest wall oscillation) are commonly used past infancy due to their convenience, as are a variety of oscillatory positive expiratory pressure devices (such as Acapella and Aerobika) and other controlled breathing techniques (e.g., autogenic drainage ). Routine aerobic exercise appears to slow the rate of decline of pulmonary function, and benefit has also been documented with weight training. No one airway clearance technique can be shown to be superior to any other, so all modes should be considered in the development of an airway clearance prescription. Adherence to daily therapy is important but rarely achieved; therefore airway clearance technique plans are individualized for each patient.
Antibiotic Therapy
Antibiotics are the mainstay of therapy designed to control progression of lung infection. The goal is to reduce the intensity of endobronchial infection and to delay progressive lung damage. The usual guidelines for acute chest infections, such as fever, tachypnea, or chest pain, are often absent. Consequently, all aspects of the patient's history and examination, including anorexia, weight loss, and diminished activity, must be used to guide the frequency and duration of therapy. Antibiotic treatment varies from intermittent short courses of 1 antibiotic to nearly continuous treatment with 1 or more antibiotics. Dosages for some antibiotics are often 2-3 times the amount recommended for minor infections because patients with CF have proportionately more lean body mass and higher clearance rates for many antibiotics than other individuals. In addition, it is difficult to achieve effective drug levels of many antimicrobials in respiratory tract secretions.
Oral Antibiotic Therapy
Indications for oral antibiotic therapy in a patient with CF include the presence of respiratory tract symptoms, physical signs, or changes in pulmonary function testing or chest x-ray. Treatment is guided by identification of pathogenic organisms in respiratory tract cultures and in vitro sensitivity testing. Common organisms, including S. aureus (MRSA or MSSA), nontypeable H. influenzae, P. aeruginosa; B. cepacia and other Gram-negative rods, are encountered with increasing frequency. The usual course of therapy is 2 wk, and maximal doses are recommended. Table 432.6 lists useful oral antibiotics. The quinolones are the only broadly effective oral antibiotics for Pseudomonas infection, but resistance against these agents may emerge. Macrolides may reduce the virulence properties of P. aeruginosa, such as biofilm production, and contribute antiinflammatory effects. Long-term therapy with azithromycin 3 times a week improves lung function in patients with chronic P. aeruginosa infection.
Table 432.6
Antimicrobial Agents for Cystic Fibrosis Lung Infection
| ROUTE | ORGANISMS | AGENTS | DOSAGE (mg/kg/24 hr) | NO. DOSES/24 hr |
|---|---|---|---|---|
| Oral | Staphylococcus aureus | Dicloxacillin | 25-50 | 4 |
| Linezolid | 20 | 2 | ||
| Cephalexin | 50 | 4 | ||
| Clindamycin | 10-30 | 3-4 | ||
| Amoxicillin-clavulanate | 25-45 | 2-3 | ||
| Haemophilus influenzae | Amoxicillin | 50-100 | 2-3 | |
| Pseudomonas aeruginosa | Ciprofloxacin | 20-30 | 2-3 | |
| Burkholderia cepacia | Trimethoprim-sulfamethoxazole | 8-10* | 2-4 | |
| Empirical | Azithromycin | 10, day 1; 5, days 2-5 | 1 | |
| Erythromycin | 30-50 | 3-4 | ||
| Intravenous | S. aureus | Nafcillin | 100-200 | 4-6 |
| Vancomycin | 40 | 3-4 | ||
| P. aeruginosa | Tobramycin | 8-12 | 1-3 | |
| Amikacin | 15-30 | 2-3 | ||
| Ticarcillin | 400 | 4 | ||
| Piperacillin | 300-400 | 4 | ||
| Ticarcillin-clavulanate | 400 † | 4 | ||
| Piperacillin-tazobactam | 240-400 ‡ | 3 | ||
| Meropenem | 60-120 | 3 | ||
| Imipenem-cilastatin | 45-100 | 3-4 | ||
| Ceftazidime | 150 | 3 | ||
| Aztreonam | 150-200 | 4 | ||
| B. cepacia | Chloramphenicol | 50-100 | 4 | |
| Meropenem | 60-120 | 3 | ||
| Aerosol | Tobramycin (inhaled) | 300 § | 2 | |
| Aztreonam (inhaled) | 75 | 3 |
* Quantity of trimethoprim.
† Quantity of ticarcillin.
‡ Quantity of piperacillin.
§ In mg per dose.
Aerosolized Antibiotic Therapy
Aerosolized antibiotics are often used as part of daily therapy when the airways are infected with P. aeruginosa . Aerosolized tobramycin inhalation solution or powder, or aztreonam inhalation solution used as a suppressive therapy (on 1 mo, off 1 mo), may reduce symptoms, improve pulmonary function, and decrease the occurrence of pulmonary exacerbations. Although these therapies are sometimes used in acute pulmonary exacerbations, the evidence to support this application is limited.
Another important indication for aerosolized antibiotic therapy is to eradicate P. aeruginosa in the airways after initial detection. Early infection may be cleared for mo to several yr in this way, although eventual reinfection is common. Other antibiotics have been used via inhalation, including liposomal amikacin and levofloxacin for P. aeruginosa , and there was no inferiority of efficacy compared with inhaled tobramycin.
Intravenous Antibiotic Therapy
For the patient who has not responded to oral antibiotics and intensive home measures with return of signs, symptoms, and FEV1 to baseline, intravenous antibiotic therapy is indicated. This therapy is usually initiated in the hospital but is sometimes completed on an ambulatory basis if the likelihood of complete adherence to the therapeutic regimen is good. The ideal duration of treatment is unknown; although many patients show improvement within 7 days, many CF physicians believe that it is usually advisable to extend the period of treatment to at least 14 days. Permanent intravenous access can be provided for long-term or frequent courses of therapy in the hospital or at home. Thrombophilia screening should be considered before the use of totally implantable intravenous devices or for recurring problems with venous catheters.
Table 432.6 lists commonly used intravenous antibiotics. In general, treatment of Pseudomonas infection is thought to require 2-drug therapy. A 3rd agent may be given for optimal coverage of S. aureus or other organisms. Aminoglycosides are usually effective when given every 24 hr to minimize toxicity and optimize convenience. Some CF physicians use peak and trough levels to guide dosing, but most clinical pharmacists recommend measuring levels at other times, commonly 2 and 12 hr, to use pharmacokinetic calculations to guide dosing. Changes in therapy should be guided by lack of improvement more than by culture results; sensitivities do not always predict response to therapy, and this may be due to the presence of other organisms that are not detected by culture methods. If patients do not show improvement, complications such as right heart failure, asthma, or infection with viruses, A. fumigatus (especially ABPA) (see Chapter 237 ), nontuberculous mycobacteria (see Chapters 217 and 399 ), or other unusual organisms should be considered. B. cepacia complex and acinetobacter are Gram-negative rods that may be particularly refractory to antimicrobial therapy. Infection control in both the outpatient and inpatient medical setting is critically important to prevent nosocomial spread of resistant bacterial organisms between patients.
Bronchodilator Therapy
Reversible airway obstruction occurs in many children with CF, sometimes in conjunction with frank asthma or allergic bronchopulmonary aspergillosis. Reversible obstruction is conventionally defined as improvement of ≥12% in FEV1 or FVC after inhalation of a bronchodilator. In many patients with CF, these may improve by only 5–10% (physiologic response), but subjects may report subjective benefit.
Antiinflammatory Agents
Corticosteroids are useful for the treatment of allergic bronchopulmonary aspergillosis and severe asthma occasionally encountered in children with CF. Prolonged systemic corticosteroid treatment of CF lung disease reduces the decline in lung function modestly but causes predictably prohibitive side effects. Inhaled corticosteroids have theoretical appeal, but there are contradictory and weak data regarding efficacy unless the patient has clinically diagnosable asthma. Ibuprofen, given chronically in high doses adjusted to achieve a peak serum concentration of 50-100 µg/mL, is associated with a slowing of disease progression, particularly in younger patients with mild lung disease. However, there are concerns regarding side effects of nonsteroidal antiinflammatory drugs, so this therapy has not gained broad acceptance. Macrolide antibiotics have an antiinflammatory effect, and 3 days/wk azithromycin has been shown to reduce the likelihood of development of pulmonary exacerbations, especially in patients with chronic Pseudomonas airway infection, so this is a commonly used therapy.
Cystic Fibrosis Transmembrane Conductance Regulator Modulator Therapies
A major breakthrough in CF therapy is ivacaftor, a small molecule potentiator of the CFTR mutation, G551D (present in ∼5% of patients). Ivacaftor activates the CFTR-G551D mutant protein, a class III CFTR mutation that results in protein localized to the plasma membrane but loss of chloride channel function (Fig. 432.11 ). Ivacaftor therapy resulted in improvement in FEV1 by an average of 10.6%, decreased the frequency of pulmonary exacerbations by 55%, decreased sweat chloride by an average of 48 mEq/L, and increased weight gain by an average of 2.7 kg. Ivacaftor is approved for patients older than 2 yr of age with class III and class IV mutations.
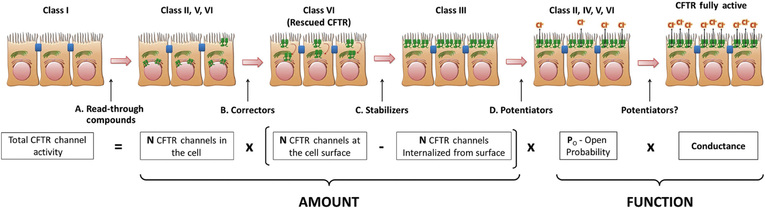
The combination of ivacaftor with lumacaftor, a corrector that stabilizes misfolded F508del and enables trafficking of the mutant molecule to the apical cell membrane where it is potentiated by ivacaftor, is available for patients older than 6 yr of age who are homozygous for the F508del mutation (see Fig. 432.11 ). This medication is associated with smaller increments in pulmonary and nutritional outcomes but is an important proof-of-concept treatment.
Tezacaftor and ivacaftor is another combination indicated for patients ≥ 12 yr with 1 or 2 Phe508del alleles. This combination improves predicted FEV1 and overall well-being (Table 432.7 ). VX-445 combined with tezacaftor-ivacaftor adds another CFTR correction agent; the triple combination improves predicted FEV1 and reduces sweat chloride levels.
Table 432.7
Cystic Fibrosis Transmembrane Regulator Modulators for Cystic Fibrosis
| DRUG | FDA-APPROVED INDICATION | FORMULATIONS | USUAL DOSAGE |
|---|---|---|---|
| Ivacaftor | ≥ 12 mo with a responsive mutation 1 | 150 mg tabs; 50, 75 mg granule packets 2 | ≥ 6 years: 150 mg q12 hr 3 |
| Lumacaftor/ivacaftor | ≥ 2 yr, F508del-homozygous | 100/125, 200/125 mg tabs; 100/125, 150/188 mg granule packets 2 |
6-11 yr: 200/250 mg q12 hr ≥ 12 yr: 400/250 mg q12 hr 4 |
| Tezacaftor/ivacaftor | ≥ 12 yr, F508del-homozygous or F508del-heterozygous with another responsive mutation 1 | 100/150 mg tabs co-packaged with ivacaftor 150 mg tabs | ≥ 12 yr: 100/150 mg tab qAM, then 150 mg ivacaftor qPM |
1 Responsive mutations are those in which chloride transport is expected to increase to at least 10% of untreated normal over baseline with drug therapy, based on clinical or in vitro data.
2 The granules should be mixed with 5 mL of room-temperature or cold soft food or liquid and consumed within 1 hr.
3 In patients 12 mo to 6 yr old, the recommended dosage is 50 mg every 12 hr for those weighing <14 kg, and 75 mg every 12 hr for those weighing ≥ 14 kg.
4 In patients 2-5 yr old, the recommended dosage is 100/125 mg every 12 hr for those weighing <14 kg, and 150/188 mg every 12 hr for those weighing ≥ 14 kg.
Modified from The Medical Letter on Drugs and Therapeutics: Tezacaftor/Ivacaftor (Symdeko) for cystic fibrosis; Med Lett 60(1558):174–176, 2018 (Table 3, p. 175).
Other Therapies
Attempts to clear recalcitrant atelectasis and airway plugging with bronchopulmonary lavage and direct installation of various medications are sometimes used in exceptional cases; there is no evidence for sustained benefit from repeated procedures. Expectorants such as iodides and guaifenesin do not effectively assist with the removal of secretions from the respiratory tract. Inspiratory muscle training can enhance maximum oxygen consumption during exercise, as well as FEV1 .
Treatment of Pulmonary Complications
Atelectasis
Lobar atelectasis occurs relatively infrequently; it may be asymptomatic and noted only at the time of a routine chest radiograph. Aggressive intravenous therapy with antibiotics and increased chest PT directed at the affected lobe may be effective. If there is no improvement in 5-7 days, bronchoscopic examination of the airways may be indicated. If the atelectasis does not resolve, continued intensive home therapy is indicated because atelectasis may resolve during a period of wk or mo.
Hemoptysis
Endobronchial bleeding usually reflects airway wall erosion into hypertrophied bronchial vessels secondary to infection. Although more common in patients with advanced disease, it is sometimes seen in adolescents with relatively mild lung disease. Blood streaking of sputum is particularly common. Small-volume hemoptysis (<20 mL) is usually viewed as a need for intensified antimicrobial therapy and chest PT. Massive hemoptysis, defined as total blood loss of ≥250 mL in a 24-hr period, is rare in the 1st decade and occurs in <1% of adolescents, but it requires close monitoring and the capability to replace blood losses rapidly. Bronchoscopy rarely reveals the site of bleeding. Bronchial artery embolization can be useful to control persistent, significant hemoptysis.
Pneumothorax
Pneumothorax (see Chapter 439 ) is encountered uncommonly in children and teenagers with CF, although it may lead to significant compromise in lung function and occasionally may be life threatening. The episode may be asymptomatic but is often attended by chest and shoulder pain, shortness of breath, or hemoptysis. A small air collection that does not grow can be observed closely. Chest tube placement with or without pleurodesis is often the initial therapy. Intravenous antibiotics are also begun on admission. Video-assisted thoracoscopic surgery (VATS) with plication of blebs, apical pleural stripping, and basal pleural abrasion should be considered if the air leak persists. Surgical intervention is usually well tolerated even in cases of advanced lung disease. The thoracotomy tube is removed as soon as possible. Previous pneumothorax with or without pleurodesis is not a contraindication to subsequent lung transplantation.
Allergic Bronchopulmonary Aspergillosis
Allergic bronchopulmonary aspergillosis occurs in 5–10% of patients with CF and may manifest as wheezing, increased cough, shortness of breath, and marked hyperinflation, or most commonly, a decrease in FEV1 that does not respond to antibiotic therapy (see Chapters 237 and 399 ). In some patients, a chest radiograph shows new, focal infiltrates. A very elevated total serum immunoglobulin E (IgE) level (>1,000) is usually the initial indication of the diagnosis. The presence of rust-colored sputum, the recovery of Aspergillus organisms from the sputum, a positive skin test for A. fumigatus , the demonstration of specific IgE and IgG antibodies against A. fumigatus, or the presence of eosinophils in a fresh sputum sample supports the diagnosis. Treatment is directed at controlling the inflammatory reaction with oral corticosteroids. Oral antifungals are usually reserved for patients who relapse after initial steroid treatment. For refractory cases, omalizumab, humanized monoclonal anti-IgE, has been effective.
Nontuberculous Mycobacteria Infection
See Chapter 244 .
Injured airways with poor clearance may be colonized by Mycobacterium avium- complex but also Mycobacterium abscessus , Mycobacterium chelonae , and Mycobacterium kansasii. Distinguishing endobronchial colonization (frequent) from invasive infection (infrequent) is challenging. Persistent fevers and new infiltrates or cystic lesions coupled with the finding of acid-fast organisms on sputum smear suggest infection. Infection with these organisms, or at least its recognition, had become increasing common. Treatment is prolonged and requires multiple antimicrobial agents. Symptoms may improve, but the nontuberculous mycobacteria are not usually cleared from the lungs.
Bone and Joint Complications
Hypertrophic osteoarthropathy causes elevation of the periosteum over the distal portions of long bones and bone pain, overlying edema, and joint effusions. Acetaminophen or ibuprofen may provide relief. Control of lung infection usually reduces symptoms. Intermittent arthropathy unrelated to other rheumatologic disorders occurs occasionally, has no recognized pathogenesis, and usually responds to nonsteroidal antiinflammatory agents. Back pain or rib fractures from vigorous coughing may require pain management to permit adequate airway clearance. These and other fractures may stem from diminished bone mineralization, the result of reduced vitamin D absorption, corticosteroid therapy, diminished weight-bearing exercises, and perhaps other factors. There may be a bone phenotype in CF that is unrelated to therapies or nutritional status and may be due to CFTR dysfunction.
Sleep-Disordered Breathing
Particularly with advanced pulmonary disease and during chest exacerbations, individuals with CF may experience more sleep arousals, less time in rapid eye movement sleep, nocturnal hypoxemia, hypercapnia, and associated neurobehavioral impairment. Nocturnal hypoxemia may hasten the onset of pulmonary hypertension and right-sided heart failure. Efficacy of specific interventions for this complication of CF has not been systematically assessed. Prompt treatment of airway symptoms and nocturnal oxygen supplementation or bilevel positive airway pressure support should be considered in selected cases, especially in patients with advanced lung disease.
Acute Respiratory Failure
Acute respiratory failure (see Chapter 89 ) rarely occurs in patients with mild to moderate lung disease and is usually the result of a severe viral or other infectious illness. Because patients with this complication can regain their previous status, intensive therapy is indicated. In addition to aerosol, postural drainage, and intravenous antibiotic treatment, oxygen is required to raise the arterial PaO 2 . An increasing PCO 2 may require ventilatory assistance. Endotracheal or bronchoscopic suction may be necessary to clear airway inspissated secretions and can be repeated daily. Right-sided heart failure should be treated vigorously. High-dose steroids have been anecdotally reported to be of benefit in this setting. Recovery is often slow. Intensive intravenous antibiotic therapy and postural drainage should be continued for 1-2 wk after the patient has regained baseline status.
Chronic Respiratory Failure
Patients with CF acquire chronic respiratory failure after prolonged deterioration of lung function. Although this complication can occur at any age, it is seen most frequently in adult patients. Because a long-standing PaO 2 <50 mm Hg promotes the development of right-sided heart failure, patients usually benefit from low-flow oxygen to raise arterial PO 2 to ≥55 mm Hg. Increasing hypercapnia may prevent the use of optimal fraction of inspired oxygen. Most patients improve somewhat with intensive antibiotic and pulmonary therapy measures and can be discharged from the hospital. Low-flow oxygen therapy is needed at home, especially with sleep. Noninvasive ventilatory support can improve gas exchange and has been documented to enhance quality of life. Ventilatory support may be particularly useful for patients awaiting lung transplantation. These patients usually display pulmonary hypertension and cor pulmonale, and this complication should be treated. Caution should be exercised to avoid ventilation-suppressing metabolic alkalosis that results from CF-related chloride depletion and, in many cases, from diuretic-induced bicarbonate retention. Chronic pain (headache, chest pain, abdominal pain, and limb pain) is frequent at the end of life and responds to judicious use of analgesics, including opioids. Dyspnea has been ameliorated with nebulized fentanyl.
Lung transplantation is an option for end-stage lung disease that is increasingly offered (see Chapter 470 ). Criteria for referral continue to be a subject of investigation and ideally include estimates of longevity with and without transplant based on lung function and exercise tolerance data. Survival and quality of life after lung transplantation is better in patients with CF than other chronic lung diseases, probably due to the relatively younger age of recipients with CF, but the current estimated 5-yr survival is about 50%, somewhat reduced compared with that of other solid organ transplants. Because of bronchiolitis obliterans (see Chapter 422.1 ) and other complications, transplanted lungs cannot be expected to function for the lifetime of a recipient, and repeat transplantation is increasingly common. The demand for donor lungs exceeds the supply, and waiting lists and duration of waits continue to be a problem.
Pulmonary Hypertension and Cor Pulmonale
Individuals with long-standing, advanced pulmonary disease, especially those with severe hypoxemia (PaO 2 < 50 mm Hg), often acquire pulmonary hypertension and chronic right-sided heart failure. Evidence for concomitant left ventricular dysfunction is often found. The arterial PO 2 should be maintained at >50 mm Hg, if possible, and hypercarbia corrected with noninvasive ventilation or intubation if necessary. Intensive pulmonary therapy, including intravenous antibiotics, is most important. Adjunctive therapy with salt restriction, diuretics, and pulmonary vasodilators may be indicated. The prognosis for heart failure is poor, but a number of patients survive for ≥5 yr after the appearance of heart failure. Heart-lung transplantation may be an option (see preceding section).
Nutritional Therapy
Up to 90% of patients with CF have loss of exocrine pancreatic function leading to inadequate digestion and absorption of fats and proteins. They require dietary adjustment and augmentation, pancreatic enzyme replacement, and supplementary vitamins. In general, children with CF need to exceed the usual required daily caloric intake to grow. Daily supplements of the fat-soluble vitamins are required.
Diet
Historically, at the time of diagnosis, many infants presented with nutritional deficits; this situation has changed because of newborn screening, but even at 2-4 wk it is not uncommon to see that weight gain has begun to fall off the standard curve.
Most children with CF have a higher-than-normal caloric need because of malabsorption despite the use of pancreatic enzyme supplementation. Encouragement to eat high-calorie foods is important and often begins with more concentrated, high-calorie formulas in the 1st yr. Even so, most mothers can breastfeed successfully. It is vitally important to promote adequate weight gain in the early years, both because of a clear relationship to later lung function and also because early deficiencies make later catch up growth more difficult. Not infrequently, parent–child interactions at feeding time are maladaptive, and behavioral interventions can improve caloric intake. The liberal use of appetite stimulants, especially cyproheptadine, in early childhood, makes the struggle a bit easier. Poorly controlled lung disease increases metabolism and decreases appetite and needs to be considered when efforts to improve weight gain are unsuccessful.
Maintenance of good weight gain and body mass index in the 1st yr of life leads to better long-term preservation of lung function, but there is a strong correlation between body mass index and FEV1 that persists through all ages in people with CF. Better nutrition also leads to improved quality of life and psychologic well-being and provides better reserves when weight loss occurs in association with intermittent acute pulmonary exacerbations.
Malabsorption is an important contributor to nutritional deficiencies, and it is important to ensure that pancreatic enzyme dosing is adequate and consistently being taken correctly with all meals and feedings. Appetite stimulants when cyproheptadine is not successful may include megestrol, oxandrolone, dronabinol, antidepressants such as mirtazaoine, and even growth hormone. CF-related diabetes needs to be ruled out.
When all these therapies fail, weight stabilization or gain can be achieved with nocturnal feeding via nasogastric tube or gastrostomy tube. These are most commonly resorted to in infants and adolescents, the 2 age groups that have the most difficulty with weight gain due to high normal demands.
Pancreatic Enzyme Replacement
Pancreatic exocrine replacement therapy given with ingested food reduces but does not fully correct stool fat and nitrogen losses. Current products are enteric-coated, pH-sensitive enzyme microspheres that come in capsules and given to children before they can swallow by opening the capsule and mixing the beads in small amounts of acidic foods such as applesauce. Strengths ranging from 3 to 40,000 IU of lipase/capsule are available. Administration of excessive doses has been linked to fibrosing colonopathy and colonic strictures, so recommendations are for enzyme dosing to stay below 2,500 lipase units/kg/meal in most circumstances. Snacks should also be covered. Some individuals require proton pump inhibitor therapy to correct acid pH in the duodenum which is due to lack of exocrine pancreatic secretions; neutralization of duodenal pH permits activation of enteric-coated pancreatic exocrine replacement therapy granules.
Vitamin and Mineral Supplements
Because pancreatic insufficiency results in malabsorption of fat-soluble vitamins (A, D, E, K), vitamin supplementation is recommended. Several vitamin preparations containing all 4 vitamins for patients with CF are available. They should be taken daily. Despite this supplementation, vitamin D deficiency is common and should be treated with doses of cholecalciferol (vitamin D3) rather than ergocalciferol (vitamin D2) in the range of 1,000 units/kg/wk. Salt supplementation is also needed during infancy and is started at the time of diagnosis.
Treatment of Intestinal Complications
Meconium Ileus
When meconium ileus (see Chapter 102 ) is suspected, diatrizoate (Gastrografin) enemas with reflux of contrast material into the ileum not only confirm the diagnosis but may also result in the passage of meconium and clearing of the obstruction. Children in whom this procedure fails require operative intervention. Children who have had meconium ileus are at greater risk for nutritional deficiency and more likely to develop problems with DIOS when older. Infants with meconium ileus should be assumed to have CF unless proven otherwise.
Distal Intestinal Obstruction Syndrome and Other Causes of Abdominal Symptoms
Despite appropriate pancreatic enzyme replacement, a number of patients accumulate fecal material in the terminal portion of the ileum and in the cecum, which may result in partial or complete obstruction. For intermittent symptoms, pancreatic enzyme replacement should be continued or even increased, and stool hydrators such as polyethylene glycol (MiraLAX) should be given. If this fails or symptoms are more severe, large-volume bowel lavage with a balanced salt solution containing polyethylene glycol may be taken by mouth or by nasogastric tube. When there is complete obstruction, a diatrizoate enema, accompanied by large amounts of intravenous fluids, can be therapeutic.
Rectal Prolapse
See Chapter 371.5 .
Although uncommon, rectal prolapse occurs most often in infants with CF and less frequently in older children with the disease. It was much more frequently seen in the past among undiagnosed young children with steatorrhea, malnutrition, and repetitive cough. The prolapsed rectum can usually be replaced manually by continuous gentle pressure with the patient in the knee-chest position. Sedation may be helpful. To prevent an immediate recurrence, the buttocks can be temporally taped closed. Adequate pancreatic enzymes, stool softener, and control of pulmonary infection result in improvement. Occasionally, a patient may continue to have rectal prolapse and may require sclerotherapy or surgery.
Hepatobiliary Disease
Liver function abnormalities associated with biliary cirrhosis can be improved by treatment with ursodeoxycholic acid. The ability of bile acids to prevent progression of cirrhosis has not been clearly documented. Portal hypertension with esophageal varices, hypersplenism, or ascites occurs in ≤8% of children with CF (see Chapter 394 ).
Obstructive jaundice in newborns with CF needs no specific therapy once the etiology has been established. End-stage liver disease is an indication for liver transplantation in children with CF (see Chapter 395 ).
Pancreatitis
Recurrent pancreatitis is seen primarily in patients with pancreatic sufficiency, and it can lead to the development of pancreatic insufficiency. Patients can be treated with pancreatic enzyme therapy and a low-fat diet (in well-nourished patients) to rest the pancreas. Further treatment of this disorder is discussed in Chapter 378 .
Cystic Fibrosis-Related Hyperglycemia and Diabetes
Onset of hyperglycemia occurs most frequently after the 1st decade. Approximately 20% of young adults are treated for hyperglycemia, although the incidence of CF-related diabetes may be up to 50% in CF adults. Ketoacidosis is rarely encountered. The pathogenesis includes both impaired insulin secretion and insulin resistance. Routine screening consisting of an annual 2-hr oral glucose tolerance test is recommended in children older than 10 yr of age, although some cases may begin earlier. Glucose intolerance with blood sugars that remain less than 200 is usually not treated unless nutrition is compromised or lung function seems affected. When treatment is indicated, insulin treatment should be instituted. The development of significant hyperglycemia favors acquisition of P. aeruginosa and B. cepacia in the airways and may adversely affect pulmonary function, especially in women. Thus careful control of blood glucose level is an important goal. Long-term vascular complications of diabetes can occur, providing an additional rationale for good control of blood glucose levels.
Other Complications
Nasal Polyps
Nasal polyps (see Chapter 406 ) occur in 15–20% of patients with CF and are most prevalent in the 2nd decade of life. Local corticosteroids and nasal decongestants occasionally provide some relief. When the polyps completely obstruct the nasal airway, rhinorrhea becomes constant or widening of the nasal bridge is noticed, surgical removal of the polyps is indicated; polyps may recur promptly or after a symptom-free interval of months to years. Polyps inexplicably stop developing in many adults.
Rhinosinusitis
Opacification of paranasal sinuses is universal in CF and is not an indication for intervention. Acute or chronic sinus-related symptoms are treated initially with antimicrobials, with or without maxillary sinus aspiration for culture. Functional endoscopic sinus surgery has anecdotally provided benefit.
Salt Depletion
Salt losses from sweat in patients with CF can be high, especially in warm arid climates. Children should have free access to salt, especially when thirsty in hot weather. Salt supplements are often prescribed to newborns and to children who live in hot weather climates. Hypochloremic alkalosis should be suspected in any patient who feels unwell in hot weather or who has had symptoms of gastroenteritis, and prompt fluid and electrolyte therapy should be instituted as needed.
Surgery
Patients with good or excellent pulmonary status can tolerate general anesthesia without any intensive pulmonary measures before the procedure but should be adherent to their usual prescribed airway clearance therapy. Those with moderate or severe pulmonary infection usually do better with a 1- to 2-wk course of intensive antibiotic treatment and increased airway clearance before surgery. If this approach is impossible, prompt intravenous antibiotic therapy is indicated once it is recognized that major surgery is required. General anesthesia may provide an opportunity to perform bronchoscopy to evaluate the airway and obtain good cultures, and this should be considered in any child with CF who will undergo surgery for any indication.
After major surgery, cough should be encouraged, and airway clearance treatments should be reinstituted as soon as possible, usually within 24 hr.
Prognosis
CF remains a life-limiting disorder, although survival has dramatically improved (Fig. 432.12 ). With exceptions, most children remain relatively healthy into adolescence or adulthood. The slow progression of lung disease eventually does reach disabling proportions. Life table data indicate a median cumulative survival of more than 40 yr, and the expectation is younger children with the disease have a life expectancy far in excess of this estimate. Outcomes are variable and related to CFTR mutation class, modifier genes, biologic and chemical exposures, disease management, and socioeconomic status.
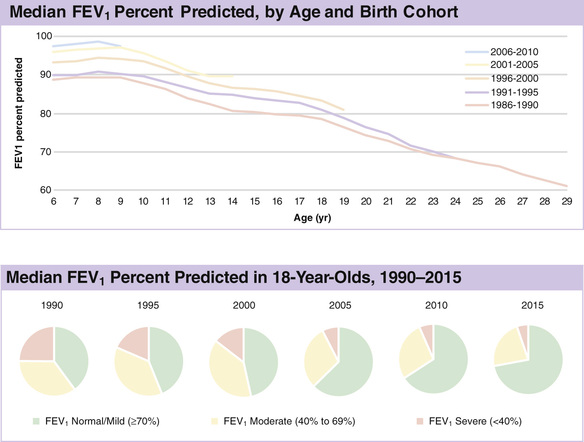
Children with CF should not be restricted in their activities. A high percentage eventually attend and graduate from college. Most adults with CF find satisfactory employment, and an increasing number marry. Transitioning care from pediatric to adult care centers by 21 yr of age is an important objective and requires a thoughtful, supportive approach involving both the pediatric and internal medicine specialists.
With increasing life span for patients with CF, a new set of psychosocial considerations has emerged, including dependence-independence issues, self-care, peer relationships, sexuality, reproduction, substance abuse, educational and vocational planning, medical care costs and other financial burdens, and anxiety concerning health and prognosis. Anxiety and depression are prevalent, as in any other chronic disease, and impact quality of life and disease self-management; screening for both is now part of comprehensive care. Many of these issues are best addressed in an anticipatory fashion, before the onset of psychosocial dysfunction. With appropriate medical and psychosocial support, children and adolescents with CF generally cope well. Achievement of an independent and productive adulthood is a realistic goal for many.