Other Distal Airway Diseases
Bronchiolitis Obliterans
Steven R. Boas
Epidemiology
Bronchiolitis obliterans (BO) is a histopathologic diagnosis characterized by chronic obstructive lung disease of the bronchioles and smaller airways, resulting from an insult to the lower respiratory tract leading to inflammation and fibrosis of the small airways. In the nontransplant patient, BO most commonly occurs in the pediatric population after respiratory infections, particularly adenovirus (see Chapter 289 ), but also Mycoplasma pneumoniae (see Chapter 250 ), measles (see Chapter 273 ), Legionella pneumophila (see Chapter 235 ), influenza (see Chapter 285 ), and pertussis (see Chapter 224 ); other causes include inflammatory diseases (juvenile idiopathic arthritis, systemic lupus erythematosus [see Chapter 183 ], scleroderma [see Chapter 185 ], Stevens-Johnson syndrome [see Chapter 177 ]), and inhalation of toxic fumes or particulate exposure (NO2 , incinerator fly ash, NH3 , diacetyl flavorings from microwave popcorn, papaverine, fiberglass) (Table 422.1 ). Postinfection obliterans may be more common in the southern hemisphere and among persons of Asian descent. BO is also commonly seen in post lung or bone marrow transplant recipients.
Table 422.1
Etiology of Bronchiolitis Obliterans
| POSTINFECTION |
| POSTTRANSPLANTATION |
| CONNECTIVE TISSUE DISEASE |
| TOXIC FUME INHALATION |
| CHRONIC HYPERSENSITIVITY PNEUMONITIS |
| ASPIRATION |
| DRUGS |
| STEVENS-JOHNSON SYNDROME |
From Moonnumakal SP, Fan LL: Bronchiolitis obliterans in children, Curr Opin Pediatr 20:272–278, 2008.
Bronchiolitis obliterans syndrome (BOS) is a clinical diagnosis related to graft deterioration after transplantation defined as a progressive decline in lung function based on FEV1. The airway obstruction is generally irreversible. BOS is considered once other causes of airway obstruction are excluded. BOS is recognized as a long-term complication of both lung and bone marrow transplantation with more than one third of survivors of lung transplantation developing this disorder. Risk factors for the development of BOS include the presence of CMV pneumonitis, aspergillus colonization, primary graft dysfunction, gastroesophageal reflux, and community-acquired respiratory viruses, as well as prolonged transplantation ischemic time.
Pathogenesis
After the initial insult, inflammation affecting terminal bronchioles, respiratory bronchioles, and alveolar ducts can result in the obliteration of the airway lumen (Fig. 422.1 ). Epithelial damage resulting in abnormal repair is characteristic of BO. Complete or partial obstruction of the airway lumen can result in air trapping or atelectasis. Parenchymal involvement is not seen. Bronchiolitis obliterans organizing pneumonia (BOOP) or what has also been termed cryptogenic organizing pneumonia is a histopathologic diagnosis. Although it is similar to many of the histologic features of BO, BOOP is also characterized by extension of the inflammatory process from distal alveolar ducts into alveoli with proliferation of fibroblasts (parenchymal involvement).

Clinical Manifestations and Diagnosis
Cough, fever, cyanosis, dyspnea, chest pain, and respiratory distress followed by initial improvement may be the initial signs of BO. In this phase, BO is easily confused with pneumonia, bronchitis, or bronchiolitis. Progression of the disease can ensue, with increasing dyspnea, chronic cough, sputum production, and wheezing. Physical examination findings are usually nonspecific and can include wheezing, hypoxemia, and crackles. Chest radiographs may be relatively normal compared with the extent of physical findings but can demonstrate hyperlucency and patchy infiltrates. Occasionally, a Swyer-James syndrome (unilateral hyperlucent lung; see Chapter 420 ) develops. Pulmonary function tests demonstrate variable findings but typically show signs of airway obstruction with a variable degree of bronchodilator response although more commonly irreversible. Exercise testing shows reduced exercise capacity and impaired oxygen consumption. Ventilation-perfusion scans reveal a typical moth-eaten appearance of multiple matched defects in ventilation and perfusion. High-resolution chest CT often demonstrates patchy areas or a mosaic pattern of hyperlucency, air trapping, and bronchiectasis (Fig. 422.2 ). Table 422.2 provides an overview of CT findings of BO and related disorders. Physical and radiologic signs can wax and wane over weeks or months. Open lung biopsy or transbronchial biopsy remains the best means of establishing the diagnosis of BO or BOOP.
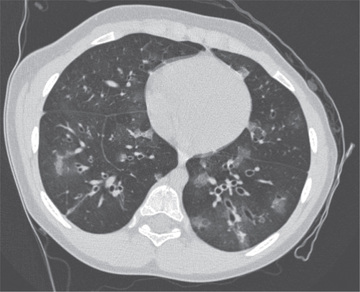
Table 422.2
High-Resolution CT Patterns in Child With Interstitial Lung Disease
| STUDIES (N) | GROUND-GLASS OPACITY | THICK SEPTA | NODULES | MOSAIC PATTERN | HONEYCOMBING | |
|---|---|---|---|---|---|---|
| Bronchiolitis obliterans | 4 | — | — | — | X | — |
| Nonspecific interstitial pneumonitis | 6 | X | — | — | — | X |
| Desquamative interstitial pneumonitis | 4 | X | — | — | — | X |
| Follicular bronchitis or neuroendocrine cell hyperplasia of infancy | 4 | X | — | — | X | — |
| Lymphocytic interstitial pneumonitis | 4 | — | — | X | — | — |
| Lymphangiomatosis | 2 | — | X | — | — | — |
| Lymphangiectasia | 2 | — | X | — | — | — |
| Pulmonary alveolar proteinosis | 2 | X | X | — | — | — |
From Long FR: Interstitial lung disease. In Slovis TL, editor: Caffey's pediatric diagnostic imaging , ed 11, Philadelphia, 2008, Mosby, Table 74.1; original data from Lynch DA, Hay T, Newell JD Jr, et al: Pediatric diffuse lung disease: diagnosis and classification using high-resolution CT, AJR Am J Roentgenol 173:713–718, 1999, and Copley SJ, Coren M, Nicholson AG, et al: Diagnostic accuracy of thin-section CT and chest radiography of pediatric interstitial lung disease, AJR Am J Roentgenol 174:549–554, 2000.
Treatment
No definitive therapy exists for BO. Administration of corticosteroids may be beneficial. Immunomodulatory agents, such as sirolimus, tacrolimus, aerosolized cyclosporine, hydroxychloroquine, and macrolide antibiotics, have been used in post–lung transplantation recipients with BO with variable success. Supportive measures with oxygen, antibiotics for secondary infections, and bronchodilators are adjunct therapies. The role of gastroesophageal reflux and its association with BO has been raised, with treatment suggested whenever the diagnosis is made. Azithromycin may be effective in patients with BOS. For BOOP, use of oral corticosteroids for up to 1 yr has been advocated as first-line therapy for symptomatic and progressive disease. Patients with asymptomatic or nonprogressive BOOP can be observed.
Prognosis
Some patients with BO experience rapid deterioration in their condition and die within weeks of the initial symptoms; most nontransplant patients survive with chronic disability. BO tends to be severe once progression ensues. In contrast to BO, a better prognosis exists for patients with BOOP, with complete recovery seen in many patients, although outcome depends on the underlying systemic disease. BOOP can relapse, especially if treatment duration is <1 yr; BOOP is amenable to repeat courses of oral corticosteroids. Unlike the more common idiopathic BOOP, progressive BOOP characterized by acute respiratory distress syndrome is rare but is aggressive in its clinical course, leading to death.
Bibliography
Aguilar PR, Michelson AP, Isakow W. Obliterative bronchiolitis. Transplantation . 2016;100:272–283.
Barker AF, Bergeron A, Rom WN, et al. Obliterative bronchiolitis. N Engl J Med . 2014;370:1820–1828.
Champs NS, Lasmar LM, Camargos PA, et al. Post-infectious bronchiolitis obliterans in children. J Pediatr (Rio J) . 2011;87(3):187–198.
Epler GR. Bronchiolitis obliterans organizing pneumonia. Arch Intern Med . 2001;161:158–164.
Fischer GB, Sarria EE, Mattiello R, et al. Post infectious bronchiolitis obliterans in children. Paediatr Respir Rev . 2010;11:233–239.
Fullmer JJ, Fan LL, Dishop MK, et al. Successful treatment of bronchiolitis obliterans in a bone marrow transplant patient with tumor necrosis factor-α blockade. Pediatrics . 2005;116:767–770.
Kurland G, Michelson P. Bronchiolitis obliterans in children. Pediatr Pulmonol . 2005;39:193–208.
Moonnumakal SP, Fan LL. Bronchiolitis obliterans in children. Curr Opin Pediatr . 2008;20:272–278.
Todd JL, Palmer SM. Bronchiolitis obliterans syndrome. Chest . 2011;140:502–508.
Weber DJ, Wilkes DS. The role of autoimmunity in obliterative bronchiolitis after lung transplantation. Am J Physiol Lung Cell Mol Physiol . 2013;304(5):L307–L311.
Follicular Bronchitis
Steven R. Boas
Follicular bronchitis is a lymphoproliferative lung disorder characterized by the presence of lymphoid follicles alongside the airways (bronchi or bronchioles) and infiltration of the walls of bronchi and bronchioles. Although the cause is unknown, an infectious etiology (viral, L. pneumophila; see Chapter 235 ) has been proposed. This disorder has been reported following lung transplant and in an HIV-positive child. It can occur in adults and children; in children, onset of symptoms generally occurs by 6 wk of age and peaks between 6 and 18 mo. Cough, moderate respiratory distress, fever, and fine crackles are common clinical findings. Fine crackles generally persist over time, and recurrence of symptoms is common. Chest radiographs may be relatively benign initially (air trapping, peribronchial thickening) but evolve into the typical interstitial pattern. Chest CT can show a fine reticular pattern, as well as bronchiectasis and centrilobular branching, but can also appear normal (see Table 422.2 ). Definitive diagnosis is made by open lung biopsy (Fig. 422.3 ). Treatment is limited, although some patients with follicular bronchitis respond to corticosteroid therapy. Prognosis is variable, with some patients having significant progression of pulmonary disease and others developing only mild obstructive airway disease. In children, it is generally associated with immunodeficiency; the differential diagnosis includes the pulmonary complications of HIV infection (see Chapter 302 ).
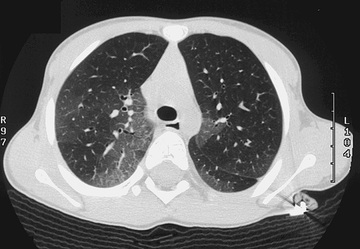
Pulmonary Alveolar Microlithiasis
Steven R. Boas
Pulmonary alveolar microlithiasis (PAM) is a rare disease characterized by the formation of lamellar concretions of calcium phosphate or “microliths” within the alveoli, creating a classic pattern on the radiograph (Fig. 422.4 ).

Epidemiology and Etiology
Although the mean age at time of diagnosis is in the mid-30s, the onset of the disease can occur in childhood and in newborns. PAM is inherited in an autosomal recessive disorder and is caused by a mutation in the type II sodium phosphate cotransporter NPT2b (SCL34A2). There are more than 15 mutations. This gene is expressed in high levels in the lungs, predominantly on the surface of alveolar type II cells. Although the precise role of this protein is unknown, it is speculated that it helps to remove phosphate generated from surfactant metabolism in the alveolar space, as well as functioning as a phosphate regulator in other organs.
In some families, progression of disease is rapid. An equal male and female incidence is noted. Although PAM is found throughout the world, there is a high incidence in Turkey and a lesser incidence in Italy, Japan, and India.
Clinical Manifestations
In early stages of the disease, patients are usually asymptomatic. When symptomatic, patients with PAM usually complain of dyspnea on exertion and nonproductive cough. Physical examination of the lungs can reveal fine inspiratory crackles and diminished breath sounds. Clubbing occurs, although this is usually a more advanced sign. Discordance between the clinical and radiographic manifestations is common. Many children are often asymptomatic on initial presentation and present with symptoms during adulthood. Complications of pneumothorax, pleural adhesions and calcifications, pleural fibrosis, apical bullae, and extrapulmonary sites of microliths have been reported (kidneys, prostate, sympathetic chain, and testes).
Diagnosis
Chest radiography typically reveals bilateral infiltrates with a fine micronodular appearance or sandstorm appearance with greater density in the lower and middle lung fields (see Fig. 422.4 ). CT of the chest shows diffuse micronodular calcified densities, with thickening of the microliths along the septa and around distal bronchioles, especially in the inferior and posterior regions (see Table 422.2 ). Diffuse uptake of technetium-99 methylene diphosphonate by nuclear scan has been reported. Open lung and transbronchial lung biopsy reveal 0.1- to 0.3-mm laminated calcific concretions within the alveoli. Although the alveoli are often normal initially, progression to pulmonary fibrosis with advancing disease usually ensues. Sputum expectoration might reveal small microliths, although this finding is not diagnostic for PAM and is not typically seen in children. Detection of calcium deposits in bronchoalveolar lavage (BAL) fluid on bronchoscopy supports the diagnosis. Pulmonary function testing reveals restrictive lung disease with impaired diffusing capacity as the disease progresses, whereas exercise testing demonstrates arterial oxygen desaturation. The diagnosis can usually be established radiographically. However, lung tissue biopsy, BAL, and detection of a mutation in the SCL34A2 gene can also be used to help confirm the diagnosis. The differential diagnosis includes sarcoidosis, miliary tuberculosis, hemosiderosis, healed disseminated histoplasmosis, pulmonary calcinosis, and metastatic pulmonary calcifications.
Treatment
No specific treatment is effective, although some clinicians have used glucocorticosteroids, etidronate disodium, and bronchopulmonary lavage with limited success. Lung transplantation has been performed for this condition without recurrence in the transplanted lung.
Prognosis
Progressive cardiopulmonary disease can ensue, leading to cor pulmonale, superimposed infections, and subsequent death in mid-adulthood. Because of the familial nature of this disease, counseling and chest radiographs of family members are indicated.
Bibliography
Castellana G, Castellana G, Gentile M, et al. Pulmonary alveolar microlithiasis: review of the 1022 cases reported worldwide. Eur Respir Rev . 2015;24(138):607–620.
Corut A, Senyigit A, et al. Mutations in SLC34A2 cause pulmonary alveolar microlithiasis and are possible associated with testicular microlithiasis. Am J Hum Genet . 2006;79:650–656.
Saito A, McCormick FX. Pulmonary alveolar microlithiasis. Clin Chest Med . 2016;37:441–448.