Renal Tubular Acidosis
Bradley P. Dixon
Renal tubular acidosis (RTA) is a disease state characterized by a non–anion gap (hyperchloremic) metabolic acidosis in the setting of a normal or near-normal glomerular filtration rate. There are four main types: proximal (type II) RTA, classic distal (type I) RTA, hyperkalemic (type IV) RTA, and a combined proximal and distal (type III). Proximal RTA results from impaired bicarbonate reabsorption and distal RTA from failure to secrete acid. Either of these defects may be inherited and persistent from birth or acquired, as is seen more commonly in clinical practice.
Normal Urinary Acidification
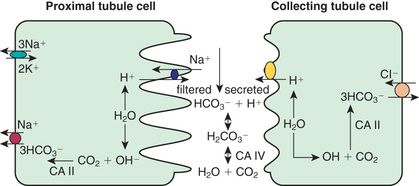
Kidneys contribute to the acid–base balance by reabsorption of filtered bicarbonate (HCO3 − ) and excretion of hydrogen ion (H+ ) produced every day. Hydrogen ion secretion from tubule cells into the lumen is key in the reabsorption of HCO3 − and the formation of titratable acid (H+ bound to buffers such as HPO4 2− ) and ammonium ions (NH4 + ). Because loss of filtered HCO3 − is equivalent to the addition of H+ to the body, all filtered bicarbonate should be absorbed before dietary H+ can be excreted. Approximately 90% of filtered bicarbonate is absorbed in the proximal tubule and the remaining 10% in the distal segments, mostly the thick ascending limb and outer medullary collecting tubule (Fig. 547.1 ). In the proximal tubule and thick ascending limb of the loop of Henle, H+ from water is secreted by the Na+ -H+ exchanger on the luminal membrane. H+ combines with filtered bicarbonate, resulting in the formation of H2 CO3 , which splits into water and CO2 in the presence of carbonic anhydrase IV. CO2 diffuses freely back into the cell, combines with OH− (from H2 O) to form HCO3 − in the presence of carbonic anhydrase II, and returns to the systemic circulation via an Na+ -HCO3 − cotransporter situated at the basolateral membrane of the cell. In the collecting tubule, H+ is secreted into the lumen by H+ ATPase (adenosine triphosphatase) and HCO3 − is returned to the systemic circulation by the HCO3 – -Cl− exchanger located on the basolateral membrane. The H+ secreted proximally and distally in excess of the filtered HCO3 − is excreted in the urine either as titratable acid (H2 PO4 − ) or as NH4 + .
Proximal (Type II) Renal Tubular Acidosis
Bradley P. Dixon
Pathogenesis
Proximal RTA can be inherited and persistent from birth or occur as a transient phenomenon during infancy. Although rare, it may be primary and isolated. Proximal RTA usually occurs as a component of global proximal tubular dysfunction or Fanconi syndrome, which is characterized by low-molecular-weight proteinuria, glycosuria, phosphaturia, aminoaciduria, and proximal RTA. Table 547.1 outlines the causes of proximal RTA (pRTA) and Fanconi syndrome. Many of these causes are inherited disorders. In addition to cystinosis and Lowe syndrome, autosomal recessive and dominant pRTA are addressed further in this section. Other inherited forms of Fanconi syndrome include galactosemia (see Chapter 105.2 ), hereditary fructose intolerance (see Chapter 105.3 ), tyrosinemia (see Chapter 103.2 ), and Wilson disease (see Chapter 384.2 ). Dent disease, or X-linked nephrolithiasis, is discussed in Chapter 549.3 . In children, an important form of secondary Fanconi syndrome is exposure to medications such as ifosfamide, a component of many treatment regimens for Wilms tumor and other malignancies.
Table 547.1
Disorders With Dysfunction of Renal Acidification—Defective HCO3 − Reclamation: Proximal Renal Tubular Acidosis
From DuBose TD Jr: Disorders of acid-base balance. In Skorecki K, Chertow GM, Marsden PA, et al (eds): Brenner & Rector's the kidney, 10/e, Philadelphia, 2016, Elsevier, 2016, Table17-7.
Autosomal Recessive Disease
Isolated autosomal recessive pRTA is caused by mutations in the gene encoding the sodium bicarbonate cotransporter NBC1. It manifests with ocular abnormalities (band keratopathy, cataracts, and glaucoma, often leading to blindness), short stature, enamel defects of the teeth, intellectual impairment, and occasionally basal ganglia calcification along with pRTA. An autosomal dominant pattern of inheritance has been identified in a single pedigree with nine members presenting with hyperchloremic metabolic acidosis, a normal ability to acidify urine, normal renal function, and growth retardation.
Cystinosis
Cystinosis is a systemic disease caused by a defect in the metabolism of cysteine that results in accumulation of cystine (an oxidized form of cysteine in which two cysteine molecules are joined together by their sulfhydryl groups through a disulfide bond) crystals in most of the major organs of the body, notably the kidney, liver, eye, and brain. It occurs at an incidence of 1 : 100,000 to 1 : 200,000. In certain populations, such as French Canadians, the incidence is much higher. At least three clinical patterns have been described. Young children with the most severe form of the disease (infantile or nephropathic cystinosis) present in the 1st or 2nd year of life with severe tubular dysfunction and growth failure. If the disease is not treated, the children develop end-stage renal disease by the end of their 1st decade. A milder form of the disease manifests in adolescents and is characterized by less-severe tubular abnormalities and a slower progression to renal failure. A benign adult form with ocular involvement but no renal involvement also exists.
Cystinosis is caused by mutations in the CTNS gene, which encodes the protein cystinosin. Cystinosin is thought to be an H+ -driven lysosomal cystine transporter. Genotype–phenotype studies demonstrate that patients with severe nephropathic cystinosis carry mutations that lead to complete loss of cystinosin function. Patients with milder clinical disease have mutations that lead to the expression of partially functional protein. Patients with nephropathic cystinosis present with clinical manifestations reflecting their pronounced tubular dysfunction and Fanconi syndrome, including polyuria and polydipsia, growth failure, and rickets. Fever, caused by dehydration or diminished sweat production, is common. Patients are typically fair skinned and blond because of diminished pigmentation. Ocular presentations include photophobia, retinopathy, and impaired visual acuity. Patients also can develop hypothyroidism, hepatosplenomegaly, and delayed sexual maturation. With progressive tubulointerstitial fibrosis, renal insufficiency is invariant.
The diagnosis of cystinosis is suggested by the detection of cystine crystals in the cornea and confirmed by measurement of increased leukocyte cystine content. Prenatal testing is available for at-risk families.
Treatment of cystinosis is directed at correcting the metabolic abnormalities associated with Fanconi syndrome or chronic renal failure. In addition, specific therapy is available with cysteamine, which converts cystine to cysteine and a cysteine–cysteamine heterodimer. This facilitates lysosomal transport and decreases tissue cystine. Oral cysteamine does not achieve adequate levels in ocular tissues, so additional therapy with cysteamine eyedrops is required. Early initiation of the drug can prevent or delay deterioration of renal function. Patients with growth failure that does not improve with cysteamine might benefit from treatment with growth hormone. Kidney transplantation is a viable option in patients with renal failure. With prolonged survival, additional complications may become evident, including central nervous system abnormalities, muscle weakness, swallowing dysfunction, and pancreatic insufficiency. It is unclear whether long-term cysteamine therapy will decrease these complications.
Lowe Syndrome
Lowe syndrome (oculocerebrorenal syndrome of Lowe) is a rare X-linked disorder characterized by congenital cataracts, mental retardation, and Fanconi syndrome. The disease is caused by mutations in the OCRL1 gene, which encodes the phosphatidylinositol polyphosphate 5-phosphatase protein. The abnormalities seen in Lowe syndrome are thought to be caused by abnormal transport of vesicles within the Golgi apparatus. Kidneys show nonspecific tubulointerstitial changes. Thickening of glomerular basement membrane and changes in proximal tubule mitochondria are also seen.
Patients with Lowe syndrome typically present in infancy with cataracts, progressive growth failure, hypotonia, and Fanconi syndrome. Significant low-molecular-weight proteinuria is common. Blindness and renal insufficiency often develop. Characteristic behavioral abnormalities are also seen, including tantrums, stubbornness, stereotypy (repetitive behaviors), and obsessions. There is no specific therapy for the renal disease or neurologic deficits. Cataract removal is generally required.
Clinical Manifestations of Proximal Renal Tubular Acidosis and Fanconi Syndrome
Patients with isolated, sporadic, or inherited pRTA present with growth failure in the 1st year of life. Additional symptoms can include polyuria, dehydration (from sodium loss), anorexia, vomiting, constipation, and hypotonia. Patients with primary Fanconi syndrome have additional symptoms, secondary to phosphate wasting, such as rickets. Those with systemic diseases present with additional signs and symptoms specific to their underlying disease. Urinalysis in patients with isolated pRTA is generally unremarkable. The urine pH is acidic (<5.5) because distal acidification mechanisms are intact in these patients. Urinary studies in patients with Fanconi syndrome demonstrate varying degrees of phosphaturia, aminoaciduria, glycosuria, uricosuria, and elevated urinary sodium or potassium. Depending on the nature of the underlying disorder, laboratory evidence of chronic renal insufficiency, including elevated serum creatinine, may be present.
Distal (Type I) Renal Tubular Acidosis
Bradley P. Dixon
Pathogenesis
Distal RTA can be sporadic or inherited. It can also occur as a complication of inherited or acquired diseases of the distal tubules. Primary or secondary causes of distal RTA can result from damaged or impaired functioning of one or more transporters or proteins involved in the acidification process, including the H+ /ATPase, the HCO3 − /Cl− anion exchangers, or the components of the aldosterone pathway. Because of impaired hydrogen ion excretion, the urine pH cannot be reduced to < 5.5, despite the presence of severe metabolic acidosis. Loss of sodium bicarbonate distally, owing to lack of H+ to bind to in the tubular lumen (see Fig. 547.1 ), results in increased chloride absorption and hyperchloremia. Inability to secrete H+ is compensated for by increased K+ secretion distally, leading to hypokalemia. Hypercalciuria is usually present and can lead to nephrocalcinosis or nephrolithiasis. Chronic metabolic acidosis also impairs urinary citrate excretion. Hypocitraturia further increases the risk of calcium deposition in the tubules. Bone disease is common, resulting from mobilization of organic components from bone to serve as buffers to chronic acidosis.
Clinical Manifestations
Distal RTA shares features with those of pRTA, including non–anion gap metabolic acidosis and growth failure; distinguishing features of distal RTA include nephrocalcinosis and hypercalciuria. The phosphate and massive bicarbonate wasting characteristic of pRTA is generally absent. Table 547.2 lists the causes of primary and secondary distal RTA. Although inherited forms are rare, three specific inherited forms of distal RTA have been identified, including an autosomal recessive form associated with sensorineural deafness.
Table 547.2
Disorders With Dysfunction of Renal Acidification—Selective Defect in Net Acid Excretion: Classic Distal Renal Tubular Acidosis
| PRIMARY DISORDERS | |
| Familial | |
| Autosomal dominant | |
| AE1 gene | |
| Autosomal recessive | |
| With deafness (rdRTA1 or ATP6V1B1 gene) | |
| Without deafness (rdRTA2 or ATP6V0A4 ) | |
| Sporadic | |
| ENDEMIC DISORDERS | |
| Northeastern Thailand | |
| DISORDERS SECONDARY TO SYSTEMIC DISORDERS | |
| Autoimmune Diseases | |
| Hyperglobulinemic purpura | Fibrosing alveolitis |
| Cryoglobulinemia | Chronic active hepatitis |
| Sjögren syndrome | Primary biliary cirrhosis |
| Thyroiditis | Polyarteritis nodosa |
| HIV nephropathy | |
| Hypercalciuria and Nephrocalcinosis | |
| Primary hyperparathyroidism | Vitamin D intoxication |
| Hyperthyroidism | Idiopathic hypercalciuria |
| Medullary sponge kidney | Wilson disease |
| Fabry disease | Hereditary fructose intolerance |
| X-linked hypophosphatemia | |
| DRUG- AND TOXIN-INDUCED DISEASE | |
| Amphotericin B | Toluene |
| Cyclamate | Mercury |
| Hepatic cirrhosis | Vanadate |
| Ifosfamide | Lithium |
| Foscarnet | Classic analgesic nephropathy |
| TUBULOINTERSTITIAL DISEASES | |
| Balkan nephropathy | Kidney transplantation |
| Chronic pyelonephritis | Leprosy |
| Obstructive uropathy | Jejunoileal bypass with hyperoxaluria |
| Vesicoureteral reflux | |
| DISORDERS ASSOCIATED WITH GENETICALLY TRANSMITTED DISEASES | |
| Ehlers-Danlos syndrome | Hereditary elliptocytosis |
| Sickle cell anemia | Marfan syndrome |
|
Medullary cystic disease Hereditary sensorineural deafness |
Jejunal bypass with hyperoxaluria |
| Osteopetrosis with carbonic anhydrase II deficiency (mixed proximal and distal RTA type III) | Carnitine palmitoyltransferase deficiency |
From DuBose TD Jr: Disorders of acid-base balance In Skorecki K, Chertow GM, Marsden PA, et al (eds): Brenner & Rector's the kidney, 10/e, Philadelphia, 2016, Elsevier, 2016, Table 17-9.
Medullary sponge kidney is a relatively rare sporadic disorder in children, although not uncommon in adults. It is characterized by cystic dilation of the terminal portions of the collecting ducts as they enter the renal pyramids. On ultrasound studies, patients often have medullary nephrocalcinosis. Although patients with this condition typically maintain normal renal function through adulthood, complications include nephrolithiasis, pyelonephritis, hyposthenuria (inability to concentrate urine), and distal RTA. Associations of medullary sponge kidney with Beckwith-Wiedemann syndrome or hemihypertrophy have been reported.
Hyperkalemic (Type IV) Renal Tubular Acidosis
Bradley P. Dixon
Pathogenesis
Type IV RTA occurs as the result of impaired aldosterone production (hypoaldosteronism) or impaired renal responsiveness to aldosterone (pseudohypoaldosteronism). Acidosis results because aldosterone has a direct effect on the H+ /ATPase responsible for hydrogen secretion. In addition, aldosterone is a potent stimulant for potassium secretion in the collecting tubule; consequently, lack of aldosterone results in hyperkalemia. This further affects the acid–base status by inhibiting ammoniagenesis and, thus, H+ excretion. Aldosterone deficiency typically occurs as a result of adrenal gland disorders such as Addison disease or some forms of congenital adrenal hyperplasia. In children, aldosterone unresponsiveness is a more common cause of type IV RTA. This can occur transiently, during an episode of acute pyelonephritis or acute urinary obstruction, or chronically, particularly in infants and children with a history of obstructive uropathy. The latter patients can have significant hyperkalemia, even in instances when renal function is normal or only mildly impaired. Rare examples of inherited forms of type IV RTA have been identified (Table 547.3 ).
Table 547.3
Disorders With Dysfunction of Renal Acidification—Generalized Abnormality of Distal Nephron With Hyperkalemia
ATPase, adenosine triphosphatase; CCT, cortical collecting tubule; PHA I, PHA II, pseudohypoaldosteronism types 1 and 2.
From DuBose TD Jr: Disorders of acid-base balance. In Skorecki K, Chertow GM, Marsden PA, et al (eds): Brenner & Rector's the kidney, 10/e, Philadelphia, 2016, Elsevier, 2016, Table 17-11.
Clinical Manifestations
Patients with type IV RTA can present with growth failure in the 1st few years of life. Polyuria and dehydration (from salt wasting) are common. Rarely, patients (especially those with pseudohypoaldosteronism type 1) present with life-threatening hyperkalemia. Patients with obstructive uropathies can present acutely with signs and symptoms of pyelonephritis, such as fever, vomiting, and foul-smelling urine. Laboratory tests reveal a hyperkalemic non–anion gap metabolic acidosis. Urine may be alkaline or acidic. Elevated urinary sodium levels with inappropriately low urinary potassium levels reflect the absence of aldosterone effect.
Diagnostic Approach to Renal Tubular Acidosis
The first step in the evaluation of a patient with suspected RTA is to confirm the presence of a normal anion gap metabolic acidosis, identify electrolyte abnormalities, assess renal function, and rule out other causes of bicarbonate loss such as diarrhea (see Chapter 55 ) (Table 547.4 ). Metabolic acidosis associated with diarrheal dehydration is extremely common, and acidosis generally improves with correction of volume depletion. Patients with protracted diarrhea can deplete their total-body bicarbonate stores and can have persistent acidosis despite apparent restoration of volume status. In instances where a patient has a recent history of severe diarrhea, full evaluation for RTA should be delayed for several days to permit adequate time for reconstitution of total-body bicarbonate stores. If acidosis persists beyond a few days in this setting, additional studies are indicated.
Table 547.4
Contrasting Features and Diagnostic Studies in Renal Tubular Acidosis
| FINDING | TYPE OF RENAL TUBULAR ACIDOSIS | ||
|---|---|---|---|
| Proximal | Classic Distal | Generalized Distal Dysfunction | |
| Plasma [K+ ] | Low | Low | High |
| Urine pH with acidosis | <5.5 | >5.5 | <5.5 or >5.5 |
| Urine net charge | Negative | Positive | Positive |
| Fanconi lesion | Present with acquired pRTA | Absent | Absent |
| Fractional bicarbonate excretion | >10–15% during alkali therapy | 2–5% | 5–10% |
| U−BPco2 | Normal | Low | Low |
| Response to therapy | Least responsive | Responsive | Less responsive |
| Associated features | Fanconi syndrome | Nephrocalcinosis/hyperglobulinemia | Renal insufficiency |
ATPase, adenosine triphosphatase; pRTA, proximal renal tubular acidosis; U−BPCO 2 , urine minus blood CO2 pressure.
From DuBose TD Jr: Disorders of acid-base balance. In Skorecki K, Chertow GM, Marsden PA, et al (eds): Brenner & Rector's the kidney, 10/e, Philadelphia, 2016, Elsevier, 2016, Table 17-17.
Serum electrolytes, blood urea nitrogen, calcium, phosphorus, creatinine, and pH should be obtained by venipuncture. Traumatic blood draws (such as heel-stick specimens), small volumes of blood in adult-size specimen collection tubes, or a prolonged specimen transport time at room temperature can lead to falsely low bicarbonate levels, often in association with an elevated serum potassium value. True hyperkalemic acidosis is consistent with type IV RTA, whereas the finding of normal or low potassium suggests type I or II. The blood anion gap should be calculated using the formula [Na+ ] − [Cl− + HCO3 − ]. Values of < 12 demonstrate the absence of an anion gap. Values of > 20 indicate the presence of an anion gap. If such an anion gap is found, then other diagnoses (lactic acidosis, diabetic ketoacidosis, inborn errors of metabolism, ingested toxins) should be investigated. If tachypnea is noted, evaluation of an arterial blood gas may be appropriate to evaluate the possibility of a mixed acid–base disorder primarily involving respiratory and metabolic components. A detailed history, with particular attention to growth and development, recent or recurrent diarrheal illnesses, a family history of mental retardation, failure to thrive, end-stage renal disease, infant deaths, or miscarriages is essential. The physical examination should determine growth parameters and volume status as well as the presence of any dysmorphic features suggesting an underlying syndrome.
Once the presence of a non–anion gap metabolic acidosis is confirmed, the urine pH can help distinguish distal from proximal causes. A urine pH < 5.5 in the presence of acidosis suggests pRTA, whereas patients with distal RTA typically have a urine pH > 6.0. The urine anion gap ([urine Na+ + urine K+ ] − urine Cl− ) is sometimes calculated to confirm the diagnosis of distal RTA. A positive gap suggests a deficiency of ammoniagenesis and, thus, the possibility of a distal RTA. A negative gap is consistent with proximal tubule bicarbonate wasting (or gastrointestinal bicarbonate wasting). A urinalysis should also be obtained to determine the presence of glycosuria, proteinuria, or hematuria, suggesting more global tubular damage or dysfunction. Random or 24 hr urine calcium and creatinine measurements will identify hypercalciuria. Renal ultrasonography should be performed to identify underlying structural abnormalities such as obstructive uropathies, as well as to determine the presence of nephrocalcinosis (Fig. 547.2 ).
Treatment and Prognosis
The mainstay of therapy in all forms of RTA is bicarbonate replacement. Patients with pRTA often require large quantities of bicarbonate, up to 20 mEq/kg/24 hr, in the form of sodium bicarbonate or sodium citrate solution (Bicitra or Shohl solution). The base requirement for distal RTAs is generally in the range of 2-4 mEq/kg/24 hr, although individual patients’ requirements can vary. Patients with Fanconi syndrome usually require phosphate supplementation. Patients with distal RTA should be monitored for the development of hypercalciuria. Those with symptomatic hypercalciuria (recurrent episodes of gross hematuria), nephrocalcinosis, or nephrolithiasis can require thiazide diuretics to decrease urine calcium excretion. Patients with type IV RTA can require chronic treatment for hyperkalemia with sodium–potassium exchange resin (i.e., sodium polystyrene sulfonate).
The prognosis of RTA depends to a large extent on the nature of any existing underlying disease. Patients with treated isolated proximal or distal RTA generally demonstrate improvement in growth, provided serum bicarbonate levels can be maintained in the normal range. Patients with systemic illness and Fanconi syndrome can have ongoing morbidity with growth failure, rickets, and signs and symptoms related to their underlying disease.
Bibliography
Alper SL. Familial renal tubular acidosis. J Nephrol . 2012;23(Suppl 16):S57–S76.
Batlle D, Haque SK. Genetic causes and mechanisms of distal renal tubular acidosis. Nephrol Dial Transplant . 2012;27:3691–3704.
Fry AC, Karet FE. Inherited renal acidoses. Physiology (Bethesda) . 2007;22:202–211.
Gil-Pena H, Mejía N, Santos F. Renal tubular acidosis. J Pediatr . 2014;164(4):691–698.
Hoorn EJ. Renal tubular disorders: from proteins to patients. Clin Biochem . 2011;44:503–504.
Magen D, Berger L, Coady MJ, et al. A loss-of-function mutation in NaPi-IIa and renal Fanconi's syndrome. N Engl J Med . 2010;362(12):1102–1108.
Nesterova G, Gahl WA. Cystinosis: the evolution of a treatable disease. Pediatr Nephrol . 2013;28:51–59.
Prie D, Friedlander G. Genetic disorders of renal phosphate transport. N Engl J Med . 2010;362:2399–2409.
Rao KI, Hesselink J, Trauner DA. Chiari I malformation in nephrotic cystinosis. J Pediatr . 2015;167:1126–1129.
Santos F, Gil-Pena H, Alvarez-Alvarez S. Renal tubular acidosis. Curr Opin Pediatr . 2017;29:206–210.
Veys KR, Elmonem MA, Arcolino FO, et al. Nephropathic cystinosis: an update. Curr Opin Pediatr . 2017;29:168–178.
Walsh SB, Unwin RJ. Renal tubular disorders. Clin Med . 2012;12:476–479.
Wappner RS. Lowe syndrome. In GeneClinics: Clinical Genetic Information Resource (database online) . http://www.geneclinics.org/ .
Rickets Associated With Renal Tubular Acidosis
Bradley P. Dixon
Rickets may be present in primary RTA, particularly in pRTA due to the added features of hypophosphatemia and phosphaturia from generalized proximal tubular dysfunction. Bone demineralization without overt rickets usually is detected in distal (type I) RTA. This metabolic bone disease may be characterized by bone pain, growth retardation, osteopenia, and, occasionally, pathologic fractures.
Bone demineralization in distal RTA probably relates to dissolution of bone because the calcium carbonate in bone serves as a buffer against the metabolic acidosis due to the hydrogen ions retained by patients with RTA.
Administration of sufficient bicarbonate to reverse acidosis reverses bone dissolution and the hypercalciuria that is common in distal RTA. Proximal RTA is treated with both bicarbonate and oral phosphate supplements to heal rickets. Doses of phosphate similar to those used in familial hypophosphatemia or Fanconi syndrome may be indicated. Vitamin D is required to offset the secondary hyperparathyroidism that complicates oral phosphate therapy. Following therapy, growth in patients with type II (proximal) RTA is greater than in patients with primary Fanconi syndrome.