Hyperparathyroidism
Daniel A. Doyle
Excessive production of parathyroid hormone (PTH) can result from a primary defect of the parathyroid glands such as an adenoma or hyperplasia (primary hyperparathyroidism).
More often, the increased production of PTH is compensatory, usually aimed at correcting hypocalcemic states of diverse origins (secondary hyperparathyroidism). In vitamin D–deficient rickets and the malabsorption syndromes, intestinal absorption of calcium is deficient but hypocalcemia and tetany may be averted by increased activity of the parathyroid glands. In pseudohypoparathyroidism, PTH levels are elevated because a mutation in the Gs α protein interferes with response to PTH. Early in chronic renal disease, hyperphosphatemia results in a reciprocal fall in the calcium concentration with a consequent increase in PTH, but in advanced stages of renal failure, production of 1,25(OH)2 D3 is also decreased, leading to worsening hypocalcemia and further stimulation of PTH. In some instances, if stimulation of the parathyroid glands has been sufficiently intense and protracted, the glands continue to secrete increased levels of PTH for months or years after kidney transplantation, with resulting hypercalcemia.
Etiology
Childhood hyperparathyroidism is uncommon. Onset during childhood is usually the result of a single benign adenoma . It usually becomes manifested after 10 yr of age. There have been a number of kindreds in which multiple members have hyperparathyroidism transmitted in an autosomal dominant fashion. Most of the affected family members are adults, but children have been involved in approximately 30% of the pedigrees. Some affected patients in these families are asymptomatic, and disease is detected only by careful study. In other kindreds, hyperparathyroidism occurs as part of the constellation known as the multiple endocrine neoplasia (MEN) syndromes (see Chapter 587 ) or of the hyperparathyroidism–jaw tumor syndrome.
Neonatal severe hyperparathyroidism is a rare disorder. Symptoms develop shortly after birth and consist of anorexia, irritability, lethargy, constipation, and failure to thrive. Radiographs reveal subperiosteal bone resorption, osteoporosis, and pathologic fractures. Symptoms may be mild, resolving without treatment, or can have a rapidly fatal course if diagnosis and treatment are delayed. Histologically, the parathyroid glands show diffuse hyperplasia. Affected siblings have been observed in some kindreds, and parental consanguinity has been reported in several kindreds. Most cases have occurred in kindreds with the clinical and biochemical features of familial hypocalciuric hypercalcemia. Infants with neonatal severe hyperparathyroidism may be homozygous or heterozygous for the mutation in the Ca2+ -sensing receptor gene, whereas most persons with 1 copy of this mutation exhibit autosomal dominant familial hypocalciuric hypercalcemia.
MEN type I (see Chapter 587 ) is an autosomal dominant disorder characterized by hyperplasia or neoplasia of the endocrine pancreas (which secretes gastrin, insulin, pancreatic polypeptide, and occasionally glucagon), the anterior pituitary (which usually secretes prolactin), and the parathyroid glands. In most kindreds, hyperparathyroidism is usually the presenting manifestation, with a prevalence approaching 100% by 50 yr of age and occurring only rarely in children younger than 18 yr of age. With appropriate DNA probes, it is possible to detect carriers of the gene with 99% accuracy at birth, avoiding unnecessary biochemical screening programs.
The gene for MEN type I is on chromosome 11q13; it appears to function as a tumor-suppressor gene and follows the two-hit hypothesis of tumor development. The first mutation (germinal) is inherited and is recessive to the dominant allele; this does not result in tumor formation. A second mutation (somatic) is required to eliminate the normal allele, which then leads to tumor formation.
Hyperparathyroidism–jaw tumor syndrome is an autosomal dominant disorder characterized by parathyroid adenomas and fibro-osseous jaw tumors. Affected patients can also have polycystic kidney disease, renal hamartomas, and Wilms tumor. Although the condition affects adults primarily, it has been diagnosed as early as age 10 yr.
MEN type II may also be associated with hyperparathyroidism (see Chapter 587 ).
Transient neonatal hyperparathyroidism has occurred in a few infants born to mothers with hypoparathyroidism (idiopathic or surgical) or with pseudohypoparathyroidism. In each case, the maternal disorder had been undiagnosed or inadequately treated during pregnancy. The cause of the condition is chronic intrauterine exposure to hypocalcemia with resultant hyperplasia of the fetal parathyroid glands. In the newborn, manifestations involve the bones primarily, and healing occurs between 4 and 7 mo of age.
Clinical Manifestations
At all ages, the clinical manifestations of hypercalcemia of any cause include muscle weakness, fatigue, headache, anorexia, abdominal pain, nausea, vomiting, constipation, polydipsia, polyuria, weight loss, and fever. When hypercalcemia is of long duration, calcium may be deposited in the renal parenchyma (nephrocalcinosis), with progressively diminished renal function. Renal calculi can develop and can cause renal colic and hematuria. Osseous changes can produce pain in the back or extremities, disturbances of gait, genu valgum, fractures, and tumors. Height can decrease from compression of vertebrae; the patient can become bedridden. Detection of completely asymptomatic patients is increasing with the advent of automated panel assays that include serum calcium determinations.
Abdominal pain is occasionally prominent and may be associated with acute pancreatitis. Parathyroid crisis can occur, manifested by serum calcium levels >15 mg/dL and progressive oliguria, azotemia, stupor, and coma. In infants, failure to thrive, poor feeding, and hypotonia are common. Cognitive impairment, convulsions, and blindness can occur as sequelae of long-standing hypercalcemia. Psychiatric manifestations include depression, confusion, dementia, stupor, and psychosis.
Laboratory Findings
The serum calcium level is elevated; 39 of 45 children with adenomas had levels >12 mg/dL. The hypercalcemia is more severe in infants with parathyroid hyperplasia; concentrations ranging from 15 to 20 mg/dL are common, and values as high as 30 mg/dL have been reported. Even when the total serum calcium level is borderline or only slightly elevated, ionized calcium levels are often increased. The serum phosphorus level is reduced to approximately 3 mg/dL or less, and the level of serum magnesium is low. The urine can have a low and fixed specific gravity, and serum levels of nonprotein nitrogen and uric acid may be elevated. In patients with adenomas who have skeletal involvement, serum phosphatase levels are elevated, but in infants with hyperplasia the levels of alkaline phosphatase may be normal even when there is extensive involvement of bone.
Serum levels of intact PTH are elevated, especially in relation to the level of calcium. Calcitonin levels are normal. Acute hypercalcemia can stimulate calcitonin release, but with prolonged hypercalcemia, hypercalcitoninemia does not occur.
The most consistent and characteristic radiographic finding is resorption of subperiosteal bone, best seen along the margins of the phalanges of the hands. In the skull, there may be gross trabeculation or a granular appearance resulting from focal rarefaction; the lamina dura may be absent. In more advanced disease, there may be generalized rarefaction, cysts, tumors, fractures, and deformities. Approximately 10% of patients have radiographic signs of rickets. Radiographs of the abdomen can reveal renal calculi or nephrocalcinosis.
Differential Diagnosis
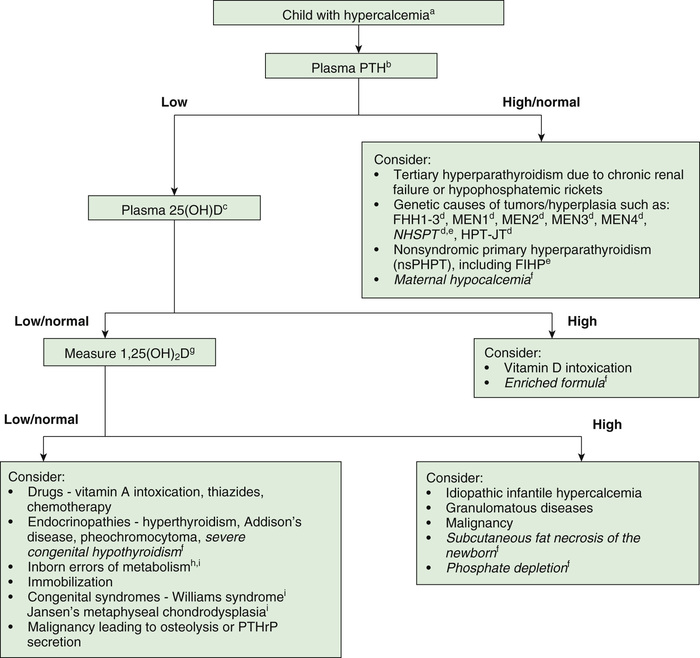
Other causes of hypercalcemia can result in a similar clinical pattern and must be differentiated from hyperparathyroidism (Table 591.1 and Fig. 591.1 ). A low serum phosphorus level with hypercalcemia is characteristic of primary hyperparathyroidism; elevated levels of PTH are also diagnostic. With hypercalcemia of any cause except hyperparathyroidism and familial hypocalciuric hypercalcemia, PTH levels are suppressed. Pharmacologic doses of corticosteroids lower the serum calcium level to normal in patients with hypercalcemia from other causes but generally do not affect the calcium level in patients with hyperparathyroidism.
Table 591.1
Adapted from Lietman SA, Germain-Lee EL, Levine MA: Hypercalcemia in children and adolescents. Curr Opin Pediatr 22:508–515, 2010; Benjamin RW, Moats-Staats BM, Calikoglu A, et al: Hypercalcemia in children. Pediatr Endocrinol Rev 5:778–784, 2008; Davies JH: A practical approach to the problems of hypercalcaemia. Endocr Dev 16:93–114, 2009.
Treatment
Surgical exploration is indicated in all instances. All glands should be carefully inspected; if an adenoma is discovered, it should be removed; very few instances of carcinoma are known in children. Most neonates with severe hypercalcemia require total parathyroidectomy; less-severe hypercalcemia remits spontaneously in others. Still others have been treated successfully with bisphosphonates and calcimimetics. The patient should be carefully observed postoperatively for the development of hypocalcemia and tetany; intravenous administration of calcium gluconate may be required for a few days. The serum calcium level then gradually returns to normal, and, under ordinary circumstances, a diet high in calcium and phosphorus must be maintained for only several months after operation.
CT, real-time ultrasonography, and subtraction scintigraphy using sestamibi/technetium-pertechnetate alone and in combination have proved effective in localizing a single adenoma versus diffuse hyperplasia in 50–90% of adults. Parathyroid surgeons often rely on intraoperative selective venous sampling with intraoperative assay of PTH for localizing and removing the source of increased PTH secretion.
Prognosis
The prognosis is good if the disease is recognized early and there is appropriate surgical treatment. When extensive osseous lesions are present, deformities may be permanent. A search for other affected family members is indicated.
Other Causes of Hypercalcemia
Daniel A. Doyle
Familial Hypocalciuric Hypercalcemia (Familial Benign Hypercalcemia)
Patients with familial hypocalciuric hypercalcemia are usually asymptomatic, and the hypercalcemia is identified by chance during routine investigation for other conditions. The parathyroid glands are normal, PTH levels are inappropriately normal, and subtotal parathyroidectomy does not correct the hypercalcemia. Serum levels of magnesium are high normal or mildly elevated. The ratio of calcium-to-creatinine clearance is usually decreased despite hypercalcemia.
The disorder is inherited in an autosomal dominant manner and is caused by a mutant gene on chromosome 3q2. Penetrance is near 100%, and the disorder can be diagnosed early in childhood by serum and urinary calcium concentrations. Detection of other affected family members is important to avoid inappropriate parathyroid surgery. The defect is an inactivating mutation in the Ca2+ -sensing receptor gene. This G-protein–coupled receptor senses the level of free Ca2+ in the blood and triggers the pathway to increase extracellular Ca2+ in the face of hypocalcemia. This receptor functions in the parathyroid and kidney to regulate calcium homeostasis; inactivating mutations lead to an increased set point with respect to serum Ca2+ , resulting in mild to moderate hypercalcemia in heterozygotes.
Granulomatous Diseases
Hypercalcemia occurs in 30–50% of children with sarcoidosis and less often in patients with other granulomatous diseases such as tuberculosis. Levels of PTH are suppressed, and levels of 1,25(OH)2 D3 are elevated. The source of ectopic 1,25(OH)2 D3 is the activated macrophage, through stimulation by interferon-α from T lymphocytes, which are present in abundance in granulomatous lesions. Unlike renal tubular cells, the 1α-hydroxylase in macrophages is unresponsive to homeostatic regulation. Oral administration of prednisone (2 mg/kg/24 hr) lowers serum levels of 1,25(OH)2 D3 to normal and corrects the hypercalcemia.
Hypercalcemia of Malignancy
Hypercalcemia often occurs in adults with a wide variety of solid tumors but is identified much less often in children. It has been reported in infants with malignant rhabdoid tumors of the kidney or congenital mesoblastic nephroma and in children with neuroblastoma, medulloblastoma, leukemia, Burkitt lymphoma, dysgerminoma, and rhabdomyosarcoma. Serum levels of PTH are rarely elevated. In most patients, the hypercalcemia associated with malignancy is caused by elevated levels of parathyroid hormone–related peptide and not PTH. Rarely, tumors produce 1,25(OH)2 D3 or PTH ectopically.
Miscellaneous Causes of Hypercalcemia
Hypercalcemia can occur in infants with subcutaneous fat necrosis. Levels of PTH are normal. In one infant, the level of 1,25(OH)2 D3 was elevated and biopsy of the skin lesion revealed granulomatous infiltration, suggesting that the mechanism of the hypercalcemia was akin to that seen in patients with other granulomatous disease. In another infant, although the level of 1,25(OH)2 D3 was normal, PTH was suppressed, suggesting the hypercalcemia was not related to PTH. Treatment with prednisone is effective.
Hypophosphatasia, especially the severe infantile form, is usually associated with mild to moderate hypercalcemia (see Chapter 724 ). Serum levels of phosphorus are normal, and those of alkaline phosphatase are subnormal. The bones exhibit rachitic-like lesions on radiographs. Urinary levels of phosphoethanolamine, inorganic pyrophosphate, and pyridoxal 5′-phosphate are elevated; each is a natural substrate to a tissue-nonspecific (liver, bone, kidney) alkaline phosphatase enzyme. Missense mutations of the tissue-nonspecific alkaline phosphatase enzyme gene result in an inactive enzyme in this autosomal recessive disorder.
Idiopathic hypercalcemia of infancy is manifested by failure to thrive and hypercalcemia during the 1st yr of life, followed by spontaneous remission. Serum levels of phosphorus and PTH are normal. The condition has been defined as resulting from increased absorption of calcium from decreased degradation of 1,25(OH)2 D3 . Mutations in the CYP24A1 gene that encodes 25-hydroxyvitamin D 24-hydroxylase, the key enzyme in 1,25(OH)2 D3 degradation, cause excessive levels of the active vitamin D metabolite, which, in turn, causes hypercalcemia in a subset of infants who receive supplemental vitamin D. An excessive rise in the level of 1,25(OH)2 D3 in response to PTH administration has been reported years after the hypercalcemic phase.
Approximately 10% of patients with Williams syndrome also inconsistently exhibit associated infantile hypercalcemia. The phenotype consists of feeding difficulties, slow growth, elfin facies (small mandible, prominent maxilla, upturned nose), renovascular disorders, and a gregarious “cocktail party” personality. Cardiac lesions include supravalvular aortic stenosis, peripheral pulmonic stenosis, aortic hypoplasia, coronary artery stenosis, and atrial or ventricular septal defects. Nephrocalcinosis can develop if hypercalcemia persists. The IQ score of 50-70 is curiously accompanied by enhanced quantity and quality of vocabulary, auditory memory, and social use of language. A contiguous gene deletion syndrome with a submicroscopic deletion at chromosome 7q11.23, which includes deletion of one elastin allele, occurs in 90% of patients and seems to account for the vascular problems. Definitive diagnosis can be established by specific fluorescence in situ hybridization. The hypercalcemia and central nervous system symptoms may be caused by deletion of adjacent genes. Hypercalcemia has been successfully controlled with either prednisone or calcitonin.
Hypervitaminosis D resulting in hypercalcemia from drinking milk that has been incorrectly fortified with excessive amounts of vitamin D has been reported. Not all patients with hypervitaminosis D develop hypercalcemia. Affected infants can manifest failure to thrive, nephrolithiasis, poor renal function, and osteosclerosis. Serum levels of 25(OH)D are a better indicator of hypervitaminosis D than levels of 1,25(OH)2 D3 because 25(OH)D has a longer half-life.
Prolonged immobilization can lead to hypercalcemia and occasionally to decreased renal function, hypertension, and encephalopathy. Children who have hypophosphatemic rickets and undergo surgery with subsequent long-term immobilization are at risk for hypercalcemia and should therefore have their vitamin D supplementation decreased or discontinued.
Jansen-type metaphyseal chondrodysplasia is a rare genetic disorder characterized by short-limbed dwarfism and severe but asymptomatic hypercalcemia (see Chapter 714 ). Circulating levels of PTH and parathyroid hormone–related peptide are undetectable. These patients have an activating PTH–parathyroid hormone–related peptide receptor mutation that results in aberrant calcium homeostasis and abnormalities of the growth plate.
Bibliography
Alos N, Eugene D, Fillion M, et al. Pamidronate: treatment for severe hypercalcemia in neonatal subcutaneous fat necrosis. Horm Res . 2006;65(6):289–294.
Bilezikian JP, Banderia L, Khan A, Cusano NE. Hyperparathyroidism. Lancet . 2018;391:168–176.
Carpten JD, Robbins CM, Villablanca A, et al. HRPT2, encoding parafibromin, is mutated in hyperparathyroidism–jaw tumor syndrome. Nat Genet . 2002;32(4):676–680.
Domico MB, Huynh V, Anand SK, et al. Severe hyperphosphatemia and hypocalcemic tetany after oral laxative administration in a 3-month-old infant. Pediatrics . 2006;118(5):e1580–e1583.
Duan K, Mete O. Familial hyperparathyroidism syndromes. Diagn Histopathol . 2016;22(3):92–100.
Fraser WD. Hyperparathyroidism. Lancet . 2009;374:145–156.
Goudet P, Dalac A, LeBras M, et al. MEN1 disease occurring before 21 years old: a 160-patient chohort study from the Groupe d'etude des Tumeurs Endocrines. J Clin Endocrinol Metab . 2015;100(4):1568–1577.
Kollars J, Zarroug AE, van Heerden J, et al. Primary hyperparathyroidism in pediatric patients. Pediatrics . 2005;115:974–980.
Letavernier E, Rodenas A, Guerrot D, et al. Williams-Beuren syndrome hypercalcemia: Is TRPC3 a novel mediator in calcium homeostasis? Pediatrics . 2012;129:e1626–e1630.
Lietman SA, Germain-Lee EL, Levine MA. Hypercalcemia in children and adolescents. Curr Opin Pediatr . 2010;22:508–515.
Marcocci C, Cetani F. Primary hyperparathyroidism. N Engl J Med . 2011;365:2389–2396.
Nesbit MA, Hannan FM, Howles SA, et al. Mutations affecting G-protein subunit α in hypercalcemia and hypocalcemia. N Engl J Med . 2013;368:2476–2486.
Obermannova B, Banghova K, Sumnik Z, et al. Unusually severe phenotype of neonatal primary hyperparathyroidism due to a heterozygous inactivating mutation in the CASR gene. Eur J Pediatr . 2009;168(5):569–591.
Pallan S, Rahman MO, Khan AA. Diagnosis and management of primary hyperparathyroidism. BMJ . 2012;344:55–60.
Schlingmann KP, Kaufmann M, Weber S, et al. Mutations in the CYP24A1 and idiopathic infantile hypercalcemia . N Engl J Med . 2011;365:410–421.
Sitges-Serra A, Bergenfelz A. Clinical update: sporadic primary hyperparathyroidism. Lancet . 2007;370:468–470.
Stokes VJ, Nielsen MF, Hannan FM, Thaller RV. Hypercalcemic disorders in children. J Bone Miner Res . 2017;32(11):2157–2170.