Neurodegenerative Disorders of Childhood
Jennifer M. Kwon
Neurodegenerative disorders of childhood encompass a large, heterogeneous group of diseases from specific genetic and biochemical defects. Children with suspected neurodegenerative disorders were once subjected to brain and rectal (neural) biopsies, but with modern neuroimaging techniques and specific biochemical and molecular diagnostic tests, these invasive procedures are rarely necessary. The most important component of the diagnostic investigation continues to be a thorough history and physical examination. The hallmark of a neurodegenerative disease is regression and progressive deterioration of neurologic function with loss of speech, vision, hearing, or locomotion, often associated with seizures, feeding difficulties, and impairment of intellect. The age of onset, rate of progression, and principal neurologic findings determine whether the disease affects primarily the white or the gray matter. Upper motor neuron signs and progressive spasticity are the hallmarks of white matter disorders; convulsions and intellectual and visual impairments that occur early in the disease course are the hallmarks of gray matter disorders. A precise history confirms regression of developmental milestones, and the neurologic examination localizes the process within the nervous system. Although the outcome of a neurodegenerative condition is usually fatal and available therapies are often limited in effect, it is important to make the correct diagnosis so that genetic counseling may be offered and prevention strategies can be implemented. Bone marrow transplantation and other novel therapies may prevent the progression of disease in certain individuals who are either presymptomatic or very early in their disease course. For all conditions in which the specific genetic defect is known, prevention by prenatal diagnosis (chorionic villus sampling or amniocentesis) is possible as is carrier detection. Table 617.1 summarizes selected inherited neurodegenerative and metabolic disorders by their usual age of onset.
Table 617.1
Selected Metabolic Conditions Associated With Developmental Regression
| AGE AT ONSET (yr) | CONDITIONS | COMMENTS |
|---|---|---|
| <2, often with hepatomegaly or hepatic effects | Fructose intolerance | Vomiting, hypoglycemia, poor feeding, failure to thrive (when given fructose) |
| Galactosemia | Lethargy, hypotonia, icterus, cataract, hypoglycemia (when given lactose) | |
| Glycogenosis (glycogen storage disease) types I-IV | Hypoglycemia, cardiomegaly (type II) | |
| Mucopolysaccharidosis types I and II | Coarse facies, stiff joints | |
| GM1 gangliosidosis | Coarse facies, macroglossia, cherry- red spot in macula | |
| Niemann-Pick disease, infantile type | Gray matter disease, failure to thrive | |
| Zellweger syndrome | Hypotonia, high forehead, flat facies | |
| Gaucher disease (neuronopathic form) | Extensor posturing, irritability | |
| Carbohydrate-deficient glycoprotein syndromes | Dysmyelination, cerebellar hypoplasia | |
| <2, without hepatomegaly | Krabbe disease | Irritability, extensor posturing, optic atrophy, and blindness |
| Rett syndrome | Girls with deceleration of head growth, loss of hand skills, hand wringing, impaired language skills, gait apraxia | |
| Maple syrup urine disease | Poor feeding, tremors, myoclonus, opisthotonos | |
| Phenylketonuria | Light pigmentation, microcephaly | |
| Menkes kinky hair disease | Hypertonia, irritability, seizures, abnormal hair | |
| Tay-Sachs disease, GM2 gangliosidoses | Seizures, cherry-red spot of macula, increased startle response | |
| Subacute necrotizing encephalopathy of Leigh disease | White matter disease, basal ganglia, brainstem lesions | |
| Canavan disease | White matter disease, macrocephaly | |
| Neurodegeneration with brain iron accumulation disease (see Table 617.4 ) | Cerebellar atrophy, optic atrophy, iron accumulation in basal ganglia, movement disorder | |
| 2-5 | Niemann-Pick disease types III and IV | Hepatosplenomegaly, gait difficulty |
| Wilson disease | Liver disease, Kayser-Fleischer ring; deterioration of cognition is late | |
| Neuronal ceroid lipofuscinosis | Gray matter disease | |
| Mitochondrial encephalopathies (e.g., myoclonic epilepsy with ragged red fibers [MERRF]) | Gray matter disease | |
| Ataxia–telangiectasia | Basal ganglia disease | |
| Neurodegeneration with brain iron accumulation syndrome | Basal ganglia disease | |
| Metachromatic leukodystrophy | White matter disease | |
| Adrenoleukodystrophy | White matter disease, behavior problems, deteriorating school performance, vision loss | |
| 5-15 | Adrenoleukodystrophy | Same as for adrenoleukodystrophy in 2 to 5 yr olds |
| Neuronal ceroid lipofuscinosis, juvenile and adult forms | Gray matter disease | |
| Refsum disease | Peripheral neuropathy, ataxia, retinitis pigmentosa | |
| Sialidosis II, juvenile form | Cherry-red macula, myoclonus, ataxia, coarse facies |
Adapted from Kliegman RM, Greenbaum LA, Lye PS: Practical strategies in pediatric diagnosis and therapy, 2e, Philadelphia, 2004, WB Saunders, p. 542.
Sphingolipidoses
Jennifer M. Kwon
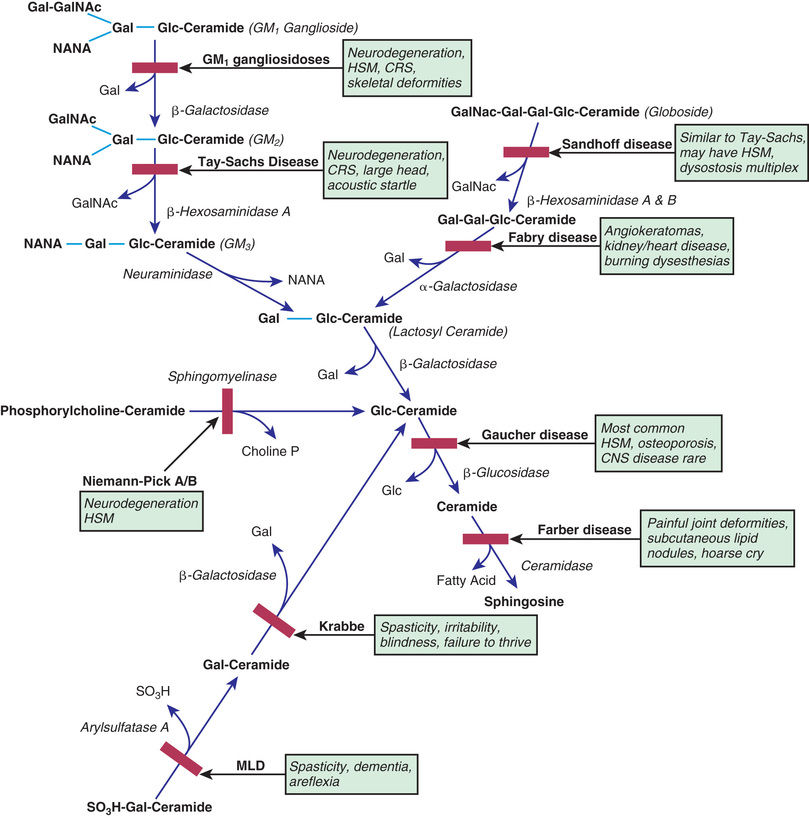
The sphingolipidoses are characterized by intracellular storage of lipid substrates resulting from defective catabolism of the sphingolipids comprising cellular membranes (Fig. 617.1 ). The sphingolipidoses are subclassified into six categories: Niemann-Pick disease, Gaucher disease, GM1 gangliosidosis, GM2 gangliosidosis, Krabbe disease, and metachromatic leukodystrophy. Niemann-Pick disease and Gaucher disease are discussed in Chapter 104.4 .
Gangliosidoses
See also Chapter 104.4 .
Gangliosides are glycosphingolipids, normal constituents of the neuronal and synaptic membranes. The basic structure of a GM1 ganglioside consists of an oligosaccharide chain attached to a hydroxyl group of ceramide and sialic acid bound to galactose. The gangliosides are catabolized by sequential cleavage of the sugar molecules by specific exoglycosidases. Abnormalities in catabolism result in an accumulation of the ganglioside within the cell. Defects in ganglioside degradation can be classified into two groups: the GM1 gangliosidoses and GM2 gangliosidoses.
GM1 Gangliosidoses
The three subtypes of GM1 gangliosidoses are classified according to age at presentation: infantile (type 1), juvenile (type 2), and adult (type 3). The condition is inherited as an autosomal recessive trait and results from a marked deficiency of acid β-galactosidase. This enzyme may be assayed in leukocytes and cultured fibroblasts. The acid β-galactosidase gene has been mapped to chromosome 3p22.3. Prenatal diagnosis is possible by measurement of acid β-galactosidase in or direct molecular testing of cultured amniotic cells.
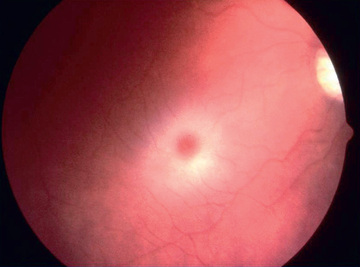
Infantile GM1 gangliosidosis presents at birth or during the neonatal period with anorexia, poor sucking, and inadequate weight gain. Development is globally delayed, and generalized seizures are prominent. The phenotype is striking and shares many characteristics with Hurler syndrome. The facial features are coarse, the forehead is prominent, the nasal bridge is depressed, the tongue is large (macroglossia), and the gums are hypertrophied. Hepatosplenomegaly is present early in the course as a result of accumulation of foamy histiocytes, and kyphoscoliosis is evident because of anterior beaking of the vertebral bodies. The neurologic examination is dominated by apathy, progressive blindness, deafness, spastic quadriplegia, and decerebrate rigidity. A cherry-red spot in the macular region is visualized in approximately 50% of cases. The cherry-red spot is characterized by an opaque ring (sphingolipid-laden retinal ganglion cells) encircling the normal red fovea (Fig. 617.2 ). Children rarely survive beyond age 2-3 yr, and death may be from aspiration pneumonia.
Juvenile GM1 gangliosidosis has a delayed onset beginning about 1 yr of age. The initial symptoms consist of incoordination, weakness, ataxia, and regression of language. Thereafter, convulsions, spasticity, decerebrate rigidity, and blindness are the major findings. Unlike the infantile type, this type is not usually marked by coarse facial features and hepatosplenomegaly. Radiographic examination of the lumbar vertebrae may show minor beaking. Children rarely survive beyond 10 yr of age. Adult GM1 gangliosidosis is a slowly progressive disease consisting of spasticity, ataxia, dysarthria, and a gradual loss of cognitive function.
GM2 Gangliosidoses
The GM2 gangliosidoses are a heterogeneous group of autosomal recessive inherited disorders that consist of several subtypes, including Tay-Sachs disease (TSD), Sandhoff disease, juvenile GM2 gangliosidosis, and adult GM2 gangliosidosis. TSD is most prevalent in the Ashkenazi Jewish population and has an approximate carrier rate of 1 in 30 Jews in the United States. TSD is caused by mutations in the HEXA gene located on chromosome 15q23. Affected infants appear normal until approximately 6 mo of age, except for a marked startle reaction to noise that is evident soon after birth. Affected children then begin to lag in developmental milestones, and by 1 yr of age, they lose the ability to stand, sit, and vocalize. Early hypotonia develops into progressive spasticity, and relentless deterioration follows, with convulsions, blindness, deafness, and cherry-red spots in almost all patients (see Fig. 617.2 ). Macrocephaly becomes apparent by 1 yr of age and results from the 200- to 300–fold normal content of GM2 ganglioside deposited in the brain. Few children live beyond 3-4 yr of age, and death is usually associated with aspiration or bronchopneumonia. A deficiency of the isoenzyme hexosaminidase A is found in tissues of patients with TSD. An accurate and inexpensive carrier detection test is available (serum or leukocyte hexosaminidase A), and this has been an effective tool in the defined population of Ashkenazi Jews. Targeted screening is responsible for the fact that currently, the rare children with TSD born in the United States are most commonly born to non-Jewish parents who are not routinely screened.
Sandhoff disease is very similar to TSD in the mode of presentation, including progressive loss of motor and language milestones beginning at 6 mo of age. Seizures, cherry-red spots, macrocephaly, and doll-like facies are present in most patients; however, children with Sandhoff disease may also have splenomegaly. The visual evoked potentials (VEPs) are normal early in the course of Sandhoff disease and TSD but become abnormal or absent as the disease progresses. The auditory brainstem responses show prolonged latencies. The diagnosis of Sandhoff disease is established by finding deficient levels of hexosaminidases A and B in serum and leukocytes. Children usually die by 3 yr of age. Sandhoff disease is caused by mutations in the HEXB gene located on chromosome 5q13.
Juvenile GM2 gangliosidosis develops in mid-childhood, initially with clumsiness followed by ataxia. Signs of spasticity, athetosis, loss of language, and seizures gradually develop. Progressive visual loss is associated with optic atrophy, but cherry-red spots rarely occur in juvenile GM2 gangliosidosis. A deficiency of hexosaminidase is variable (total deficiency to near normal) in these patients. Death occurs around 15 yr of age.
Adult GM2 gangliosidosis is characterized by a myriad of neurologic signs, including slowly progressive gait ataxia, spasticity, dystonia, proximal muscle atrophy, and dysarthria. Generally, visual acuity and intellectual function are unimpaired. Hexosaminidase A activity alone or hexosaminidases A and B activity is reduced significantly in the serum and leukocytes.
Krabbe Disease (Globoid Cell Leukodystrophy)
Krabbe disease (KD) is a rare autosomal recessive neurodegenerative disorder characterized by severe myelin loss and the presence of globoid bodies in the white matter. The gene for KD (GALC) is located on chromosome 14q24.3-q32.1. The disease results from a marked deficiency of the lysosomal enzyme galactocerebroside β-galactosidase (GALC). KD is a disorder of myelin destruction rather than abnormal myelin formation. Normally, myelination begins in the 3rd trimester, corresponding with a rapid increase of GALC activity in the brain. In patients with KD, galactocerebroside cannot be metabolized during the normal turnover of myelin because of deficiency of GALC. When galactocerebroside is injected into the brains of experimental animals, a globoid cell reaction ensues. It is postulated that a similar phenomenon occurs in humans; nonmetabolized galactocerebroside stimulates the formation of globoid cells that reflect the destruction of oligodendroglial cells. Because oligodendroglial cells are responsible for the elaboration of myelin, their loss results in myelin breakdown, thus producing additional galactocerebroside and causing a vicious circle of myelin destruction.
The symptoms of KD become evident in the first few months of life and include excessive irritability and crying, unexplained episodes of hyperpyrexia, vomiting, and difficulty feeding. In the initial stage of KD, children are often treated for colic or milk allergy with frequent formula changes. Generalized seizures may appear early in the course of the disease. Alterations in body tone with rigidity and opisthotonos and visual inattentiveness as a result of optic atrophy become apparent as the disease progresses. In the later stages of the illness, blindness, deafness, absent deep-tendon reflexes, and decerebrate rigidity constitute the major physical findings. Most patients die by 2 yr of age. MRI and magnetic resonance spectroscopy are useful for evaluating the extent of demyelination in KD. Umbilical cord blood (stem cell) transplantation from unrelated donors in asymptomatic babies may favorably alter the natural history but will not help patients who already have neurologic symptoms.
Late-onset KD has been described beginning in childhood or adolescence. Patients present with optic atrophy and cortical blindness, and their condition may be confused with adrenoleukodystrophy. Slowly progressive gait disturbances, including spasticity and ataxia, are prominent. As with classic KD, globoid cells are abundant in the white matter, and leukocytes are deficient in GALC. An examination of the cerebrospinal fluid shows an elevated protein content, and the nerve conduction velocities are markedly delayed as a result of segmental demyelination of the peripheral nerves.
Metachromatic Leukodystrophy
This disorder of myelin metabolism is inherited as an autosomal recessive trait and is characterized by a deficiency of arylsulfatase A activity. The ARSA gene is located on chromosome 22q13.33. The absence or deficiency of arylsulfatase A leads to accumulation of cerebroside sulfate within the myelin in both the central and peripheral nervous systems because of the inability to cleave sulfate from galactosyl-3-sulfate ceramide. The excessive cerebroside sulfate is thought to cause myelin breakdown. Prenatal diagnosis of metachromatic leukodystrophy (MLD) is made by assaying of arylsulfatase A activity in chorionic villi or cultured amniotic fluid cells. Cresyl violet applied to tissue specimens produces metachromatic staining of the sulfatide granules, giving the disease its name. Some individuals with low arylsulfatase A enzyme activity are clinically normal and have a pseudodeficiency state that can only be confirmed by additional genetic or biochemical tests. Those affected with MLD are generally classified according to age of onset: late infantile, juvenile, and adult.
Late infantile MLD begins with insidious onset of gait disturbances between 1 and 2 yr of age. The child initially appears awkward and frequently falls, but locomotion is gradually impaired significantly and support is required to walk. The extremities are hypotonic, and the deep-tendon reflexes are absent or diminished. Within the next several months, the child can no longer stand, and deterioration in intellectual function becomes apparent. The speech is slurred and dysarthric, and the child appears dull and apathetic. Visual fixation is diminished, nystagmus is present, and examination of the retina shows optic atrophy. Within 1 yr from the onset of the disease, the child is unable to sit unsupported, and progressive decorticate postures develop. Feeding and swallowing are impaired because of pseudobulbar palsies, and a feeding gastrostomy is required. Patients ultimately become stuporous and die of aspiration or bronchopneumonia by age 5-6 yr. Neurophysiologic evaluation shows slowing of peripheral nerve conduction velocities and progressive changes in the VEPs, auditory brainstem responses, and somatosensory evoked potentials. CT and MRI images of the brain indicate diffuse symmetric attenuation of the cerebellar and cerebral white matter, and examination of the cerebrospinal fluid shows an elevated protein content. Bone marrow transplant or lentiviral hematopoietic stem cell gene therapy is a promising experimental therapy for the management of late infantile MLD patients identified very early in the course of their disease.
Juvenile MLD has many features in common with late infantile MLD, but the onset of symptoms is delayed to 5-10 yr of age. Deterioration in school performance and alterations in personality may herald the onset of the disease. This is followed by incoordination of gait, urinary incontinence, and dysarthria. Muscle tone becomes increased, and ataxia, dystonia, or tremor may be present. In the terminal stages, generalized tonic-clonic convulsions are prominent and are difficult to control. Patients rarely live beyond mid-adolescence.
Adult MLD occurs from the 2nd to 6th decades. Abnormalities in memory, psychiatric disturbances, and personality changes are prominent features. Slowly progressive neurologic signs, including spasticity, dystonia, optic atrophy, and generalized convulsions, lead eventually to a bedridden state characterized by decorticate postures and unresponsiveness.
Bibliography
Parenti G, Andria G, Ballabio A. Lysosomal storage diseases: from pathophysiology to therapy. Annu Rev Med . 2015;66:471–486.
Platt FM. Sphingolipid lysosomal storage disorders. Nature . 2014;510:68–75.
Rosenberg JB, Kaminsky SM, Aubourg P, et al. Gene therapy for metachromatic leukodystrophy. J Neuro Res . 2014;94:1169–1179.
Sessa M, Lorioli L, Fumagalli F, et al. Lentiviral haemopoietic stem-cell gene therapy in early-onset metachromatic leukodystrophy: an ad-hoc analysis of a non-randomised, open-label, phase 1 /2 trial. Lancet . 2016;388:476–486.
Neuronal Ceroid Lipofuscinoses
Jennifer M. Kwon
The neuronal ceroid lipofuscinoses (NCLs) are a group of inherited, neurodegenerative, lysosomal storage disorders characterized by visual loss, progressive dementia, seizures, motor deterioration, and early death. The NCLs are so named because of the intracellular accumulation of fluorescent lipopigments ceroid and lipofuscin. They comprise a genetically and phenotypically heterogeneous group of disorders (currently there are at least nine NCL types) that have traditionally been subclassified by age of onset, among other clinical features. They differ from one another in the associated ultrastructural patterns of the inclusions as seen by electron microscopy. Evaluation of neuronal biopsies (either brain, rectal, conjunctival, or skin) was once required for diagnosis. With the advent of enzymatic and molecular testing methods, clinicians can make specific NCL diagnoses using less-invasive methods (Table 617.2 ).
Table 617.2
Clinical and Genetic Characteristics of the Neuronal Ceroid Lipofuscinoses
| NCL TYPE | GENE* | PROTEIN | AGE OF ONSET | CLINICAL PRESENTATION |
|---|---|---|---|---|
| Congenital | CLN10 | Cathepsin ‡ | Birth (but can present later, even in adults) |
Severe seizures, blindness, rigidity, early death Can also present similar to late-infantile forms |
| Infantile | CLN1 | Palmitoyl-protein thioesterase-1 (PPT1) ‡ | 6-24 mo | Early onset, often rapid progression of seizures; cognitive and motor decline with visual loss |
| Variant infantile | CLN1 | 3 yr to adulthood |
Chronic course Initial visual loss followed then by slow mental and motor decline and seizures |
|
| Late infantile | CLN2 | Tripeptidyl peptidase-1 (TPP1) ‡ | 2-8 yr | Seizures, often severe and intractable; cognitive and motor decline; and visual loss |
| CLN5 | Partially soluble protein | |||
| CLN6 | Membrane protein | |||
| CLN7 | Membrane protein | |||
| CLN8 | Membrane protein | 5-10 yr | Severe epilepsy, progressive with mental retardation | |
| Juvenile | CLN3 | Membrane protein | 4-10 yr |
Visual loss is usually the initial presenting complaint Also have mental and motor disorders and seizures |
* Note that all of the NCL genes have the prefix CLN. The adult form (also called Kufs disease, with locus CLN4, caused by mutations in DNAJC5 ) is not well characterized and is not included in the table. Genetic testing is available for all listed genes.
‡ Enzyme testing is available.
Infantile-type neuronal ceroid lipofuscinosis (INCL, Haltia-Santavuori) begins in the 1st yr of life with myoclonic seizures, intellectual deterioration, and blindness. Optic atrophy and brownish discoloration of the macula are evident on examination of the retina, and cerebellar ataxia is prominent. The electroretinogram typically shows small-amplitude or absent waveforms. Death occurs during childhood. The infantile form is caused by recessive mutations of the gene for the lysosomal enzyme palmitoyl-protein thioesterase-1 (PPT1) on chromosome 1p32. A number of cell types in INCL patients show characteristic intracellular fine granular osmiophilic deposits discernible by electron microscopy.
A subset of children with PPT1 enzyme deficiency has a much less severe course with clinical features resembling those of the juvenile-onset NCL patients. Clinically, these variant INCL patients have a course that is often quite distinct from the typical, classic rapidly degenerating infantile form, yet they have PPT1 deficiency and granular osmiophilic deposits on pathology. There is no clear CLN1 genotype that predicts severity of phenotype.
Late infantile-type neuronal ceroid lipofuscinosis (LINCL, Jansky-Bielschowsky) generally presents with myoclonic seizures beginning between 2 and 4 yr of age in a previously normal child. Dementia and ataxia are combined with a progressive loss of visual acuity and microcephaly. Examination of the retina shows marked attenuation of vessels, peripheral black bone spicule pigmentary abnormalities, optic atrophy, and a subtle brown pigment in the macular region. The electroretinogram and VEP are abnormal early in the course of disease. The autofluorescent material is deposited in neurons, fibroblasts, and secretory cells. Electron microscopic examination of the storage material in skin or conjunctival biopsy material typically shows curvilinear profiles. LINCL can be caused by autosomal recessive mutations of several different genes: CLN2 gene, which codes for a tripeptidyl peptidase-1 (TPP1) that is essential for the degradation of cholecystokinin-8, as well as the CLN5, CLN6, and CLN8 genes, which code for membrane proteins that have not been completely characterized. CLN8 is also known as the locus of northern epilepsy syndrome, which is often called progressive epilepsy with cognitive impairment. CLN2 has been treated with intraventricular cerliponase alfa with less decline in language and motor function but serious side effects.
Juvenile-type neuronal ceroid lipofuscinosis (JNCL, Spielmeyer-Vogt or Batten disease) is the most common form of NCL disease and is generally caused by autosomal-recessive mutations in CLN3 . (Patients who present clinically with JNCL but have PPT1 or TPP1 deficiency are said to have variant INCL or LINCL, respectively.) Children affected with JNCL tend to develop normally for the 1st 5 yr of life. Their initial symptom is usually progressive visual loss and their retinal pigmentary changes often result in an initial diagnosis of retinitis pigmentosa. The funduscopic changes are similar to those for the late infantile type. After disease onset, there may be a rapid decline with changes in cognition and personality, motor incoordination, and seizures. Myoclonic seizures are not as prominent as in LINCL, but parkinsonism can develop and impair ambulation. Patients die in their late twenties to early thirties. In JNCL caused by CLN3, the electron microscopy of tissues show deposits called fingerprint profiles, and routine light microscopy of a peripheral blood smear may show lymphocyte vacuoles.
Bibliography
Cooper JD, Tarczyluk MA, Nelvagal HR. Towards a new understanding of NCL pathogenesis. Biochim Biophys Acta . 2015;1852:2256–2261.
Geraets RD, Koh SY, Hastings ML, et al. Moving towards effective therapeutic strategies for neuronal ceroid lipofuscinosis. Orphanet J Rare Dis . 2016;11:40.
Nickel M, Simonati A, Jacoby D, et al. Disease characteristics and progression in patients with late-infantile neuronal ceroid lipofuscionsis type 2 (CLN2) disease: an observational cohort study. Lancet Child Adolesc Health . 2018;2:582–590.
Nita DA, Mole SE, Minassian BA. Neuronal ceroid lipofuscinoses. Epileptic Disord . 2016;18(Suppl 2):S73–S88.
Schulz A, Ajayi T, Specchio N, et al. Study of intravenous cerliponase alfa for CLN2 disease. N Engl J Med . 2018;378(20):1898–1906.
Sialidosis
Jennifer M. Kwon
Sialidosis is the result of lysosomal sialidase deficiency, secondary to autosomal recessive mutations in the sialidase (α-neuraminidase, NEU1 ) gene on chromosome 6p21.3. The accumulation of sialic acid–oligosaccharides with markedly increased urinary excretion of sialic acid–containing oligosaccharides is associated with clinical presentations that range from the milder sialidosis type I to the more severe sialidosis type II associated with both neurologic and somatic features.
Sialidosis type I, the cherry-red spot myoclonus syndrome, usually presents in the 2nd decade of life, when a patient complains of visual deterioration. Inspection of the retina shows a cherry-red spot, but, unlike patients with TSD, visual acuity declines slowly in individuals with cherry-red spot myoclonus syndrome. Myoclonus of the extremities is gradually progressive and often debilitating and eventually renders patients nonambulatory. The myoclonus is triggered by voluntary movement, touch, and sound and is not controlled with anticonvulsants. Generalized convulsions responsive to antiepileptic drugs occur in most patients.
Sialidosis type II patients present at a younger age and have cherry-red spots and myoclonus, as well as somatic involvement, including coarse facial features, corneal clouding (rarely), and dysostosis multiplex, producing anterior beaking of the lumbar vertebrae. Type II patients may be further subclassified into congenital and infantile (childhood) forms, depending on the age at presentation. Examination of lymphocytes shows vacuoles in the cytoplasm, biopsy of the liver demonstrates cytoplasmic vacuoles in Kupffer cells, and membrane-bound vacuoles are found in Schwann cell cytoplasm, all attesting to the multiorgan nature of sialidosis type II. No distinctive neuroimaging findings or abnormalities in electrophysiologic studies are noted in this group of disorders. Patients with sialidosis have been reported to live beyond the 5th decade.
Some cases of what appears to be sialidosis type II are the result of combined deficiencies of β-galactosidase and α-neuraminidase resulting from deficiency of protective protein/cathepsin A that prevents premature intracellular degradation of these two enzymes. These patients have galactosialidosis and they are clinically indistinguishable from those with sialidosis type II. Consequently, patients who have features of sialidosis type II with marked urinary excretion of oligosaccharides should be tested for protective protein/cathepsin A deficiency as well as sialidase deficiency.
Bibliography
Borden EJ, Annuziata I, d'Azzo A. Lysosomal multienzyme complex: pros and cons of working together. Cell Mol Life Sci . 2014;71:2017–2032.
Caciotti A, Di Rocco M, Filocamo M, et al. Type II sialidosis: review of the clinical spectrum and identification of a new splicing defect with chitotriosidase assessment in two patients. J Neurol . 2009;256:1911–1915.
d'Azzo A, Machado E, Annunziata I. Pathogenesis, emerging therapeutic targets and treatment in sialidosis. Expert Opin Orphan Drugs . 2015;3:491–504.
Ramachandran N, Girard JM, Turnbull J, et al. The autosomal recessively inherited progressive myoclonus epilepsies and their genes. Epilepsia . 2009;50(Suppl 5):29–36.
Miscellaneous Neurodegenerative Disorders
Jennifer M. Kwon
Pelizaeus-Merzbacher Disease
Pelizaeus-Merzbacher disease (PMD) is an X-linked recessive disorder characterized by nystagmus and abnormalities of myelin. PMD is caused by mutations in the proteolipid protein (PLP1) gene, on chromosome Xq22, which is essential for CNS myelin formation and oligodendrocyte differentiation. Mutations in the same gene can cause familial spastic paraparesis (progressive spastic paraparesis type 2, SPG2). PLP1 mutations causing disease include point mutations, deletions, gene duplications, and other gene dosage changes.
Clinically, classic PMD is recognized by nystagmus and roving eye movements, along with head nodding during infancy. Developmental milestones are delayed; ataxia, choreoathetosis, and spasticity ultimately develop. Optic atrophy and dysarthria are associated findings, and death occurs in the 2nd or 3rd decade. The major pathologic finding is a loss of myelin with intact axons, suggesting a defect in the function of oligodendroglia. An MRI scan shows a symmetric pattern of delayed myelination. It is now recognized that a broad spectrum of phenotypes, including SPG2 and peripheral nerve abnormalities, can also result from mutations in the PLP1 gene.
Other PMD-like, hypomyelinating leukodystrophies continue to be identified and should be considered in the differential diagnosis of PMD. These include Allan-Herndon-Dudley syndrome and the TUBB4A related disorders
Alexander Disease
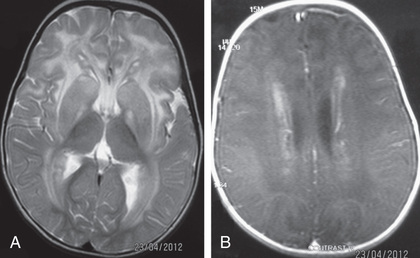
This is a rare disorder that causes progressive macrocephaly and leukodystrophy. Alexander disease is caused by dominant mutations in the glial fibrillary acidic protein (GFAP) gene, on chromosome 17q21, and cases are usually sporadic in families. Pathologic examination of the brain discloses deposition of eosinophilic hyaline bodies called Rosenthal fibers in astrocyte processes. These accumulate in a perivascular distribution throughout the brain. In the classic infantile form of Alexander disease, degeneration of white matter is most prominent frontally. The diagnosis may be suggested by MRI (Fig. 617.3 ) and MR spectroscopy demonstrating abnormal metabolic substrates. Affected children develop progressive loss of intellect, spasticity, and unresponsive seizures causing death by 5 yr of age. However, there are milder forms that present later in life and may not have the characteristic frontal predominance or megalencephaly.
Canavan Spongy Degeneration
See Chapter 103.15 .
Other Leukodystrophies
Metabolic and degenerative disorders can present with significant cerebral white matter changes, such as some mitochondrial disorders (see Chapters 104.1 and 616.2 ) and glutaric aciduria type 1 (see Chapter 103.14 ). In addition, the broader use of brain MRI has brought to light new leukodystrophies.
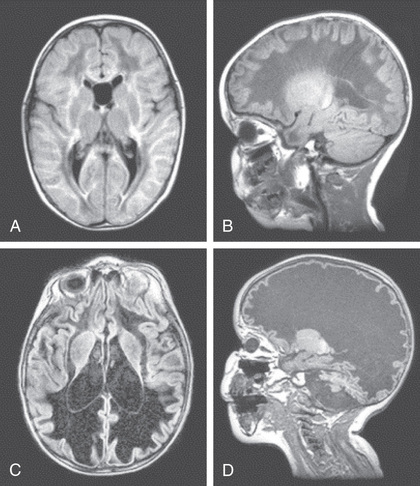
One example is vanishing white matter disease or childhood ataxia with central nervous system (CNS) hypomyelination characterized by ataxia and spasticity (Fig. 617.4 ). Some patients also have optic atrophy, seizures, and cognitive deterioration. The age of presentation and the rapidity of decline in leukodystrophies can be quite variable. In the early-onset forms, decline is usually rapid and followed quickly by death; in the later-onset forms, mental decline is usually slower and milder. Interestingly, acute demyelination in these disorders can be triggered by fever or fright. The diagnosis of vanishing white matter disease or childhood ataxia with CNS hypomyelination is based on clinical findings, characteristic abnormalities on cranial MRI, and autosomal recessive mutations in one of five causative genes (EIF2B1, EIF2B2, EIF2B3, EIF2B4, and EIF2B5) encoding the five subunits of the eucaryotic translation initiation factor, eIF2B. An approach to leukodystrophies based on MRI findings is noted in Fig. 617.5 , and the diagnostic evaluation is noted in Table 617.3 .
 and
and  yr. The first MRI (A, B)
was obtained soon after the onset of symptoms. The initial FLAIR image (A)
shows diffuse abnormality and partial cystic degeneration of the cerebral white matter, whereas the follow-up FLAIR image (C)
shows that all of the cerebral white matter has been replaced by fluid. The initial T1-weighted sagittal image (B)
shows the typical stripe-like pattern within the abnormal white matter, whereas the follow-up image (D)
shows that all of the cerebral white matter has disappeared and that only the cerebral cortex and ependymal lining are preserved. Surprisingly, the absent white matter looks swollen with stretching of the overlying cortex in the broad gyri. The cerebellum has become highly atrophic.
(From Van der Knaap MS, Valk J: Magnetic resonance of myelination and myelin disorders, 3e. Heidelberg, 2004, Springer.)
yr. The first MRI (A, B)
was obtained soon after the onset of symptoms. The initial FLAIR image (A)
shows diffuse abnormality and partial cystic degeneration of the cerebral white matter, whereas the follow-up FLAIR image (C)
shows that all of the cerebral white matter has been replaced by fluid. The initial T1-weighted sagittal image (B)
shows the typical stripe-like pattern within the abnormal white matter, whereas the follow-up image (D)
shows that all of the cerebral white matter has disappeared and that only the cerebral cortex and ependymal lining are preserved. Surprisingly, the absent white matter looks swollen with stretching of the overlying cortex in the broad gyri. The cerebellum has become highly atrophic.
(From Van der Knaap MS, Valk J: Magnetic resonance of myelination and myelin disorders, 3e. Heidelberg, 2004, Springer.)
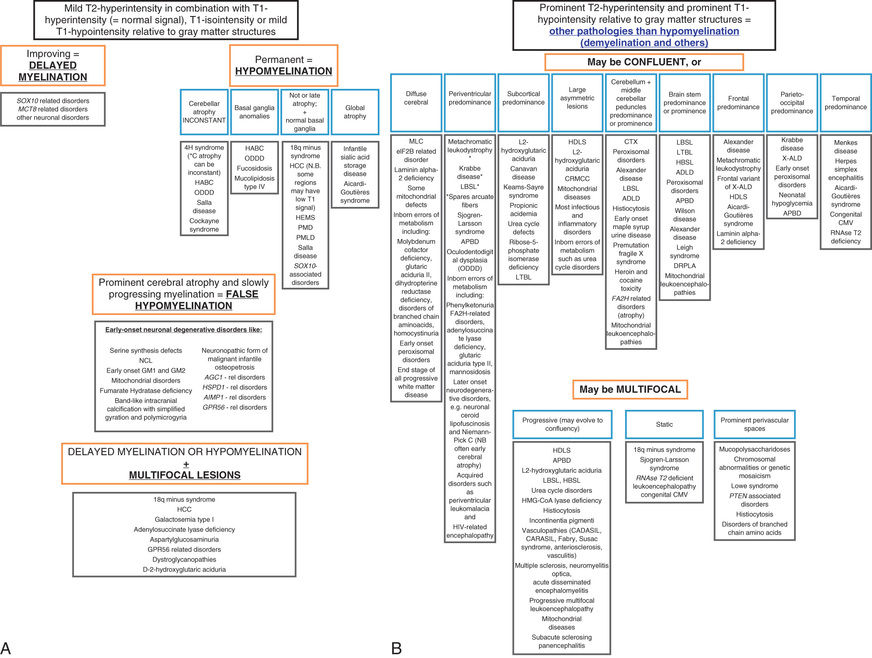
Table 617.3
| CLINICAL/LABORATORY TEST * | DIAGNOSTIC TARGET |
|---|---|
| Brain and spinal MRI (± gadolinium, ± MRS) | Establish white matter disease; ± evidence of leaky blood brain–barrier and metabolite accumulation (mitochondrial disorders, Canavan disease, Sjögren-Larson syndrome, peroxisomal biogenesis disorders) |
| Ophthalmologic exam | Document ophthalmologic signs in several leukodystrophies |
| Head CT | Assess for calcifications |
| Plasma very-long-chain fatty acids | X-linked adrenoleukodystrophy and adrenomyeloneuropathy and peroxisomal biogenesis disorders |
| Lysosomal enzymes (leukocytes) | Metachromatic leukodystrophy, Krabbe disease, multiple sulfatase deficiency, galactosialidosis, sialidosis |
| Blood lactate, pyruvate, amino acids | Mitochondrial disorders |
| Lumbar puncture (cell count, protein, ± CSF neopterin, ± interferon-alpha) | Nonspecific marker of demyelination; ± pleocytosis and markers for Aicardi-Goutières syndrome |
| Urine sulfatides | Metachromatic leukodystrophy, multiple sulfatase deficiency |
| Urine organic acids | L-2-hydroxyglutarate; N-acetyl aspartic acid for Canavan disease; Krebs cycle intermediates (mitochondrial disorders) |
| Neurophysiologic studies (BAER EMG/NCV, VEP, SSEP) | Characterize involvement of cranial and peripheral nerves, optic tracts, and spinal tracts |
| Genetic analyses | As indicated for each leukodystrophy or genetic leukoencephalopathy |
* Additional tests may be indicated for patients with certain distinctive clinical presentations or extraneurologic features suggestive of one or more specific leukodystrophies.
BAER, brainstem auditory evoked response test; CSF, cerebrospinal fluid; CT, computed tomography; EMG, electromyogram; MRI, magnetic resonance imaging; MRS, magnetic resonance spectroscopy; NCV, nerve conduction velocity test; SSEP, somatosensory evoked potential test; VEP, visual evoked potential test.
From Parikh S, Bernard G, Leventer RJ, et al: A clinical approach to the diagnosis of patients with leukodystrophies and genetic leukoencephalopathies, Mol Gen Metab 114:501-515, 2018, Table 6.
Menkes Disease
Menkes disease (kinky hair disease) is a progressive neurodegenerative condition inherited as an X-linked recessive trait. The Menkes gene, ATP7A, on Xq21.1, codes for a copper-transporting, P-type adenosine triphosphatase, and mutations in the protein are associated with low serum copper and ceruloplasmin levels, as well as a defect in intestinal copper absorption and transport. Symptoms begin in the first few months of life and include hypothermia, hypotonia, and generalized myoclonic seizures. The facies are distinctive, with chubby, rosy cheeks and kinky, colorless, friable hair. Microscopic examination of the hair shows several abnormalities, including trichorrhexis nodosa (fractures along the hair shaft) and pili torti (twisted hair). Feeding difficulties are prominent and lead to failure to thrive. Severe cognitive impairment and optic atrophy are constant features of the disease. Neuropathologic changes include tortuous degeneration of the gray matter and marked changes in the cerebellum with loss of the internal granule cell layer and necrosis of the Purkinje cells. Death can occur by 3 yr of age in untreated patients. Very rarely does Menkes disease manifest in females, and when it does, symptoms are milder.
Copper histidine therapy may be effective in preventing neurologic deterioration in some patients with Menkes disease, particularly when treatment is begun in the neonatal period or, preferably, with the fetus. These presymptomatic children are currently identified because of a family history of an affected brother. Copper is essential in the early stages of CNS development, and its absence probably accounts for the neuropathologic changes. Infants diagnosed presymptomatically in the 1st 10 days of life can be started on an experimental protocol of daily copper–histidine subcutaneous injections (as of 2017, only available at NIH under a program supervised by Dr. Stephen Kaler). Optimal response to copper–histidine injection treatment appears to occur only in patients who are identified in the newborn period and whose mutations permit residual copper-transport activity.
The occipital horn syndrome, a skeletal dysplasia caused by different mutations in the same gene as that involved in Menkes disease, is a relatively mild disease. The two diseases are often confused, because the biochemical abnormalities are identical. Resolution of the uncertainty about treatment of patients with Menkes disease will require careful genotype–phenotype correlation, along with further clinical trials of copper therapy.
Rett Syndrome
This syndrome is not strictly speaking a degenerative disease but is a disorder of early brain development marked by a period of developmental regression and deceleration of brain growth after a relatively normal neonatal course. It is an X-linked disease that occurs predominantly in females. The frequency is approximately 1 in 15,000-22,000 children. Rett syndrome is caused by mutations in the MeCP2 gene on Xq28, which codes for a transcription factor that binds to methylated CpG islands and silences transcription. Development may proceed normally until 1 yr of age, when regression of language and motor milestones and acquired microcephaly become apparent. An ataxic gait or fine tremor of hand movements is an early neurologic finding. Most children develop peculiar sighing respirations with intermittent periods of apnea that may be associated with cyanosis. The hallmark of Rett syndrome is repetitive hand-wringing movements and a loss of purposeful and spontaneous use of the hands; these features may not appear until 2-3 yr of age. Autistic behavior is a typical finding in all patients. Generalized tonic-clonic convulsions occur in the majority but may be well controlled by anticonvulsants. Feeding disorders and poor weight gain are common. After the initial period of neurologic regression, the disease process appears to plateau, with persistence of the autistic behavior. Cardiac arrhythmias may result in sudden, unexpected death at a rate that is higher than the general population. Generally, females survive into adulthood.
Postmortem studies show significantly reduced brain weight (60–80% of normal) with a decrease in the number of synapses, associated with a decrease in dendritic length and branching. The phenotype may be related to failure to suppress expression of genes that are normally silent in the early phases of postnatal development. Although very few males survive with the classic Rett syndrome phenotype, genotyping of boys without the classic Rett syndrome phenotype but with intellectual disability and other atypical neurologic features has detected a significant number with mutations in MeCP2. Mutations in MeCP2 have been demonstrated in normal female carriers, females with Angelman syndrome, and males with fatal encephalopathy, Klinefelter (47 XXY) syndrome, and familial X-linked cognitive impairment. Males may present with a Rett-like syndrome if they have an MECP2 duplication.
Some females have an atypical Rett phenotype associated with severe myoclonic seizures in infancy, slowing of head growth, and developmental arrest and have mutations in another X-linked gene encoding for cyclin-dependent kinase–like 5 (CDKL5), which may interact with MeCP2 and other proteins regulating gene expression.
Neurodegeneration With Brain Iron Accumulation
Neurodegeneration with brain iron accumulation represents multiple, age-of-onset–dependent disorders characterized by extrapyramidal symptoms and intellectual deterioration and regression, with iron deposition in the basal ganglia. There is significant phenotypic variability of these disorders; however, a characteristic finding on MRI demonstrates symmetric T2-signal homogeneous hypointensity. Common neurodegeneration with brain iron accumulation disorders are distinguished in Table 617.4 and an approach to their diagnosis is noted in Fig. 617.6 . Clinical features, which are highly variable, may include dystonia, parkinsonism, ataxia, spasticity, psychiatric symptoms, and intellectual impairment. Treatment should focus on the specific disorder and is usually symptomatic relief rather than curative. Iron chelation has been attempted without major long-term benefit.
Table 617.4
Overview of Neurodegeneration With Brain Iron Accumulation Conditions and Genes (if Known)
| CONDITION (ACRONYM) | SYNONYM | GENE | CHROMOSOMAL POSITION | LB PATHOLOGY | CHILDHOOD-ONSET VARIANT | LATE-ONSET VARIANT | ||
|---|---|---|---|---|---|---|---|---|
| AGE OF ONSET | CLINICAL PRESENTATION | AGE OF ONSET | CLINICAL PRESENTATION | |||||
| PKAN | NBIA1 | PANK2 | 20p13 | No | Early childhood, around age 3 | Typical PKAN | Teens or early adulthood | Atypical PKAN |
| PLAN | NBIA2, PARK14 | PLA2G6 | 22q12 | √ | Infancy | Infantile neuroaxonal dystrophy | Teens or early adulthood | Dystonia parkinsonism |
| FAHN | SPG35 | FA2H | 16q23 | Not known | Childhood | Leukodystrophy, hereditary spastic paraplegia | Adulthood (age range up to 30 yr) | May resemble idiopathic Parkinson disease |
| MPAN | — | C19orf12 | 19q12 | √ | — | Pyramidal extrapyramidal syndrome | ||
| Kufor-Rakeb disease | PARK9 | ATP13A2 | 1p36 | √ | Childhood- teens | Parkinsonism, pyramidal tract signs, eye movement disorder | ||
| BPAN | SENDA syndrome | WDR45 | Xp11.23 | Not known | Childhood | Encephalopathy with psychomotor regression, then static | Then: 20s to 30s | Sudden onset progressive dystonia parkinsonism |
| Aceruloplasminemia | — | CP | 3q23 | No | — | — | 50s (range: 16-70) | Extrapyramidal, diabetes, dementia |
| Neuroferritinopathy | — | FTL | 19q13 | No | — | — | 40s | Chorea, dystonia, dementia |
| Idiopathic late-onset cases | — | Probably heterogeneous | Probably heterogeneous | Heterogeneous | — | — | Heterogeneous | Parkinsonism; it may resemble idiopathic Parkinson disease |
√, Present; BPAN, beta-propeller–associated neurodegeneration; CP, ceruloplasmin; FA2H, fatty acid 2-hydroxylase; FAHN, fatty acid 2-hydroxylase–associated neurodegeneration; FTL, ferritin light chain; MPAN, mitochondrial membrane–associated neurodegeneration; NBIA, neurodegeneration with brain iron accumulation; PANK2, pantothenate kinase 2; PKAN, pantothenate kinase–associated neurodegeneration; PLA2G6, phospholipase A2; PLAN, PLA2G6-associated neurodegeneration; SENDA, static encephalopathy of childhood with neurodegeneration in adulthood; SPG, spastic paraplegia.
From Schneider SA, Zorzi G, Nardocci N: Pathophysiology and treatment of neurodegeneration with brain iron accumulation in the pediatric population, Curr Treat Option Neurol 15:652-667, 2013, Table 1.

Bibliography
Arber CE, Li A, Houlden H, Wray S. Insights into molecular mechanisms of disease in neurodegeneration with brain iron accumulation:unifying theories. Neuropathol Appl Neurobiol . 2016;42:220–241.
Charzewska A, Wierzba J, Izycka-Swieszewska E, et al. Hypomyelinating leukodystrophies—a molecular insight into the white matter pathology. Clin Genet . 2016;90:293–304.
Helman G, Van Haren K, Escolar ML, Vanderver A. Emerging treatments for pediatric leukodystrophies. Pediatr Clin North Am . 2015;62:649–666.
Kaler SG, et al. ATP7A-related copper transport disorders. Pagon RA, Adam MP, Ardinger HH. GeneReviews® [internet]. Seattle (WA) . University of Washington: Seattle; 2003:1993–2017 [updated August 18, 2016].
Kaufmann WE, Stallworth JL, Everman DB, Skinner SA. Neurobiologically-based treatments in rett syndrome: opportunities and challenges. Expert Opin Orphan Drugs . 2016;4:1043–1055.
Leonard H, Cobb S, Downs J. Clinical and biological progress over 50 years in rett syndrome. Nat Rev Neurol . 2017;13:37–51.
Meyer E, Kurian MA, Hayflick SJ. Neurodegeneration with brain iron accumulation: genetic diversity and pathophysiological mechanisms. Annu Rev Genomics Hum Genet . 2015;16:257–279.
Ma M, Adams HR, Seltzer LE, et al. Phenotype differentiation of FOXG1 and MECP2 disorders: a new method for characterization of developmental encephalopathies. J Pediatr . 2016;178:233–240.
Møller LB, Lenartowicz M, Zabot MT, et al. Clinical expression of menkes disease in females with normal karyotype. Orphanet J Rare Dis . 2012;7:6.
Osorio JM, Rowitch DH, Tesar P, et al. Concise review: stem cell-based treatment of pelizaeus merzbacher disease. Stem Cells . 2017;35:311–315.
Parikh S, Bernard G, Leventer RJ, et al. A clinical approach to the diagnosis of patients with leukodystrophies and genetic leukoencephalopathies. Mol Gen Metab . 2018;114:501–515.
Ramocki MB, Tavyev YJ, Peters SU. The MECP2 duplication syndrome. Am J Med Genet . 2010;152A:1079–1088.
Smpokou P, Samanta M, Berry GT, et al. Menkes disease in affected females: the clinical disease spectrum. Am J Med Genet . 2014;167A:417–420.
Srivastava S, Naidu S, et al. Alexander disease. Pagon RA, Adam MP, Ardinger HH. GeneReviews® [internet]. Seattle (WA) . University of Washington: Seattle; 2002:1993–2017 [updated January 8, 2015].
Tumer Z. An overview and update of ATP7A mutation leading to menkes disease and occipital horn syndrome. Hum Mutat . 2013;34:417–429.
van der Knaap MS, Pronk JC, Scheper GC. Vanishing white matter disease. Lancet Neurol . 2006;5:413–423.
Wortham NC, Proud CG. eIF2B: recent structural and functional insights into a key regulator of translation. Biochem Soc Trans . 2015;43:1234–1240.