Demyelinating Disorders of the Central Nervous System
Cheryl Hemingway
Acquired demyelinating disorders of the central nervous system (CNS) collectively are rare disorders occurring with an annual incidence of 0.5-1.66 per 100,000 children. They present with neurologic dysfunction caused by immune-mediated attacks on the white matter insulating the brain, optic nerves, and spinal cord. The white matter insulation is formed by myelin contained within oligodendrocytes wrapping around nerve axons. In contrast to genetically determined leukodystrophies (sometimes called dysmyelinating disorders) that produce disrupted white matter, acquired demyelinating disorders generally target normally formed white matter.
There have been significant advances in our understanding of the pathogenesis of demyelination together with a growing interest in the role of B cells and CNS antibodies in demyelination. There are two IgG antibodies recognized as playing an important role in demyelination, aquaporin 4-antibody (AQP4-Ab) and myelin oligodendrocyte glycoprotein antibody (MOG-Ab). The aquaporins, plasma membrane water-transporting proteins, are expressed in astrocytes and involved in water movement, cell migration, and neuroexcitation. Myelin oligodendrocyte glycoprotein is exclusively expressed in the CNS, and although it is only a minor component of the myelin sheath, its location on the outermost lamellae and on the cell surface of oligodendrocytes makes it available for antibody binding. Increased awareness of the importance of these antibodies, together with available disease-modifying treatments (DMTs) has made accurate diagnosis in demyelinating disorders crucial.
Pediatric demyelinating syndromes are characterized clinically by (1) localization of neurologic deficits (i.e., a single site, such as the spinal cord [transverse myelitis, TM], optic nerves [optic neuritis, ON], or brainstem versus a polyregional demyelination); (2) the presence or absence of encephalopathy; (3) the disease course (i.e., monophasic versus repeated attacks involving either the same region or new CNS regions); and (4) the presence or absence of specific antibodies.
MRI of the brain and spine is useful to characterize both symptomatic and clinically silent demyelinating lesions, aid in the diagnosis of demyelinating syndrome, and predict the likelihood of further recurrence. Serial MRIs may be needed to confirm the diagnosis and can be used to monitor the treatment response and guide the escalation of a DMT. The presence of oligoclonal bands (OCBs) in cerebrospinal fluid (CSF) analysis is used to confirm the diagnosis of multiple sclerosis (MS) (Table 618.1 ); their absence may suggest an alternative diagnosis. However, OCBs are seen in other inflammatory CNS diseases. Additional studies, including an autoimmune profile, antibody testing, metabolic testing, genetic testing, catheter angiography, and sometimes even brain biopsy, may be required to evaluate for mimics of demyelination, such as systemic rheumatologic disorders, mitochondrial disorders, primary CNS angiitis, infection, neoplasm, and genetic conditions such as leukodystrophies (Tables 618.2 and 618.3 ).
Table 618.1
Acute Demyelinating Disorders of the Central Nervous System
Table 618.2
Table 618.3
MR Imaging Red Flags for the Diagnosis of Children with Acquired Demyelinating Syndromes
| MR imaging | Leptomeningeal enhancement |
SVcPACNS Infection Tumor HLH |
Leptomeningeal enhancement is not a feature of MS in adults; it has emerged as a red flag for vasculitic or malignant processes in the pediatric cohort. |
| Lesion expansion |
Tumor Lymphoma PML Sarcoidosis |
Increased size of T2 lesions on serial imaging is well recognized in MS, although this should always prompt consideration of malignancy. Increasing size of a white matter–predominant lesion without lesion enhancement in a patient treated with immunosuppressant therapy (or a patient with known HIV) should prompt consideration of PML. PML is a risk for MS patients exposed to more intense immunosuppressive therapies. | |
| Hemorrhage |
ANE Stroke Cerebellitis AHLE Large-vessel CNS vasculitis SVcPACNS |
Although susceptibility-weighted imaging reveals tiny microfoci of hemosiderin in MS patients, hemorrhage large enough to be visible on conventional MRI sequences is not a feature of ADS or MS, and should prompt consideration of disorders in which the cerebral vasculature is specifically involved. |
ADS, acquired demyelinating syndrome; AHLE, acute hemorrhagic leukoencephalitis; CNS, central nervous system; HIV, human immunodeficiency virus; HLH, hemophagocytic lymphohistiocytosis; MS, multiple sclerosis; PML, progressive multifocal leukoencephalopathy; SVcPACNS, small-vessel childhood primary angiitis of the central nervous system.
From O'Mahony J, Shroff M, Banwell B: Mimics and rare presentations of pediatric demyelination, Neuroimaging Clin North Am 23:321-336, 2013, Table 2.
The majority of children presenting with an episode of demyelination are monophasic; they do not relapse. Monophasic demyelinating disorders of childhood include acute disseminated encephalomyelitis (ADEM), optic neuritis (ON), and transverse myelitis (TM); relapsing forms of demyelination include MS and neuromyelitis optica spectrum disorder (NMOSD).
Acute Disseminated Encephalomyelitis
Cheryl Hemingway
ADEM is an inflammatory, demyelinating event of early childhood presenting with an acute onset of polyfocal neurologic deficits, accompanied by encephalopathy and changes compatible with demyelination on brain MRI (see Table 618.1 ).
Epidemiology
Although ADEM can occur at any age, most series report a mean age of between 5 and 8 yr with a slight male predominance. The reported incidence ranges from 0.1-0.6 per 100,000 per year in the pediatric population. ADEM is usually monophasic, but recurrence can occur; if the recurrence is 3 mo or longer after the first episode, the condition is termed multiphasic disseminated encephalomyelitis (MDEM). Up to 50% of cases of ADEM have been found to be associated with MOG-Ab positivity in the serum (see Chapter 618.6 ), and almost all cases of MDEM are MOG-Ab positive; there is thus a strong likelihood that as MOG-Ab testing becomes more available, cases of non-MOG-Ab–positive MDEM will become exceptionally rare. An episode of ADEM can also be followed by non-ADEM demyelination in a new location. In this scenario, if the MOG-Ab is negative, MS may be diagnosed. If ADEM is followed by a relapse in a specific location, such as the optic nerve (ON), then ADEM-ON is diagnosed. If the ON and spinal cord are involved, then NMOSD (see Table 618.1 ); the latter two are frequently associated with MOG-Ab positivity.
Pathogenesis
Molecular mimicry induced by infectious exposure or vaccine has been thought to trigger production of CNS autoantigens, although causality has never been proven. Many patients experience a transient febrile illness in the month prior to ADEM onset. Preceding infections associated with ADEM include influenza, Epstein-Barr virus, cytomegalovirus, varicella, enterovirus, measles, mumps, rubella, herpes simplex, and Mycoplasma pneumoniae. Postvaccination ADEM has been reported following immunizations for rabies, smallpox, measles, mumps, rubella, Japanese encephalitis B, pertussis, diphtheria–polio–tetanus, and influenza, although the risk of ADEM postvaccination is significantly lower than following the infection itself.
Clinical Manifestations
Initial symptoms of ADEM may include lethargy, fever, headache, vomiting, meningeal signs, and seizures, including status epilepticus. Encephalopathy is the hallmark of ADEM, ranging from changes in behavior and persistent irritability to coma. Focal neurologic deficits can be difficult to ascertain in the obtunded or very young child, but common neurologic signs in ADEM include visual loss, cranial neuropathies, ataxia, and motor and sensory deficits, plus bladder/bowel dysfunction with concurrent spinal cord demyelination. The clinical course is usually rapidly progressive over days. Intensive care unit admission may be required, particularly for patients with brainstem dysfunction or raised intracranial pressure.
Neuroimaging
Head CT scanning may be normal or show hypodense regions. Cranial MRI, the imaging study of choice, typically exhibits bilateral, large, multifocal, and sometimes confluent, edematous mass-like T2 lesions with variable enhancement within white and gray matter of the cerebral hemispheres, cerebellum, and brainstem. Deep gray matter structures (e.g., thalami, basal ganglia) are often involved, although this may not be specific to ADEM (Figs. 618.1 and 618.2 ). The spinal cord may have an abnormal T2 signal or enhancement, with or without clinical signs of myelitis. MRI lesions of ADEM typically appear to be of similar age, but their evolution may lag behind the clinical presentation. Serial MRI imaging 3-12 mo following ADEM shows improvement and often complete resolution of T2 abnormalities, although residual gliosis may remain.
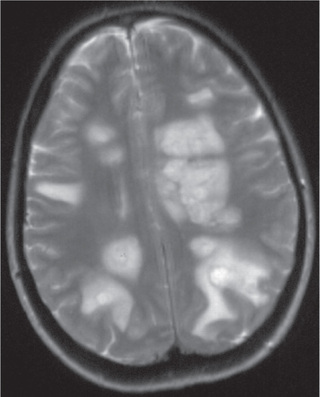
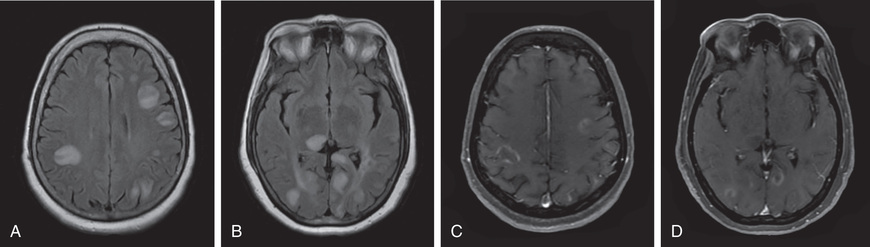
Severe involvement may progress to an acute hemorrhagic leukoencephalopathy (Weston-Hurst disease) with large lesions, edema, mass affect, and a polymorphonucleated cell pleocytosis (in contrast to lymphocytic pleocytosis in the CSF noted in typical ADEM).
Laboratory Findings
There is no biologic marker for ADEM, and laboratory findings can vary widely. CSF studies are often normal or can exhibit pleocytosis with lymphocytic or monocytic predominance. CSF protein can be elevated, especially on repeat studies. Elevated CSF immune globulin production can be present, but true OCB positivity is rare. Electroencephalograms often show generalized slowing, consistent with encephalopathy, although polyregional demyelination of ADEM can also cause focal slowing or epileptiform discharges.
Differential Diagnosis
ADEM is a clinical diagnosis supported by MRI, CSF, and serum findings. The differential diagnosis for ADEM is broad, and empirical antibiotic and antiviral treatment should be considered while infectious evaluations are pending. Follow-up MRI examinations 3-12 mo after ADEM should show improvement; new or enlarging T2 lesions should prompt reevaluation for other etiologies, such as MS, antibody-associated disorders, leukodystrophies, tumor, vasculitis, or mitochondrial, metabolic, or rheumatologic disorders (Table 618.4 and see Tables 618.1 to 618.3 ).
Table 618.4
| ADEM WITH OR WITHOUT MOG-AB | MS | |
|---|---|---|
| Age and sex |
<10 yr Boys and girls equal |
>10 yr Female preponderance |
| Seizures | + | − |
| Encephalopathy | + | − |
| Fever/vomiting | + | − |
| Family history | No | 20% |
| Optic neuritis | Bilateral | Unilateral |
| Manifestations | Polysymptomatic | Monosymptomatic |
| CSF |
Pleocytosis (lymphocytosis) OCBs negative |
Acellular OCBs positive |
| MRI | Large, fluffy, poorly demarcated T2 lesions involving white and gray matter | Ovoid T2 lesions involving juxtacortical, periventricular, or infratentorial areas or spinal lesions; T1 hypointense lesions |
| MRI follow-up after 30 days | No new lesions | New lesions seen |
+ , More likely to be present; −, less likely to be present; ADEM, acute disseminated encephalomyelitis; CSF, cerebrospinal fluid; MS, multiple sclerosis, MOG-Ab, myelin oligodendrocyte glycoprotein antibody; OCBs, oligoclonal bands.
Treatment
Although there are no randomized controlled trials to compare acute treatments for ADEM or other demyelinating disorders of childhood, high-dose intravenous steroids are commonly employed (typically, methylprednisolone 20-30 mg/kg per day for 5 days with a maximum dose of 1000 mg per day) followed by an oral prednisolone taper of 1-2 mg/kg/day (maximum 40-60 mg/day) over 4-6 wk. Other treatment options include intravenous immunoglobulin (usually 2 g/kg administered over 2-5 days) or plasmapheresis (typically 5-7 exchanges administered every other day) for refractory or severe cases. There is no consensus about the timing of these treatments for ADEM.
Prognosis
Most children experience full motor recovery after ADEM, but residual defects can be seen, and cognitive deficits or behavioral changes are not uncommon. Recovery starts within days to weeks, but symptoms can fluctuate.
Bibliography
Duman Ö, Yürekli VA, Gencpinar P, et al. Unusual clinical cases that mimic acute disseminated encephalomyelitis. Acta Clin Croat . 2015;54(3):371–377.
Krupp LB, et al. International pediatric multiple sclerosis study group criteria for pediatric multiple sclerosis and immune-mediated central nervous system demyelinating disorders: revisions to the 2007 definitions. Mult Scler . 2013;19:1261–1267.
Pohl D, Alper G, Van Haren K, et al. Acute disseminated encephalomyelitis: updates on an inflammatory CNS syndrome. Neurology . 2016;87:S38–S45.
Optic Neuritis
Cheryl Hemingway
Optic neuritis (ON) is defined as inflammation of one or both optic nerves. It presents with visual dysfunction. It can be idiopathic and can occur together with other systemic inflammatory conditions or inflammatory conditions of the CNS, such as ADEM, MS, or NMOSD.
Epidemiology and Clinical Presentation
ON is one of the most common of the acquired demyelinating syndromes and accounts for a quarter of all demyelinating presentations in childhood. The typical presentation is unilateral or bilateral visual loss over hours to days, abnormal color vision, visual field loss, and sometimes a relative afferent pupillary defect. The visual loss can be quite severe, with the majority of children at 20/200VA or worse. Periocular pain or pain with eye movement and, at times, a headache are common features. Bilateral ON is more common in younger children and is often associated with a preceding viral infection. Unilateral ON can also be followed in time by bilateral involvement. Funduscopic examination in some patients reveals optic nerve head swelling (papillitis), but in others inflammation occurs in the retrobulbar optic nerve portion and thus the appearance of the optic nerve is normal. Optic nerve pallor is often noted following an initial episode or in those with relapsing ON.
Diagnostic Evaluation
Electrodiagnostics, in particular visual evoked potentials (VEPs), may be helpful, with the VEPs often detecting prolonged latency. In the younger child, VEPs may also detect clinically silent episodes of ON in the opposite eye. Optical coherence tomography (OCT) can detect structural retinal change, such as retinal nerve fiber layer (RNFL) thinning, and may be helpful in monitoring the young child.
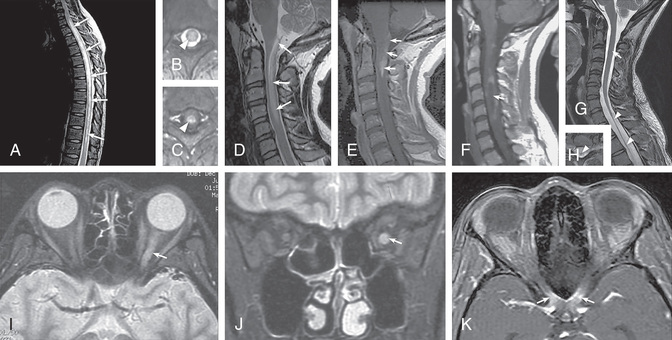
MRI of the orbits may be normal but usually shows, on T1-weighted images, thickened optic nerves, with an increased signal on T2-weighted scans (Fig. 618.3F and G ). Longitudinally extensive ON involving the chiasm is thought to be more commonly associated with antibody-mediated demyelination (see Fig. 618.3C ).
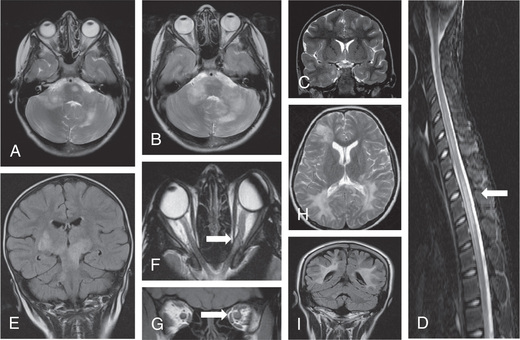
CSF analysis for OCBs is not always indicated, but this together with an MRI of the brain is very useful for predicting the risk of MS. In the face of a normal MRI brain scan and negative OCBs, the risk of developing MS is extremely low.
There are a number of conditions that can both mimic and be associated with ON. A detailed ophthalmologic review is essential, and depending on the history and clinical findings, investigations may be needed to exclude systemic rheumatologic disorders (e.g., systemic lupus erythematosus [SLE], sarcoidosis, Behçet disease), infectious diseases (viral disease, Lyme disease, syphilis, tuberculosis), mitochondrial disorders (e.g., Leber hereditary optic neuropathy), vascular events, or toxic, nutritional, or metabolic disorders. Antibody testing in serum for both AQP4-Ab and MOG-Ab is recommended to ensure that prophylactic treatment can be provided if indicated (e.g., AQP4-Ab positive) or to provide counseling on the risk of recurrence (MOG-Ab positivity).
Treatment
No randomized controlled trials have been conducted for pediatric ON, but the standard of care, based on clinical experience and adult trials, is high-dose intravenous steroids (typically methylprednisolone 20-30 mg/kg per day for 3-5 days, with a maximum dose of 1000 mg per day). In adults, the Optic Neuritis Treatment Trial (ONTT) showed that steroid administration led to a faster recovery, but no differences were seen in the long-term visual outcome. As with other severe episodes of demyelination, further treatment options include intravenous immunoglobulin (usually 2 g/kg administered over 2-5 days) or plasmapheresis (typically 5-7 exchanges administered every other day); there is no clear evidence of their benefit and no consensus about when to use them in isolated ON. Trials in adults have concentrated on neuroprotection; phenytoin has a beneficial effect on RNFL thinning in acute ON.
Prognosis
Reassuringly, full recovery of high-contrast visual acuity (HCVA) usually occurs in children, although irreversible damage is often detected in the structural integrity and may be evidenced by RNFL thinning on OCT, defective color vision, and impairments in low-contrast visual acuity (LCVA). Pediatric patients with AQP4-antibody–associated optic nerve demyelination are thought to more commonly be left with long-term visual disability than patients with other causes of ON.
Bibliography
Absoud M, Cummins C, Desai N, et al. Childhood optic neuritis clinical features and outcome. Arch Dis Child . 2011;96:860–862.
Bonhomme GR, Waldman AT, Balcer LJ, et al. Pediatric optic neuritis: brain MRI abnormalities and risk of multiple sclerosis. Neurology . 2009;72:881–885.
Bonhomme GR, Mitchell EB. Treatment of pediatric optic neuritis. Curr Treat Options Neurol . 2012;14:93–102.
Eyre M, Hameed A, Wright S, et al. Retinal nerve fibre layer thinning is associated with worse visual outcome after optic neuritis in children with a relapsing demyelinating syndrome. Dev Med Child Neurol . 2018 [Epub ahead of print].
Perez-Cambrodi RJ, Cubillana AGH, et al. Optic neuritis in pediatric population: a review in current tendencies of diagnosis and management. J Optom . 2014;7(3):125–130.
Waldman AT, Stull LB, Galetta SL, et al. Pediatric optic neuritis and risk of multiple sclerosis. J AAPOS . 2011;15:441–446.
Transverse Myelitis
Cheryl Hemingway
Transverse myelitis (TM) is a condition characterized by rapid development of both motor and sensory deficits at any level of the spinal cord. TM presents acutely as either partial or complete cord involvement with bilateral signs and in adults and older children with a clear sensory level. TM has multiple causes and can be idiopathic or secondary to either an immune-mediated condition (postinfectious or antibody driven) or as a result of direct infection (infectious myelitis). In TM, evidence of spinal cord inflammation can be demonstrated by an MRI-documented–enhancing lesion, CSF pleocytosis (>10 cells), or an increased immunoglobulin G (IgG) index. The progression is rapid, and the time to maximal disability is more than 4 hr and sooner than 21 days.
Epidemiology
TM is more common in adults but is estimated to affect around 2 per million children per year. A bimodal age distribution is observed in those younger than 5 yr and older than 10 yr. Although they represent a small subset, children 5 yr of age and younger develop spinal cord dysfunction over hours to a few days. They often have a history of an infectious disease, possibly of viral or mycoplasmal origin, or of an immunization within the few weeks preceding the development of their neurologic difficulties. The clinical loss of function is often severe and may seem complete. Although a slow recovery (weeks to months) is common in these cases, it is likely to be incomplete. The likelihood of independent ambulation in young children is approximately 40%. The pathologic findings of perivascular infiltration with mononuclear cells imply an infectious or inflammatory basis. Overt necrosis of the spinal cord may be seen and may be tied to specific etiologies, including infectious etiologies such as enterovirus infection.
In older children, the syndrome may be different, and outcomes may vary by etiology. Although the onset is also rapid, with a nadir in neurologic function occurring between 2 days and 2 wk, recovery is more rapid and more likely to be complete. In a small but important number of cases, necrosis and irreversible injury may occur. The condition can be associated with underlying etiologies, including systemic vasculitic entities (e.g., SLE), antibody-mediated CNS disorders (e.g., AQP4-Ab or MOG-Ab-associated NMOSD), infectious etiologies (e.g., mycoplasma, enterovirus), or idiopathic disease. Pathologic or imaging examinations show acute inflammation with demyelination in some cases. There is no sex or familial predisposition in those with idiopathic TM.
Acute flaccid myelitis is an idiopathic (probably caused by enterovirus D68 or D71) disorder presenting with paralysis or weakness, CSF pleocytosis, and MRI demonstrating myelitis with abnormalities often of the anterior horn gray matter. Paralysis may be asymmetric and is not usually accompanied by a sensory deficit; cranial nerve involvement may include facial weakness, dysarthria, and dysphagia.
Clinical Manifestations
TM is often preceded within the previous 1-3 wk by a mild nonspecific illness, minimal trauma, or perhaps an immunization. Discomfort or overt pain in the neck or back, depending on the level of the lesion, is common. Depending on its severity, the condition progresses to numbness, anesthesia, ataxia, areflexia, and motor weakness in the truncal and appendicular musculature at or distal to the lesion. Paralysis begins as flaccidity (paraparesis, tetraparesis), but over a few weeks, spasticity develops and is evidenced by hyperreflexia and clonus. Rarely is the weakness unilateral. Unilaterality suggests the presence of a hemicord lesion, associated most commonly with MS, and, as such, should raise suspicion for this disorder, particularly in adolescents with this presentation. Urinary retention is a common and early finding; incontinence occurs later in the course. Although most have sensory loss manifesting as anesthesia, paresthesia, or allodynia, early sensory findings may be isolated to the posterior column, emphasizing the importance of the evaluation of vibratory sensation. Other findings may include priapism or respiratory compromise, as well as spinal shock and subsequent autonomic dysreflexia.
Diagnostic Evaluation
Because acute TM is a diagnosis of exclusion, a thorough evaluation should be completed in all cases. The differential diagnosis includes, amongst other conditions, Guillain-Barré syndrome, demyelinating disorders, systemic rheumatologic conditions, meningitis and infectious myelitis, spinal cord infarction, arteriovenous malformations, trauma, mass lesions, bony and intervertebral disc distortion, abscess, and spine and spinal cord tumors (Table 618.5 ).
Table 618.5
Clinical and Radiologic Mimics of Transverse Myelitis
Modified from Thomas T, Branson HM: Childhood transverse myelitis and its mimics, Neuroimaging Clin North Am 23:267–278, 2013, Box 11.
MRI with and without contrast enhancement is essential to rule out a mass lesion requiring neurosurgical intervention. In both conditions, T1-weighted images of the spine at the anatomic level of involvement may be normal or may show distention of the spinal cord. In the infantile form, T2-weighted images show high signal intensity that extends over multiple segments. In the adolescent form, the high signal is often centrally located, involving the gray matter and the neighboring white matter. It may be limited to one or two segments but frequently extends over multiple segments. A limited degree of contrast enhancement after the administration of gadolinium is expected, especially in the infantile form, and denotes an inflammatory condition. Cervical and cervicothoracic lesions represent the majority of acute TM lesions. Axial cuts of the spinal cord are invaluable and can help to establish potential etiologies. Hemicord involvement may indicate MS. Holocord involvement with typical brain and optic nerve involvement may indicate NMOSD. If the involvement is predominantly of the gray matter, it may indicate a vasculitic or infectious process, including SLE or enterovirus infection. Nerve root enhancement is occasionally seen and should raise suspicion for a mixed picture (central and peripheral demyelination) or anterior horn cell involvement (Fig. 618.4 ). In up to 6% of presentations, MRI with 1.5T and 3T may not show spinal cord lesions. Repeat imaging at 7 days may show atrophy in these cases. MRI of the brain is also indicated. Evidence of other foci of demyelination is seen in at least 40% of patients, and depending on the lesion localization, MS, NMOSD, SLE, and enterovirus-associated acute flaccid myelitis should be considered. In patients with encephalopathy, ADEM must be considered.
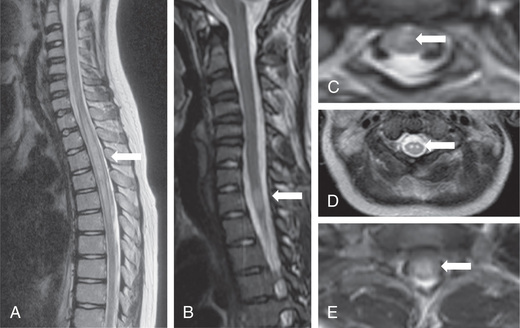
After exclusion of a mass lesion on MRI, a lumbar puncture is indicated. The number of mononuclear cells is usually elevated. The level of CSF protein may be elevated or normal. CSF should be analyzed for cells, protein, immunoglobulin index, OCBs, and infectious etiologies. The presence of inflammatory cells is essential for the diagnosis of TM.
Because one of the most important possibilities for this condition is neuromyelitis optica spectrum disorder (NMOSD), the serum of all patients should be analyzed for both AQP-4 and MOG antibodies. Older children with the condition should also have serum studies sent for other autoimmune disorders, especially SLE.
Treatment
There are no standards for the treatment of TM. Available evidence suggests that modulation of the immune response may be effective in decreasing the severity and duration of the condition. The use of high-dose steroids, particularly methylprednisolone, is the initial approach to treatment of the childhood forms of TM. If there is a poor response to high-dose steroids, other therapeutic approaches for acute intervention include intravenous immunoglobulin and plasma exchange. If the TM is secondary to an underlying antibody-driven disorder, treatments such as rituximab or cyclophosphamide can be considered. Long-term prophylactic therapy is recommended for children with recurrent forms of the disease (Table 618.6 ).
Table 618.6
| NUMBER OF LESIONS WITH OBJECTIVE CLINICAL EVIDENCE | ADDITIONAL DATA NEEDED FOR A DIAGNOSIS OF MULTIPLE SCLEROSIS | |
|---|---|---|
| ≥2 clinical attacks | ≥2 | None* |
| ≥2 clinical attacks | 1 (as well as clear-cut historical evidence of a previous attack involving a lesion in a distinct anatomic location † ) | None* |
| ≥2 clinical attacks | 1 | Dissemination in space demonstrated by an additional clinical attack implicating a different CNS site or by MRI ‡ |
| 1 clinical attack | ≥2 | Dissemination in time demonstrated by an additional clinical attack or by MRI OR demonstration of CSF-specific OCBs ‡ |
| 1 clinical attack | 1 |
Dissemination in space demonstrated by an additional clinical attack implicating a different CNS site or by MRI AND Dissemination in time demonstrated by an additional clinical attack or by MRI OR demonstration of CSF-specific OCBs ‡ |
* No additional tests are required to demonstrate dissemination in space and time. However, unless MRI is not possible, brain MRI should be obtained in all patients in whom the diagnosis of MS is being considered. In addition, spinal cord MRI or CSF examination should be considered in patients with insufficient clinical and MRI evidence supporting MS, with a presentation other than a typical clinically isolated syndrome or with atypical features. If imaging or other tests (e.g., CSF) are undertaken and are negative, caution needs to be taken before making a diagnosis of MS, and alternative diagnoses should be considered.
† Clinical diagnosis based on objective clinical findings for two attacks is most secure. Reasonable historical evidence for one past attack, in the absence of documented objective neurologic findings, can include historical events with symptoms and evolution characteristic for a previous inflammatory demyelinating attack; at least one attack, however, must be supported by objective findings. In the absence of residual objective evidence, caution is needed.
‡ The presence of CSF-specific OCBs does not demonstrate dissemination in time per se but can substitute for the requirement for demonstration of this measure.
If the 2017 McDonald criteria are fulfilled and there is no better explanation for the clinical presentation, the diagnosis is MS. If MS is suspected by virtue of a clinically isolated syndrome but the 2017 McDonald criteria are not completely met, the diagnosis is possible MS. If another diagnosis arises during the evaluation that better explains the clinical presentation, the diagnosis is not MS.
From Thompson AJ, Banwell BL, Barkhof F, et al: Diagnosis of multiple sclerosis: 2017 revisions of the McDonald criteria. Lancet 17:162-173, 2018.
Prognosis
Older children with acute TM have a better outcome than adults, with nearly 50% making a good recovery by 2 yr. This may reflect the higher likelihood of MOG-Ab–associated disorders in the older child. The most common sequelae in the remaining 50% are sensory problems and bladder dysfunction.
The treatment of acute flaccid myelitis has included steroids and IVIG; despite these therapies, patients often have an incomplete recovery.
Bibliography
Absoud M, et al. Pediatric transverse myelitis. Neurology . 2016;87:S46–S52.
Anderson EW, Kornberg AJ, Freeman JL, et al. Acute flaccid myelitis in childhood: a retrospective cohort study. Eur J Neurol . 2017;24(8):1077–1083.
Banwell B, Dale RC. Understanding risk of relapse and risk of disability after childhood transverse myelitis. Neurology . 2015;84:332–334.
Deiva K, Absoud M, Hemingway C, et al. Acute idiopathic transverse myelitis in children. Neurology . 2015;84:341–349.
Greenberg BM. Treatment of acute transverse myelitis and its early complications. Continuum (Minneap Minn) . 2011;17:733–743.
Mécharles S, Herrmann C, Poullain P, et al. Acute myelitis due to zika virus infection. Lancet . 2016;387:1481.
Messacar K, Schreiner TL, Van Haren K, et al. Acute flaccid myelitis: a clinical review of US cases 2012-2015. Ann Neurol . 2016;80:326–338.
Nelson GR, Bonkowsky JL, Doll E, et al. Recognition and management of acute flaccid myelitis in children. Pediatr Neurol . 2016;55:17–21.
Outteryck O, Baille G, Hodel J, et al. Extensive myelitis associated with anti-NMDA receptor antibodies. BMC Neurol . 2013;13:211.
Sorte DE, Poretti A, Newsome SD, et al. Longitudinally extensive myelopathy in children. Pediatr Radiol . 2015;45:244–257.
Tyler KL. Rationale for the evaluation of fluoxetine in the treatment of enterovirus D68-associated acute flaccid myelitis. JAMA Neurol . 2015;72(5):493–494.
Multiple Sclerosis
Cheryl Hemingway
Multiple sclerosis (MS) is a chronic demyelinating autoimmune disorder of the brain, spinal cord, and optic nerves characterized by a relapsing–remitting course of neurologic events without encephalopathy separated in time (i.e., more than one episode of at least 24 hr at least 30 days apart) and space (i.e., in more than one CNS region) (Table 618.6 ). When presenting for the first time in those under 18 yr, it is known as pediatric-onset MS (POMS). Recurrent events lead to progressive accumulation of both physical and cognitive disability and brain atrophy.
Epidemiology and Risk Factors
POMS is rare, with an estimated incidence in northern countries such as the United Kingdom and Canada of 1-2 per million under 16 yr of age. Around 10% of MS patients report in retrospect that they experienced their first symptoms before the age of 18 yr. Before puberty, the condition appears to affect males and females equally, but after puberty there is almost a 2 : 1 female predominance. Almost always, POMS presents as the relapsing-remitting form, and features suggestive of primary progressive MS should prompt careful evaluation for alternative conditions (Table 618.7 ).
Table 618.7
Differential Diagnosis of Multiple Sclerosis: Selected Disorders with a Progressive Course
| CLINICAL FEATURES | MRI FINDINGS | CSF FINDINGS | OTHER INVESTIGATIONS | |
|---|---|---|---|---|
| HTLV1-associated myelopathy | Progressive myelopathy; residence or travel to an endemic area (especially West Indies or Japan) | Spinal cord atrophy (thoracic more than cervical); T2-hyperintense brain lesions in some patients | OCBs sometimes present | CSF HTLV1 antibody testing |
| Dural arteriovenous fistula | Subacute, progressive myelopathy | Extensive spinal cord T2 hyperintensity often extending to conus, with or without gadolinium enhancement; dilated veins over dorsal surface of cord (often subtle); brain MRI normal | OCBs absent | Spinal angiography |
| Nutritional myelopathy (vitamin B12 or copper deficiency) | Subacute progressive myelopathy or myeloneuropathy; optic atrophy (severe vitamin B12 deficiency); anemia or pancytopenia | T2 hyperintensity of upper cervical cord classically affecting posterior columns; brain MRI normal | OCBs absent | Serum B12 , methylmalonic acid; serum copper levels, ceruloplasmin |
| Primary lateral sclerosis (or upper motor neuron predominant ALS) | Spastic quadriparesis or hemiparesis; with or without bulbar involvement; with or without development of lower motor neuron signs | MRI normal or showing T2 hyperintensity in corticospinal tracts | OCBs absent | Electromyography looking for lower motor neuron involvement |
| Leukodystrophies: adrenomyeloneuropathy; Krabbe disease; Alexander disease; hereditary diffuse leukoencephalopathy with axonal spheroids | Progressive myelopathy (adrenomyeloneuropathy, Krabbe disease); bulbar symptoms, ataxia (Alexander disease); early cognitive impairment (hereditary diffuse leukoencephalopathy with axonal spheroids) | Highly variable; diffuse, symmetric T2 hyperintensity sparing subcortical U fibers; with posterior hemispheric predominance (adrenomyeloneuropathy); spinal cord MRI normal or showing atrophy | OCBs absent | Very-long-chain fatty acids (adrenomyeloneuropathy); genetic testing available for some leukodystrophies |
| Hereditary spastic paraplegia (especially SPG5 ) | Slowly progressive myelopathy (spasticity greater than weakness) with or without other neurologic symptoms and family history | Spinal cord atrophy; supratentorial and infratentorial white matter lesions (SPG5); atrophy of corpus callosum | OCBs absent | Genetic testing |
| Spinocerebellar ataxias | Progressive cerebellar ataxia, with or without other neurologic symptoms and family history | Early, prominent cerebellar findings, with or without spinal cord, atrophy | OCBs absent | Genetic testing |
CSF, cerebrospinal fluid; HTLV1, human T-lymphotropic virus type 1; OCB, oligoclonal band; ALS, amyotrophic lateral sclerosis.
From Brownlee WJ, Hardy TA, Fazekas F, Miller DH: Multiple sclerosis 1: diagnosis of multiple sclerosis: progress and challenges. Lancet 389:1336-1346, 2017, Table 2, p. 1341.
In adults, a possible complex interplay of environmental (e.g., sunlight, low vitamin D status, obesity, and toxins), infectious (e.g., Epstein-Barr virus exposure), and genetic factors (e.g., HLADRB1*15:01, obesity) are thought to influence MS susceptibility. Studies in pediatrics have so far confirmed the role of some but not all of the above factors, and it may be that environmental factors in pediatric MS are more important than genetic factors.
Pathogenesis
Immune system dysregulation involving T and B lymphocytes triggers inflammation, axonal demyelination, axonal loss, and regeneration within both white and gray matter. Inflammatory infiltrates within actively demyelinating lesions of relapsing-remitting MS are targets for DMTs.
Clinical Manifestations
Presenting symptoms in pediatric MS are polyregional in more than half of patients and include focal sensory loss or other paresthesia (39–63%); cerebellar symptoms such as ataxia or dysarthria (44–55%); unilateral or, less often, bilateral pain on eye movements and reduced visual acuity (ON) (36–38%); brainstem symptoms in 30–31%, and in 29–50%, motor deficits, including focal deficits, hemiparesis, paraparesis, and bowel/bladder dysfunction (from TM or other spinal lesions). Encephalopathy is not seen apart from when there is significant brainstem involvement.
Imaging and Laboratory Findings
Brain MRI exhibits typically discrete, ovoid, asymmetric T2 lesions in cerebral white matter, particularly in the periventricular regions, as well as juxtacortical, cortical, brainstem, cerebellar, and, less commonly, the deep gray matter (Fig. 618.5 ). Spine MRI typically, when involved, shows partial-width cord lesions restricted to 1-2 spine segments. Longitudinally extensive lesions are more likely to occur in NMOSD (associated with MOG-Ab and AQP4-Ab) than in MS. CSF may be normal or exhibit mild lymphocytosis, particularly in younger children. CSF OCBs are positive in CSF but not in serum (type 2 pattern) in more than 90% of pediatric MS patients, but are usually negative (type 1 pattern) or present in both CSF and serum (type 4 pattern) in NMOSD. Abnormal evoked potential studies can localize disruptions in visual, auditory, or somatosensory pathways.
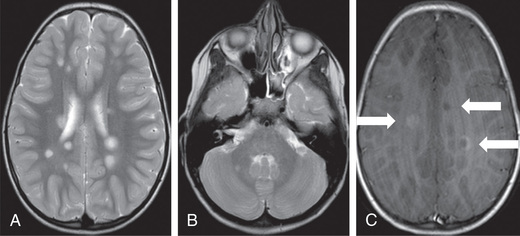
Diagnosis and Differential Diagnosis
Pediatric MS can usually be diagnosed following two demyelinating episodes without encephalopathy localizing to distinct CNS regions, lasting longer than 24 hr and separated by more than 30 days, provided no other plausible explanation exists. MS diagnostic criteria use an MRI to serve as a surrogate for recurrent demyelination, enabling MS diagnosis after the first event. For adults and children the initial MRI may be sufficient to diagnose MS if it demonstrates dissemination in space (≥2 T2 lesions involving juxtacortical, periventricular, infratentorial, or spine regions) and time (presence of gadolinium-enhancing lesion and nonenhancing T2 lesion in same scan). Alternatively, MS can be diagnosed with a follow-up MRI at any time interval exhibiting accumulation of T2 or gadolinium-enhancing lesions in the brain or spine. The 2017 McDonald diagnostic criteria allow the presence of intrathecal OCBs to substitute for dissemination in time (see Table 618.1 ). Challenges may arise in distinguishing a first attack of pediatric MS from other acquired demyelinating syndromes, in particular those associated with known antibodies (e.g., AQP4-Ab or MOG-Ab) or ADEM (Table 618.8 and see Table 618.4 ).
Table 618.8
Differential Diagnosis of Multiple Sclerosis: Clinical, MRI, and Serologic Findings of the Main Disorders That Can Resemble Relapsing-Remitting Disease
| NEUROLOGIC FEATURES | MRI FEATURES | BLOOD TEST AND CSF FINDINGS | |
|---|---|---|---|
| Acute disseminated encephalomyelitis (typically found in children) | Similar to MS symptoms but encephalopathy is typical; also multifocal symptoms | Large spectrum from small punctate lesions to tumefactive lesions with mass effect, in the supratentorial or infratentorial white matter, bilateral, and asymmetric; involvement of cerebral cortex, deep gray matter, brainstem, and spinal cord; enhancement | CSF pleocytosis; serum antibody to MOG |
| Neuromyelitis optica spectrum disorders | Concomitant or concurrent (severe) ON and TM; nausea and vomiting; paroxysmal tonic spasms | Longitudinally extensive spinal cord lesion (>3 vertebral segments); optic chiasmal involvement; pencil-thin ependymal enhancement and cloud-like enhancement | Serum antibody to AQP4 and to MOG; sometimes, mild pleocytosis; CSF OCBs infrequent |
| Neurosarcoidosis | Cranial nerve involvement (primarily facial and optic nerve); headache; raised intracranial pressure; meningitis; seizures; myelopathy | Meningeal enhancement with pituitary, hypothalamic, and cranial nerve involvement; brain white matter lesions; simultaneous enhancement of all lesions | Raised serum and CSF ACE (not sensitive or specific for sarcoidosis); CSF OCBs sometimes present |
| CNS vasculitis | Confusion, headache, personality change; seizures; stroke-like symptoms | Ischemic, multiple lesions; predominance of lesions at corticosubcortical junction; intracranial hemorrhage; meningeal enhancement; simultaneous enhancement of all lesions; microbleeds | Serum antineutrophil cytoplasmic antibodies; CSF OCBs sometimes present |
| Susac syndrome | Visual loss; sensorineural hearing loss; encephalopathy; headache; memory loss; behavioral disturbances | Focal and small lesions in supratentorial and infratentorial regions (both white matter and gray matter); involvement of corpus callosum (snowball lesions); leptomeningeal enhancement | CSF OCBs usually absent |
| Hypoxic-ischemic vasculopathies (in particular small-vessel disorder) | Stroke events; cognitive decline; focal neurologic signs; gait disturbance | Punctate and peripheral white matter lesions, sparing U fibers; symmetric and confluent periventricular lesions; lacunar infarcts; involvement of central transverse fibers in pons; microbleeds | Serum testing for vascular risk factors (diabetes, hypercholesterolemia); CSF OCBs absent |
| Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL) | Migraine; stroke events; psychiatric problems and dementia | Temporal pole lesions; external capsule and U-fiber lesions; microbleeds | CSF OCBs absent; testing for NOTCH3 gene mutation |
| Connective tissue disorders (SLE, Sjögren syndrome, antiphospholipid antibodies syndrome) | Optic nerve, brain, and spinal cord involvement; neuropsychiatric symptoms; seizures; ischemic episodes | Brain infarcts and hemorrhage; basal ganglia lesions; punctate (subcortical) lesions; spinal cord lesions; cerebral venous sinus thrombosis; parotid gland involvement in Sjögren syndrome | Serum antinuclear antibody; extractable nuclear antigens (in particular, anti SS-A(Ro) and SS-B(La) antibodies for Sjögren syndrome, and anti-Sm for SLE); CSF OCBs usually absent |
| Neuro-Behçet disease | Brainstem syndrome; myelopathy; meningoencephalitis | Large brainstem lesions; basal ganglia, subcortical white matter, and spinal cord lesions; gadolinium enhancement; cerebral venous sinus thrombosis | HLA-B5; CSF pleocytosis; CSF OCBs usually absent |
| Chronic lymphocytic inflammation with pontine perivascular enhancement responsive to steroids (CLIPPERS) | Cranial nerve dysfunction and long-tract signs; symptoms referable to brainstem or cerebellar dysfunction; spinal cord syndrome; cognitive dysfunction | Multiple punctate, patchy, and linear regions of gadolinium enhancement relatively confined to pons; lesions also involving cerebellum, basal ganglia, supratentorial white matter, brainstem, and spinal cord | CSF OCBs sometimes present |
| Fabry disease | Stroke events; vertigo | Posterior infarcts; multiple white matter lesions with pulvinar involvement (T1 hypointense lesions) | Reduced activity of GLA enzyme; analysis of GLA gene |
| Leber hereditary optic neuropathy | Bilateral sequential optic neuropathies with poor visual recovery; more common in men than women | Normal or might show white matter lesions (Harding disease) | OCBs absent; genetic testing |
Infectious diseases are not included in this table but should be considered, especially in cases of atypical demyelinating lesions. CSF, cerebrospinal fluid; ACE, angiotensin-converting enzyme; GLA, α galactosidase A; OCB, oligoclonal band.
Modified from Thompson AJ, Baranzini SE, Geurts J, et al: Multiple sclerosis, Lancet 391:1622-1636, 2018, Table 3, pp. 1628-1629.
Treatment
Relapses causing functional disability may be treated with, 20-30 mg/kg/day (maximum 1000 mg/day) for 3-5 days, with or without prednisolone taper. DMTs reduce the relapse frequency and T2 lesion load, mainly by targeting the inflammatory response that predominates during the relapsing-remitting phase of MS. There are now a large number of available treatment options, including injectable treatments, oral medications, and infusions. They range from immunomodulating to immunosuppressing options, and the choice and sequencing are becoming highly specialized (Table 618.9 ). Almost all DMT use in pediatrics is off label, and a number of randomized controlled trials are currently in progress. The only medication with U.S. FDA approval at the present time is fingolimod, following completion of a 2 yr randomized, double-blind, controlled trial between oral fingolimod and intramuscular interferon-beta-1α. This trial demonstrated that in those between 10 and 18 yr of age with POMS, fingolimod reduced the annualized relapse rate by 82% when compared with interferon-beta-1α. This efficacy is greater than that seen in adults, possibly due to the greater inflammatory nature of POMS. This is one of the reasons that prompt initiation of treatment is recommended for all those diagnosed with POMS.
Table 618.9
Overview of Available and Emerging Therapies Used in Pediatric Multiple Sclerosis and Other Relapsing Demyelinating Disorders
| MEDICATION AND ROUTE OF ADMINISTRATION | MEDICATION CLASS | MECHANISM IN MS | COMMONLY REPORTED OR SERIOUS SIDE EFFECTS | EFFICACY |
|---|---|---|---|---|
| FIRST-LINE THERAPIES APPROVED FOR MS IN ADULTS | ||||
|
Interferon-β-1a and β-1b (subcutaneous or intramuscular injection on alternate days, 3 times weekly, weekly or bimonthly depending on preparation) |
Immunomodulator | Modulates T cells and cytokine production | Injection site reaction; flu-like symptoms; headache, muscle aches, transaminitis; leukopenia; tissue necrosis at injection site (rare) | ~33% decrease in ARR and slows progression of disability |
|
Glatiramer acetate (daily or 3 times weekly, subcutaneous injection) |
Immunomodulator | Modulates T-cell response by altering antigen presentation | Injection site reactions; transient flushing, chest tightness and shortness of breath. Lipodystrophy at injection sites | ~33% decrease in ARR and slows progression of disability |
|
Dimethyl fumarate (DMF) (oral medication 12 hourly with food i.e, twice a day) |
Immunomodulator | Unclear mechanism but modulates cytokine production and decreases lymphocyte count. Neuroprotectant; antioxidant | Flushing reaction; GI upset; headache; proteinuria, leukopenia. Rare reports of PML in those with severe prolonged lymphopenia | Reduces number of relapses by ~ 50% compared with placebo in adults. Pediatric trials ongoing |
| SECOND-LINE THERAPIES APPROVED FOR MS IN ADULTS | ||||
| Teriflunomide | Immunomodulator | Impairs DNA proliferation via pyrimidine synthesis inhibition and decreases T and B cells | Infections; headaches; diarrhea; transaminitis; alopecia; teratogenicity | Pediatric trials ongoing |
|
Natalizumab (infusion over 2-3 hr every 4 wk) |
Monoclonal antibody | Targets α4 -integrin on vascular endothelium and prevents T- and B-cell migration into CNS and other tissues | Infusion reactions with headache, dizziness, rash; rare anaphylaxis. May affect liver function. Risk of PML able to be stratified by JC virus status, length of treatment, and previous treatments. Immune reconstitution syndrome after discontinuation; melanoma | Reduces number of relapses by ~,70% in adults |
|
Fingolimod (daily oral medication: first dose, cardiac monitoring required and need to ensure good compliance because of risks of first-dose bradycardia and heart block) |
Immunomodulator | Modulates sphingosine-1-phosphate receptors; causes T-cell sequestration in lymphoid compartments | First-dose bradycardia; cardiac arrhythmia; systemic viral infection; persistent lymphopenia with risk of severe herpetic and varicella infection; macular edema; transaminitis; basal cell carcinoma. Rare cases of PML | FDA approved for pediatrics May 2018 following trial showing 82% decrease in ARR compared with interferon β |
|
Alemtuzumab (infusions 2 courses: first for 5 consecutive days; second 12 mo later for 3 consecutive days) |
Monoclonal antibody | Anti-CD52 antibody target; depletes mature T cells | Infusion reactions; opportunistic infection, secondary autoimmune disorders, including thyroiditis (50% risk), immune thrombocytopenia (1%), Goodpasture syndrome. Need monthly blood tests for 4 yr after last course. | Highly effective in adults; ~ 55% decrease in ARR compared with interferons. Pediatric trial ongoing |
|
Cladribine (oral tablets 2 courses: first for 4-5 consecutive days during mo 1 and 2; second as before 12 mo later) |
Immunomodulator | Selective activity against CD4 and CD8 T cells and CD19 B cells via adenosine deaminase activity | Neutropenia, lymphopenia, infection, oral herpes, GI disorders, and rash |
Reduced relapses by ~ 58% vs placebo in adults and delay in disability progression. No evidence as yet in pediatrics. |
|
Rituximab (infusions given 2 wk apart ~ every 6 mo) |
Monoclonal antibody | Targets CD20, a marker of immature B cells; depletes B-cell populations | Infusion-related side effects; hepatitis, PML (rate undefined). | Used off label for adult MS; no efficacy assessments available in pediatric MS |
|
Ocrelizumab (infusions given 2 wk apart ~ every 6 mo) |
Monoclonal antibody | Targets CD20, a marker of immature B cells; depletes B-cell populations | Headache; infusion-related side effects; theoretic risk of PML (undefined) and, possibly, malignancy | In adult MS showed decrease in ARR of 50% compared with interferons; no evidence in pediatric MS to date |
|
Laquinimod (daily oral medication) |
Immunomodulator | Modulates T-cell and cytokine production; antiinflammatory; possibly neuroprotective | Transaminitis, back pain, headache | In adult MS ARR of 20–25% compared with placebo; no data in pediatric use to date |
| OTHER MEDICATIONS USED FOR DEMYELINATING DISORDERS | ||||
|
Azathioprine (intravenous infusion or oral tablets daily) |
Chemotherapeutic | Disrupts purine metabolism; effects include cytotoxic immune cell depletion | GI side effects, alopecia, bone marrow suppression, and blood dyscrasias, transaminitis, infections, secondary malignancy, alopecia. Increased side effects with low TPMT enzyme activity | No efficacy assessments available in pediatric MS, small retrospective studies for NMOSD |
|
Cyclophosphamide (intravenous infusion or oral tablets daily) |
Chemotherapeutic | DNA alkylation; effects include cytotoxic immune cell depletion | Hemorrhagic cystitis; bladder cancer; late-onset malignancy; infection; infertility | No efficacy assessments available in pediatric demyelination |
|
Mycophenolate mofetil (MMF) (intravenous infusion or oral tablets twice daily) |
Immunosuppressant | Disrupts purine synthesis and impairs B- and T-lymphocyte proliferation | GI side effects, alopecia, bone marrow suppression and blood dyscrasias, transaminitis, infections, secondary malignancy, alopecia. Teratogenic. | |
| Vitamin D | Vitamin/hormone | Modulates immune cell expression | Hypercalcemia and kidney stones at serum 25(OH) vitamin D level > 100 ng/mL | Prospective trials in pediatric and adult MS are currently underway |
CNS, central nervous system; ARR, annualized relapse rate; MS, multiple sclerosis; JC virus, John Cunningham virus; PML, progressive multifocal leukoencephalopathy; TPMT, thiopurine methyltransferase; GI, gastrointestinal.
The debate about DMT use for both adult and pediatric patients concerns whether one should start with the safer, less efficacious first-line agents and escalate if treatment fails, or whether remission should first be induced with the more effective treatments and then the patient maintained on safer medications. Adult trials to answer this question are underway. Currently, the more efficacious treatments are generally used only for those with highly active MS, although one could argue that with the increased inflammatory activity, higher relapse rate, and young age at which disability occurs, pediatric-onset MS in particular may benefit from high-efficacy early treatment (HEET).
Prognosis
Studies of pediatric MS prior to widespread DMT use suggested a higher relapse rate but slower rate of disease progression compared with adults. Despite this longer time to irreversible disability (20-30 yr), pediatric MS patients acquire disability at a younger age than adults owing to the earlier age of onset of disease. Similar to adults with MS, pediatric MS patients can acquire fixed neurologic deficits affecting the visual and other cranial nerves, motor and sensory function, balance, and bowel/bladder function. Children with MS have also been shown to have an overall smaller head size, brain volume, and thalamic volume in particular. This can be attributed to gray matter degeneration, and cognitive disability can be demonstrated in 30–50% of young people with pediatric-onset MS, more than that seen in adult-onset MS.
Fatigue is a major symptom in pediatric MS that can lead to a poor quality of life. It is important to address this together with other factors, such as mood, sleep quality, and sleep hygiene. Pharmacologic management of fatigue is challenging, but psychology-based therapy with cognitive behavioral therapy and pacing has been shown to be effective.
Bibliography
Absoud M, Cummins C, Chong WK, et al. Paediatric UK demyelinating disease longitudinal study (PUDDLS). BMC Pediatr . 2011;11:68.
Aubert-Broche B, Fonov V, Narayanan S, et al. Onset of multiple sclerosis before adulthood leads to failure of age-expected brain growth. Neurology . 2014;83:2140–2146.
Aliaga E, Barkhof F. MRI mimics of multiple sclerosis. Handb Clin Neurol . 2014;122:291–316.
Baumann M, Hennes EM, Schanda K. Children with multiphasic disseminated encephalomyelitis and antibodies to the myelin oligodendrocyte glycoprotein (MOG): extending the spectrum of MOG antibody positive diseases. Mult Scler . 2016;22(14):1821–1829.
Brownlee WJ, Hardy TA, Fazekas F, Miller DH. Multiple sclerosis 1: diagnosis of multiple sclerosis: progress and challenges. Lancet . 2017;389:1336–1346.
Comi G, Radaelli M, Sørensen PS. Evolving concepts in the treatment fo relapsing multiple sclerosis. Lancet . 2017;389:1347–1356.
Fadda G, Brown RA, Longoni G, et al. MRI and laboratory features and the performance of international criteria in the diagnosis of multiple sclerosis in children and adolescents: a prospective cohort study. Lancet Child Adolesc . 2018;2:191–204.
Graves JS, Barcellos LF, Shao X, et al. Genetic predictors of relapse rate in pediatric MS. Mult Scler . 2016;22:1528–1535.
Hacohen Y, Mankad K, Chong WK, et al. Diagnostic algorithm for relapsing acquired demyelinating syndromes in children. Neurology . 2017;89(3):269–287.
Hennes EM, Baumann M, Lechner C, Rostasy K. MOG spectrum disorders and role of MOG-antibodies in clinical practice. Neuropediatrics . 2018;49(1):3–11.
Huppke P, Rostasy K, Karenfort M, et al. Acute disseminated encephalomyelitis followed by recurrent or monophasic optic neuritis in pediatric patients. Mult Scler . 2013;19:941–946.
Kappos L, Bar-Or A, Cree BAC, et al. Siponimod versus placebo in secondary progressive multiple sclerosis (EXPAND): a double-blind, randomised, phase 3 study. Lancet . 2018;391:1263–1272.
Karussis D, Petrou P. The spectrum of post-vaccination inflammatory CNS demyelinating syndromes. Autoimmun Rev . 2014;13:215–224.
Kitley J, Waters P, Woodhall M, et al. Neuromyelitis optica spectrum disorders with aquaporin-4 and myelin-oligodendrocyte glycoprotein antibodies: a comparative study. JAMA Neurol . 2014;71:276–283.
McGinley M, Rossman IT. Bringing the HEET: the argument for High-efficacy early treatment for Pediatric-onset multiple sclerosis. Neurother . 2017;14:985–998.
Olsson T, Barcellos LF, Alfredsson L. Interactions between genetic, lifestyle and environmental risk factors for multiple sclerosis. Nat Rev Neurol . 2017;13:25–36.
Ontaneda D, Thompson AJ, Fox RJ, Cohen JA. Progressive multiple sclerosis: prospects for disease therapy, repair, and restoration of function. Lancet . 2017;389:1357–1366.
Rae-Grant A, Day GS, Marrie RA, et al. Comprehensive systematic review summary: disease-modifying therapies for adults with multiple sclerosis. Neurology . 2018;90:789–800.
Rae-Grant A, Day GS, Marrie RA, et al. Practice guideline recommendations summary: disease-modifying therapies for adults with multiple sclerosis. Neurology . 2018;90:777–788.
Reich DS, Lucchinetti CF, Calabresi PA. Multiple sclerosis. N Engl J Med . 2018;378(2):169–180.
Thompson AJ, Banwell BL, Barkhof F, et al. Diagnosis of multiple sclerosis: 2017 revisions of the McDonald criteria. Lancet . 2018;17:162–173.
Thompson AJ, Baranzini SE, Geurts J, et al. Multiple sclerosis. Lancet . 2018;391:1622–1636.
Neuromyelitis Optica Spectrum Disorders
Cheryl Hemingway
Neuromyelitis optica spectrum disorders (NMOSDs) classically present with episodes of ON and/or longitudinally extensive TM. The discovery of pathogenic antibodies to the astrocyte water channel protein aquaporin-4 (AQP4) and the incorporation of these antibodies into the 2015 revised diagnostic criteria for NMOSDs have not only enabled clinicians to clearly distinguish AQP4-Ab–related disorders from other demyelinating conditions but has also widened the spectrum of the group of disorders to include brainstem syndromes (e.g., area postrema syndrome) and recurrent forms of ON and TM (see Table 618.1 ). MOG-Ab has recently been identified in many of the antibody-negative presentations, with no reports of both antibodies being present in a single individual.
Epidemiology
AQP4-Ab–positive NMOSD presents usually in the older adult, with MOG-Ab NMOSD much more common in children and young people, but both can occur across a wide age spectrum. Population studies vary significantly but suggest a pediatric incidence for NMOSDs of 0.5–4.5%. AQP4-Ab–driven NMOSD is significantly more common in females than in males, with MOG-Ab–associated disorders having only a slight female preponderance. NMOSD is also more common in Asians than in blacks or whites and appears to have a higher mortality rate in individuals of African descent than in others. Although most cases of NMO are idiopathic and only occasional familial cases have been reported, there are a few known genetic risk factors, including the HLA-DRB1*0301 allele and a single-nucleotide polymorphism in CD58, which have been associated with NMOSDs in specific population groups.
Pathogenesis
The water channels, against which the AQP4-IgG antibody is directed, are most abundant on the astrocyte foot processes within the periventricular regions, brainstem, optic nerves, and spinal cord. Antibody, mainly of the IgG1 subtype, binds to the extracellular loops of the AQP4 protein activating the classical complement pathway with C5b-C9 components, leading to leukocyte attraction and degranulation and causing astrocyte death. Chemokines from dying astrocytes and activated leukocytes attract macrophages, leading to death of oligodendrocytes and neurons, with subsequent necrosis or even cavitation in affected tissues.
Clinical Manifestations
NMOSD presents most commonly with ON, TM, or an area postrema syndrome such as intractable vomiting or hiccups. The symptoms and signs of TM depend on the spinal level and completeness of the inflammatory changes. ON or TM may occur simultaneously or may be separated in time by weeks or even years. Some patients present with seizures and encephalopathy mimicking ADEM. Others exhibit endocrinopathies such as the syndrome of inappropriate antidiuretic hormone secretion, diabetes insipidus or hyperinsulinemia, disrupted puberty, or obesity. NMOSD has also been associated with other autoimmune conditions, such as SLE, Sjögren syndrome, diabetes, and thyroiditis.
Imaging and Laboratory Findings
Neuroimaging studies should include the entire spine, optic nerves if visual symptoms are present, and brain. Brain imaging can be normal, may have subtle changes in the white matter tracts, or can demonstrate large, hazy, ill-defined white matter lesions and/or gray matter involvement, such as thalamic lesions. Brain lesions frequently localize to areas of high AQP4-ab expression, such as the periaqueductal gray matter, dorsal brainstem, and diencephalon (Fig. 618.6 ). Spinal imaging may reveal short or longitudinally extensive TM; longitudinally extensive ON involving the chiasm is more common in MOG-Ab disease but can occur in both. Imaging can usually be distinguished from MS by the absence of discrete, well-defined oval lesions in the periventricular white matter, but AQP4-Ab and MOG-Ab disease are often not able to be reliably differentiated on imaging.
AQP4-Ab can be found in both the serum and CSF, with the serum being more sensitive. MOG-Ab also is more commonly positive in serum, implying extrathecal production of antibody. If there is a high clinical suspicion of an antibody-driven disorder and a negative test, it is important to recognize that there are a number of different methods of antibody testing and the sensitivity of the assays varies, so repeat testing may be indicated. CSF in patients with NMOSD often has a number of white blood cells, with a higher cell count seen in MOG-Ab. Unlike MS, CSF in NMOSD is usually negative for OCBs.
Diagnosis and Differential Diagnosis
The International Panel for NMO Diagnosis (IPND) published new criteria for NMOSD in 2015. They placed a high emphasis on the presence or absence of AQP4 antibody (see Table 618.1 ). In seropositive patients, once alternative diagnoses have been excluded, only one core clinical criterion is required: ON, TM, area postrema syndrome, narcolepsy, or diencephalic syndrome with compatible MRI lesions. If AQP4-ab negative, the diagnosis is more stringent, with two core clinical criteria required.
The differential diagnosis includes other demyelinating disorders, such as MS or ADEM; vasculitis and rheumatologic disorders including SLE, Behçet disease, and neurosarcoidosis (usually accompanied by other nonneurologic manifestations); idiopathic TM, tropical spastic paraparesis, and viral encephalomyelitis (none of which have NMO antibodies in the serum or CSF); genetic disorders such as hemophagocytic lymphohistiocytosis (HLH) or mutation in the DARS gene; metabolic causes such as biotinidase deficiency and riboflavin-responsive conditions; idiopathic causes of isolated ON, or other acute forms of monocular or binocular visual loss (Table 618.10 ; see also Chapter 649 ). Additional considerations, depending on the location of the lesions, include lymphoma, Langerhans cell histiocytosis, tuberculosis, and vitamin B12 or E deficiencies.
Table 618.10
Red Flags: Findings Atypical for NMOSD*
* These are some common or key findings that should prompt a thorough investigation for competing differential diagnoses before making a diagnosis of NMOSD.
LETM, longitudinally extensive transverse myelitis lesions; MS, multiple sclerosis; NMO, neuromyelitis optica; NMOSD, neuromyelitis optica spectrum disorders.
From Wingerchuk DM, Banwell B, Bennett JL, et al: International consensus diagnostic criteria for neuromyelitis optica spectrum disorders, Neurology 85:177-189, 2015, Table 2, p. 180.
Treatment
Treatment involves (1) acute and longer-term removal of the antibody, (2) minimizing CNS injury, and (3) treating symptoms. Initial episodes and relapses may be treated acutely with methylprednisolone, 20-30 mg/kg/day (maximum 1000 mg/day) usually for 5 days, but for a severe attack, this can be extended. An oral taper is recommended, especially if antibody results are not available at the time of discharge. If there is minimal improvement acutely, plasma exchange (PLEX) can be considered either before or after IVIG (2 g/kg over 2-5 days) and a repeat course of steroids. Rituximab can be used both acutely and to prevent further relapses.
In adult AQP4-Ab–positive NMOSD, effective DMT options include azathioprine, mycophenolate mofetil (MMF), or rituximab. Small retrospective studies in AQP4-Ab–positive pediatric NMOSD have confirmed benefit in reducing the relapse rate with both MMF and rituximab. Preliminary evidence in adults suggests that eculizumab, a monoclonal antibody against the C5 complement protein, reduces recurrences and may improve disability in patients with severe NMOSD. A pilot study of tocilizumab, an anti-IL-6 monoclonal antibody, demonstrated efficacy in AQP4-NMOSD in adults. Trials are currently ongoing with satralizumab, a humanized IL-6 monoclonal antibody, in both adults and children. Medications used for treatment of MS are either ineffective or can exacerbate relapses, again highlighting the importance of an accurate diagnosis.
Prognosis
The majority of AQP4-positive patients have a relapsing phenotype with progressive accrual of disability, whereas those with MOG-Ab-positive NMOSD can be monophasic. In the relapsing phenotypes, the relapse rate is higher in those with AQP4-Ab disorders than MOG-Ab, and some studies show a better recovery and long-term prognosis for MOG-Ab–associated disorders. Similar to adults with NMOSD, pediatric patients are often left with fixed neurologic deficits affecting the visual acuity, visual fields, color vision, motor and sensory function, balance, and bowel/bladder function, and the best outcomes are achieved with a multidisciplinary team.
Bibliography
Alvarenga MP, Fernandez O, Leyva L, et al. The HLA DRB1*03:01 allele is associated with NMO regardless of the NMO-IgG status in Brazilian patients from rio de janeiro. J Neuroimmunol . 2017;310:1–7.
Asgari N, Jarius S, Laustrup H, et al. Aquaporin-4-autoimmunity in patients with systemic lupus erythematosus: a predominantly population-based study. Mult Scler . 2018;24(3):331–339.
Banwell B, Bar-Or A, Arnold DL, et al. Clinical, environmental, and genetic determinants of multiple sclerosis in children with acute demyelination: a prospective national cohort study. Lancet Neurol . 2011;10:436–445.
Dale RC, Brilot F, Duffy LV, et al. Utility and safety of rituximab in pediatric autoimmune and inflammatory CNS disease. Neurology . 2014;83:142–150.
Hacohen Y, Absoud M, Woodhall M, et al. Autoantibody biomarkers in childhood-acquired demyelinating syndromes: results from a national surveillance cohort. J Neurol Neurosurg Psychiatry . 2014;85:456–461.
Hacohen Y, Messina S, Gan HW, et al. Endocrinopathies in paediatric-onset neuromyelitis optica spectrum disorder with aquaporin 4 (AQP4) antibody. Mult Scler . 2018;24:679–684.
Jurynczyk M, Geraldes R, Probert F, et al. Distinct brain imaging characteristics of autoantibody-mediated CNS conditions and multiple sclerosis. Brain . 2017;140:617–627.
Jurynczyk M, Messina S, Woodhall MR, et al. Clinical presentation and prognosis in MOG-antibody disease: a UK study. Brain . 2017;140:3128–3138.
Kim HJ, Paul F, Lana-Peixoto MA, et al. MRI characteristics of neuromyelitis optica spectrum disorder: an international update. Neurology . 2015;84:1165–1173.
Kim SM, Kim SJ, Lee HJ, et al. Differential diagnosis of neuromyelitis optica spectrum disorders. Ther Adv Neurol Disord . 2017;10(7):265–289.
Kitley J, Leite MI, Nakashima I, et al. Prognostic factors and disease course in aquaporin-4 antibody-positive patients with neuromyelitis optica spectrum disorder from the United Kingdom and Japan. Brain . 2012;135:1834–1849.
Kitley J, Palace J. Therapeutic options in neuromyelitis optica spectrum disorders. Expert Rev Neurother . 2016;16:319–329.
Lennon VA, WIngerchuck DM, Kryzer TA, et al. A serum autoantibody marker of neuromyelitis optica: distinction from multiple sclerosis. Lancet . 2004;364:2106–2112.
Liu J, Shi Z, Lian Z, et al. Association of CD58 gene polymorphisms with NMO spectrum disorders in a han Chinese population. J Neuroimmunol . 2017;309:23–30.
Nosadini M, Alper G, RIney CJ, et al. Rituximab monitoring and redosing in pediatric neuromyelitis optica spectrum disorder. Neurol Neuroimmunol Neuroinflamm . 2016;3:e188.
Papadopoulos MC, Verkman AS. Aquaporin water channels in the nervous system. Nat Rev Neurosci . 2013;14:265–277.
Wingerchuk DM, Banwell B, Bennett JL, et al. International consensus diagnostic criteria for neuromyelitis optica spectrum disorders. Neurology . 2015;85:177–189.
Myelin Oligodendrocyte Glycoprotein–Associated Disorders
Cheryl Hemingway
There is an increasing awareness of a group of demyelinating disorders that are associated with an IgG antibody to a glycoprotein in the outer layer of the myelin sheath, myelin oligodendrocyte glycoprotein (MOG).
Clinical Presentation
The clinical phenotype appears to be distinct from MS but overlaps with patients with ADEM and AQP4-Ab–positive NMOSD. MOG-Ab is present in more than one third of children who present with an initial episode of demyelination, in more than half of those presenting with ADEM, and in almost all of those with relapsing ADEM (MDEM). MOG autoimmunity is not only more common in the young, but it also appears to demonstrate age-dependent phenotypes, with presentation in the pediatric population more heterogeneous than that seen in adults. In adults, the majority of cases present with ON or NMOSD, whereas in pediatric patients, MOG-Abs are also detected in a range of other relapsing phenotypes, including relapsing inflammatory optic neuritis (RION), ADEM followed by optic neuritis (ADEM-ON), brainstem demyelination, and AQP4-Ab–negative NMOSD (see Table 618.1 ).
Imaging and Laboratory Findings
MRI findings are atypical for MS and can show widespread white matter involvement and increased frequency of longitudinally extensive TM, and over time they can develop into a leukodystrophy-like pattern (see Fig. 618.3 ). These findings, previously thought to be attributed to early-onset MS, are now being recognized as hallmarks of MOG-Ab–associated disease. Intrathecal OCBs are not normally present, again something previously thought attributable to early-onset MS.
Treatment
Treatment of acute attacks is similar to other demyelinating disorders and includes high-dose methylprednisolone, plasma exchange, and IVIG, depending on the severity of the presentation and the response. There is currently no clarity about DMTs that may be helpful over the long term. A complicating factor is the potentially long interval between relapses, making it difficult to determine the true efficacy of DMTs and guide early decisions as to whether or not to treat. Some studies have also demonstrated a potential worsening when MOG-Ab disorders are treated with MS medications, highlighting again the importance of an accurate clinical diagnosis.
In those who are assessed as likely to benefit from DMTs, medications such as mycophenolate mofetil and azathioprine are frequently offered, either with or without steroids. Rituximab has been used, and although there are some reports of benefit, there are also reports of severe exacerbations, particularly in those with relapsing brainstem demyelination. It is important to remember that although both AQP4 and MOG disorders are antibody driven, the former is an astrocytopathy whereas the latter is an oligodendrocytopathy, making extrapolation of treatment effects from one condition to the other unwise. In a recent large study, the only treatment consistently to have shown benefit in high-risk individuals was monthly IVIG.
Although the full spectrum of phenotypes and best treatment options are currently still being determined, it is important to consider MOG-Ab–associated disorders in the differential diagnosis and to seek expert advice on treatment when possible.
Prognosis
Although some phenotypes appear to be associated with a more benign course than, for example, AQP4-Ab demyelination, other phenotypes such as brainstem demyelination can have a very high relapse rate and be quite debilitating. Relapses can also occur many years after the first event, with intervals of more than 10 yr having been reported. Cognitive deficits are seen frequently in those with young onset and frequent relapses.
Bibliography
Cobo-Calvo A, Ruiz A, Maillart E, et al. Clinical spectrum and prognostic value of CNS MOG autoimmunity in adults: the MOGADOR study. Neurology . 2018;90(21):e1858–e1869.
Duignan S, Wright S, Rossor T, et al. Myelin oligodendrocyte glycoprotein and aquaporin-4 antibodies are highly specific in children with acquired demyelinating syndromes. Dev Med Child Neurol . 2018;60(9):958–962.
Hacohen Y, Rossor T, Mankad K, et al. Leukodystrophy-like’ phenotype in children with myelin oligodendrocyte glycoprotein antibody-associated disease. Dev Med Child Neurol . 2018;60(4):417–423.
Hacohen Y, Vincent A. Autoimmune neurological disorders-does the age matter? Eur J Paediatr Neurol . 2018;22:341–343.
Hacohen Y, Wong YY, Lechner C, et al. Disease course and treatment responses in children with relapsing myelin oligodendrocyte glycoprotein Antibody-associated disease. JAMA Neurol . 2018;75(4):478–487.
Hennes EM, Baumann M, Schanda K, et al. Prognostic relevance of MOG antibodies in children with an acquired demyelinating syndrome. Neurology . 2017;89:900–908.
Reindl M, Di Pauli F, Rostasy K, Berger T. The spectrum of MOG autoantibody-associated demyelinating diseases. Nat Rev Neurol . 2013;9:455–461.