Abnormalities of the Optic Nerve
Scott E. Olitsky, Justin D. Marsh
Optic Nerve Aplasia
This rare congenital anomaly is typically unilateral. The optic nerve, retinal ganglion cells, and retinal blood vessels are absent. A vestigial dural sheath usually connects with the sclera in a normal position, but no neural tissue is present within this sheath. Optic nerve aplasia typically occurs sporadically in an otherwise healthy person.
Optic Nerve Hypoplasia
Hypoplasia of the optic nerve is a nonprogressive condition characterized by a subnormal number of optic nerve axons with normal mesodermal elements and glial supporting tissue. In typical cases, the nerve head is small and pale, with a pale or pigmented peripapillary halo or double ring sign (Fig. 649.1 ).
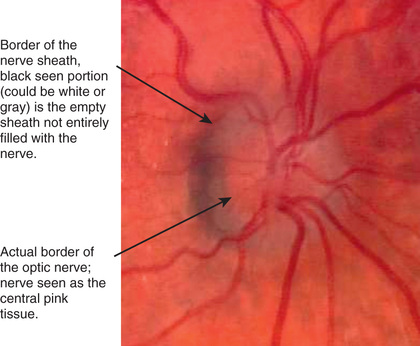
This anomaly is associated with defects of vision and of visual fields of varying severity, ranging from blindness to normal or near-normal vision. It may be associated with systemic anomalies that most commonly involve the central nervous system (CNS). Protean CNS defects such as hydranencephaly or anencephaly or more focal lesions compatible with continued development may accompany optic nerve hypoplasia, but unilateral or bilateral optic nerve hypoplasia may be found without any concomitant defects.
Optic nerve hypoplasia is a principal feature of septo-optic dysplasia of de Morsier, a developmental disorder characterized by the association of anomalies of the midline structures of the brain with hypoplasia of the optic nerves, optic chiasm, and optic tracts; typically noted are agenesis of the septum pellucidum, partial or complete agenesis of the corpus callosum, and malformation of the fornix, with a large chiasmatic cistern. Patients may have hypothalamic abnormalities and endocrine defects ranging from panhypopituitarism to isolated deficiency of growth hormone, hypothyroidism, or diabetes insipidus. Neonatal hypoglycemia and seizures are important presenting signs in affected infants.
MRI is preferred for evaluating CNS abnormalities in patients with optic nerve hypoplasia. During MRI, special attention should be directed to the pituitary infundibulum, where ectopia of the posterior pituitary may be found. Posterior pituitary ectopia appears on MRI as an absence of the pituitary infundibulum with an abnormal bright spot at the upper infundibulum area. This abnormality is present in approximately 15% of patients and suggests a posterior pituitary hormone deficiency, requiring further endocrinologic workup. Endocrine function should be watched closely in patients with optic nerve hypoplasia. The cause of optic nerve hypoplasia remains unclear.
Children with periventricular leukomalacia display an unusual form of optic nerve hypoplasia. The optic nerve demonstrates a large cup within a normal-size optic disc. This form of optic nerve hypoplasia occurs secondary to transsynaptic degeneration of optic axons caused by the primary bilateral lesion in the optic radiation (periventricular leukomalacia).
Optic Nerve Coloboma
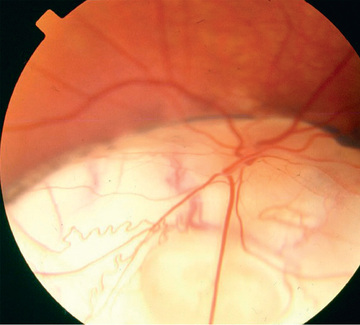
Optic nerve colobomas can be unilateral or bilateral. The visual acuity can range from normal to complete blindness. The coloboma develops secondary to incomplete closure of the embryonic fissure. The defect may produce a partial or total excavation of the optic disc (Fig. 649.2 ). Chorioretinal and iris colobomas may also occur. Optic nerve colobomas may be seen in a multitude of ocular and systemic abnormalities, including the CHARGE (C, coloboma; H, heart disease; A, atresia choanae; R, retarded growth and development and/or CNS anomalies; G, genetic anomalies and/or hypogonadism; E, ear anomalies and/or deafness) association.
Morning Glory Disc Anomaly
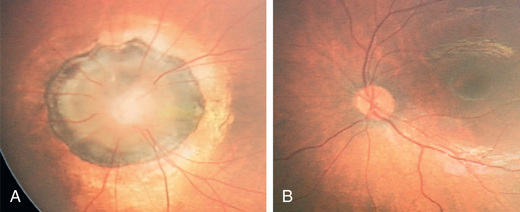
This term describes a congenital malformation of the optic nerve characterized by an enlarged, excavated, funnel-shaped disc with an elevated rim resembling a morning glory flower. White glial tissue is present in the central part of the disc (Fig. 649.3 ). The retinal vessels are abnormal and appear at the peripheral disc, coursing over the elevated pink rim in a radial fashion. Pigmentary mottling of the peripapillary region is usually seen. Most cases are unilateral. Females are affected twice as often as males. Visual acuity is usually severely reduced. Morning glory disc anomaly has been associated with basal encephalocele in patients with midfacial anomalies. Abnormalities of the carotid circulation can also be seen in patients with morning glory anomaly. Moyamoya disease is a well-described associated finding.
Tilted Disc
In this congenital anomaly, the vertical axis of the optic disc is directed obliquely, so that the upper temporal portion of the nerve head is more prominent and anterior to the lower nasal portion of the disc. The retinal vessels emerge from the upper temporal portion of the disc rather than from the nasal side. Often noted is a peripapillary crescent or conus. Associated visual field defects and myopic astigmatism may be found. Clinical recognition of the tilted disc syndrome is important to avoid confusion of its disc and visual field signs with those of papilledema and intracranial tumor.
Drusen of the Optic Nerve
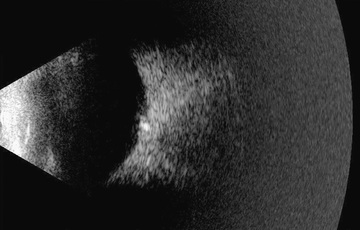
These globular, acellular bodies are thought to arise from axoplasmic derivatives of disintegrating nerve fibers. Drusen may be buried within the optic nerve, producing elevation of the optic nerve head (which can be confused with papilledema), or they may be partially or completely exposed, appearing as refractile bodies at the surface of the disc. Visual field defects and spontaneous hemorrhages of the peripapillary nerve fiber layer may occur in association with drusen. Drusen may occur as an autosomal dominant condition. B-scan ultrasonography can help to positively identify drusen suspected on clinical ophthalmic exam (Fig. 649.4 ).
Papilledema
The term papilledema is reserved to describe swelling of the nerve head secondary to increased intracranial pressure (ICP). Clinical manifestations of papilledema include edematous blurring of the disc margins, fullness or elevation of the nerve head, partial or complete obliteration of the disc cup, capillary congestion and hyperemia of the nerve head, generalized engorgement of the veins, loss of spontaneous venous pulsation, hemorrhages in the nerve fiber layer around the disc, and peripapillary exudates (see Fig. 608.2 ). In some cases edema extending into the macula may produce a fan- or star-shaped figure. In addition, concentric peripapillary retinal wrinkling (Paton lines) may be noted. Transient obscuration of vision may occur, lasting seconds and associated with postural changes. Vision, however, is usually normal in acute papilledema. Normally, when the ICP is relieved, the papilledema resolves and the disc returns to a normal or nearly normal appearance within 6-8 wk. Sustained chronic papilledema or long-standing unrelieved increased ICP may, however, lead to permanent nerve fiber damage, atrophic changes of the disc, macular scarring, and impairment of vision.
The pathophysiology of papilledema is probably as follows: elevation of intracranial subarachnoid cerebrospinal fluid (CSF) pressure, elevation of CSF pressure in the sheath of the optic nerve, elevation of tissue pressure in the optic nerve, stasis of axoplasmic flow and swelling of the nerve fibers in the optic nerve head, and secondary vascular changes and the characteristic ophthalmoscopic signs of venous stasis. Associated neuro-ophthalmic signs of increased ICP in infants and children include 6th cranial nerve palsy and attendant esotropia, lid retraction, paresis of upward gaze, tonic downward deviation of the eyes, and convergent nystagmus.
The common etiologies of papilledema in childhood are intracranial tumors and obstructive hydrocephalus, intracranial hemorrhage, the cerebral edema of trauma, meningoencephalitis, toxic encephalopathy, and certain metabolic diseases. Regardless of the cause, the optic disc signs of increased ICP in early childhood may occasionally be modified by the distensibility of the young skull. In the absence of conditions associated with early closure of sutures and early obliteration of the fontanel (craniosynostosis, Crouzon disease, and Apert syndrome), infants with increased ICP may not develop papilledema.
The differential diagnosis of papilledema includes structural changes of the disc (pseudopapilledema, pseudoneuritis, drusen, and myelinated nerve fibers), with which it may be confused, and the disc swelling of papillitis associated with optic neuritis in addition to the disc changes of hypertension and diabetes mellitus. Unless retinal hemorrhage or edema involves the macular area, the preservation of good central vision and the absence of an afferent pupillary defect (Marcus Gunn pupil) help to differentiate acute papilledema from the edema of the optic nerve head found in acute optic neuritis.
Papilledema is a neurologic emergency. It can be accompanied by other signs of increased ICP, including headaches, nausea, and vomiting. Neuroimaging should be performed; if no intracranial masses are detected; a lumbar puncture and determination of CSF pressure should follow.
Optic Neuritis
This is any inflammation or demyelinization of the optic nerve with attendant impairment of function. The process is usually acute, with rapidly progressive loss of vision. It may be unilateral or bilateral. Pain on movement or palpation of the globe may precede or accompany the onset of visual symptoms. There is decreased visual activity, decreased color vision and contrast sensitivity, a relative afferent pupillary defect, and a normal macula and peripheral retina.
When the retrobulbar portion of the nerve is affected without ophthalmoscopically visible signs of inflammation at the disc, the term retrobulbar optic neuritis is applied. When there is ophthalmoscopically visible evidence of inflammation of the nerve head, the term papillitis or intraocular optic neuritis is used. When there is involvement of both the retina and the papilla, the term optic neuroretinitis is used.
In childhood, optic neuritis may occur as an isolated condition or as a manifestation of a neurologic or systemic disease. Optic neuritis may be secondary to inflammatory diseases (systemic lupus erythematosus, sarcoidosis, Behçet disease, autoimmune optic neuritis); infections (tuberculosis, syphilis, Lyme disease, meningitis, viral encephalitis, HIV, or postinfectious disease); and toxic or nutritional disorders (methanol, ethambutol, vitamin B12 deficiency). It may signify one of the many demyelinating diseases of childhood (see Chapter 618 ). Although a significant percentage of adults who experience an episode of optic neuritis eventually develop other symptoms associated with multiple sclerosis (MS), young children with optic neuritis are seemingly at less risk (risk of MS is 19% within 20 yr). High-risk features suggestive of MS include visual acuity better than no light perception, periocular pain, acutely normal-appearing optic nerve, no retinal abnormalities, and abnormal MRI suggesting a demyelinating disease. Bilateral optic neuritis in children may be associated with acute disseminated encephalomyelitis or neuromyelitis optica (NMO or Devic disease). NMO is characterized by rapid and severe bilateral visual loss accompanied by transverse myelitis and paraplegia. Involvement of the brain stem and occasionally the cortex may be seen on MRI. NMO-specific immunoglobulin G (directed to the aquaporin 4 water channel) is the diagnostic test of choice for Devic syndrome. Optic neuritis may also be secondary to an exogenous toxin or drug, as with lead poisoning or as a complication of long-term high-dose treatment with chloramphenicol or vincristine. Extensive pediatric neurologic and ophthalmic investigation, including MRI and lumbar puncture, is usually required. Idiopathic NMO is associated with anti–aquaporin 4 antibodies, otherwise known as NMO antibodies.
In most cases of acute optic neuritis, some improvement in vision begins within 1-4 wk after onset, and vision may improve to normal or near normal within weeks or months. The course varies with cause. Although central vision may recover fully, it is common to find permanent defects in other areas of visual function (contrast sensitivity, color, brightness sense, and motion perception). Recurrences may occur especially, but not universally, in patients who go on to develop MS.
A treatment trial has demonstrated that high-dose intravenous methylprednisolone may help to speed the visual recovery in young adults, and it may prevent the development of MS in those at risk. It is unknown to what degree the results of the aforementioned trial may be extrapolated to optic neuritis in childhood.
Leber Optic Neuropathy
This entity is characterized by a sudden loss of central vision occurring in the 2nd and 3rd decades of life and primarily affects young males. A characteristic peripapillary telangiectatic microangiopathy occurs not only in the presymptomatic phase of involved eyes but also in a high number of asymptomatic offspring in the female line. Disc hyperemia and edema mark the acute phase of visual loss. One eye is usually affected before the other. Visual field loss and impaired color vision are also present. In time, progressive optic atrophy and vision loss usually ensue. The tortuous angiopathy becomes less obvious. Although visual function after the initial loss generally remains stable, a significant and sometimes complete recovery may occur in as many as 30% of affected individuals. This recovery may take place years or decades after the initial episode of acute vision loss. The peripapillary angiopathy, the lack of short-term remission, and the degree of symmetry serve to distinguish most cases of Leber disease from the optic neuritis of MS.
Leber optic neuropathy is maternally inherited and is caused by defective cytoplasmic mitochondrial DNA. Multiple point mutations in the mitochondrial DNA that lead to the development of the disorder have been found. Because of the mitochondrial nature of the disorder, skeletal and cardiac muscle disorders, including electrocardiographic abnormalities, may also be encountered in affected individuals.
Optic Atrophy
This term denotes degeneration of optic nerve axons, with attendant loss of function. The ophthalmoscopic signs of optic atrophy are pallor of the disc and loss of substance of the nerve head, sometimes with enlargement of the disc cup. The associated vision defect varies with the nature and site of the primary disease or lesion.
Optic atrophy is the common expression of a wide variety of congenital or acquired pathologic processes (Table 649.1 ). The cause may be traumatic, inflammatory, degenerative, neoplastic, or vascular; intracranial tumors and hydrocephalus are principal causes of optic atrophy in children. In some cases progressive optic atrophy is hereditary. Dominantly inherited infantile optic atrophy is a relatively mild heredodegenerative type that tends to progress through childhood and adolescence. Autosomal recessively inherited congenital optic atrophy is a rare condition that is evident at birth or develops at a very early age; the visual defect is usually profound. Behr optic atrophy is a hereditary type associated with hypertonia of the extremities, increased deep tendon reflexes, mild cerebellar ataxia, some degree of mental deficiency, and possibly external ophthalmoplegia. This disorder principally afflicts boys 3-11 yr of age. Some forms of heredodegenerative optic atrophy are associated with sensorineural hearing loss, as may occur in some children with juvenile-onset (insulin-dependent) diabetes mellitus. In the absence of an obvious cause, optic atrophy in an infant or child warrants extensive etiologic investigation.
Optic Nerve Glioma
Optic nerve glioma, more properly referred to as juvenile pilocytic astrocytoma, is the most frequent tumor of the optic nerve in childhood. This neuroglial tumor may develop in the intraorbital, intracanalicular, or intracranial portion of the nerve; the chiasm is often involved.
The tumor is a cytologically benign hamartoma that is generally stationary or only slowly progressive. The principal clinical manifestations when the tumor occurs in the intraorbital portion of the nerve are unilateral loss of vision, proptosis, and deviation of the eye; optic atrophy or congestion of the optic nerve head may occur. Chiasmal involvement may be attended by defects of vision and visual fields (often bitemporal hemianopia), increased ICP, papilledema or optic atrophy, hypothalamic dysfunction, pituitary dysfunction, and sometimes nystagmus or strabismus. Juvenile pilocytic astrocytomas occur with increased frequency in patients with neurofibromatosis (see Chapter 614.1 ).
Treatment of optic pathway gliomas is controversial. The best management is usually periodic observation with serial radiography (preferably MRI). Only symptomatic and radiographically progressing optic nerve gliomas require strong consideration for treatment. If a patient has unsightly proptosis with complete or nearly complete loss of vision of the affected eye, surgical removal may be appropriate when the tumor is confined to the intraorbital, intracanalicular, or prechiasmal portion of the nerve. When the chiasm is involved, resection is not usually indicated and radiation and chemotherapy may be necessary.
Traumatic Optic Neuropathies
Injury to the optic nerve may result from both direct and indirect trauma. Direct trauma to the optic nerve is a result of a penetrating injury to the orbit with transection or contusion of the nerve. Blunt trauma to the orbit may also lead to severe visual loss if the traumatic force is transmitted to the optic canal and causes disruption of the blood supply to the intracanalicular portion of the nerve. Treatment with high-dose corticosteroids has not proved to be effective; it has been shown that similar regimens involve an increased relative risk of death when they are given to patients who have experienced significant head injuries.