Other Inherited Disorders of Skeletal Development
Jacqueline T. Hecht, William A. Horton
Great advances in our understanding have led to delineation of the genetic basis of disorders that were previously poorly understood. Some of these conditions are now classified into a gene family based on their molecular and clinical findings.
Ellis–Van Creveld Syndrome
The Ellis–van Creveld syndrome (MIM 225500), also known as chondroectodermal dysplasia, is a skeletal and an ectodermal dysplasia. The skeletal dysplasia presents at birth with short limbs, especially the middle and distal segments, accompanied by postaxial polydactyly of the hands and sometimes of the feet (Fig. 720.1 ). Nail dysplasia and dental anomalies (including neonatal, absent, premature loss of teeth, and upper lip defects) constitute the ectodermal dysplasia. Common manifestations also include atrial septal defects and other congenital heart defects.
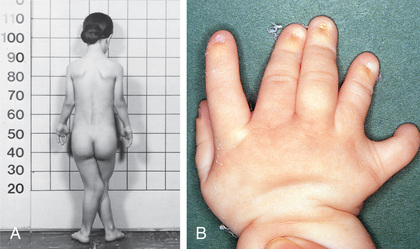
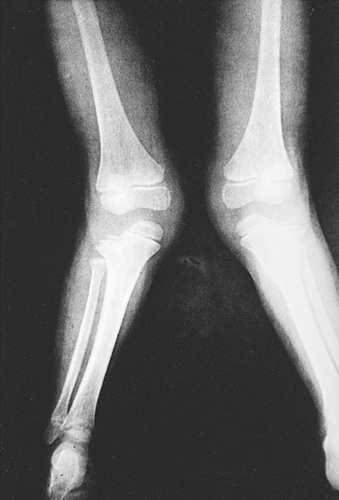
Skeletal radiographs reveal short tubular bones with clubbed ends, especially the proximal tibia and ulna (Fig. 720.2 ). Carpal bones display extra ossification centers and fusion; cone-shaped epiphyses are evident in the hands. A bony spur is often noted above the medial aspect of the acetabulum.
Ellis–van Creveld syndrome is an autosomal recessive trait that occurs most often in the Amish. Mutations have been identified in one of two genes, EVC (EVC1 ) or EVC2 (LIMBIN) , which map in a head-to-head configuration to chromosome 4p. Mutations of EVC2 are detected in the allelic condition Weyers acrofacial dysostosis (MIM 193530). EVC and EVC2 proteins are thought to influence hedgehog signaling in cilia by constitutively associating in a ring-like pattern in the ciliary transition zone and transducing extracellular signals to the nucleus via hedgehog signaling. Fgf18 may also play a significant role. This disorder is now classified under the ciliopathies with major skeletal involvement.
Approximately 30% of patients die of cardiac or respiratory problems during infancy. Life span is otherwise normal; adult heights range from 119 to 161 cm.
Asphyxiating Thoracic Dystrophy (See Also Chapter 445.3 )
Asphyxiating thoracic dystrophy (MIM 208500), or Jeune syndrome , is an autosomal recessive chondrodysplasia that resembles Ellis–van Creveld syndrome. Newborn infants present with a long, narrow thorax and respiratory insufficiency associated with pulmonary hypoplasia. Neonates often die. Other neonatal manifestations include slightly short limbs and postaxial polydactyly. This condition results from a disturbance of primary cilia, most often from mutations of the gene encoding cytoplasmic dynein 2 heavy chain 1 (DYNC2H1 ). This disorder is now classified under ciliopathies with major skeletal involvement.
Skeletal radiographs show very short ribs with anterior expansion. Tubular limb bones are short with bulbous ends; cone-shaped epiphyses occur in hand bones. The iliac bones are short and square with a spur above the medial aspect of the acetabulum (Fig. 720.3 ).
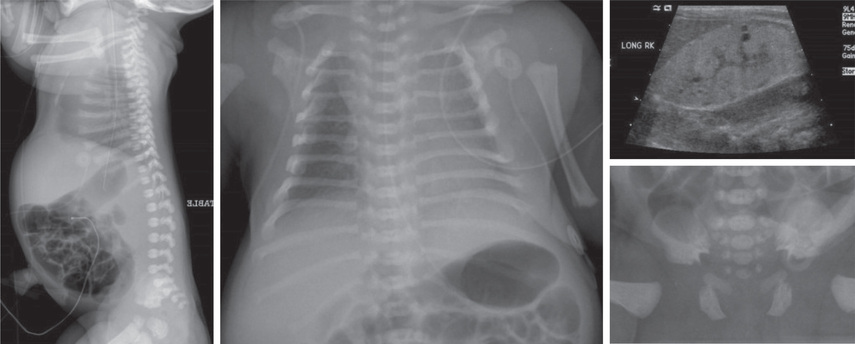
If infants survive the neonatal period, respiratory function usually improves as the rib cage grows. Surgery that produces lateral thoracic expansion improves rib growth and enhances chest wall dimensions. Progressive renal dysfunction often develops during childhood. Intestinal malabsorption and hepatic dysfunction have also been reported.
Short-Rib Polydactyly Syndromes
These conditions, which share the clinical features of constricted thoracic cage, short ribs, polydactyly, very short extremities, lethality during the newborn period and autosomal recessive inheritance, are grouped into five syndromes (SRP-2-5). Mutations that map to cilia-related genes, DYNCH2H, TT21B, WDR 19, WDR34, WDR35, IFT80, IFT140, IFT172 AND NEK1 , are found in this group of disorders.
Cartilage-Hair Hypoplasia–Anauxetic Spectrum Disorders
Cartilage-hair hypoplasia (CHH; MIM 250250) is also known as metaphyseal chondrodysplasia–McKusick type is part of a spectrum of disorders with metaphyseal involvement that includes metaphyseal dysplasia without hypotrichosis and anauxetic dysplasia. All disorders are characterized by severe disproportionate short stature, which is usually recognized at birth; the short limbs can lead to prenatal detection. They all show autosomal recessive inheritance and are caused by mutations in RMRP , a gene coding for a large untranslated RNA component of an enzyme complex involved in processing mitochondrial RNA. Loss of this gene product interferes with processing of both messenger RNA and ribosomal RNA, and correlates with the extent of bone dysplasia, whereas loss of messenger RNA processing correlates with the degree of hair hypoplasia, immunodeficiency, and hematologic abnormality. Molecular testing confirms the diagnosis, and prenatal diagnosis is available if the mutation is identified either in the patient or the parents.
Cartilage-Hair Hypoplasia
CHH is recognized during the 2nd year because of growth deficiency affecting the limbs, accompanied by flaring of the lower rib cage, a prominent sternum, and bowing of the legs. The hands and feet are short, and the fingers are very short with extreme ligamentous laxity. The hair is thin, sparse, and light colored; the nails are hypoplastic; and the skin can be hypopigmented.
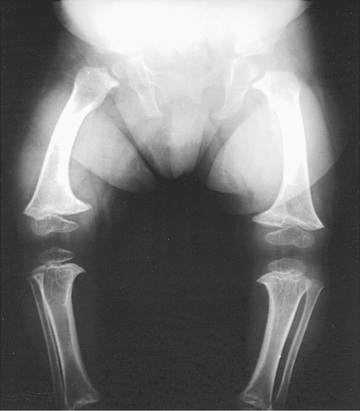
Radiographs show short tubular bones with flared, irregularly mineralized, and cupped metaphyses (Fig. 720.4 ). The knees are more affected than are the hips, and the fibula is disproportionately longer than the tibia. The metacarpals and phalanges are short and broad. Spinal radiographs reveal mild platyspondyly.
Nonskeletal manifestations associated with CHH include immunodeficiency (T-cell abnormalities, neutropenia, leukopenia, and susceptibility to varicella zoster virus infections; children also may have complications from smallpox and polio vaccinations), malabsorption, celiac disease, and Hirschsprung disease. Adults are at risk for malignancy, especially non-Hodgkin lymphoma and skin tumors. Adult height ranges from 107 to 157 cm.
The highest birth prevalence is in the Amish and Finnish populations because of founder effect. Carrier frequency in the Amish is 1 : 19 with 1 per 1,300 births affected compared to a carrier frequency of 1 : 76 and 1 in 23,000 births affected in Finland. The exact prevalence in the general population is not known, but CHH is relatively rare. However, 2 allelic conditions, metaphyseal dysplasia without hypotrichosis and anauxetic dysplasia expand the phenotypic spectrum. Children with a growth disorder and abnormal hair should be evaluated for RMRP mutations so as to not miss the diagnosis.
Metatropic Dysplasia
Metatropic dysplasia (MIM 156530) is an autosomal dominant disorder resulting from heterozygous mutations of transient receptor potential vanilloid family 4 (TRPV4), which encodes a calcium-permeable cation channel. Newborn infants present with a long narrow trunk and short extremities. A tail-like appendage sometimes extends from the base of the spine. Odontoid hypoplasia is common and may be associated with cervical instability. Kyphoscoliosis appears in late infancy and progresses through childhood, often becoming severe enough to compromise cardiopulmonary function. The joints are large and become progressively restricted in mobility, except in the hands. Contractures often develop in the hips and knees during childhood. Although severely affected infants can die at a young age from respiratory failure, patients usually survive, although they can become disabled as adults from the progressive musculoskeletal deformities. Adult heights range from 110 to 120 cm.
Skeletal radiographs show characteristic changes dominated by severe platyspondyly and short tubular bones with expanded and deformed metaphyses that exhibit a dumbbell appearance (Fig. 720.5 ). The pelvic bones are hypoplastic and exhibit a halberd appearance because of a small sacrosciatic notch and a notch above the lateral margin of the acetabulum. Metatropic dysplasia is included in the TRPV4 group.
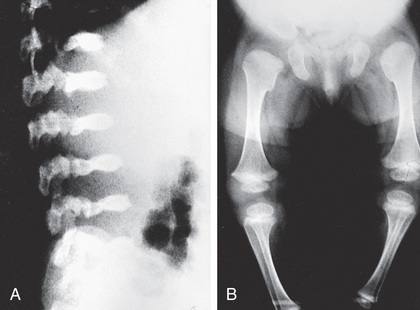
Spondylometaphyseal Dysplasia, Kozlowski Type
Kozlowski type of spondylometaphyseal dysplasia (MIM 184252) is an autosomal dominant allelic disorder to metatropic dysplasia caused by TRPV4 mutations. Mutations of TRPV4 have also been identified in autosomal dominant brachyolmia (MIM 113500), whose phenotype is dominated by progressive scoliosis and platyspondyly on x-rays and familial digital arthropathy with brachydactyly (MIM 606835), which is characterized by deforming painful osteoarthritis of the interphalangeal, metacarpophalangeal, and metatarsophalangeal joints starting after the 1st decade of life. The rest of the skeleton is unaffected.
Kozlowski type of spondylometaphyseal dysplasia manifests in early childhood with mild short stature involving mostly the trunk and a waddling gait. The hands and feet may be short and stubby. Radiographs show flattening of vertebral bodies. The metaphyses of tubular bones are widened and irregularly mineralized, especially at the proximal femur. The pelvic bones manifest mild hypoplasia. Scoliosis can develop during adolescence. The disorder is otherwise uncomplicated, and manifestations are limited to the skeleton. Adults reach heights of 130-150 cm.
Disorders Involving Filamins
Mutations of genes encoding filamin A and filamin B proteins have been detected in diverse disorders of skeletal development: filamin A mutations in otopalatodigital syndromes type 1 and 2, frontometaphyseal dysplasia, Melnick-Needles syndrome and terminal osseous dysplasia with pigmentary defects (MIM 311300, 304120, 305620, 309350, 300244) and filamin B mutations in Larsen syndrome and perinatal lethal atelosteogenesis types 1 and 3, spondylo-carpal-tarsal dysplasia and Boomerang dysplasia (MIM 150250, 108720, 108721, 272460, 112310). Filamins functionally connect extracellular to intracellular structural proteins, thereby linking cells to their local microenvironment, which is essential for skeletal development and growth.
Juvenile Osteochondroses
The juvenile osteochondroses are a heterogeneous group of disorders in which regional disturbances in bone growth cause noninflammatory arthropathies. Table 720.1 summarizes the juvenile osteochondroses. Some have localized pain and tenderness (Freiberg disease, Osgood-Schlatter disease [see Chapter 697.4 ], osteochondritis dissecans [see Chapter 697.3 ]), whereas others present with painless limitation of joint movement (Legg-Calvé-Perthes disease [see Chapter 698.3 ], Scheuermann disease [see Chapter 699.4 ]). Bone growth may be disrupted, leading to deformities. The diagnosis is usually confirmed radiographically, and treatment is symptomatic. The pathogenesis of these disorders is believed to involve ischemic necrosis of primary and secondary ossification centers. Although familial forms have been reported, these disorders usually occur sporadically.
Table 720.1
Caffey Disease (Infantile Cortical Hyperostosis)
This is a rare disorder of unknown etiology characterized by cortical hyperostosis with inflammation of the contiguous fascia and muscle. It is often sporadic, but both autosomal dominant (MIM 244460) and autosomal recessive (MIM 127000) forms have been reported. Mutations in Fam111A (family with sequence similarity 111, member A) and TBCE (tubulin-specific chaperone E) have been identified in the autosomal dominant and recessive forms, respectively. Caffey dysplasia is classified in the slender bone dysplasia group.
Prenatal and more often postnatal onsets have been described. Prenatal onset may be mild (autosomal dominant) or severe (autosomal recessive). Severe prenatal disease is characterized by typical bone lesions, polyhydramnios, hydrops fetalis, severe respiratory distress, prematurity, and high mortality. Onset in infancy (younger than 6 mo; average: 10 wk) is most common; manifestations include the sudden onset of irritability, swelling of contiguous soft tissue that precedes the cortical thickening of the underlying bones, fever, and anorexia. The swelling is painful with a wood-like induration but with minimal warmth or redness; suppuration is absent. There are unpredictable remissions and relapses; an episode can last 2 wk to 3 mo. The most common bones involved include the mandible (75%) (Fig. 720.6 ), the clavicle, and the ulna. If swelling is not prominent or visible, the diagnosis might not be evident.
Laboratory features include elevated erythrocyte sedimentation rate and serum alkaline phosphatase as well as, in some patients, increased serum prostaglandin E levels. There may be thrombocytosis and anemia. The radiographic features include soft tissue swelling and calcification and cortical hyperostosis (Fig. 720.7 ). All bones may be affected except the phalanges or vertebral bodies. The differential diagnosis includes other causes of hyperostosis such as chronic vitamin A intoxication, prolonged prostaglandin E infusion in children with ductal dependent congenital heart disease, primary bone tumors, and scurvy.

Complications are unusual but include pseudoparalysis with limb or scapula involvement, pleural effusions (rib), torticollis (clavicle), mandibular asymmetry, bone fusion (ribs or ulna and radius), and bone angulation deformities (common with severe prenatal onset). Treatment includes indomethacin and prednisone (if there is a poor response to indomethacin).
Fibrodysplasia Ossificans Progressiva
Fibrodysplasia ossificans progressiva (FOP) (MIM 135100) is a rare and severely disabling disorder characterized by progressive extraskeletal heterotopic bone formation in soft connective tissues including muscles, tendons, ligaments, fascia, and aponeuroses. With the exception of deformity of the large toes, infants are normal at birth. Episodes of painful soft-tissue swelling with inflammation usually begin in early childhood initially involving the upper back and neck, and later the entire trunk and extremities. Repeated episodes (flare-ups) slowly transform the soft tissues into bands or plates of bone that span joints and progressively limit movement and mobility. Episodes are often triggered by injury, intramuscular injections, and viral infection. Most patients are wheelchair bound by their late teens. The average life span is approximately 40 yr, with death usually resulting from complications of thoracic insufficiency.
FOP results from heterozygous activating mutations of the gene (ACVR1) encoding the bone morphogenetic protein (BMP) type I receptor, activin A receptor type I (ALK2) . Patients with classic FOP have the same missense ACVR1 mutation, which enhances BMP signaling, which, in turn, induces inflammation and aberrant endochondral ossification through mechanisms that are poorly understood. Environmental factors, such as injury, play an important role in triggering these events. ACVR1 mutations usually occur sporadically, but autosomal dominant transmission has rarely been observed. FOP is classified in the disorganized development of skeletal components group.
There is currently no definitive treatment for FOP. Supportive care includes avoidance of injury-prone physical activities, intramuscular injections including immunizations and overstretching of the jaw during dental procedures. Corticosteroids and other antiinflammatory agents reduce inflammation and pain during flare-ups, but are unable to prevent heterotopic bone formation. Studies in FOP animal models suggest that BMP type I kinase inhibitors and retinoic acid receptor γ agonists, which block chondrogenesis—the initial step in endochondral ossification—may be useful therapies in the future. An animal FOP study has indicated that mutant ALK2 responds to activin A, induces canonical BMP signaling and leads to heterotopic bone formation, providing an additional possible therapeutic target. Notably, the retinoic acid receptor γ agonist Palovarotene is being tested in an ongoing phase 2 clinical trial with FOP patients (NCT02190747).