14
Heart Failure and Cardiogenic Shock
Daniel Sedehi and Venu Menon
Background
Acute heart failure syndrome (AHFS) is a term intended to capture pathologic changes in cardiac function that are new or that destabilize an already vulnerable cardiac substrate. These alterations in myocardial, valvular, pericardial, or electrical function frequently result in admission to the emergency department (ED), where rapid recognition and correction of the destabilizing change are essential to successful therapy.1,2 AHFS now accounts for over 3 million ED visits annually—a number that has increased dramatically over the last several decades as a result of an aging population as well as significant advances in acute reperfusion therapy, neurohumoral modulation for heart failure (HF), preventive strategies for sudden cardiac death, and primary prevention strategies that limit and attenuate the consequences of cardiac injury.2–9 Nearly 80% of AHFS patients have an established diagnosis of HF; 20% of AHFS patients will present with new-onset HF. Common comorbid conditions in these patients include coronary artery disease, hypertension (HTN), diabetes mellitus, atrial fibrillation, and chronic renal dysfunction.
The 2009 American College of Cardiology/American Heart Association (ACC/AHA) guidelines define three classes of AHFS:
Class 1—volume overload: pulmonary congestion, systemic congestion
Class 2—impaired cardiac output: hypotension, organ failure, shock
Class 3—combination of classes 1 and 2
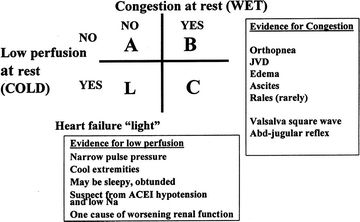
This classification of AHFS echoes a diagnostic approach developed in 199910 (Fig. 14.1). The figure, which shows fluid status on one axis and cardiac output on the other, provides a useful description of four hemodynamic profiles based on evidence of resting congestion and hypoperfusion. As the majority of AHFS presenting to the ED will fall into class 1, or “B” in Figure 14.1, this chapter focuses on the diagnostic and therapeutic strategies involved in the care of this patient population. Cardiogenic shock (CS) is addressed in the second part of the chapter.
History and Physical Exam
An essential component of the AHFS patient history is identification of possible triggers of cardiac decompensation, including dietary indiscretion, physician-prescribed adjustments in medication dosage, medication nonadherence, and usage of new medications, including over-the-counter medications such as NSAIDs. Infections, particularly of the respiratory or genitourinary tract, may also trigger decompensation.
AHFS patients with pulmonary congestion will commonly report dyspnea, orthopnea, and postural nocturnal dyspnea. Patients will often deny an inability to sleep but will admit to sleeping in a reclining chair or even upright. Many of these patients also complain of lower extremity edema, a sign of underlying right HF. Right HF can also present with nausea, vomiting, anorexia, and gradually increasing abdominal girth.
Most cases of acute HF originate in the left ventricle and result in shortness of breath, exercise intolerance, and clinical signs of pulmonary congestion. In the ED, initial assessment should focus on respiratory status and adequacy of circulation.
Observation of the patient's presenting position (e.g., sitting, recumbent) can provide important information regarding the severity of pulmonary edema; patients sitting erect or leaning on their knees in a “tripod” position are typically the most dyspneic and congested. Attention should also be paid to a patient's respiratory rate and the ability to speak in full sentences; aberrations in either of these may suggest an urgent need for ventilatory support and/or rapid correction of volume overload. The pulmonary exam should evaluate for signs of congestion, including inspiratory crackles suggestive of elevated left ventricular (LV) pressures. Percussion of posterior chest walls may also reveal pleural effusions secondary to AHFS, typically larger in the right lung.
Assessment of circulation begins with a tactile skin exam. In a “warm and wet” patient, skin temperature suggests normal peripheral vascular tone and implies there will be a positive response to a diuretic alone. In a “cold and clammy” patient, skin findings result from peripheral vasoconstriction and the body's attempt to maintain central vascular tone, blood pressure, and perfusion of vital organs; in these cases, vasodilators and inotropes may be required.
The quality and character of a patient's pulses provide additional information on circulatory adequacy. Diminished and thready pulses denote low pulse pressure and suggest low cardiac output. Irregular pulses suggest arrhythmias such as atrial fibrillation, while frequent ectopic beats may accompany electrolyte imbalances. Pulsus alternans, a clinical feature seen almost exclusively in severe LV dysfunction, is characterized by alternating forceful and weak peripheral pulses at regular beat intervals.
Jugular venous distension (JVD), a marker of fluid status, is often pronounced in patients presenting with AHFS.11 Careful examination of the neck veins—starting with the patient sitting upright and then with the bed lowered—can provide vital information about a patient's intravascular volume status. As a marker for right atrial pressure, and thereby right-sided preload, jugular venous pressure (JVP) should also be measured, using the technique described in Chapter 15. JVP also may also be assessed using the hepatojugular reflex; continued abdominal compression will result in sustained (>3 seconds) elevation of the JVP in patients with volume overload or a failing right ventricle. Assessment of inspiratory change in JVP is important as well; Kussmaul sign (a failure to decrease or even an increase in the mean JVP during inspiration) can be seen in constrictive and restrictive processes, as well as in the setting of an acute right ventricular (RV) infarction.12,13
Precordial palpation should be performed for determination of displaced cardiac apex, parasternal heaves, and thrills. A displaced apex with sustained impulse suggests either long-standing HTN or LV cavity enlargement in a dilated cardiomyopathy. Parasternal heave can accompany this in the setting of RV hypertrophy or enlargement. Any palpable murmur is abnormal and classified as a thrill or an 4/6 murmur on the Levine grading scale.
Auscultation of the heart should focus on extra heart sounds; in patients with volume overload, for example, a postsystolic sound (the S3) is a very specific marker for reduced ejection fraction.14 Murmurs and their characteristics are important to note, as a patient with 3/6 or greater holosystolic murmur over the apex radiating to the axilla may have hemodynamically significant mitral regurgitation.15 Finally, irregular heart rhythms may suggest a new destabilizing arrhythmia.
Bilateral lower extremity pitting edema may be seen in patients with AHFS and elevated JVP. Lower extremity edema may be present in other conditions (such as liver failure and nephrotic syndrome) and is not specific to HF. As the location of edema is gravity dependent, patients who are not upright (e.g., bedbound) must be examined for edema in the dependent parts of their body, such as the sacrum.
Ascites can also be seen in AHFS and is often associated with severe right-sided HF. Palpation of the liver will reveal enlargement and sometimes tenderness, both signs of hepatic congestion. Pulsatility of the liver may be observed in patients with severe tricuspid insufficiency.
Diagnostic Evaluation
Electrocardiogram
Sinus tachycardia is the most common finding on an electrocardiogram (ECG) in AHFS. This reflects compensatory response to preserve cardiac output in the setting of impaired stroke volume. Arrhythmias such as atrial fibrillation are present in approximately 20% of patients with AHFS who present to the ED. Acute coronary syndromes (ACS)—as an ST-segment elevation myocardial infarction (STEMI), a non–ST-segment elevation myocardial infarction (NSTEMI), or an unstable angina—can cause an acute decompensation in patients with underlying structural heart disease or even severe dysfunction in a previously normal heart.27
Laboratory Testing
Hyponatremia is a common finding in AHFS; up to 25% of patients will have a serum sodium of <135 mEq/L. Patients with baseline serum sodium of <137 mEq/L have been shown to have significantly shorter life expectancy than patients with a normal baseline serum sodium, and patients with a baseline serum sodium <130 mEq/L have a 15% 12-month survival rate.16 While potassium is within the normal range for most AHFS patients, potassium levels can be elevated by typical AHFS medications, including angiotensin-converting enzyme (ACE) inhibitors, angiotensin receptor blockers, or aldosterone antagonist diuretics such as spironolactone. Concomitant renal dysfunction—the result of impaired perfusion due to diminished cardiac output or venous renal congestion—can exacerbate this hyperkalemia, sometimes to dangerous levels.17,18 In patients with severe right HF, a hepatic transaminitis may also be observed.19
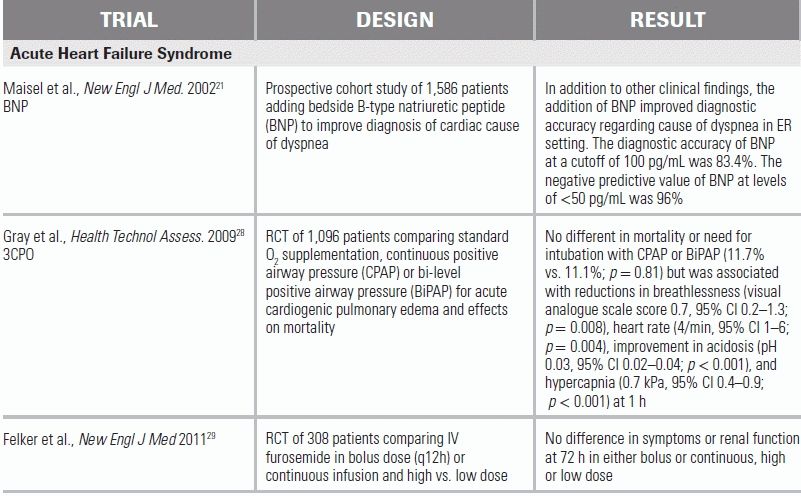
Natriuretic peptides (BNP, NT-proBNP) are normally secreted in response to ventricular distension and stretch and may be of particular use to the emergency physician (EP). Brain natriuretic peptide (BNP) and N-terminal pro–brain natriuretic peptide (NT-proBNP) act as endogenous diuretics, cardiac myocyte relaxants (lusitropic agents), and vasodilators. In the clinical setting, they have been shown to aid in the differentiation of the causes of acute dyspnea in patients with concomitant COPD and HF.20–24 Natriuretic peptides have a high negative predictive value for cardiac failure–induced dyspnea in the patient with AHFS. In a recent prospective cohort study of 1,586 patients, the diagnostic accuracy of BNP at a cutoff of 100 pg/mm was 83.4 % and the negative predictive value at levels of <50 pg/mm was 96%.21 Importantly, natriuretic peptides are insensitive in patients with marked obesity and may be elevated in patients with chronic renal failure or long-standing HF.22,23,25 In these cases, knowledge of the patient's baseline (dry weight) BNP level aids in decision making.
Chest Radiograph
Obtaining a chest radiograph (CXR) is considered standard of care in the evaluation of dyspnea, and approximately 80% of patients with AHFS will have radiographic evidence of pulmonary edema. Attention should be paid to the cardiac silhouette, as well. Enlargement can indicate chronic LV dysfunction; a globular appearance, pericardial effusion; and pericardial calcification, constriction.26 Significant pleural effusions (typically left greater than right) are often present in patients with congestive HF.
Differential Diagnosis
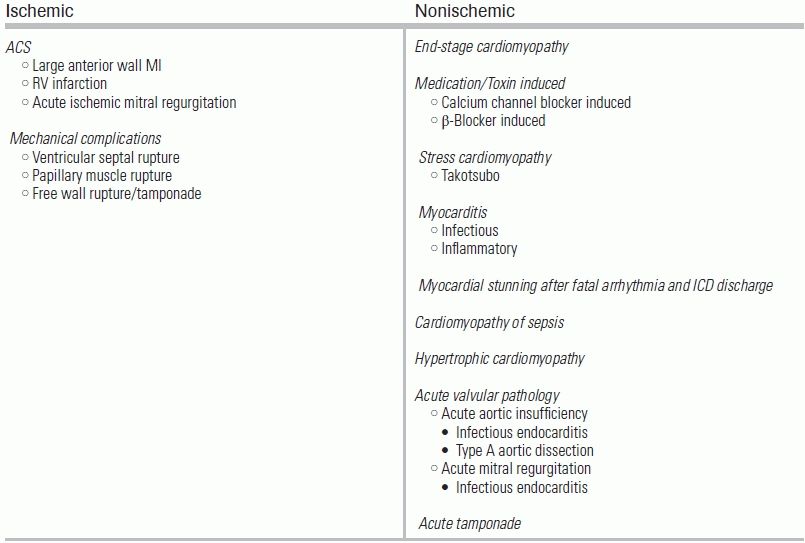
The differential diagnosis for causes of AHFS is extensive but may be broadly divided into ischemic and nonischemic (Table 14.1).
TABLE 14.1 Causes of AHFS
Management Guidelines
The initial assessment of the patient with AHFS begins with an assessment of respiratory status and the need for immediate respiratory support with either invasive or noninvasive positive pressure ventilation (NIPPV). In patients with intact mental status but severe respiratory distress, NIPPV has been shown to improve ventilation and reduce cardiac strain. A recent RCT of 2,096 patients with acute cardiogenic pulmonary edema compared standard O2 supplementation and NIPPV and showed significant reductions in breathlessness, heart rate, acidosis, and hypercapnia at 1 hour with NIPPV use.28 In patients with altered sensorium who are unable to adequately protect their airway, rapid sequence intubation should be performed immediately. In patients who are not hypoxemic, supplemental oxygen therapy is still effective at relieving dyspnea and should be provided.
In the severely hypertensive patient, sublingual nitroglycerin can favorably alter hemodynamics by decreasing afterload and preload. Sublingual administration is preferred as it rapidly increases blood levels in a bolus fashion, after which a nitroglycerin drip can be initiated.
Morphine is commonly administered for patient comfort but may produce undesirable effects, including a higher incidence of need for mechanical ventilation, intensive care unit admission, and prolonged hospital stay. Morphine has also been associated with increased mortality in patients presenting with NSTEMI.28
Most AHFS patients will benefit from diuretic therapy instituted in the ED, with the notable exception of patients with hypertrophic obstructive cardiomyopathy, whose cardiac output is dependent on adequate preload. Intravenous (IV) furosemide is the safest and most effective initial therapy, resulting in rapid diuresis. A recent RCT of patients with AHFS demonstrated IV bolus dosing of furosemide—preferred in the ED for ease of administration—to be equivalent to continuous infusion, with no significant difference in patients' assessment of symptoms or in the change in renal function.29
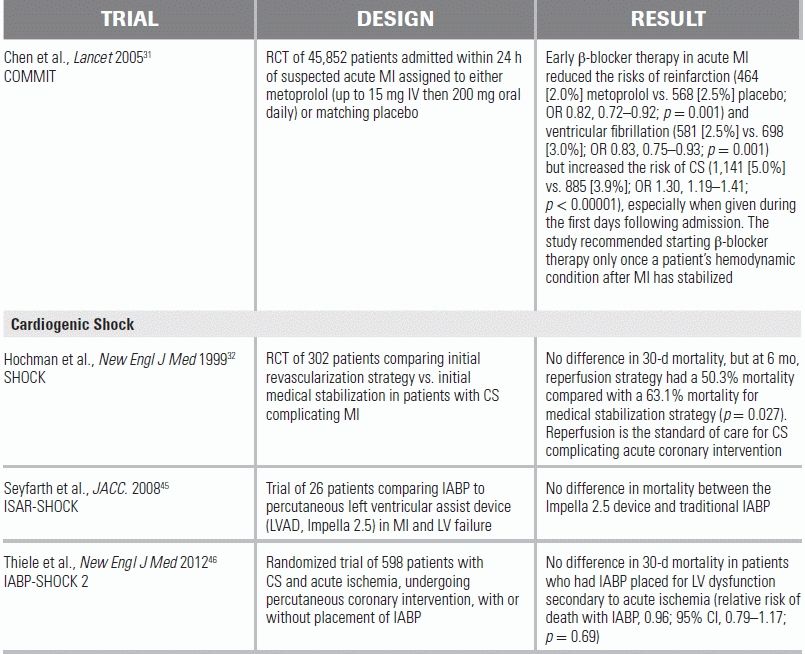
Arrhythmias can be the precipitating event in AHFS.30 For the AHFS patient presenting with a tachyarrhythmia—such as atrial fibrillation with rapid ventricular response—IV calcium channel and β-blocking agents should be avoided, as their negative inotropic effect may produce further hemodynamic deterioration. The COMMIT trial demonstrated that routine use of IV β blockade in patients with STEMI reduced the risks of reinfarction and ventricular fibrillation, but these gains were offset by increased rates of CS.31 In the absence of STEMI, consideration can be given to IV β blockade in low dosages, such as 5 to 15 mg total of IV metoprolol. In a state of volume overload, diuresis can sometimes be a more effective modality for rate control. In the patient with hemodynamic instability, immediate electrical cardioversion should be performed.
CARDIOGENIC SHOCK
Background
CS is defined as severely depressed cardiac output leading to life-threatening tissue hypoperfusion. In-hospital mortality from CS is approximately 60%, a number that has not changed dramatically over the last 20 years.32 Approximately 3% to 5% of patients with AHFS and 5% to 8% of patients with STEMI will present in CS. Of patients with CS in the setting of STEMI, the majority will have left anterior descending artery occlusions.33,34 Other risk factors for CS in the setting of myocardial infarction (MI) include advanced age, diabetes, a history of peripheral arterial disease, stroke, transient ischemic attack, coronary artery bypass grafting, and prior STEMI.35,36 The remainder of this chapter discusses the management of CS due to acute MI.
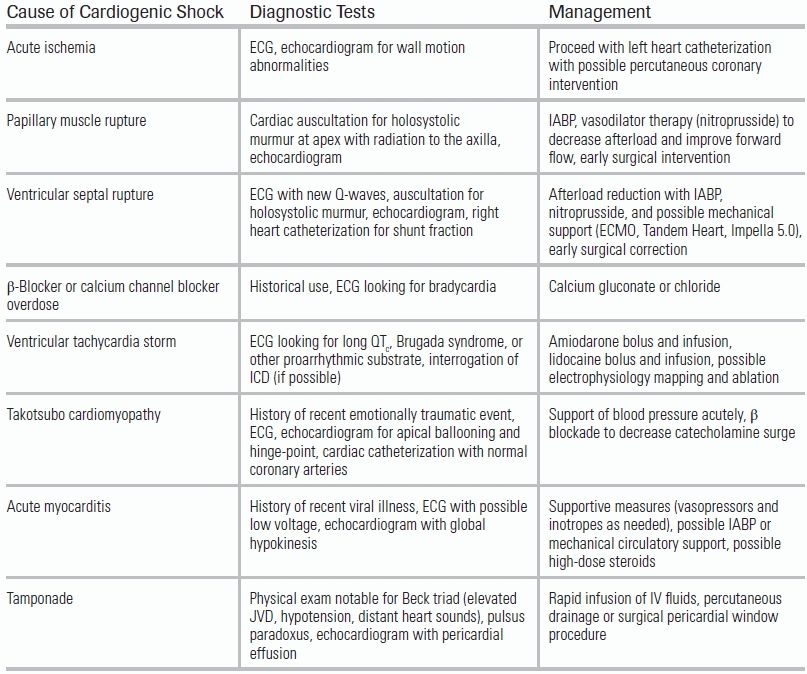
LV failure is the most common cause of CS. LV failure may result from one significant ischemic event or from a series of smaller ischemic insults over time. Subjects who are not reperfused, or who present late in the course of their MI, are particularly vulnerable. Importantly, when the degree of hemodynamic compromise is disproportionate to the area of myocardium at risk (i.e., as seen on ECG), alternate etiologies for the patient's hemodynamic instability should be sought. These include mechanical complications such as ventricular septal rupture and papillary muscle rupture—which can occur as early as the first day of infarction—as well as tamponade, acute aortic dissection, pulmonary embolism, medication effect, and occult blood loss (Table 14.2).
TABLE 14.2 Causes of Cardiogenic Shock
Physical Exam
Cold and clammy extremities are common in patients with an inadequate cardiac output and are due to the compensatory release of endogenous catecholamines that cause peripheral vasoconstriction and shunting of blood to vital organs. Diminished stroke volume may also result in a thready pulse, low pulse pressure, and a compensatory tachycardia.37,38 Importantly, attenuation of this tachycardic response may be noted in patients with chronic β-blocker use or conduction abnormalities.
Determination of volume status is a challenging but important step in the management of CS. As with any patient with HF, measurement of JVP, lung and thoracic auscultation, and assessment of extremity and dependent edema should be performed. CS remains one of the few clinical entities in which use of a pulmonary artery catheter (PAC) continues to be recommended. PAC monitoring can help confirm adequate intravascular status and enables the clinician to differentiate true CS from a mixed cardiogenic and vasodilatory process. Only experienced clinicians should perform PAC insertion, preferably in a medical or cardiac ICU.
The cardiac examination in a patient in CS should focus on the auscultation of cardiac murmurs. Holosystolic murmurs can suggest either a ventricular septal rupture or acute mitral regurgitation secondary to papillary muscle rupture. Distant heart sounds suggest a pericardial effusion and possibly tamponade secondary to free wall rupture.
Diagnostic Evaluation
Electrocardiogram
ST elevation consistent with acute ischemic injury is an indication for prompt reperfusion.32 Care must be taken to look for new arrhythmias, such as atrial fibrillation, ventricular tachycardia, or heart blocks, that can result in severe bradycardia.39
Laboratory Testing
New-onset end-organ dysfunction is a defining feature of shock. Newly elevated creatinine or hepatic enzymes suggest poor delivery of blood to the kidneys and liver. Elevated lactate levels, seen with any tissue hypoperfusion, are a quantitative measure of shock severity. Cardiac enzyme levels, as well, may be useful in helping elucidate the magnitude and timing of the ischemic event.40
Chest Radiograph
In patients with difficult physical exams, a CXR is useful in assessing for pulmonary edema and pleural effusions.27 Although not specific, a bulbous cardiac silhouette may suggest pericardial fluid or tamponade from a free wall rupture, while unilateral pulmonary edema may be seen in the setting of acute mitral regurgitation due to papillary muscle rupture.41
Echocardiogram
Echocardiography is an invaluable diagnostic tool in CS and can help identify the precipitating pathology. Acute valvular pathology, tamponade, RV infarction, takotsubo cardiomyopathy, LV wall motion abnormalities, and ventricular septal rupture—to name a few—can be ruled in or out by echocardiography.42
Management Guidelines
Primary reperfusion therapy is the mainstay of treatment in patients presenting with CS secondary to acute myocardial ischemia. The SHOCK trial demonstrated an initial early revascularization strategy—using either percutaneous coronary intervention or coronary artery bypass grafting—to be associated with a 13% decrease in mortality when compared to medical stabilization strategy (50.3% vs. 63.1%).32 In the absence of contraindications, these patients should be taken for mechanical revascularization as urgently as possible. Pharmacologic lytic therapy is ineffective in this population, as poor blood flow leads to low drug concentration in the target coronary arteries.
Mechanical complications of MIs, such as ventricular septal rupture or papillary muscle rupture, are surgical emergencies minimally amenable to medical management. Adjunctive mechanical support devices, such as an Impella, Tandem Hearts, or intra-aortic balloon pumps (IABPs), may be considered as a bridge to surgical intervention but are not definitive therapies. Although IABPs and Impellas have often been used to provide adjunctive hemodynamic support in the setting of CS, they have yet to be proven to improve clinical outcomes or decrease mortality. In the IABP SHOCK-2 trial, patients with CS due to MI in whom an early invasive strategy was planned were randomized to either IABP or medical therapy alone. No difference was seen between the patient groups in terms of mortality (relative risk with IABP, 0.96).43 As an alternative therapy for the same clinical scenario, Impella 2.5 L/min was compared with IABP in the ISAR-Shock 2 trial. Both therapies were shown to have similar effects on 30-day mortality (30-day mortality 46% in both groups).44
Vasopressors and Inotropes
In an ED setting, temporary stabilization of the patient with vasopressors or inotropes may be necessary. The use of vasopressors in CS should be limited to patients with hypotension (systolic blood pressure <80 mm Hg or mean arterial pressure <65 mm Hg). Increasing afterload provides a temporizing solution for a failing heart but will also increase cardiac oxygen consumption. An understanding of the appropriate application of these agents is important.
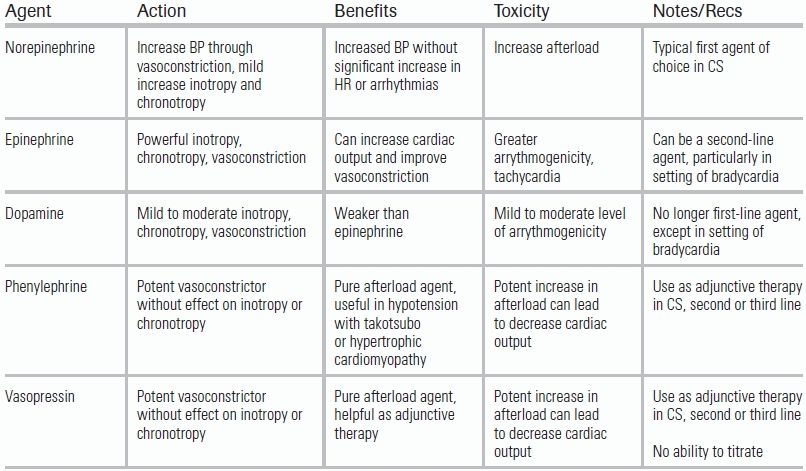
Norepinephrine is a synthetic catecholamine with vasoconstrictive as well as positive chronotropic (heart rate) and inotropic (contractility) effects. Its primary effect is stimulation of α receptors, which raises systemic vascular resistance (SVR) leading to an increase in blood pressure. Norepinephrine also increases heart rate and contractility through a weak but present agonism of β receptors. As with any α receptor agonist, or vasopressor, long-term use or sustained high doses can result in peripheral tissue ischemia.
Phenylephrine is pure α receptor agonist and vasoconstrictor; like norepinephrine, it raises blood pressure by increasing SVR. It has no direct effect on the heart but will increase blood pressure. Phenylephrine should be limited to use in patients with hypertrophic obstructive cardiomyopathy.
Epinephrine is a powerful vasoconstrictor and inotrope and acts directly on both α and β receptors. The result is a more dramatic increase in inotropy and chronotropy with a similar, or even more powerful, degree of vasoconstriction than norepinephrine. As a potent chronotropic agent, the drug is capable of inducing cardiac arrhythmias, including atrial fibrillation and ventricular fibrillation. For this reason, it is considered a second- or third-line agent in CS.
Vasopressin is an analog of antidiuretic hormone and acts on the vasopressin receptor. A powerful vasoconstrictor, it works best at a fixed dose in concert with another vasoconstrictor such as norepinephrine or phenylephrine. There is limited data for the use of vasopressin in CS, and for this reason, it is recommended only as a third- or fourth-line agent.
Dopamine is weaker version of epinephrine and has effects on the α, β, and Dopamine (D) receptors. Traditional teaching is that at increasing doses, dopamine affects different receptors; in practice, dopamine can be used simultaneously as a vasoconstrictor, inotrope, and chronotrope. Data from the SOAP II trial suggested dopamine and norepinephrine to have similar efficacy in CS but found dopamine to be associated with more adverse events, specifically arrhythmias43 (Table 14.3).
TABLE 14.3 Vasopressors and Inotropes in Cardiogenic Shock
CONCLUSION
In patients with HF and CS, early and accurate assessment of volume status and cardiac function improves outcome. Close hemodynamic monitoring is vital, and the use of intra-arterial blood pressure monitoring is strongly recommended. Early involvement of a cardiology intensivist is recommended for any unstable patient with AHFS.
LITERATURE TABLE
CI, confidence interval; OR, odds ratio.
REFERENCES
1.Gheorghiade M, Zannad F, Sopko G, et al; International Working Group on Acute Heart Failure Syndromes. Acute heart failure syndromes: current state and framework for future research. Circulation. 2005;112:3958–3968.
2.Hunt SA, Abraham WT, Chin MH, et al. 2009 Focused Update Incorporated into the ACC/AHA 2005 Guidelines for the diagnosis and management of heart failure in adults. J Am Coll Cardiol. 2009;53:e1–e90.
3.Tracy CM, Epstein AE, Darbar D, et al. 2012 ACCF/AHA/HRS focused update incorporated into the ACCF/AHA/HRS 2008 guidelines for device-based therapy of cardiac rhythm abnormalities. J Am Coll Cardiol. 2013;61:e6–e75.
4.Moss AJ, Hall WJ, Cannom DS, et al. Improved survival with an implanted defibrillator in patients with coronary disease at high risk for ventricular arrhythmia. Multicenter Automatic Defibrillator Implantation Trial Investigators. N Engl J Med. 1996;335:1933–1940.
5.The effects of tissue plasminogen activator, streptokinase, or both on coronary-artery patency, ventricular function, and survival after acute myocardial infarction. The GUSTO Angiographic Investigators. N Engl J Med. 1993;329:1615–1622.
6.A clinical trial comparing primary coronary angioplasty with tissue plasminogen activator for acute myocardial infarction. The Global Use of Strategies to Open Occluded Coronary Arteries in Acute Coronary Syndromes (GUSTO IIb) Angioplasty Substudy Investigators. N Engl J Med. 1997;336:1621–1628.
7.Van de Werf F, Ross A, Armstrong P, et al. Primary versus tenecteplase-facilitated percutaneous coronary intervention in patients with ST-segment elevation acute myocardial infarction (ASSENT-4 PCI): randomised trial. Lancet. 2006;367:569–578.
8.Bigger JT. Prophylactic use of implanted cardiac defibrillators in patients at high risk for ventricular arrhythmias after coronary-artery bypass graft surgery. Coronary Artery Bypass Graft (CABG) Patch Trial Investigators. N Engl J Med. 1997;337:1569–1575.
9.Buxton AE, Fisher JD, Josephson ME, et al. Prevention of sudden death in patients with coronary artery disease: the Multicenter Unsustained Tachycardia Trial (MUSTT). Prog Cardiovasc Dis. 1993;36:215–226.
10.Stevenson LW. Tailored therapy to hemodynamic goals for advanced heart failure. Eur J Heart Fail. 1999;1:251–257.
11.Drazner MH, Rame JE, Stevenson LW, et al. Prognostic importance of elevated jugular venous pressure and a third heart sound in patients with heart failure. N Engl J Med. 2001;345:574–581.
12.Cintron GB, Hernandez E, Linares E. Bedside recognition, incidence and clinical course of right ventricular infarction. Am J Cardiol. 1981;47:224–227.
13.Vaitkus PT, Kussmaul WG. Constrictive pericarditis versus restrictive cardiomyopathy: a reappraisal and update of diagnostic criteria. Am Heart J. 1991;122:1431–1441.
14.Johnston M, Collins SP, Storrow AB. The third heart sound for diagnosis of acute heart failure. Curr Heart Fail Rep. 2007; 4:164–168.
15.Bonow RO, Carabello BA, Chatterjee K, et al. 2008 focused update incorporated into the ACC/AHA 2006 guidelines for the management of patients with valvular heart disease. J Am Coll Cardiol. 2008;52:e1–e142.
16.Lee WH, Packer M. Prognostic importance of serum sodium concentration and its modification by converting-enzyme inhibition in patients with severe chronic heart failure. Circulation. 1986;73:257–267.
17.Juurlink DN, Mamdani MM, Lee DS, et al. Rates of hyperkalemia after publication of the randomized aldactone evaluation study. N Engl J Med. 2004;351:543–551.
18.Pitt B, Zannad F, Remme WJ, et al. The effect of spironolactone on morbidity and mortality in patients with severe heart failure. Randomized aldactone evaluation study investigators. N Engl J Med. 2000;341:709–717.
19.Matthews JC, Dardas TF, Dorsch MP. Right-sided heart failure: diagnosis and treatment strategies. Curr Treat Options Cardiovasc Med. 2008;10:329–341.
20.McCullough PA, Nowak RM, McCord J, et al. B-Type natriuretic peptide and clinical judgment in emergency diagnosis of heart failure. Circulation. 2002;106:416–422.
21.Maisel AS, Krishnaswamy P, Nowak RM, et al; Breathing Not Properly Multinational Study Investigators. Rapid measurement of B-type natriuretic peptide in the emergency diagnosis of heart failure. N Engl J Med. 2002;347:161–167.
22.Daniels LB, Clopton P, Bhalla V, et al. How obesity affects the cut-points for B-type natriuretic peptide in the diagnosis of acute heart failure. Am Heart J. 2006;151:999–1005.
23.McCullough PA, Duc P, Omland T, et al. B-type natriuretic peptide and renal function in the diagnosis of heart failure: an analysis from the breathing not properly multinational study. Am J Kidney Dis. 2003;41:571–579.
24.Januzzi JL Jr, Camargo CA, Anwaruddin S, et al. The N-terminal Pro-BNP Investigation of Dyspnea in the Emergency department (PRIDE) study. Am J Cardiol. 2005;95:948–954.
25.Tsutamoto T, Wada A, Maeda K, et al. Attenuation of compensation of endogenous cardiac natriuretic peptide system in chronic heart failure: prognostic role of plasma brain natriuretic peptide concentration in patients with chronic symptomatic left ventricular dysfunction. Circulation. 1997;96:509–516.
26.Gimlette T. Constrictive pericarditis. Br Heart J. 1959;21:9–16.
27.Killip T III, Kimball JT. Treatment of myocardial infarction in a coronary care unit. Am J Cardiol. 1967;20:457–464.
28.Gray AJ, Goodacre S, Newby DE, et al; 3CPO Study Investigators. A multicentre randomised controlled trial of the use of continuous positive airway pressure and non-invasive positive pressure ventilation in the early treatment of patients presenting to the emergency department with severe acute cardiogenic pulmonary oedema: the 3CPO trial. Health Technol Assess. 2009;13:1–106.
29.Felker GM, Lee KL, Bull DA, et al. Diuretic strategies in patients with acute decompensated heart failure. N Engl J Med. 2011;364:797–805.
30.Karasek J, Widimsky P, Ostadal P, et al. Acute heart failure registry from high-volume university hospital ED: comparing European and US data. Am J Emerg Med. 2012;30:695–705.
31.Chen ZM, Pan HC, Chen YP, et al; COMMIT (Clopidogrel and Metoprolol in Myocardial Infarction Trial) collaborative Group. Early intravenous then oral metoprolol in 45,852 patients with acute myocardial infarction: randomised placebo-controlled trial. Lancet. 2005;366:1622–1632.
32.Hochman JS, Sleeper LA, Webb JG, et al. Early revascularization in acute myocardial infarction complicated by cardiogenic shock. SHOCK Investigators. Should we emergently revascularize occluded coronaries for cardiogenic shock. N Engl J Med. 1999;341:625–634.
33.Bengtson JR, Kaplan AJ, Pieper KS, et al. Prognosis in cardiogenic shock after acute myocardial infarction in the intervencional era. J Am Coll Cardiol. 1992;20:1482–1489.
34.Webb JG, Sleeper LA, Buller CE, et al. Implications of the timing of onset of cardiogenic shock after acute myocardial infarction: a report from the SHOCK Trial Registry. J Am Coll Cardiol. 2000;36:1084–1090.
35.Zeymer U. Predictors of in-hospital mortality in 1333 patients with acute myocardial infarction complicated by cardiogenic shock treated with primary percutaneous coronary intervention (PCI) Results of the primary PCI registry of the Arbeitsgemeinschaft Leitende Kardiologische Krankenhausärzte (ALKK). Eur Heart J. 2004;25:322–328.
36.Hasdai D, Holmes DR Jr, Califf RM, et al. Cardiogenic shock complicating acute myocardial infarction: predictors of death. Am Heart J. 1999;138:21–31.
37.Menon V, Hochman JS. Management of cardiogenic shock complicating acute myocardial infarction. Heart. 2002;88(5):531–537.
38.Reynolds HR, Hochman JS. Cardiogenic shock: current concepts and improving outcomes. Circulation. 2008;117:686–697.
39.Berger PB, Ryan TJ. Inferior myocardial infarction. High-risk subgroups. Circulation. 1990;81:401–411.
40.Katus HA, Remppis A, Neumann FJ, et al. Diagnostic efficiency of troponin T measurements in acute myocardial infarction. Circulation. 1991;83:902–912.
41.Stout KK, Verrier ED. Acute valvular regurgitation. Circulation. 2009;119:3232–3241.
42.Gianni M. Apical ballooning syndrome or takotsubo cardiomyopathy: a systematic review. Eur Heart J. 2006;27:1523–1529.
43.De Backer D, Biston P, Devriendt J, et al. Comparison of dopamine and norepinephrine in the treatment of shock. N Engl J Med. 2010;362:779–789.
44.Pfeffer MA, Braunwald E, Moyé LA, et al. Effect of captopril on mortality and morbidity in patients with left ventricular dysfunction after myocardial infarction. Results of the survival and ventricular enlargement trial. The SAVE Investigators. N Engl J Med. 1992;327:669–677.
45.Seyfarth M, Sibbing D, Bauer I, et al. A randomized clinical trial to evaluate the safety and efficacy of a percutaneous left ventricular assist device versus intra-aortic balloon pumping for treatment of cardiogenic shock caused by myocardial infarction. J Am Coll Cardiol. 2008;52:1584–1588.
46.Thiele H, Zeymer U, Neumann F-J, et al. Intraaortic balloon support for myocardial infarction with cardiogenic shock. N Engl J Med. 2012;367:1287–1296.