Cirrhosis
In cirrhosis, the liver architecture is diffusely abnormal and interferes with liver blood flow and function; this leads to the clinical manifestations of portal hypertension and liver failure.
 Aetiology
Aetiology
Causes of cirrhosis are shown in Box 14.12. Alcohol is currently the most common cause in the West, but likely to be superseded by NAFLD; viral infection the most common worldwide. Young patients with cirrhosis must be investigated to exclude treatable causes (e.g. Wilson's disease).
Pathogenesis
Although the liver has a remarkable capacity to adapt to injury through tissue repair, chronic injury results in inflammation, matrix deposition, necrosis and angiogenesis, all of which lead to fibrosis (Fig. 14.21). Liver injury causes necrosis and apoptosis, releasing cell contents and reactive oxygen species (ROS). This activates hepatic stellate cells and tissue macrophages through the CC-chemokine ligand 2–CC-chemokine receptor 2 (CCL2–CCR2) axis (see p. 440). These cells phagocytose necrotic and apoptotic cells and secrete pro-inflammatory mediators, including transforming growth factor-beta (TGF-β); this leads to transdifferentiation of stellate cells to myofibroblasts and platelet-derived growth factor (PDGF), which stimulates myofibroblast proliferation. Macrophages degrade scar matrix by secretion of matrix metalloproteinases (MMPs), but this is inhibited by concurrent myofibroblast and macrophage production of tissue inhibitors of metalloproteinases (TIMPs). This results in progressive matrix deposition and scar accumulation. Increased gut permeability and hepatic lipopolysaccharide–Toll-like receptor 4 (LPS–TLR4) signalling also promotes fibrogenesis. Repetitive or chronic injury and inflammation perpetuate this process.
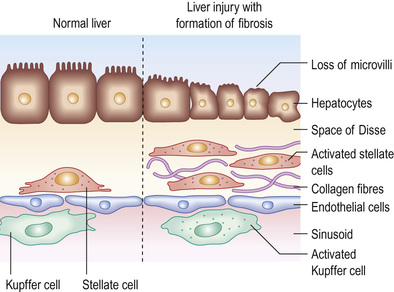
If the cause of fibrosis is eliminated (e.g. treatment of viral hepatitis), resolution (complete reversal to near-normal liver architecture) of early fibrosis can occur. In cirrhosis, regression (improvement, not reversal) occurs, which improves clinical outcomes. Antifibrotic therapies are emerging (including stem cell transplant strategies) but currently liver transplantation is the only available treatment for liver failure.
Pathology
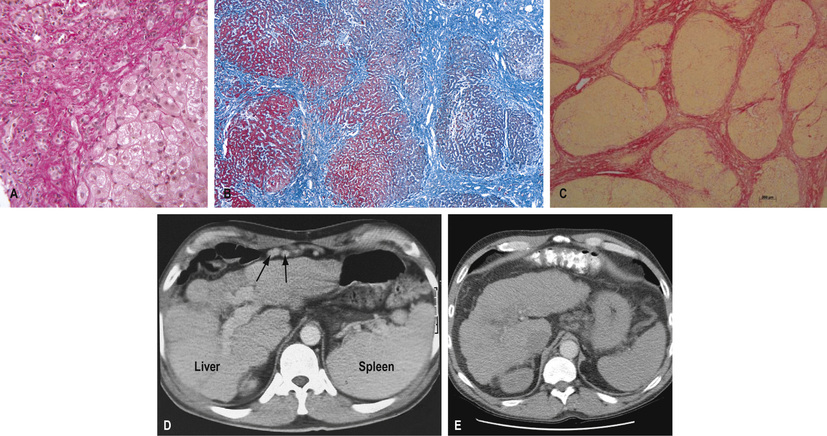
The characteristic features of cirrhosis are regenerating nodules separated by fibrous septa, and loss of lobular architecture within the nodules (Fig. 14.22A–C). Two types are described:
• Micronodular cirrhosis. Regenerating nodules are usually <3 mm in size with uniform involvement of the liver; often caused by alcohol or biliary tract disease.
• Macronodular cirrhosis. The nodules are of variable size and normal acini may be seen within larger nodules; often caused by chronic viral hepatitis.
A mixed picture with small and large nodules occurs occasionally.
Symptoms and signs are described on pages 447–448.
 Investigations
Investigations
Investigations aim to assess the severity and type of liver disease.
Severity
• Liver function. Serum albumin and prothrombin time are the best indicators of liver function; the outlook is poor if the albumin level is <28 g/L. The degree to which the prothrombin time is prolonged is commensurate with disease severity.
• Liver biochemistry. This may be normal, depending on the severity of disease. In most cases, there is a slight elevation in the serum ALP and serum aminotransferases. In decompensated cirrhosis, all biochemistry is deranged.
• Serum electrolytes. A low sodium indicates severe liver disease due to a defect in free water clearance or excess diuretic therapy.
• Serum creatinine. An elevated concentration >130 µmol/L is a marker of poor prognosis.
• Biomarkers. In the Enhanced Liver Fibrosis (ELF™) test, which assesses fibrosis, a value of <7.7 indicates none to mild, 7.7–9.8 moderate, and ≥9.8 severe.
In addition, a serum α-fetoprotein of >200 ng/mL is strongly suggestive of HCC.
Type
This can be determined by:
• copper and caeruloplasmin (see p. 479)
• α1-antitrypsin (see pp. 479–480).
Serum copper and serum α1-antitrypsin should always be measured in young cirrhotics. Total iron-binding capacity (TIBC) and ferritin should be measured to exclude hereditary haemochromatosis; genetic markers are also available (see pp. 477–479).
Imaging
• Ultrasound examination can demonstrate changes in the size and shape of the liver. Fatty change and fibrosis produce a diffusely increased echogenicity. In established cirrhosis, there may be marginal nodularity of the liver surface and distortion of the arterial vascular architecture. The patency of the portal and hepatic veins can be evaluated. Ultrasound is useful for detecting HCC.
• Transient elastography is increasingly used to avoid liver biopsy (see p. 446). Technical limitations preclude its use in patients with ascites or morbid obesity, but it is suitable for most.
• CT scanning (see p. 446) is also helpful. Figure 14.22D–E show hepatosplenomegaly and the dilated collaterals seen in chronic liver disease. Arterial phase-contrast-enhanced scans are useful for detecting of HCC.
• Endoscopy is performed for the detection and treatment of varices and portal hypertensive gastropathy. Colonoscopy is occasionally carried out for colopathy.
• MRI scanning is useful in the diagnosis of both malignant and benign tumours such as haemangiomas. MR angiography can demonstrate the vascular anatomy, and MR cholangiography the biliary tree.
Liver biopsy
This remains the ‘gold standard’ for confirming the type and severity of liver disease. Adequate samples, in terms of length and number of complete portal tracts, are necessary for diagnosis and staging/grading of chronic viral hepatitis; in macronodular cirrhosis, the core of liver may fragment, causing sampling errors. Immunocytochemical stains can identify viruses, bile ducts, angiogenic structures and oncogenic markers. Chemical measurement of iron and copper is necessary to confirm a diagnosis of iron overload or Wilson's disease. Digital image analysis of Picrosirius red staining can be used to quantitate collagen in biopsy specimens (Fig. 14.22C).
Management
Management is that of the complications of decompensated cirrhosis. Patients should undergo 6-monthly ultrasound to screen for the early development of HCC (see p. 485), as all therapeutic strategies work best with small, single tumours.
Treatment of the underlying cause may arrest or reverse cirrhosis. Patients with compensated cirrhosis should lead a normal life. Those at risk should receive hepatitis A and B vaccination. The only dietary restriction is to reduce salt intake (≤2 g sodium per day). Alcohol should be avoided, as should aspirin and non-steroidal anti-inflammatory drugs (NSAIDs), which may precipitate gastrointestinal bleeding or renal impairment.
Prognosis
Prognosis is extremely variable. In general, the 5-year survival rate is approximately 50%, depending on the stage at which diagnosis is made. Poor prognostic indicators are shown in Box 14.13. Development of any complication usually worsens the prognosis.
 Box 14.13
Box 14.13
Poor prognostic indicators in cirrhosis
There are a number of prognostic classifications based on modifications of Child's grading (A, B and C; Box 14.14) and the Model for End-stage Liver Disease (MELD; Box 14.15), based on serum bilirubin, creatinine and INR, which is widely used as a predictor of mortality in patients awaiting liver transplantation. Modifications of the MELD score (e.g. UKELD) are used in some countries.
Box 14.14
Scoring systems in cirrhosis: modified Child–Pugh classification
| Score | 1 | 2 | 3 |
| Ascites | None | Mild | Moderate/severe |
| Encephalopathy | None | Mild | Marked |
| Bilirubin (µmol/L) | <34 | 34–50 | >50 |
| Albumin (g/L) | >35 | 28–35 | <28 |
| Prothrombin time (seconds over normal) | <4 | 4–6 | >6 |
Add above scores for your patient for survival figures below: | |||

Box 14.15
Scoring systems in cirrhosis
Model of End-stage Liver Disease (MELD)
3.8 × LN (bilirubin in mg/dL) + 9.6 × LN (creatinine in mg/dL) + 11.2 × LN (INR) + 6.4*
To convert:
MELD scores (with no complications):
INR, International Normalized Ratio; LN, natural logarithm.
*Check online
Acute-on-chronic liver failure
Acute-on-chronic liver failure (ACLF) refers to the condition of patients hospitalized for an acute complication of cirrhosis accompanied by organ failure(s); it has a high short-term mortality. ACLF is believed to be distinct from traditional decompensated cirrhosis, based not only on the presence of organ failure(s) and high mortality rate, but also on younger age, alcohol aetiology, higher prevalence of certain precipitating events (e.g. bacterial infections or active alcohol excess), and a higher level of systemic inflammation. It is estimated to occur in 31% of patients hospitalized with an acute complication of cirrhosis.
Liver assist devices
As the liver has great potential to regenerate, extracorporeal liver support devices may allow patients with liver failure to be bridged to recovery or even liver transplantation. Current approaches include the use of biological devices that contain hepatocytes and those that function as detoxification devices, and artificial liver support systems. However, no device is currently available routinely.
Gut–liver axis
The liver is exposed to gut-derived bacterial components that have little consequence in health, as an effective gut barrier limits the amount of bacterial components transported to the liver. In advanced liver disease, the intestinal barrier function is compromised due to changes in gut motility, an increase in intestinal permeability, and suppression of gut immunological functions. This leads to bacteria and components entering the portal circulation and being transported to the liver, activating Toll-like receptors and producing an inflammatory response. This cross-talk between the intestinal microbiota and the liver is referred to as the gut–liver axis; it is seen as a key pathophysiological mechanism in the progression of liver disease and development of the complications of cirrhosis. Antibiotics and non-selective β-blockers intercept the gut–liver axis by blocking bacterial translocation, which is likely to account for their beneficial effects in reducing portal pressure, variceal haemorrhage and spontaneous bacterial peritonitis. However, absorbable antibiotics will lead to the selection of resistant bacteria. Rifaximin, a poorly absorbed antibiotic used for encephalopathy, specifically affects the gut flora and has a low risk for inducing resistance; it may therefore have a role in this indication.
Liver transplantation
This is the only established treatment for advanced liver disease. Shortage of donors is a major problem in all developed countries and in some, such as Japan or South Korea, living related donors form the majority.
Indications
These include:
• Acute liver disease: acute hepatic failure of any cause (see p. 463).
• Chronic liver disease: usually for complications of cirrhosis that are no longer responsive to therapy.
The timing of transplantation depends on donor availability. All patients with end-stage cirrhosis (Child's grade C; MELD score ≥20; UKELD score ≥49) and those with debilitating symptoms should be referred to a transplant centre. In addition, specific extrahepatic complications of cirrhosis, even with preserved liver function, such as hepatopulmonary syndrome and porto-pulmonary hypertension, can be reversed by transplantation.
• Primary biliary cholangitis. Patients should be transplanted when their serum bilirubin is persistently >100 µmol/L or when they have symptoms such as intractable pruritus.
• Chronic hepatitis B if HBV DNA-negative or levels are falling with therapy. Following transplantation, recurrence of hepatitis is prevented by hepatitis B immunoglobulin and nucleoside analogues in combination (see pp. 243–244).
• Chronic hepatitis C. This is the most common indication. Universal HCV re-infection occurs with varying severity and cirrhosis occurs in 10–20% at 5 years. The new antiviral drugs are highly likely to improve these figures greatly.
• Autoimmune hepatitis. In patients who have failed to respond to medical treatment, the disease can recur.
• Alcoholic liver disease. Well-motivated patients who have stopped drinking without improvement of liver disease are offered a transplant, with frequent counselling. It has been shown that transplantation may represent life-saving treatment in patients with severe alcoholic hepatitis who are not responding to medical therapy. However, further studies are awaited.
• Primary metabolic disorders. Examples are Wilson's disease, hereditary haemochromatosis and α1-antitrypsin deficiency.
• NASH cirrhosis. Now one of the most common causes of chronic liver disease in developed countries, this is likely to become the most frequent indication for transplantation.
• Other conditions, such as primary sclerosing cholangitis (PSC), polycystic liver disease, and metabolic diseases such as primary oxaluria.
Contraindications
Absolute contraindications include active sepsis outside the hepatobiliary tree, malignancy outside the liver, liver metastases (except neuroendocrine), and a lack of psychological commitment on the part of the patient.
Relative contraindications are mainly anatomical considerations that make surgery more difficult, such as extensive splanchnic venous thrombosis. With exceptions, patients aged 70 years or over are not usually given a transplant. In HCC, the recurrence rate is high, unless there are fewer than three small (<3 cm) lesions or a solitary nodule of <5 cm (Milan criteria).
Preparation for surgery
Pre-transplant work-up includes confirmation of the diagnosis, ultrasound and cross-sectional imaging, and radiological demonstration of the hepatic arterial and biliary trees, as well as assessment of cardiorespiratory and renal status. In view of the ethical and financial implications of transplantation, regular psychosocial, and possibly psychiatric, support is vital.
The donor should be ABO-compatible (HLA matching is not necessary) and have no evidence of active sepsis, malignancy, or HIV, HBV or HCV infection. Younger donors (<50 years) experience better graft function. The donor liver is cooled and stored on ice; its preservation time may be up to 20 hours. The recipient operation takes approximately 8 hours and rarely requires a large blood transfusion. Cadaveric donor livers (from heart-beating or non-heart-beating donors) may consist of whole graft, split grafts (for two recipients) or reduced grafts. Live donors may be healthy individuals or patients with, for example, familial amyloid polyneuropathy, whose livers can be transplanted into others (domino transplant). The mortality of right lobe donors is between 1 in 200 and 1 in 400.
The operative mortality is low and most postoperative deaths occur in the first 3 months. Sepsis and haemorrhage can be serious complications. Opportunistic infections occur owing to immunosuppression. Various immunosuppressive agents have been used, but tacrolimus – alone or in combination with azathioprine or mycophenolate mofetil, steroids, sirolimus and microemulsified ciclosporin are the most common.
Rejection
• Acute or cellular rejection usually occurs 5–10 days post transplant; it can be asymptomatic or there may be a fever. On biopsy, a pleomorphic portal infiltrate is seen with prominent eosinophils, bile duct damage and endothelialitis of the blood vessels. This responds well to immunosuppressive therapy.
• Chronic ductopenic rejection is seen between 6 weeks and 9 months post transplant, with disappearing bile ducts (vanishing bile duct syndrome, VBDS), and an arteriopathy with narrowing and occlusion of the arteries. Early ductopenic rejection is rarely reversed by immunosuppression and often requires retransplantation.
Prognosis
Elective liver transplantation in low-risk patients has a 90% 1-year survival and a 70–85% 5-year survival. Patients require life-long immunosuppression, although doses can be reduced over time without significant problems. Future strategies to reduce immunosuppression requirements after transplant may include infusion of autologous regulatory T cells. HCV cirrhosis, PSC and HCC are conditions in which long-term survival after transplantation is compromised by disease recurrence.
Complications and effects of cirrhosis
These are shown in Box 14.16.
 Portal hypertension
Portal hypertension
The portal vein is formed by the union of the superior mesenteric and splenic veins. Normal pressure is 5–8 mmHg with only a small gradient across the liver to the hepatic vein, in which blood is returned to the heart via the inferior vena cava. Portal hypertension is classified according to the site of obstruction:
• Prehepatic – blockage of the portal vein before the liver
• Intrahepatic – distortion of the liver architecture, either pre-sinusoidal (e.g. schistosomiasis) or post-sinusoidal (e.g. cirrhosis)
As portal pressure rises above 10–12 mmHg, the compliant venous system dilates and collaterals form within the systemic venous system. The main sites of collaterals are the gastro-oesophageal junction, rectum, left renal vein, diaphragm, retroperitoneum and the anterior abdominal wall via the umbilical vein.
The collaterals at the gastro-oesophageal junction (varices) are superficial and tend to rupture. Portosystemic anastomoses at other sites rarely cause symptoms. Rectal varices are found frequently (30%) if looked for and can be differentiated from haemorrhoids, which are lower in the anal canal. The microvasculature of the gut becomes congested, giving rise to portal hypertensive gastropathy and colopathy, in which there is punctate erythema and erosions, which can bleed.
Pathophysiology
Following liver injury and fibrogenesis, the contraction of activated myofibroblasts (mediated by endothelin, nitric oxide and prostaglandins) contributes to increased resistance to blood flow. This increased resistance leads to portal hypertension and opening of portosystemic anastomoses in both pre-cirrhotic and cirrhotic livers. Neoangiogenesis also occurs. The hyperdynamic circulation of cirrhosis (caused by nitric oxide, cannabinoids and glucagon) leads to peripheral and splanchnic vasodilatation. This, combined with plasma volume expansion due to sodium retention (see ‘Ascites’, pp. 472–473), has a significant effect in maintaining portal hypertension.
Aetiology
The most common cause is cirrhosis (Box 14.17). Others are described below.
 Box 14.17
Box 14.17
Causes of portal hypertension
Prehepatic causes
Extrahepatic blockage due to portal vein thrombosis can be caused by congenital portal venous abnormalities, neonatal sepsis of the umbilical vein, or inherited prothrombotic conditions, such as factor V Leiden or primary myeloproliferative disorders with or without JAK2 mutations (see p. 97).
Patients present with gastrointestinal bleeding, often at a young age. They have normal liver function, and prognosis following bleeding is therefore excellent.
The portal vein blockage can be identified by ultrasound with Doppler imaging; CT and MR angiography are also used.
Treatment for variceal bleeding is usually repeated endoscopic therapy or non-selective beta-blockade. Splenectomy is only performed if there is isolated splenic vein thrombosis. Anticoagulation prevents further thrombosis and intestinal infarction, and does not increase the risk of bleeding.
Intrahepatic causes
Cirrhosis is by far the most common cause but others include:
• Non-cirrhotic portal hypertension is characterized by mild portal tract fibrosis on liver histology. The aetiology is unknown, but arsenic, vinyl chloride, antiretroviral therapy and other toxins have been implicated. A similar disease is frequently found in India. The liver lesion does not progress and prognosis is good.
• Schistosomiasis with extensive pipe-stem fibrosis in endemic areas such as Egypt and Brazil. Often, there is concomitant liver disease, such as HCV infection, which was transmitted by non-sterile equipment.
• Other causes include congenital hepatic fibrosis, and nodular regenerative hyperplasia and partial nodular transformation.
Post-hepatic causes
Prolonged severe heart failure with tricuspid incompetence or constrictive pericarditis can cause portal hypertension. The Budd–Chiari syndrome is described on page 482.
Clinical features
Patients are often asymptomatic, the only clinical evidence being splenomegaly, although features of chronic liver disease may exist (see pp. 447–448).
Variceal haemorrhage
Approximately 90% of patients with cirrhosis will develop gastro-oesophageal varices over 10 years, but only one-third of these will bleed. Bleeding is likely to occur with large varices, or those with red signs at endoscopy, and in severe liver disease.
Management
Management can be divided into:
Despite the therapeutic techniques available, prognosis ultimately depends on the severity of the underlying liver disease; overall 6-week mortality from variceal haemorrhage is 15–25%, reaching 50% in Child's grade C.
Initial management of acute variceal bleeding
See Figure 14.23, and also the general management of gastrointestinal haemorrhage on page 385.
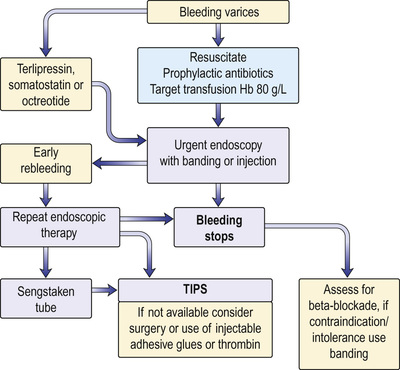
Resuscitation
• Assess the patient: pulse, blood pressure and conscious state.
• Insert a large-bore intravenous line and obtain blood for group and crossmatching, haemoglobin, prothrombin time/INR, urea, electrolytes, creatinine, liver biochemistry and blood cultures.
• Restore blood volume with plasma expanders or, if possible, blood transfusion. See the treatment of shock for more detail (pp. 1156–1161). Prompt correction, but not over-correction, of hypovolaemia is necessary in cirrhosis patients, as their baroreceptor reflexes are diminished. A target haemoglobin of 80 g/L is sufficient and this lessens the likelihood of early rebleeding.
• Monitor for alcohol withdrawal. Give intravenous thiamine.
• Start prophylactic antibiotics. These treat and prevent infection, and reduce early rebleeding and mortality.
Urgent endoscopy
Endoscopy (Fig. 14.24) should be performed to confirm the diagnosis and to exclude bleeding from other sites (e.g. gastric ulceration) and portal hypertensive gastropathy/gastric antral vascular ectasia (GAVE). The latter describes chronic gastric congestion, punctate erythema and gastric erosions, which may contribute to chronic anaemia. Portal hypertensive gastropathy and GAVE are distinct entities; management of portal hypertensive gastropathy is centred on reduction in portal pressures with β-blockers, whereas treatment of GAVE is endoscopic and uses various ablative techniques.
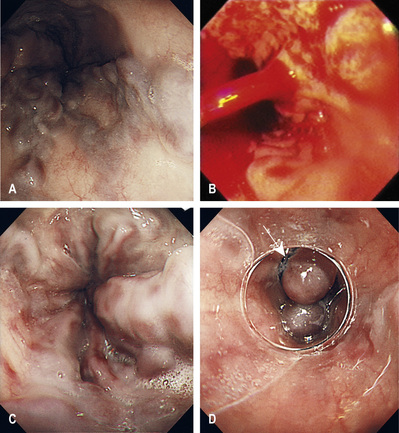
Variceal banding or injection sclerotherapy
Banding of oesophageal varices is performed by mounting a band on the tip of the endoscope, sucking the varix into the end of the scope, and dislodging the band over the varix using a trip-wire mechanism.
Between 15% and 20% of bleeding comes from gastric varices, which are associated with a greater mortality (up to 40%); endoscopic injection of cyanoacrylate is the best treatment.
Overall, haemostasis is achieved in 80–90% of patients. Best practice is to perform the endoscopy with the patient under general anaesthetic and to provide appropriate airway support.
Injection sclerotherapy is now rarely performed for oesophageal varices.
Other measures
This is used to restrict portal inflow by splanchnic arterial constriction and has shown benefit when used in combination with endoscopic techniques.
• Terlipressin. This is the only vasoconstrictor proven to reduce mortality. The dose is 2 mg 6-hourly, reducing to 1 mg 4-hourly after 48 h if a prolonged regimen is required (up to 5 days). Terlipressin should not be given to patients with ischaemic heart disease. The patient may complain of abdominal colic, and may defecate and have facial pallor owing to generalized vasoconstriction. If haemostasis has been achieved at endoscopy, there is probably little added benefit with this therapy and treatment should be tapered accordingly.
• Somatostatin. This has few side-effects. An infusion of 250–500 µg/h reduces bleeding and is reserved for patients with contraindications to terlipressin. A recent prospective, multicentre study showed that haemostatic effects and safety at day 5 of treatment did not differ significantly between terlipressin, somatostatin and octreotide when utilized as adjuvants to endoscopic treatment.
Balloon tamponade
Balloon tamponade is used if endoscopic therapy has failed or if there is exsanguinating haemorrhage. The usual balloon tube is a four-lumen Sengstaken–Blakemore, which should be left in place for no more than 12 hours and removed in the endoscopy room prior to endoscopy. The tube is passed into the stomach and the gastric balloon inflated with air and pulled back. It should be positioned in close apposition to the gastro-oesophageal junction to prevent the cephalad variceal blood flow to the bleeding point. The oesophageal balloon should only be inflated if bleeding is not controlled by the gastric balloon alone.
Haemostasis is achieved in up to 90%. However, the balloon may cause serious complications, such as aspiration pneumonia, oesophageal rupture and mucosal ulceration. The procedure is also very unpleasant for the patient.
A self-expanding covered metal stent (Danis), which has a wire loop to enable removal, and is introduced orally or endoscopically, can be placed over the varices. This is effective and has the advantages that it does not impair swallowing, cannot be removed by uncooperative patients, and allows post-endoscopic investigation. The stent is removed 7 days after insertion. It is currently only used in specialist centres but early results are encouraging.
Additional management of the acute episode
• Measures to prevent encephalopathy. Portosystemic encephalopathy (PSE) can be precipitated by a large bleed (blood contains protein). Management is as outlined on page 474.
• Nursing. Patients require high-dependency/intensive care nursing. They should remain nil by mouth until bleeding has stopped.
• Reduction in acid secretion. Ranitidine may be preferable to PPIs, as it lessens the risk of Clostridium difficile infection; PPIs are widely used, however. Sucralfate 1 g four times daily can reduce oesophageal ulceration following endoscopic therapy.
Management of an acute rebleed
Rebleeding occurs in about 15–20% within 5 days. The source should be established by endoscopy and is sometimes due to ulceration or slippage of a ligation band. Endoscopy should be performed once only to control rebleeding. If haemostasis cannot be achieved, then transjugular intrahepatic portocaval shunting will be necessary.
Transjugular intrahepatic portocaval shunt
Transjugular intrahepatic portocaval shunt (TIPS) is used when bleeding cannot be controlled either acutely or following a rebleed. Under X-ray guidance, a guidewire is passed from the jugular vein into the liver and the portal vein. After balloon expansion of the tract between the hepatic and portal veins, an expandable, covered metal shunt is placed over the wire to form a channel between the systemic and portal venous systems. It reduces the hepatic sinusoidal and portal vein pressure by creating a total shunt. There is an increased risk of portal systemic encephalopathy. Stent stenosis or thrombosis is far less frequent with ‘covered’, compared to ‘bare’, stents. Collaterals arising from the splenic or portal veins can be selectively embolized. These reduce rebleeding rates compared to endoscopic techniques, but do not improve survival and increase encephalopathy.
Emergency surgery
This is performed rarely when other measures fail or if TIPS is not available. Oesophageal transection and ligation of the feeding vessels to the bleeding varices is most commonly performed; an alternative is acute portosystemic shunt surgery.
Prevention of recurrent variceal bleeding (secondary prophylaxis)
The risk of bleeding recurring without prophylaxis is 60–80% over a 2-year period, with an approximate mortality of 20% per episode.
Prophylactic long-term measures
Oral propranolol or carvedilol to reduce the resting pulse rate by 25% decreases portal pressure. Portal inflow is reduced by a decrease in cardiac output (β1) and by blockade of β2 vasodilator receptors on the splanchnic arteries, leaving an unopposed vasoconstrictor effect. Significant reduction of hepatic venous pressure gradient (HVPG; measured by hepatic vein catheterization) is associated with very low rates of rebleeding, particularly if <12 mmHg. Data have emerged demonstrating that additional α1-adrenergic blockade, causing vasodilatation with carvedilol, may increase the number of patients with a reduction in hepatic venous pressure gradient compared to propranolol.
Endoscopic treatment
Repeated courses of banding at 2-weekly intervals lead to obliteration of varices. This markedly reduces rebleeding, most instances occurring before the varices have been fully obliterated. Between 30% and 40% of varices return per year, so follow-up endoscopy should be performed at 1–3 months after obliteration and then every 6–12 months. Complications of banding include oesophageal ulceration, mediastinitis and, rarely, strictures.
Combination therapy reduces overall bleeding compared to endoscopic therapy alone but with no overall improvement in mortality. A pragmatic approach is therefore to give combination therapy to those who can tolerate beta-blockade, and band ligation alone to those who cannot.
Surgery
• Surgical portosystemic shunting is associated with an extremely low risk of rebleeding and is used if TIPS is not available. Hepatic encephalopathy is a significant complication. The ‘shunts’ are usually an end-to-side portocaval anastomosis or a selective distal splenorenal shunt (Warren shunt).
• Devascularization procedures, including oesophageal transection, do not produce encephalopathy, and can be used when there is splanchnic venous thrombosis.
• Liver transplantation (see pp. 468–469) is the best option when there is poor liver function.
Prophylactic measures (primary prophylaxis)
Patients with cirrhosis and significant varices that have not bled should be prescribed non-selective β-blockers. This reduces the chances of upper gastrointestinal bleeding by approximately 50%, may increase survival, and is cost-effective. If there are contraindications, variceal banding is an option.
Ascites
Ascites, fluid within the peritoneal cavity, is a common complication of cirrhosis. Several factors underlie its pathogenesis:
• Sodium and water retention results from peripheral arterial vasodilatation (secondary to nitric oxide, atrial natriuretic peptide and prostaglandins), which causes a reduction in the effective blood volume. This reduction activates the sympathetic nervous system and the renin–angiotensin system, promoting salt and water retention (see Fig. 9.3).
• Portal hypertension exerts a local hydrostatic pressure, leading to increased hepatic and splanchnic production of lymph, and transudation of fluid into the peritoneal cavity.
• Low serum albumin (due to poor liver function) may further contribute by reducing plasma oncotic pressure.
In patients with ascites, urine sodium excretion rarely exceeds 5 mmol in 24 hours. Loss of sodium from extrarenal sites accounts for approximately 30 mmol in 24 hours. Under these circumstances, a normal daily sodium intake of 120–200 mmol results in a positive sodium balance of approximately 90–170 mmol in 24 hours (equivalent to 600–1300 mL of fluid retained).
Clinical features
Abdominal swelling may develop over days or several weeks. Precipitating factors include a high-sodium diet or development of an HCC or splanchnic vein thrombosis. Mild abdominal pain and discomfort are common but, if more severe, should raise the suspicion of spontaneous bacterial peritonitis (see below). Respiratory distress and difficulty eating accompany tense ascites.
The presence of fluid is confirmed clinically by demonstrating shifting dullness. Many patients also have peripheral oedema. A pleural effusion (usually right-sided) may infrequently be found and arises from the passage of ascites through congenital diaphragmatic defects.
Investigations
A diagnostic aspiration of 10–20 mL of fluid should be obtained for:
• Cell count. A neutrophil count >250 cells/mm3 is indicative of an underlying (usually spontaneous) bacterial peritonitis.
• Protein measurement. A high serum–ascites albumin gradient of >11 g/L suggests portal hypertension, and a low gradient <11 g/L is associated with non-liver disease-related abnormalities of the peritoneum, such as neoplasia (Box 14.18).
Box 14.18
The serum–ascites albumin gradient
(Modified from Chung RT, Iafrate AJ, Amrein PC et al. Case records of the Massachusetts General Hospital. New England Journal of Medicine 2006; 354:2166–2175.)
The differential diagnosis of ascites is listed in Box 14.19.
Box 14.19
Causes of ascites according to type of ascitic fluid
Management
The aim is both to reduce sodium intake and to increase renal sodium excretion, producing a net reabsorption of fluid from the ascites into the circulating volume. The maximum rate at which ascites can be mobilized is 500–700 mL in 24 hours (see below).
• Serum electrolytes, creatinine and estimated glomerular filtration rate (eGFR). Check on alternate days; weigh the patient and measure urinary output daily.
• Bed rest. This will cause a diuresis by improving renal perfusion, but is rarely helpful.
• Dietary sodium restriction. It is possible to reduce sodium intake to 40 mmol in 24 h and still maintain an adequate protein and calorie intake with a palatable diet.
• Drugs. Many contain significant amounts of sodium (up to 50 mmol daily). Examples include antacids and antibiotics (particularly penicillins and cephalosporins). Sodium-retaining drugs (NSAIDs, corticosteroids) should be avoided.
• Fluid restriction. This is unnecessary unless the serum sodium is <128 mmol/L (see below).
• Diuretics. The diuretic of choice is the aldosterone antagonist, spironolactone, starting at 100 mg daily. Chronic administration produces gynaecomastia. Eplerenone 25 mg once daily does not cause gynaecomastia.
The aim of diuretic therapy should be to produce a net loss of fluid approaching 700 mL in 24 hours (0.7 kg weight loss, or 1.0 kg if peripheral oedema is present). Although 60% of patients respond on this regimen, the spironolactone can be increased gradually to 400 mg daily if necessary, providing there is no hyperkalaemia. A loop diuretic, such as furosemide 20–40 mg or bumetanide 0.5 mg or 1 mg daily, is added if response is poor. These loop diuretics have several potential disadvantages, including hyponatraemia, hypokalaemia and volume depletion.
Diuretics should be temporarily discontinued if a rise in serum creatinine occurs, representing over-diuresis and hypovolaemia, or if there is hyperkalaemia or worsening encephalopathy. Hyponatraemia almost always represents haemodilution secondary to a failure to clear free water (usually a marker of reduced renal perfusion), and diuretics should be stopped if the sodium falls below 128 mmol/L. Vaptans (see p. 164), vasopressin V2-receptor antagonists that increase free water clearance, have a small beneficial effect on hyponatraemia and ascites but do not affect mortality, complications of cirrhosis or renal failure; routine use in cirrhosis cannot be recommended.
Paracentesis
This is used to relieve symptomatic tense ascites or when diuretic therapy is insufficient to control accumulation of fluid. The main complications are hypovolaemia and renal dysfunction (post-paracentesis circulatory dysfunction), predominantly due to an accentuation of the arteriolar vasodilatation already present in these patients; this is more likely with >5 L removal and worse liver function. In patients with normal renal function and without hyponatraemia, this is overcome by infusing albumin (8 g/L of ascitic fluid removed). In practice, up to 20 L can be removed over 4–6 hours, with albumin infusion.
Shunts
A TIPS may be inserted to treat resistant ascites, providing there is no spontaneous portosystemic encephalopathy and there is minimal disturbance of renal function. Frequency of paracentesis and diuretic use is reduced and nutrition is enhanced. Survival may also improve.
A peritoneo-bladder conduit, by means of an implantable, rechargeable, battery-powered pump, has been developed for use in patients with advanced cirrhosis and resistant ascites (alfapump®). This removes ascites from the peritoneal cavity into the urinary bladder, to be eliminated through urination. Early studies have shown a reduction in the need for large-volume paracentesis but several complications, including pain and infection, occur.
Spontaneous bacterial peritonitis
Spontaneous bacterial peritonitis (SBP) represents a serious complication that is an indication for referral for transplant assessment; it occurs in up to 18% of patients with ascites who have undergone a decompensation. The infecting organisms gain access to the peritoneum by haematogenous spread; most are Escherichia coli, Klebsiella or enterococci. The condition should be suspected in any patient with ascites who deteriorates, as pain and pyrexia are frequently absent. Diagnostic aspiration should always be performed. A raised neutrophil count in ascites is sufficient evidence alone to start immediate treatment. A broad-spectrum antibiotic is used, with subsequent alteration according to culture results in combination with infusions of human albumin solution. Evidence has emerged that non-selective β-blockers should be stopped following diagnosis of SBP, as prescription increases the risk of renal impairment. Mortality is 10–15%. Recurrence is common (70% within a year) and secondary prevention – for example, with norfloxacin 400 mg daily – prolongs survival. Primary prophylaxis of SBP in patients with ascites protein <10 g/L or severe liver disease may prevent hepatorenal syndrome and improve survival.
Portosystemic encephalopathy
Portosystemic encephalopathy (PSE) is a chronic neuropsychiatric syndrome that is secondary to cirrhosis. Acute encephalopathy can occur in acute hepatic failure (see p. 462). PSE can arise in portal hypertensive patients due to spontaneous ‘shunting’, or in those with surgical or TIPS shunts.
Pathogenesis
In cirrhosis, the portal blood bypasses the liver via collaterals, and ‘toxic’ metabolites pass directly to the brain to produce encephalopathy. Ammonia-induced alteration of brain neurotransmitter balance, especially at the astrocyte–neurone interface, is considered to be the leading pathophysiological mechanism. Ammonia is produced by the breakdown of protein by intestinal bacteria. Other implicated substances are free fatty acids and mercaptans; accumulation of false neurotransmitters (octopamine) or activation of the γ-aminobutyric acid (GABA) inhibitory neurotransmitter system may also be responsible. Increased blood levels of aromatic amino acids (tyrosine and phenylalanine) and reduced branched-chain amino acids (valine, leucine and isoleucine) also occur. The factors precipitating PSE are shown in Box 14.20.
Box 14.20
Factors precipitating portosystemic encephalopathy
• Gastrointestinal haemorrhage
• Infection, including spontaneous bacterial peritonitis
• Fluid and electrolyte disturbance due to diuretic therapy or paracentesis
• Drugs (e.g. any central nervous system depressant)
TIPS, transjugular intrahepatic portocaval shunt.
Clinical features
An acute onset often has a precipitating factor (Box 14.20). The patient becomes increasingly drowsy and comatose.
Chronically, there is a disorder of personality, mood and intellect, with a reversal of normal sleep rhythm. These changes may fluctuate and a collateral history should be obtained. The patient is irritable, confused and disorientated, and has slow, slurred speech. General features include nausea, vomiting and weakness. There is hyper-reflexia and increased tone. Coma occurs as the encephalopathy becomes more marked. Convulsions are very rare, but if they do occur, other causes must be considered.
Signs include:
• fetor hepaticus (a sweet smell to the breath)
• a coarse flapping tremor seen when the hands are outstretched and wrists hyperextended (asterixis)
• constructional apraxia, with the patient being unable to write or draw a five-pointed star, for example
• decreased mental function, which can be assessed by using the serial sevens test or a trail-making (or connection) test.
Investigations
Diagnosis is clinical.
Additional investigations
• EEG shows a decrease in the frequency of the normal α-waves (8–13 Hz) to 1.5–3 Hz. These changes occur before coma supervenes.
• Visual evoked responses (see p. 823) also detect subclinical encephalopathy.
• Arterial blood ammonia can be useful for the differential diagnosis of coma and for following a patient with PSE, but is not always available.
Management
• Identify and remove the possible precipitating cause, such as cerebral depressant drugs, constipation or electrolyte imbalance due to over-diuresis.
• Give purgation and enemas to empty the bowels of nitrogenous substances. Lactulose (10–30 mL three times daily) is an osmotic purgative that reduces the colonic pH and limits ammonia absorption. Lactilol (β-galactoside sorbitol 30 g daily) is metabolized by colonic bacteria and is comparable in efficacy.
• Maintain nutrition, if necessary via a fine-bore nasogastric tube, and do not restrict protein for more than 48 h.
• Give antibiotics. Rifaximin is a poorly absorbed semisynthetic antibiotic based on rifamycin that has a beneficial effect on secondary prevention of PSE. Metronidazole (200 mg four times daily) may be effective acutely. Neomycin should be avoided.
• Stop or reduce diuretic therapy.
• Give intravenous fluids as necessary (beware too much sodium).
• Increase protein in the diet to the limit of tolerance as encephalopathy improves.
Prognosis
Acute encephalopathy in acute hepatic failure has a very poor prognosis associated with that of the disease itself. In cirrhosis, chronic PSE adversely affects prognosis but the course is very variable. Very rarely with chronic portosystemic shunting, an organic syndrome with cerebellar signs or choreoathetosis develops, as well as myelopathy leading to a spastic paraparesis due to demyelination. These patients require referral to a liver transplant centre.
Renal failure (hepatorenal syndrome)
The hepatorenal syndrome typically occurs in patients with advanced cirrhosis, portal hypertension, jaundice and ascites. The urine output is low with a low urinary sodium concentration, a maintained capacity to concentrate urine (i.e. intact tubular function) and almost normal renal histology. The renal failure is therefore described as ‘functional’. It is often precipitated by over-vigorous diuretic therapy, NSAIDs, diarrhoea, paracentesis and infection, particularly spontaneous bacterial peritonitis.
The mechanism is similar to that of ascites, with extreme peripheral vasodilatation leading to decreased effective blood volume and consequent hypotension (see pp. 472–473). This causes increased plasma renin, aldosterone, noradrenaline (norepinephrine) and vasopressin, leading to renal vasoconstriction. There is an increased pre-glomerular vascular resistance that causes blood to be directed away from the renal cortex. This leads to a reduced glomerular filtration rate and plasma renin remains high. Salt and water retention occurs, with reabsorption of sodium from the renal tubules.
Eicosanoids have been incriminated in the pathogenesis, supported by precipitation of the syndrome by inhibitors of prostaglandin synthase, such as NSAIDs.
Diuretic therapy should be stopped and intravascular hypovolaemia corrected, preferably with albumin. Terlipressin or noradrenaline with intravenous albumin improves renal function in approximately 50%. Liver transplantation is the best option. In patients who are candidates for transplantation, haemodialysis can be used as a bridging option but is frequently difficult to perform, and survival is generally limited by the severity of the hepatic failure.
Hepatopulmonary syndrome
This is hypoxaemia in patients with advanced liver disease due to intrapulmonary vascular dilatation with no evidence of primary pulmonary disease. The patients have features of cirrhosis with spider naevi and clubbing, as well as cyanosis. Most are asymptomatic, but with more severe disease, patients are breathless on standing. Transthoracic echocardiography shows intrapulmonary shunting, and arterial blood gases confirm hypoxaemia. These changes are improved with liver transplantation.
Porto-pulmonary hypertension
This occurs in 1–2% of patients with cirrhosis and portal hypertension. It may respond to medical therapy (e.g. intravenous epoprostenol, or oral bosentan and sildenafil). Severe pulmonary hypertension is a contraindication to liver transplantation.
Primary hepatocellular carcinoma
This is discussed on page 485.
Types of cirrhosis
Alcoholic cirrhosis
This is discussed in the section on alcoholic liver disease (see pp. 480–482).
Primary biliary cholangitis
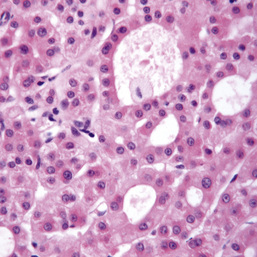
Primary biliary cholangitis (PBC; Fig. 14.25) is a chronic disorder with progressive destruction of small bile ducts, leading to cirrhosis. Women aged 40–50 years constitute 90% of patients. PBC is diagnosed increasingly frequently in its milder forms. The prevalence is approximately 7.5 per 100 000, with a 1–6% increase in first-degree relatives. PBC has been called ‘chronic non-suppurative destructive cholangitis’, a term more descriptive of the early lesion, which emphasizes the fact that true cirrhosis occurs only in the later stages.
Aetiology
The aetiology is unknown but an immunological basis is well described. Serum anti-mitochondrial antibodies (AMA) are found in almost all patients. The mitochondrial antigen M2 is specific to PBC and five M2-specific antigens have been identified. The presence of AMA in high titre is unrelated to the clinical or histological picture and its role in pathogenesis is unclear. Antibodies against nuclear antigens, e.g. anti-gp210, are present in 50% of patients and correlate with progression towards liver failure.
It seems likely that an environmental factor acts on a genetically predisposed host via molecular mimicry, initiating autoimmunity. E. coli and Novosphingobium aromaticivorans antibodies are present in high titre.
Synthesis of IgM is increased, thought to be due to failure of the switch from IgM to IgG antibody synthesis. No specific associated class II major histocompatibility complex (MHC) loci have been found.
Clinical features
Asymptomatic patients are discovered on routine examination or screening and may have hepatomegaly, a raised serum alkaline phosphatase or autoantibodies.
Pruritus is often the earliest symptom. Fatigue, which is frequently disabling, may accompany the pruritus, particularly in progressive cases. When jaundice appears, hepatomegaly is usually present. Pigmented xanthelasma on eyelids or deposits of cholesterol in the creases of the hands may be seen.
Associated disorders
Autoimmune disorders (e.g. Sjögren syndrome, scleroderma, thyroid disease) occur with increased frequency. Keratoconjunctivitis sicca (dry eyes and mouth) is seen in 70% of cases. Renal tubular acidosis, membranous glomerulonephritis, coeliac disease and interstitial pneumonitis are also associations.
Investigations
• Mitochondrial antibodies – measured routinely by ELISA (in titres >1 : 160) – are present in over 95% of patients; M2 antibody is 98% specific. Other non-specific antibodies (e.g. anti-nuclear factor and smooth muscle) may also be present.
• High serum alkaline phosphatase is often the only liver biochemistry abnormality.
• Serum cholesterol is raised.
• Ultrasound can show a diffuse alteration in liver architecture.
• Liver biopsy shows characteristic histological features of a portal tract infiltrate, mainly of lymphocytes and plasma cells; approximately 40% have granulomas. Most of the early changes are in zone 1. Later, there is damage to and loss of small bile ducts with ductular proliferation. Portal tract fibrosis and, eventually, cirrhosis are seen.
Hepatic granulomas are also seen in sarcoidosis, tuberculosis, schistosomiasis, drug reactions, brucellosis and parasitic infestation (e.g. strongyloidiasis).
Differential diagnosis
The classical picture presents little difficulty with diagnosis (and can be confirmed by biopsy, although this is only necessary in doubtful cases). A group of patients with the histological changes of PBC but the serology of autoimmune hepatitis are termed as having autoimmune cholangitis and respond to steroids and azathioprine.
In the jaundiced patient, extrahepatic biliary obstruction should be excluded by ultrasound or MRCP.
Management
• Ursodeoxycholic acid (10–15 mg/kg) improves bilirubin and aminotransferase levels. It should be given early in the asymptomatic phase, as these patients benefit, whereas no benefit is achieved in advanced disease.
• Steroids improve biochemical and histological disease but cause osteoporosis and other side-effects, and so should not be used.
• Malabsorption of fat-soluble vitamins (A, D and K) occurs and supplementation is required.
• Bisphosphonates are required for osteoporosis. Despite raised serum lipid concentrations, PBC is not associated with an increased cardiovascular disease risk and strategies for prevention of vascular events should be tailored to the individual.
• Pruritus is difficult to control; colestyramine is helpful, although unpalatable. Rifampicin, and naloxone and naltrexone (opioid antagonists) have been shown to be of benefit. Intractable pruritus can be relieved by plasmapheresis or a molecular absorbent recirculating system (MARS).
• The lack of effective medical therapy has made PBC a major indication for liver transplantation (see p. 468).
• Fatigue is common and can be severely debilitating; there is no proven therapy and transplantation does not improve symptoms. Modafinil, used for narcolepsy, has shown promise but has yet to be evaluated in randomized studies; it may cause significant side-effects and has addictive potential.
Complications
The complications are those of cirrhosis. In addition, osteoporosis and, rarely, osteomalacia and a polyneuropathy occur.
Prognosis
Prognosis is very variable. Asymptomatic patients and those presenting with pruritus only will survive for more than 20 years. Symptomatic patients with jaundice have a more rapidly progressive course and may die of liver failure or bleeding varices within 5 years. Liver transplantation should therefore be offered when the serum bilirubin is persistently above 100 µmol/L.
Primary sclerosing cholangitis
Primary sclerosing cholangitis (PSC) is a chronic cholestatic liver disease characterized by fibrosing inflammatory destruction of both the intra- and extrahepatic bile ducts. In 75% of patients, PSC is associated with inflammatory bowel disease (usually ulcerative colitis); it is not unusual for PSC to predate the onset of inflammatory bowel disease. The causes are unknown but genetic susceptibility to PSC is associated with the HLA-A1-B8-DR3 haplotype. The autoantibody pANCA (anti-neutrophil cytoplasmic antibody) is found in the serum of 60% of cases. Seventy per cent of patients are men and the average age of onset is approximately 40 years. Secondary PSC is seen in patients with HIV and cryptosporidium (see p. 350) and may follow ketamine misuse.
Clinical features
With increasing screening of patients with inflammatory bowel disease, PSC is detected at an asymptomatic phase with abnormal liver biochemistry, usually a raised serum alkaline phosphatase. Symptomatic presentation is usually with fluctuating pruritus, jaundice and cholangitis.
Diagnosis
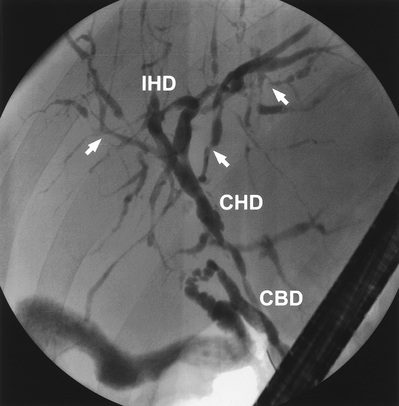
The typical biliary changes associated with PSC may be identified by MRCP. The cholangiogram characteristically shows irregularity of calibre of both intra- and extrahepatic ducts, although either may be involved alone (Fig. 14.26).
Pathology
Histology can be contributory; it shows inflammation of the intrahepatic biliary radicles with associated scar tissue, classically described as have the appearance of ‘onion skin’. These changes range from minor inflammatory infiltrates to the level of established cirrhosis.
Management
PSC is a slowly progressive lesion (symptoms and biochemical tests may fluctuate), which ultimately leads to liver cirrhosis and associated decompensation. Recurrent cholangitis may be a feature before the onset of cirrhosis. Cholangiocarcinoma occurs in up to 15% of patients (see pp. 485–486 and 498).
The only proven treatment is liver transplantation. The bile acid, ursodeoxycholic acid, has been evaluated extensively but there is no evidence of benefit. High-dose therapy (30 mg/kg) may be deleterious. In a small minority, the dominant lesion is sited in the extrahepatic ducts (Fig. 14.26). Such lesions may be amenable to endoscopic biliary intervention with balloon dilatation and temporary stent placement (see p. 497).
Secondary biliary cirrhosis
Cirrhosis can result from prolonged (for months) large duct biliary obstruction. Causes include bile duct strictures, gallstones and sclerosing cholangitis. An ultrasound examination and MRCP, sometimes followed by ERCP or percutaneous transhepatic cholangiography (where the ducts are cannulated under ultrasound guidance through the skin) if cannulation is difficult, is performed to outline the ducts, and any remedial cause is treated.
Hereditary haemochromatosis
Hereditary haemochromatosis (HH; see also p. 445) is an inherited disease characterized by excess iron deposition in various organs, leading to eventual fibrosis and functional organ failure. There are four main types of inherited disorders:
• type 1 HFE: the HFE gene (mutation C282Y) is the most common and is on chromosome 6
• type 2A: juvenile HJV gene (mutation G320V)
type 2B: juvenile HAMP gene (mutation 93delG)
All are transmitted by an autosomal recessive gene, apart from the ferroportin iron overload, which is dominantly transmitted.
HH has a prevalence in Caucasians (homozygotes) of 1 in 400, but very variable phenotypic expression and a heterozygote (carrier) frequency of 1 in 10. It is the most common single-gene disorder in Caucasians.
Aetiology
Between 85% and 90% of patients with overt HH are homozygous for the Cys 282 Tyr (C282Y mutation): that is, type 1 HFE. A second mutation (His 63 Asp, H63D) occurs in about 25% of the population and is in complete linkage disequilibrium with Cys 282 Tyr.
Another form of haemochromatosis (type 3) occurs in southern Europe and is associated with TfR2, a transferrin receptor isoform. The other types, ferroportin-related (type 4) and juvenile forms (types 2A and 2B), are much rarer.
Dietary intakes of iron and chelating agents (ascorbic acid) may be relevant. Iron overload may be present in alcoholics; alcohol excess per se does not cause HH, although there is a history of excess alcohol intake in 25% of patients.
Mechanism of damage
The HFE gene protein interacts with the transferrin receptor 1, which is a mediator in intestinal iron absorption (see Fig. 16.8). Iron is taken up by the mucosal cells inappropriately, exceeding the binding capacity of transferrin.
Hepcidin, a protein synthesized in the liver (Fig. 14.27), is central to the control of iron absorption; it is increased in iron deficiency states and decreased with iron overload. The mutations described above disrupt hepcidin expression, thereby internalizing ferroportin and leading to uninhibited iron overload.

Hepatic expression of the hepcidin gene is decreased in HFE haemochromatosis, facilitating liver iron overload. Excess iron is then gradually taken up by the liver and other tissues over a long period. It seems likely that it is the iron itself that precipitates fibrosis.
Pathology
In symptomatic patients, the total body iron content is 20–40 g, compared with 3–4 g in a normal person. The iron content is particularly increased in the liver (Fig. 14.28) and pancreas (50–100 times normal) but is also increased in other organs (e.g. the endocrine glands, heart and skin).

In established cases, the liver shows extensive iron deposition and fibrosis. Early in the disease, iron is deposited in the periportal hepatocytes (in pericanalicular lysosomes). Later, it is distributed widely throughout all acinar zones, biliary duct epithelium, Kupffer cells and connective tissue. Cirrhosis is a late feature.
Clinical features
The course of the disease depends on a number of factors, including gender, dietary iron intake, presence of associated hepatotoxins (especially alcohol) and genotypes. Overt clinical manifestations occur more frequently in men; the reduced incidence in women is probably explained by physiological blood loss and a smaller dietary intake of iron. Most affected individuals present in the fifth decade. The classic triad of bronze skin pigmentation (due to melanin deposition), hepatomegaly and diabetes mellitus is present only in cases of gross iron overload.
Hypogonadism secondary to pituitary dysfunction is the most common endocrine feature. Deficiency of other pituitary hormones is also found, but symptomatic endocrine deficiencies, such as loss of libido, are very rare. Cardiac manifestations, particularly heart failure and arrhythmias, are common, especially in younger patients. Calcium pyrophosphate is deposited asymmetrically in both large and small joints (chondrocalcinosis), leading to an arthropathy. The exact relationship of chondrocalcinosis to iron deposition is uncertain.
Complications
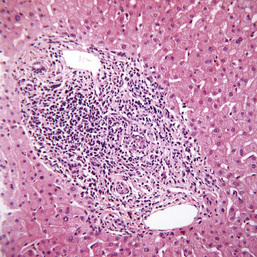
Some 30% of people with cirrhosis will develop primary hepatocellular carcinoma (HCC; Fig. 14.29). HCC has been described only very rarely in non-cirrhotic patients in whom the excess iron stores have been removed. Early diagnosis is vital.
Investigations
Homozygotes
• Serum iron is elevated (>30 µmol/L) in 90% with a reduction in the TIBC and a transferrin saturation of >45%.
• Serum ferritin is elevated (usually >500 µg/L or 240 nmol/L).
• Liver biochemistry is often normal, even with established cirrhosis.
Heterozygotes
Heterozygotes may have normal biochemical tests or modest increases in serum iron transferrin saturation (>45%) or serum ferritin (usually >400 µg/L).
Genetic testing
If iron studies are abnormal, genetic testing is performed.
Liver biopsy
This is not required for diagnosis, but is useful to establish the extent of tissue damage, assess tissue iron, and measure the hepatic iron concentration (>180 µmol/g dry weight of liver indicates haemochromatosis).
Mild degrees of parenchymal iron deposition in patients with other forms of cirrhosis, particularly if due to alcohol, can often cause confusion with true homozygous HH.
Magnetic resonance imaging
MRI shows a dramatic reduction in the signal intensity of the liver and pancreas owing to the paramagnetic effect of ferritin and haemosiderin. A highly T2-weighted, gradient recalled echo (GRE) technique detects all clinically relevant liver iron overload (>60 µmol/g of liver). In secondary iron overload (haemosiderosis), which involves the reticuloendothelial cells, the pancreas is spared, enabling distinction between these two conditions.
Management
Venesection
This prolongs life and may reverse tissue damage; the risk of malignancy still remains if cirrhosis is present. All patients should have excess iron removed as rapidly as possible. This is achieved using venesection of 500 mL performed twice weekly for up to 2 years, i.e. 160 units with 250 mg of iron per unit, which equals 40 g removed. During venesection, serum iron and ferritin and the mean corpuscular volume (MCV) should be monitored. These fall only when available iron is depleted. Three or four venesections per year are required to prevent re-accumulation of iron. Serum ferritin should remain within the normal range.
Manifestations of the disease usually improve or disappear, except for diabetes, testicular atrophy and chondrocalcinosis. The requirements for insulin often diminish in diabetic patients. Testosterone replacement is often helpful.
In the rare patient who cannot tolerate venesection (because of severe cardiac disease or anaemia), chelation therapy with desferrioxamine, either intermittently or continuously by infusion, has been successful in removing iron.
Screening
In all cases of HH, all first-degree family members must be screened to detect early and asymptomatic disease. HFE mutation analysis is performed with measurement of transferrin saturation and serum ferritin.
In the general population, serum iron and transferrin saturation are the best and cheapest tests available.
Wilson's disease (progressive hepatolenticular degeneration)
Dietary copper is normally absorbed from the stomach and upper small intestine. Copper is transported to the liver loosely bound to albumin; in the liver, it is incorporated into apocaeruloplasmin, forming caeruloplasmin, a glycoprotein synthesized in the liver, and secreted into the blood. The remaining copper is normally excreted in the bile and excreted in faeces.
Wilson's disease is a very rare inborn error of copper metabolism that results in copper deposition in various organs, including the liver, the basal ganglia of the brain and the cornea. It is potentially treatable and all young patients with liver disease must be screened for this condition.
Aetiology
Wilson's disease is an autosomal recessive disorder with a molecular defect within a copper-transporting ATPase encoded by a gene (designated ATP7B) located on chromosome 13. It affects between 1 in 30 000 and 1 in 100 000 individuals. Over 300 mutations have been identified, the most frequent being His 1069 Gly (H1069Q), found in approximately 50% of Caucasian patients; compound heterozygotes are common. This mutation is rare in India and Asia. Wilson's disease occurs worldwide, particularly in countries where consanguinity is common. There is a failure of both incorporation of copper into procaeruloplasmin, which leads to low serum caeruloplasmin, and biliary excretion of copper. There is a low serum caeruloplasmin in over 80% of patients but this is not the cause of the copper deposition. The precise mechanism for the failure of copper excretion is not known.
Pathology
The liver histology is not diagnostic and varies from that of chronic hepatitis to macronodular cirrhosis. Stains for copper show a periportal distribution but this can be unreliable (see below). The basal ganglia are damaged and show cavitation, the kidneys show tubular degeneration, and erosions are seen in bones.
Clinical features
Children usually present with hepatic problems, whereas young adults have more neurological problems, such as tremor, dysarthria, involuntary movements and eventually dementia. The liver disease varies from episodes of acute hepatitis, especially in children (which can go on to acute hepatic failure), to chronic hepatitis or cirrhosis.
Typical signs are those of chronic liver disease with neurological signs of basal ganglia involvement (see p. 855). A specific sign is the Kayser–Fleischer ring, caused by copper deposition in Descemet's membrane in the cornea. It appears as a greenish-brown pigment at the corneoscleral junction and frequently requires slit-lamp examination for identification. It may be absent in young children.
Investigations
• Serum copper and caeruloplasmin are usually reduced but can be normal.
• Urinary copper is usually increased to 100–1000 µg in 24 h (1.6–16 µmol); normal levels <40 µg (0.6 µmol).
• Liver biopsy aids diagnosis, which depends on measurement of the amount of copper in the liver (>250 µg/g dry weight), although high levels of copper are also found in the liver in chronic cholestasis.
• Haemolysis and anaemia may be present.
• Genetic analysis is limited but selected exons are screened according to population groups.
Management
• Lifetime treatment with penicillamine, 1–1.5 g daily, is effective in chelating copper. If treatment is started early, clinical and biochemical improvement can occur. Urinary copper levels should be monitored and the drug dose adjusted downwards after 2–3 years. Serious side-effects of the drug occur in 10% and include skin rashes, leucopenia, skin changes and renal damage.
• Trientine (1.2–1.8 g/day) and zinc acetate (150 mg/day) are used as maintenance therapy and for asymptomatic cases. All siblings and children of patients should be screened (ATP7B mutation analysis is useful) and treatment with zinc is given, even in the asymptomatic if there is evidence of copper accumulation. A diet low in copper (i.e. excluding chocolate and peanuts) is advised.
Prognosis
Early diagnosis and effective treatment have improved the outlook. Neurological damage is, however, permanent. Acute hepatic failure or decompensated cirrhosis should be treated by liver transplantation.
Alpha1-antitrypsin deficiency
A deficiency of alpha1-antitrypsin (α1-AT; see also p. 1081) is sometimes associated with liver disease and pulmonary emphysema (particularly in smokers). Part of a family of serine protease inhibitors, or serpin superfamily, α1-AT is a glycoprotein. Deficiency of α1-AT is a genetic disorder and 1 in 10 northern Europeans carries an abnormal gene.
The protein is a 394-amino acid 52 kDa acute phase protein that is synthesized in the liver and constitutes 90% of the serum α1-globulin seen on electrophoresis. Its main role is to inhibit the proteolytic enzyme, neutrophil elastase.
The gene is located on chromosome 14. The genetic variants of α1-AT are characterized by their electrophoretic mobilities as medium (M), slow (S) or very slow (Z). The normal genotype is protease inhibitor MM (PiMM), the homozygote for Z is PiZZ, and the heterozygotes are PiMZ and PiSZ. S and Z variants are due to a single amino acid replacement of glutamic acid at positions 264 and 342 of the polypeptide, respectively. This results in decreased synthesis and secretion of the protein by the liver as protein–protein interactions occur between the reactive centre loop of one molecule and the β-pleated sheet of a second (loop sheet polymerization).
How this causes liver disease is uncertain. It is postulated that failure of secretion of the abnormal protein leads to an accumulation in the liver, causing liver damage.
Clinical features
The majority of patients with clinical disease are homozygotes with a PiZZ phenotype. Some may present in childhood and a few require transplantation. Approximately 10–15% of adult patients will develop cirrhosis, usually over the age of 50 years, and 75% will have respiratory problems. Approximately 5% of patients die of their liver disease. Heterozygotes (e.g. PiSZ or PiMZ) may develop liver disease but the risk is small.
Investigations
• Serum α1-antritrypsin is low, at 10% of the normal level in the PiZZ phenotypes, and 60% of normal in the S variant.
Histologically, periodic acid–Schiff (PAS)-positive, diastase-resistant globules that contain α1-AT are seen in periportal hepatocytes. Fibrosis and cirrhosis can be present.
Management
There is no treatment, apart from dealing with the complications of liver disease. Patients with hepatic decompensation should be assessed for liver transplantation, and should stop smoking (see p. 1081).
Alcoholic Liver Disease
This section describes the pathology and clinical features of alcoholic liver disease. The amounts needed to produce liver damage, alcohol metabolism, and other clinical effects of alcohol are described on pages 217–218.
Ethanol is metabolized in the liver by two pathways, resulting in an increase in the NADH/NAD ratio. The altered redox potential causes increased hepatic fatty acid synthesis with decreased fatty acid oxidation; both events lead to hepatic accumulation of fatty acid, which is then esterified to glycerides.
The changes in oxidation–reduction also impair carbohydrate and protein metabolism and are the cause of the centrilobular necrosis of the hepatic acinus that is typical of alcohol damage. TNF-α release from Kupffer cells causes the release of reactive oxygen species, leading, in turn, to tissue injury and fibrosis.
Acetaldehyde is formed by the oxidation of ethanol, and its effect on hepatic proteins may well be a factor in producing liver cell damage. The exact mechanism of alcoholic hepatitis and cirrhosis is unknown, but since only 10–20% of people who drink heavily will develop cirrhosis, a genetic predisposition is recognized. Immunological mechanisms have also been proposed, with the release of cytokines, particularly IL-8, which is a neutrophil chemoattractant; infiltration with neutrophils is a feature of alcoholic hepatitis.
Alcohol can enhance the effects of the toxic metabolites of drugs (e.g. paracetamol) on the liver, as it induces microsomal metabolism via the microsomal ethanol oxidizing system (MEOS; see p. 217).
Pathology
Alcohol can produce a wide spectrum of liver disease from fatty change to hepatitis and cirrhosis.
Fatty liver
The metabolism of alcohol invariably produces fat in the liver (Fig. 14.30), mainly in zone 3. This is minimal with small amounts of alcohol, but with larger amounts the cells become swollen with fat (steatosis). There is no liver cell damage. The fat disappears on stopping alcohol. Steatosis is also seen in non-alcoholic fatty liver disease (NAFLD) (see p. 465).
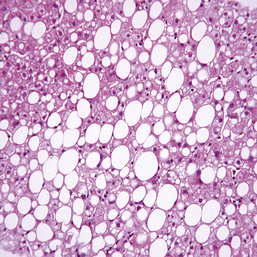
In some cases, collagen is laid down around the central hepatic veins (perivenular fibrosis) and this can sometimes progress to cirrhosis without a preceding hepatitis. Alcohol directly affects stellate cells, transforming them into collagen-producing myofibroblast cells (see p. 440).
Alcoholic hepatitis
In addition to fatty change, there is infiltration by polymorphonuclear leucocytes and hepatocyte necrosis, mainly in zone 3. Dense cytoplasmic inclusions called Mallory bodies are sometimes seen in hepatocytes and giant mitochondria are also a feature. Mallory bodies are suggestive of, but not specific for, alcoholic damage, as they can be found in other liver disease, such as Wilson's disease and primary biliary cholangitis. If alcohol consumption continues, alcoholic hepatitis may progress to cirrhosis.
Alcoholic cirrhosis
This is classically of the micronodular type but a mixed pattern is also seen accompanying fatty change, and evidence of pre-existing alcoholic hepatitis may be present.
Clinical features
Fatty liver
There are often no symptoms or signs. Vague abdominal symptoms of nausea, vomiting and diarrhoea are due to the more general effects of alcohol on the gastrointestinal tract. Hepatomegaly, sometimes huge, can occur, together with other features of chronic liver disease.
Alcoholic hepatitis
The clinical features vary in degree:
• The patient may be well, with few symptoms, the hepatitis only being apparent on the liver biopsy in addition to fatty change.
• Mild to moderate symptoms of ill-health, occasionally with mild jaundice, may occur. Signs include all the features of chronic liver disease. Liver biochemistry is deranged and the diagnosis is made on liver histology.
• In the severe case, often superimposed on alcoholic cirrhosis, the patient is ill, with jaundice and ascites. Abdominal pain is frequently present, and a high fever is associated with the liver necrosis. On examination, there is deep jaundice, hepatomegaly, sometimes splenomegaly, and ascites with ankle oedema. The signs of chronic liver disease are also present.
Alcoholic cirrhosis
This represents the final stage of liver disease from alcohol use. Nevertheless, patients can be very well with few symptoms. On examination, there are usually signs of chronic liver disease (p. 448). The diagnosis is confirmed by liver biopsy.
The patient usually presents with one of the complications of cirrhosis. In many cases, there are features of alcohol dependency (see pp. 921–922), as well as evidence of involvement of other systems, such as polyneuropathy.
Investigations
Fatty liver
An elevated MCV often indicates heavy drinking. Liver biochemistry shows mild abnormalities with elevation of both serum aminotransferase enzymes. The γ-GT level is a useful test for determining whether the patient is taking alcohol. With severe fatty infiltration, marked changes in all liver biochemical parameters can occur. Ultrasound or CT will demonstrate fatty infiltration, as will liver histology. Elastography (p. 446) can be used to estimate the degree of fibrosis.
Alcoholic hepatitis
Investigations show a leucocytosis with markedly deranged liver biochemistry and elevated:
A low serum albumin may also be found. Rarely, hyperlipidaemia with haemolysis (Zieve syndrome) may occur.
Liver biopsy, if required, is performed by the transjugular route because of the prolonged prothrombin time.
Alcoholic cirrhosis
Investigations are as for cirrhosis in general.
Management and prognosis
General management
All patients should stop drinking alcohol. Delirium tremens (a withdrawal symptom) is treated with diazepam. Intravenous thiamine should be given empirically to prevent Wernicke–Korsakoff encephalopathy. Bed rest is necessary, along with a diet high in protein and vitamin supplements. Dietary protein sometimes needs to be limited because of encephalopathy. Patients must be advised to participate in alcohol cessation programmes. The likelihood of abstention is dependent on many factors, particularly social and family ones.
Fatty liver
The patient is advised to stop drinking alcohol; the fat will disappear and the liver biochemistry usually returns to normal. Small amounts of alcohol can be drunk subsequently, as long as patients are aware of the problems and can control their consumption.
Alcoholic hepatitis
In severe cases, the patient requires admission to hospital. Nutrition must be maintained with enteral feeding, if necessary, and vitamin supplementation given. Steroid therapy has been widely used in patients with a discriminant function score of >32 but a recent multicentre UK study, which included over 1000 patients, suggested that there was no survival benefit.
Discriminant function (DF)
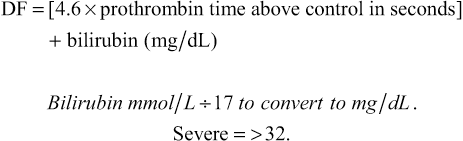
The response to steroid therapy can also be evaluated by the Lille score (>0.45 indicates poor response to steroids, which can therefore be stopped) and the Glasgow score (Boxes 14.21 and 14.22). A Glasgow score of >9 indicates that steroids are necessary because at >9 the 28-day mortality is 75%, while at <9 it is 50%. The MELD score (see p. 467) is also used but does not indicate which patients need steroid therapy.
Box 14.21
Lille score for alcoholic hepatitis
(Calculator at http://www.lillemodel.com)
R = 3.19 − (0.101 × age in years) + (0.147 × albumin on admission in g/L) + (0.0165 × change in bilirubin level from day 0 to day 7 in µmol/L) − (0.206 × renal insufficiency [0 if absent, 1 if present]a ) − (0.0065 × bilirubin day 0 in µmol/L) − (0.0096 × INR)
A score of <0.16 indicates a 96% chance of survival at 28 days; ≥0.56 indicates a 55% chance of survival at 28 days.
Score = EXP(−R)/[1+EXP(−R)]
Box 14.22
Glasgow alcoholic hepatitis score
| Score | 1 | 2 | 3 |
| Age | <50 | >50 | |
| White cell count (× 109/L) | <15 | >15 | |
| Urea (nmol/L) | <5 | >5 | |
| Bilirubin (µmol/L) | <125 | 125–250 | >250 |
| INR | <1.5 | 1.5–2.0 | >2.0 |
| Poor prognosis = total score >9. | |||

Infection must be excluded or concomitantly treated. Treatment for encephalopathy and ascites is commenced. Antifungal prophylaxis should also be used.
Patients are advised to stop drinking for life, as this is undoubtedly a pre-cirrhotic condition. The prognosis is variable and, despite abstinence, the liver disease is progressive in many patients. Granulocyte-colony stimulating factor (G-CSF) for 5 days improves 90-day survival in preliminary studies.
In severe cases, the mortality is at least 50%; with a prothrombin time twice normal, progressive encephalopathy and acute kidney injury, the mortality approaches 90%. Early transplantation for patients with severe alcoholic hepatitis has a survival rate of 78%, compared with 32% of those not transplanted. Unfortunately, many return to drinking.
Alcoholic cirrhosis
The management of cirrhosis is described on page 467. Again, all patients are advised to stop drinking for life. Abstinence from alcohol results in an improvement in prognosis, with a 5-year survival of 90%, but with continued drinking this falls to 60%. In advanced disease (i.e. jaundice, ascites and haematemesis) the 5-year survival rate falls to 35%, most of the deaths occurring in the first year. Liver transplantation results in good survival; recurrence of cirrhosis due to recidivism is rare. Patients often sign a contract with their clinicians regarding their abstinence, both before and after transplantation.
A trial of abstention to establish whether liver disease can improve is mandatory, but transplantation should not be denied if the patient continues to deteriorate. Specific follow-up regarding alcohol use is recommended.
HCC is a complication, particularly in men.
Budd–Chiari Syndrome
In this condition, there is obstruction to the venous outflow of the liver owing to occlusion of the hepatic vein. In one-third of patients the cause is unknown, but specific causes include hypercoagulability states (e.g. paroxysmal nocturnal haemoglobinuria, polycythaemia vera) or thrombophilia (see p. 575), taking the contraceptive pill, or leukaemia. Other causes include occlusion of the hepatic vein owing to posterior abdominal wall sarcomas, renal or adrenal tumours, hepatocellular carcinoma, hepatic infections (e.g. hydatid cyst), congenital venous webs, radiotherapy or trauma to the liver.
Clinical features
The acute form presents with abdominal pain, nausea, vomiting, tender hepatomegaly and ascites (a fulminant form occurs particularly in pregnant women). In the chronic form, there is enlargement of the liver (particularly the caudate lobe), mild jaundice, ascites, a negative hepatojugular reflux, and splenomegaly with portal hypertension.
Investigations
Investigations show a high protein content in the ascitic fluid and characteristic liver histology with centrizonal congestion, haemorrhage, fibrosis and cirrhosis. Ultrasound, CT or MRI will demonstrate hepatic vein occlusion with diffuse abnormal parenchyma on contrast enhancement. The caudate lobe is spared because of its independent blood supply and venous drainage. There may be compression of the inferior vena cava. Pulsed Doppler sonography or colour Doppler is useful, as it shows abnormalities of flow in the hepatic vein. Thrombophilia screening is mandatory. Multiple defects of coagulation occur. Thrombosis of the portal vein is present in 2% of patients.
Differential diagnosis
A similar clinical picture can be produced by inferior vena caval obstruction, right-sided cardiac failure or constrictive pericarditis, and appropriate investigations should be performed.
Management
In the acute situation, thrombolytic therapy can be given. Ascites should be treated, as should any underlying cause (e.g. polycythaemia). Congenital webs should be treated radiologically or resected surgically. A TIPS is the treatment of choice, as caval compression does not prejudice the efficacy of TIPS. Surgical portocaval shunts are reserved for those who fail this treatment, providing there is no caval obstruction or severe caval compression when a caval stent can be inserted. Liver transplantation is the first-choice treatment for chronic Budd–Chiari syndrome and for the fulminant form. Life-long anticoagulation is mandatory following TIPS and transplantation.
Prognosis
The prognosis depends on the aetiology but some patients can survive for several years.
Hepatic Sinusoidal Obstruction Syndrome
Hepatic sinusoidal obstruction syndrome (SOS; previously known as veno-occlusive disease) is due to injury of the hepatic veins and presents clinically like the Budd–Chiari syndrome. It was originally described in Jamaica, where the ingestion of toxic pyrrolizidine alkaloids in bush tea (made from plants of the genera Senecio, Heliotropium and Crotalaria) caused damage to the hepatic veins. It can be seen in other parts of the world. It also occurs as a complication of chemotherapy and total body irradiation, used before allogeneic bone marrow transplantation. The development of SOS after bone marrow transplantation carries a high mortality. Treatment is supportive, with control of ascites and hepatocellular failure. TIPS has been used in a few cases. Defibrotide is recommended for prophylactic use before bone marrow transplantation in children and adults with a high risk of SOS (e.g. pre-existing hepatic disease or prior treatment with gemtuzumab ozogamicin).
Fibropolycystic Diseases
These diseases are usually inherited and lead to the presence of cysts or fibrosis in the liver, kidney and occasionally the pancreas, and other organs.
Polycystic disease of the liver
Multiple cysts can occur in the liver as part of autosomal dominant polycystic disease of the kidney (see pp. 789–790). These cysts are usually asymptomatic but occasionally cause abdominal pain and distension. Liver function is normal and complications such as oesophageal varices are very rare. The prognosis is excellent and depends on the kidney disease.
Solitary cysts
These are usually found by chance during imaging and are mainly asymptomatic.
Congenital hepatic fibrosis
In this rare condition, the liver architecture is normal but there are broad collagenous fibrous bands extending from the portal tracts. Congenital hepatic fibrosis is often inherited as an autosomal recessive condition but can also occur sporadically. It usually presents in childhood with hepatosplenomegaly, and portal hypertension is common. It may present later in life and can be misdiagnosed as cirrhosis.
A wedge biopsy of the liver may be required to confirm the diagnosis. The outlook is good and the condition should be distinguished from cirrhosis. Patients who bleed do well after endoscopic therapy of varices (or a portocaval anastomosis) because of their good liver function.
Congenital intrahepatic biliary dilatation (Caroli's disease)
In this rare, non-familial disease there are saccular dilatations of the intrahepatic or extrahepatic ducts. It can present at any age (although it usually does so in childhood) with fever, abdominal pain and recurrent attacks of cholangitis with Gram-negative septicaemia. Jaundice and portal hypertension are absent. Diagnosis is by ultrasound, percutaneous transhepatic cholangiography and MRCP. There is an increased risk of biliary malignancy.
Liver Abscess
Pyogenic abscess
Pyogenic abscesses are uncommon; they may be single or multiple. The most common used to be a portal pyaemia from intra-abdominal sepsis (e.g. appendicitis or perforations), but now the aetiology is not known in many cases. In the elderly, biliary sepsis is a common cause. Other causes include trauma, bacteraemia and direct extension from, for example, a perinephric abscess.
The organism found most commonly is E. coli. Streptococcus milleri and anaerobic organisms such as Bacteroides are often seen. Other organisms include Enterococcus faecalis, Proteus vulgaris and Staphylococcus aureus. Often the infection is mixed.
Clinical features
Some patients are not acutely ill and present with malaise lasting several days or even months. Others can present with fever, rigors, anorexia, vomiting, weight loss and abdominal pain. In these patients, a Gram-negative septicaemia with shock can occur. On examination, there may be little to find. Alternatively, the patient may be toxic, febrile and jaundiced. In such patients, the liver is tender and enlarged, and there may be signs of a pleural effusion or a pleural rub in the right lower chest.
Investigations
Patients who are not acutely ill are often investigated as a case of ‘pyrexia of unknown origin’ (PUO) and most investigations will be normal. Often, the only clue to the diagnosis is a raised serum alkaline phosphatase.
• Serum bilirubin is raised in 25% of cases.
• Normochromic normocytic anaemia may occur, usually accompanied by a polymorphonuclear leucocytosis.
• Serum alkaline phosphatase, ESR and CRP are often raised.
• Serum vitamin B12 is very high, as it is stored in and subsequently released from the liver.
Imaging
Ultrasound is useful for detecting abscesses. A CT scan may be of value in complex and multiple lesions (Fig. 14.31). A chest X-ray will show elevation of the right hemidiaphragm with a pleural effusion in the severe case. Depending on age, imaging of the colon may be necessary to find the source of the infection.
Management
Aspiration of the abscess should be attempted under ultrasound control. Antibiotics should initially cover Gram-positive, Gram-negative and anaerobic organisms until the causative organism is identified.
Further drainage via a large-bore needle under ultrasound control or surgically may be necessary if resolution is difficult or slow. Any underlying cause must also be treated.
Prognosis
The overall mortality depends on the nature of the underlying pathology but has been reduced to approximately 16% with needle aspiration and antibiotics. A unilocular abscess in the right lobe has the best prognosis. Scattered multiple abscesses have a very high mortality, with only 1 in 5 patients surviving.
Amoebic abscess
This condition occurs worldwide and must be considered in patients travelling from endemic areas. Entamoeba histolytica (see pp. 305–306) can be carried from the bowel to the liver in the portal venous system, leading to portal inflammation, with the development of multiple microabscesses and, eventually, single or multiple large abscesses.
Clinically, the onset is usually gradual but may be sudden. There is fever, anorexia, weight loss and malaise. There is often no history of dysentery. On examination, the patient looks ill and has tender hepatomegaly and signs of an effusion or consolidation in the base of the right side of the chest. Jaundice is unusual.
Investigations
These are as for pyogenic abscess, plus:
• Serological tests for amoeba (e.g. haemagglutination, amoebic complement fixation test, ELISA). These are always positive, particularly if there are bowel symptoms; they remain positive after a clinical cure and therefore do not indicate current disease. A repeat negative test, however, is good evidence against an amoebic abscess.
• Diagnostic aspiration of fluid looking like anchovy sauce.
Management
Metronidazole 800 mg three times daily is given for 10 days. Aspiration is used in patients failing to respond, in those with multiple and sometimes large abscesses, and in those with abscesses in the left lobe of the liver or impending rupture.
Complications
Complications include rupture, secondary infection and septicaemia.
Other Infections of the Liver
Schistosomiasis
Schistosoma mansoni and S. japonicum affect the liver, but S. haematobium rarely does so (see also pp. 312–314). During their life cycle, the ova reach the liver via the venous system and obstruct the portal branches, producing granulomas, fibrosis and inflammation, but not cirrhosis.
Clinical features and investigations
Clinically, there is hepatosplenomegaly and portal hypertension, which is particularly severe with S. mansoni. In Egypt, there is frequently concomitant chronic hepatitis C infection.
Investigations show a raised serum alkaline phosphatase, and ova can be found in the stools (centrifuged deposits) and in rectal and liver biopsies. Skin tests and other immunological tests often give false results and may also be positive because of past infection.
Management
Treatment is with praziquantel, but fibrosis still remains with a potential risk of portal hypertension, characteristically pre-sinusoidal due to intense portal fibrosis.
Hydatid disease
Cysts caused by Echinococcus granulosus are single or multiple. They usually occur in the lower part of the right lobe. The cyst has three layers: an outside layer derived from the host, an intermediate laminated layer, and an inner germinal layer that buds off brood capsules to form daughter cysts (see also pp. 315–316).
Clinical features and investigations
Clinically, there may be no symptoms or there may be a dull ache and swelling in the right hypochondrium.
Investigations show a peripheral eosinophilia in 30% of cases and usually a positive hydatid complement fixation test or haemagglutination (85%). Plain abdominal X-ray may show calcification of the outer coat of the cyst. Ultrasound and CT scan demonstrate cysts and may show diagnostic daughter cysts within the parent cyst (Fig. 14.32).
Management
Medical treatment (e.g. with albendazole 10 mg/kg, which penetrates into large cysts) results in cysts becoming smaller. Puncture, aspiration, injection, re-aspiration (PAIR) has been used since the 1980s. Fine-needle aspiration is undertaken under ultrasound control with chemotherapeutic cover. Surgery can be performed with removal of the cyst intact, if possible, after first sterilizing the cyst with alcohol, saline or cetrimide. Chronic calcified cysts can be left; there have been no well-designed clinical trials for any therapy.
Complications and prognosis
These include rupture into the biliary tree or other organs, or intraperitoneally, with spread of infection. The prognosis without any complications is good, although there is always a risk of rupture. Preventative measures include deworming of pet dogs and preventing pets from eating infected carcasses, as well as veterinary control programmes.
Echinococcus multilocularis causes alveolar echinococcosis and is almost exclusively a hepatic disease, with a high mortality if not treated. Early diagnosis enables radical surgery and then continued chemosuppression.
Acquired immunodeficiency syndrome
The liver is often involved in AIDS and is a significant cause of morbidity or mortality. HIV (see also pp. 331–355) itself is not the cause of the liver abnormalities. Clinical hepatomegaly is common (60% of patients). The following are seen, although less frequently in areas where anti-retroviral therapy (ART) is available:
• Pre-existing/coincidental viral hepatitis. The hepatitis (HBV, HCV, HDV) progresses more rapidly and is a leading cause of death.
• Neoplasia. Kaposi's sarcoma and non-Hodgkin's lymphoma may be seen, and there is an increased risk of HCC.
• Opportunistic infection (e.g. Mycobacterium tuberculosis, M. avium-intracellulare, Cryptococcus, Candida albicans, toxoplasmosis).
• Secondary sclerosing cholangitis (see p. 350).
• Non-cirrhotic portal hypertension associated with anti-retroviral therapy.
Liver Disease in Pregnancy
See pages 1303–1304.
Liver Tumours
Secondary liver tumours
The most common liver tumour is a secondary (metastatic) tumour, particularly from the gastrointestinal tract (from the distribution of the portal blood supply), breast or bronchus. Secondary liver tumours are usually multiple.
Clinical features
These are variable but usually include weight loss, malaise, upper abdominal pain and hepatomegaly, with or without jaundice.
Diagnosis
Ultrasound is the primary investigation, with CT or MRI to define metastases and look for a primary. The serum alkaline phosphatase is almost invariably raised.
Management
This will depend on the site of the primary and the burden of liver metastases. The best results are obtained in colorectal cancer in patients with few hepatic metastases. If the primary tumour is removed and hepatic resection is performed, reasonable survival rates are possible. Chemotherapy is used, particularly with breast cancer (see p. 634). Radiofrequency ablation of the metastases is an alternative to surgery. Thermal therapy and cryotherapy are also used.
Primary malignant tumours
Primary liver tumours may be benign or malignant, but the most common are malignant.
Hepatocellular carcinoma
Hepatocellular carcinoma (HCC) is the fifth most common cancer worldwide.
Aetiology
Carriers of HBV and HCV have an extremely high risk of developing HCC. In areas where HBV is prevalent, 90% of patients with this cancer are positive for HBV. Cirrhosis is present in approximately 80% of these patients. The development of HCC is related to the integration of viral HBV DNA into the genome of the host hepatocyte (see p. 456), and to the degree of viral replication (>10 000 copies/mL). The risk of HCC in HCV is higher than in HBV (even higher with both HBV and HCV), despite no viral integration. Unlike HBV infection, cirrhosis is always present in HIV. Primary liver cancer is also associated with other forms of cirrhosis, such as alcoholic cirrhosis, non-alcoholic fatty liver disease (NAFLD)-associated cirrhosis, and haemochromatosis. Males are affected more than females. Other aetiological factors are aflatoxin (a metabolite of a fungus found in groundnuts) and androgenic steroids, and there is a weak association with the contraceptive pill.
Pathology
The tumour either is single or occurs as multiple nodules throughout the liver. Histologically, it consists of cells resembling hepatocytes. It can metastasize via the hepatic or portal veins to the lymph nodes, bones and lungs.
Clinical features
The clinical features include weight loss, anorexia, fever, an ache in the right hypochondrium and ascites. The rapid development of these features in a cirrhotic patient is suggestive of HCC. On examination, an enlarged, irregular, tender liver may be felt. Increasingly, due to surveillance, HCC is found without symptoms in patients with cirrhosis.
Investigations
• Routine liver biochemistry, full blood count, urea and electrolytes.
• Serum α-fetoprotein may be raised, but is normal in at least a third of patients.
• Ultrasound scans show filling defects in 90% of cases.
• Enhanced CT scans (Fig. 14.33) identify HCC but it is difficult to confirm the diagnosis in lesions smaller than 1 cm. An MRI can help to delineate lesions further.

• Tumour biopsy (see Fig. 14.29), particularly under ultrasonic guidance, is now used less frequently for diagnosis as imaging techniques show characteristic appearances (hypervascularity of the nodule and lack of portal vein washout) and because seeding along the biopsy tract can occur.
Management and prognosis
See page 638.
Prevention
Persistent HBV infection, usually acquired after perinatal infection, is a high risk factor for HCC in many parts of the world, such as South-east Asia. Widespread vaccination against HBV is being used and this has reduced the annual incidence of HCC in Taiwan.
Cholangiocarcinoma
Cholangiocarcinomas are increasing in incidence and can be extrahepatic (see p. 498) or intrahepatic (see p. 638). Intrahepatic adenocarcinomas arising from the bile ducts account for approximately 10% of primary tumours of the liver and biliary tract. They are not associated with cirrhosis or hepatitis B. In the Far East, they may be associated with infestation with Clonorchis sinensis or Opisthorchis viverrini. The clinical features are similar to those of primary HCC, except that jaundice is frequent with hilar tumours, and cholangitis is more common. There is an increased association with inflammatory bowel disease and primary sclerosing cholangitis (see pp. 476–477).
Surgical resection is rarely possible and patients usually die within 6 months. Transplantation is contraindicated, outside of specialized protocols.
Benign tumours
The most common benign tumour is a haemangioma. It is usually small and single but can be multiple and large. Haemangiomas are usually found incidentally on ultrasound, CT or MRI, and have characteristic appearances. They require no treatment.
Hepatic adenomas are associated with oral contraceptives. They can present with abdominal pain or intraperitoneal bleeding. Resection is required only for symptomatic patients, those with tumours >5 cm in diameter, and in those in whom discontinuation of oral contraception does not result in shrinkage of the tumour. Immunohistochemical characteristics are helpful in indicating malignant potential, which is far more common in men.
Miscellaneous Conditions of the Liver
Hepatic mitochondrial injury syndromes
These syndromes – in which there is mitochondrial damage with inhibition of β-oxidation of fatty acids – can be categorized as having the following causes:
• Genetic, with abnormalities that include medium-chain acyl-coenzyme A dehydrogenase deficiency, leading to microsteatosis.
• Toxins leading to liver failure, which include aflatoxin and cerulide (produced by Bacillus cereus); the latter causes food poisoning (see p. 277).
• Drugs (e.g. i.v. tetracycline, valproic acid and nucleoside reverse-transcriptase inhibitors), which can produce a fatal microsteatosis.
• Idiopathic, the best known being fatty liver of pregnancy (see p. 1304) and Reye syndrome. The latter, caused by inhibition of β-oxidation and uncoupling of oxidative phosphorylation in mitochondria, leads in children to an acute encephalopathy and diffuse microvesicular fatty infiltration of the liver. Aspirin ingestion and viral infections have been implicated as precipitating agents. Mortality is about 50%, usually due to cerebral oedema.
Idiopathic adult ductopenia
This unexplained condition is characterized by pruritus and cholestatic jaundice. Histology of the liver shows a decrease in intrahepatic bile ducts in at least 50% of the portal tracts, together with the features of cholestasis and marked fibrosis or cirrhosis. In most, the disease is progressive and the only treatment is liver transplantation.
Indian childhood cirrhosis
This condition of children is seen in the Indian subcontinent. The cause is unknown. Eventually, there is development of a micronodular cirrhosis with excess copper in the liver.
Hepatic porphyrias
These are dealt with on page 1290.
Cystic fibrosis
Cystic fibrosis (see also pp. 1088–1089) mainly affects the lung and pancreas, but patients can develop fatty liver, cholestasis and cirrhosis. The aetiology of the liver involvement is unclear.
Coeliac disease
Abnormal liver biochemical tests are common in coeliac disease (see also p. 396) and return to normal with a gluten-free diet. A tissue transglutaminase test should be performed if hepatic causes are not found when investigating abnormal liver biochemistry.
Drugs and the Liver
Drug metabolism
The liver is the major site of drug metabolism. Drugs are converted from fat-soluble to water-soluble substances that can be excreted in the urine or bile. This metabolism of drugs is mediated by a group of mixed-function enzymes (see p. 19).
Drug hepatotoxicity
Many drugs impair liver function. When abnormal liver biochemical tests are found, drugs should always be considered as a cause, particularly when other causes have been excluded. Damage to the liver by drugs is usually classified as being either predictable (or dose-related) or non-predictable (not dose-related) (see p. 22). However, there is considerable overlap and at least six mechanisms may be involved in the production of damage:
1. disruption of intracellular calcium homeostasis
2. disruption of bile canalicular transport mechanisms
3. formation of non-functioning adducts (enzyme–drug), which may then lead to
4. presentation on the surface of the hepatocyte as new immunogens (attacked by T cells)
6. inhibition of mitochondrial function, which prevents fatty acid metabolism and accumulation of both lactate and reactive oxygen species.
The predominant mechanism or combination of mechanisms determines the type of liver injury: that is, hepatitic, cholestatic or immunological (skin rashes, fever and arthralgia, i.e. serum-sickness syndrome). Eosinophilia and circulating immune complexes and antibodies are occasionally detected.
When a small amount of hepatotoxic drug whose effect is dose-dependent (e.g. paracetamol) is ingested, a large proportion of it undergoes conjugation with glucuronide and sulphate, while the remainder is metabolized by microsomal enzymes to produce toxic derivatives that are immediately detoxified by conjugation with glutathione. If larger doses are ingested, the former pathway becomes saturated and the toxic derivative is produced at a faster rate. Once the hepatic glutathione is depleted, large amounts of the toxic metabolite accumulate and produce damage (see p. 79).
The ‘predictability’ of drugs to produce damage can, however, be affected by metabolic events preceding their ingestion. For example, chronic alcohol users may become more susceptible to liver damage because of the enzyme-inducing effects of alcohol, or ill or starving patients may become susceptible because of the depletion of hepatic glutathione produced by starvation. Many other factors, such as environmental or genetic effects, may be involved in determining the ‘susceptibility’ of certain patients to certain drugs.
The incidence of drug hepatotoxicity is 14 per 100 000 population with a 6% mortality. It is the most common cause of acute liver failure in the USA. Liver transplantation is used.
Hepatitic damage
The type of damage produced by various drugs is shown in Box 14.23. Most reactions occur within 3 months of starting the drug. Monitoring liver biochemistry in patients on long-term treatment, such as anti-tuberculosis therapy, is mandatory. If a drug is suspected of causing hepatic damage, it should be stopped immediately. Liver biopsy is of limited help in confirming the diagnosis, but occasionally, hepatic eosinophilia or granulomas may be seen. Diagnostic challenge with subtherapeutic doses of the drug is sometimes required after the liver biochemistry has returned to normal, to confirm the diagnosis.
Box 14.23
Liver damage produced by some drugs
| Types of liver damage | Drugs | |
| Zone 3 necrosis | ||
| Zone 1 necrosis | Ferrous sulphate | |
| Microvesicular fat | Sodium valproate | Tetracyclines |
| Steatohepatitis | Nifedipine | |
| Fibrosis | ||
| Vascular | ||
| Sinusoidal dilatation | Contraceptive drugs | Anabolic steroids |
| Peliosis hepatis | ||
| Veno-occlusive | Pyrrolizidine alkaloids (Senecio in bush tea) | Cytotoxics – cyclophosphamide |
| Acute hepatitis | ||
| Chronic hepatitis | ||
| General hypersensitivity | ||
| Canalicular cholestasis | ||
| Biliary sludge | Ceftriaxone | |
| Sclerosing cholangitis | Hepatic arterial infusion of 5-fluorouracil | |
| Hepatic tumours | Pills with high hormone content (adenomas) | |
| Hepatocellular carcinoma | Contraceptive pill | Danazol |
| Nodular regenerative hyperplasia | Azathioprine | Some anti-retroviral therapies |
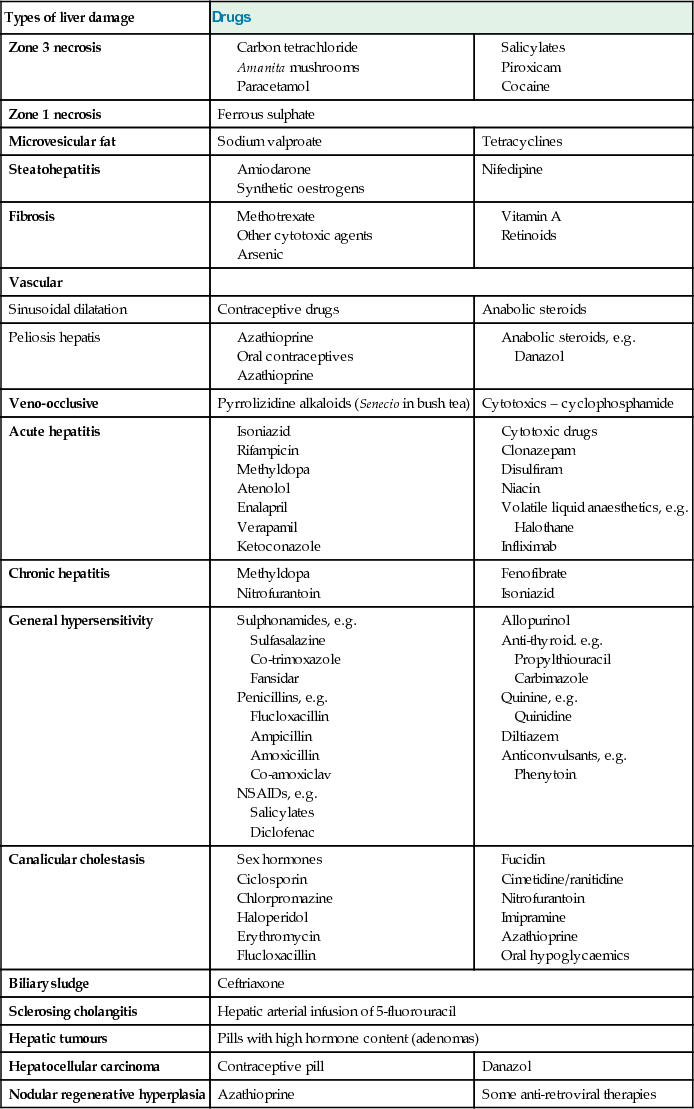
Individual drugs
In high doses, paracetamol produces liver cell necrosis (see above). The toxic metabolite binds irreversibly to liver cell membranes. Overdosage is discussed on pages 79–80.
Volatile liquid anaesthetics
Halothane, which was the first available drug in this class, is now not used in the UK because it produced hepatitis in patients with repeated exposures. Both sevoflurane and isoflurane also cause hepatotoxicity in those patients sensitized to halogenated anaesthetics; however, the risk is smaller than with halothane and remote with desflurane.
Steroid compounds
Cholestasis is caused by natural and synthetic oestrogens, as well as methyltestosterone. These agents interfere with canalicular biliary flow by blocking MRP2 and MDR3 (see Fig. 14.4) and cause a pure cholestasis.
Cholestasis is rare with the contraceptive pill because of the low dosage used. However, the contraceptive pill is associated with an increased incidence of gallstones, hepatic adenomas (rarely, HCCs), the Budd–Chiari syndrome and peliosis hepatis. The latter condition, which also occurs with anabolic steroids, consists of dilatation of the hepatic sinusoids to form blood-filled lakes.
Phenothiazines
Phenothiazines (e.g. chlorpromazine) can produce a cholestatic picture owing to a hypersensitivity reaction. This occurs in 1% of patients, usually within 4 weeks of starting the drug. Typically, it is associated with a fever and eosinophilia. Recovery occurs on stopping the drug.
Anti-tuberculous chemotherapy
Isoniazid produces elevated aminotransferases in 10–20% of patients. Hepatic necrosis with jaundice occurs in a smaller percentage. The hepatotoxicity of isoniazid is related to its metabolites and is dependent on acetylator status. Rifampicin produces hepatitis, usually within 3 weeks of starting the drug, particularly in patients on high doses. Pyrazinamide produces abnormal liver biochemical tests and, rarely, liver cell necrosis.
Amiodarone
This leads to a steatohepatitis histologically, and liver failure if the drug is not stopped in time.
Sodium valproate
This causes mitochondrial injury with microvesicular steatosis. Intravenous carnitine should be used as an antidote.
Drug prescribing for patients with liver disease
The metabolism of drugs is impaired in severe liver disease (with jaundice and ascites), as the removal of many drugs depends on liver blood flow and the integrity of the hepatocyte. In general, therefore, the effect of drugs is prolonged by liver disease and also by cholestasis. This is further accentuated by portosystemic shunting, which diminishes the first-pass extraction of drugs. With hypoproteinaemia, there is decreased protein binding of some drugs, and bilirubin competes with many drugs for the binding sites on serum albumin. In patients with portosystemic encephalopathy, care must be taken in prescribing drugs with a central depressant action, such as narcotics, including codeine and anxiolytics. Other common drugs to be avoided in cirrhosis include angiotensin-converting enzyme (ACE) inhibitors (which cause hepatorenal failure) and NSAIDs (which cause bleeding).