Chapter 21
Structure and Mechanism of Voltage-Gated Ion Channels
Chapter Outline
II. Introduction: How Is Ion Channel Structure Studied?
III. Biochemistry of Ion Channels: Purification and Characterization of Voltage-Gated Channels
IIIA. Electric Fish Are a Rich Source of Ion Channels
IIIB. Toxins and Drugs as Markers for Ion Channels during Purification
IIIC. Isolation of Channel Molecules Based on Fractionation Procedures
IIID. Information about Channel Structure from Channel Purification
IIIE. Reconstitution of Purified Proteins Confirms their Identity as Channels
IIIF. Limitations of Biochemical Characterization of Channels
IV. Channel Structure Investigation through Manipulation of DNA Sequences Encoding Channel Polypeptides
IVA. Primary Structure of Ion Channels Determined Using Recombinant DNA Technology
IVB. Analysis of the Primary Structure of Large Polypeptide Voltage-Gated Channels
IVC. How Is Channel Structure Formed?
IVD. Sequence Homology among the Transmembrane Domains of Ion Channels
IVE. How Are Topographical Predictions for Channel Structure Tested?
IVF. The Genetic Approach Used to Identify Nucleic Acid Clones Coding for K+ Channels
IVG. K+ Channels are Homologous to a Single Internal Repeat of Na+ Channels
V. Molecular Mechanisms of Channel Function: How Does One Investigate Them?
VA. Choosing Interesting Sites and Segments as Targets for Mutagenesis
VB. Use of Expression Systems in Mutagenesis Studies
VC. Domains Involved in Voltage-Dependent Activation
VD. Limitations of Mutagenesis in the Study of Channel Mechanisms
VE. The Mechanism of Channel Inactivation
VF. Other Gating-Related Domains in Voltage-Sensitive Channels
VI. Isoforms of Voltage-Gated Channels as Part of a Large Superfamily
I Summary
The structure, mechanism and expression of ion channels are currently intense areas of research interest. The first advances in these areas were made by the application of biochemical techniques to the problems of channel purification. Such studies have told us the size of intact channels and their subunit composition and have yielded clues regarding functionally important domains (such as pores, gates, modulation sites and non-protein modifications). For Na+ and Ca2+ channels, attention has been focused on a large polypeptide (so-called α peptide) found in each purified preparation. These α proteins apparently have all the molecular apparatus required for channel operation, while smaller associated subunits may play other roles in channel modulation, synthesis or cellular localization. Purified material has also been important in the construction of probes to identify clones of channel-encoding DNA fragments.
Recent work has applied recombinant DNA technology to determine the primary amino acid sequence of channels through cloning. These sequences have revealed widespread sequence homology among the superfamily of voltage-gated ion channels, e.g. between Na+ and Ca2+ channel α polypeptides. Furthermore, the large α proteins have four domains of homology within their sequences, suggesting that pore formation occurs via a common “staves of a barrel” architecture. K+ channel sequences, determined via a genetic approach, are relatively shorter and they partially correspond to a single internal repeat of the larger α peptides. Hence, K+ channels are thought to consist of tetramers with each subunit contributing to the pore wall. Other analyses of amino acid sequences have given clues regarding the mechanistically and structurally important domains of ion channels. One analysis was based on thermodynamic (hydropathy) considerations to identify possible membrane-spanning domains. In addition, the sequences were subjected to an empirical analysis that predicts regions of secondary structure, such as helices and sheets. From such predictions detailed models of channel structure and function have been developed. These models have been tested using immunological and mutagenesis approaches and candidate domains for ion selectivity, pore wall formation, gating and modulation have been identified. However, detailed knowledge of the molecular mechanisms of channel function is still limited.
Finally, molecular studies have revealed an astonishing diversity of ion channels at all levels of organization. In particular, the genome of most organisms can express multiple channel isotypes, sometimes coexisting within the same living cell. Much remains to be learned about channel function and the role of channel isotypes in the biology of both simple and complex organisms.
II Introduction: How Is Ion Channel Structure Studied?
Previous chapters have described how the flow of ions across cell membranes is the basis for electrical excitability and signaling in the nervous system. These flows are controlled by a special class of macromolecules known as ion channels that form gated pores in the cell membrane. The structure of ion channels and the current thinking about how these structures form transmembrane pores that gate to open and closed states in response to changes in membrane voltage are discussed in this chapter. Subsequent chapters will describe other important aspects of ion channels, such as interactions with drugs and toxins, modulation by intracellular messengers and specific ion channel types found in intracellular organelles and involved in synaptic transmission.
What are the questions that should be addressed in this chapter? Most fundamentally, we wish to know how these macromolecular structures give rise to the important functional properties of ion channels, namely, the formation of transmembrane pores, the selective transport of specific ions and the ability to open and close the pore; i.e. how do ion channels function as molecular mechanisms? As we will see, these considerations also lead into a brief consideration of the structural diversity among ion channels and the origin and possible purposes of such diversity.
Cellular ion channels are basically proteins. In the earliest structural studies, biochemical methods were employed to purify channel molecules from excitable tissues. This approach has been essential to the current state of knowledge of ion channel structure. However, ion channels have certain physicochemical properties that have limited the amount of information obtained solely through biochemical characterization. This bottleneck has been broken recently through the application of recombinant DNA methodologies, which have uncovered the primary structures of numerous ion channel types, while providing a powerful means to study higher order structure and function. However, this latter approach has its own limitations and, ultimately, we will need to “see” more directly the actual structure of the channels to understand more fully their molecular mechanisms of action. In this regard, it is encouraging that significant progress has been made recently in elucidating the structure of purified channels using x-ray crystallographic techniques.
III Biochemistry of Ion Channels: Purification and Characterization of Voltage-Gated Channels
Before the advent of recombinant DNA methods, ion channels were extensively studied by first purifying channels from an excitable tissue and then characterizing the purified molecules by a combination of chemical and physical techniques. While this biochemical approach has fallen somewhat out of use in favor of DNA cloning techniques, it seems certain to re-emerge as a necessary adjunct to structural studies that now seem feasible (see Section VH). Thus, a brief introduction to channel biochemistry seems appropriate here. The first voltage-gated channel to be purified was the Na+ channel from the electric eel, Electrophorus electricus, and the account of how this was achieved will serve to illustrate the basic rationale for the biochemical approach (see Miller et al., 1983).
The basic procedure for purification of membrane-associated proteins (Fig. 21.1) is as follows. First, an enriched fraction of membranes is usually prepared by disrupting the tissue and its cells mechanically and subsequently separating the insoluble fraction containing membranes and connective tissue components from the soluble fraction consisting of cytoplasmic protein. Although relatively non-specific, this step also often gives substantial enrichment of ion channel preparations. Next, the membranes are solubilized through the use of detergents. This step disperses the membranes into minute droplets of lipid, protein and detergent known as micelles. This is often the most problematic step of a purification, because the micellar fraction must be dispersed well enough so that no more than one protein molecule is present in each micelle. Too much detergent often results in irreversible denaturation of channels, hence preventing their identification during the purification process. Thus, the solubilization process must be optimized and often many different detergents at various concentrations are tested under a plethora of conditions before the right combination is found. Finally, the dispersed micelles are fractionated based on their physical properties or affinities for certain compounds until a pure sample of channel protein is isolated.
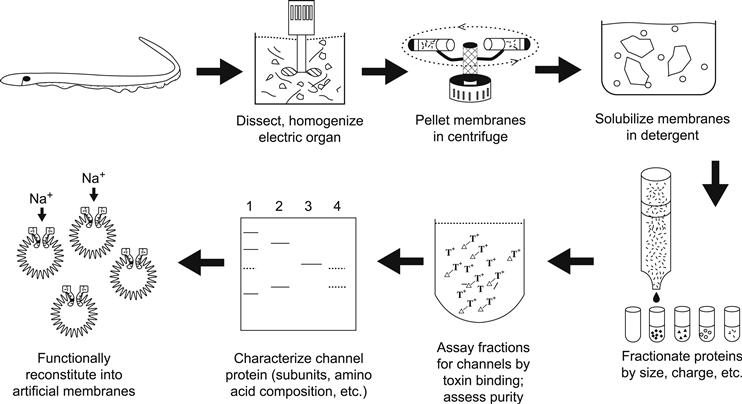
FIGURE 21.1 Scheme used for the biochemical isolation of Na+ channels from electric tissue of the eel Electrophorus electricus. Purification protocols devised for other ion channels utilize similar general principles but differ in details.
IIIA Electric Fish Are a Rich Source of Ion Channels
The first requirement for purification of any protein is to have a rich source. Actually, this can be a serious difficulty, since ion channels are present in very small amounts in most excitable tissues. The rarity of channels might at first seem paradoxical in view of the ubiquity of excitability phenomena in nearly all cell membranes. However, the reader should recall that only minute numbers of channels are needed to mediate the flow of ions required to cause the changes in transmembrane voltage during electrical excitation (Table 21.1).
TABLE 21.1. Sodium Channel Densities in Selected Excitable Tissues
| Tissue | Surface Density Channels/μm2 | Tissue Density (μg channel/g tissue) |
| Mammalian | ||
| Vagus nerve (unmyelinated) | 110 | 28 |
| Node of Ranvier | 2100 | — |
| Skeletal muscle (various) | 2066–557 | 6–14 |
| Other Animals | ||
| Squid giant axon | 166–533 | ≈1 |
| Frog sartorius muscle | 280 | 6 |
| Electric eel electroplax (excitable surface) | 550 | 38 |
| Garfish olfactory nerve | 35 | 96 |
| Lobster walking leg nerve | 90 | 24 |
Fortunately, there exist some highly specialized “freaks” of nature that use large ionic currents for purposes other than signaling and stimulus transduction. These are the strongly electric fish, which are capable of generating powerful electrical discharges to stun prey and defend themselves. The electric organs of these animals contain large numbers of ion channels that are present at high density on the surface of the excitable cells of the electric organ (see Chapter 48). As a result, the first two ion channels to be purified were the acetylcholine receptor channel of the electric ray Torpedo and the Na+ channel of the electric eel Electrophorus.
IIIB Toxins and Drugs as Markers for Ion Channels during Purification
Having a rich source of channels does not guarantee a successful isolation of channels. One must also have a very specific and sensitive assay for the presence of solubilized channels to follow the process of fractionation. Unfortunately, unlike enzymes or other chemically active proteins, the activity of ion channels is to transport ions across membranes. Since this barrier is disrupted during the first stage of purification, the compartments essential to channel function no longer exist and hence the electrically detectable flow of ions through channels no longer occurs. Instead, one may use the binding of powerful neurotoxins to channels to assess the degree of purity attained during fractionation.
For Na+ channels, there exist highly selective inhibitors of sodium transport known as guanidinium toxins. The most commonly used of these are tetrodotoxin (TTX), obtained from certain species of toxic puffer fish, and saxitoxin (STX), a product of certain dinoflagellates that “bloom” during oceanic “red tides”. Either toxin may be radioactively labeled and these substances very specifically interact with Na+ channels from a wide variety of sources with high affinity. To assess purity, one may thus compare the number of toxin-binding sites in a fraction with the amount of protein present.
IIIC Isolation of Channel Molecules Based on Fractionation Procedures
With a good source of channels, efficient solubilization procedures and a specific and sensitive assay, one may proceed to attempts at purification. For Na+ channels, fractionation based on charge, followed by size fractionation of the mixed channel/lipid/detergent micelle, has resulted in highly purified preparations of Na+ channels. Alternatively, in some protocols, channel proteins have been specifically purified using affinity chromatography, in which channels are selectively retained by a column to which channel-binding substances, such as drugs, toxins or antibodies, are attached.
Skeletal muscle Ca2+ channels have also been extensively purified and characterized using much the same approaches as those described for Na+ channels. In this case, it has been the L-type channels that were isolated, using radiolabeled drugs of the dihydropyridine class to tag channels irreversibly in the intact muscle membrane. This allowed the investigators to isolate the channel protein by following the fractionation of the bound radiolabeled drug (Sharp et al., 1987).
IIID Information about Channel Structure from Channel Purification
There is much to be learned about channel structure from the physicochemical characteristics of the purified molecule. Some of this information forms the basis for separating the molecule from others in membrane extracts (such as size or charge differences), as described previously, whereas other measurements may be done on the purified material itself. Thus, for Na+ channels, it was found that the channel is very negatively charged. Furthermore, in detergent solution, the channel appears quite large; in micellar form, its size is on the order of several million molecular weight. Both characteristics are important clues regarding the chemical composition of Na+ channels.
The subunit composition of a channel is an especially important characteristic to be determined from the purified material. For a number of enzymes, separate functions have been attributed to different subunits; hence breaking down the structural entities represents an important basis for further structure–function correlations. In brief, one usually determines the subunit composition of the purified material by analyzing it using denaturing electrophoresis, in which the intersubunit bonds are broken and each subunit separated according to size in an electric field. The most commonly used technique is sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE), in which each polypeptide species appears as a separate band on staining the polyacrylamide sieve used to separate polypeptides. Unfortunately, this is often more difficult than it appears, since (1) one is often not sure whether different bands on a gel are contaminants or true subunits and (2) the stoichiometry of subunits is difficult to establish because of variations in the intrinsic staining intensity among various polypeptides. As a result, considerable effort is expended by various researchers to determine (and argue about!) subunit composition using a wide variety of approaches. For the voltage-gated Na+ channel, a single large polypeptide apparently accounts for pore formation, ion selectivity and gating. This protein appears on SDS-PAGE as a broad band at high molecular weight.
Given the large size of the eel electric organ, enough protein can be purified to allow study of its chemical composition. Not too surprisingly, Na+ channels have an elevated proportion of hydrophobic amino acids compared to soluble globular proteins, likely reflecting domains of the channel that insert into the lipid membrane. However, the channel molecule appears to be much more hydrophobic than can be accounted for from its amino acid composition. Thus, further chemical analysis has found that a large number of lipid molecules are bound to the eel protein. Finally, it has been found that Na+ channels are modified by the presence of extensive domains of carbohydrate. This glycosylation makes up about 30% of the molecule (by weight).
Na+ channels have also been purified from mammalian brain and muscle tissue. Like the eel channel, they consist primarily of heavily glycosylated large polypeptides. However, unlike the electric organ channel, they are found in association with one or several smaller polypeptides that have been thought to be channel subunits (Table 21.2). The large polypeptide common to Na+ channels from all sources is referred to as alpha (α) while smaller subunits have been designated as beta (muscle) or β1 and β2 (brain). At present, the role of these accessory polypeptides in channel function is not certain, although recent evidence suggests that they may aid in channel expression and modulate channel gating (Isom et al., 1992).
TABLE 21.2. Subunit Composition of Voltage-Gated Cation Channels
| Channel/tissue | Subunit | Molecular Weighta |
| Na+ | ||
| Eel electric organ | α | 260 (208) |
| Rat brain | α | 260 (220) |
| β1 | 36 (23) | |
| β2 | 33 | |
| Rat skeletal muscle | α | 260 (209) |
| β | 36 | |
| Chick heart | α | 235 |
| Ca2+ | ||
| L-type, rabbit skeletal muscle | α-1 | 170 (212) |
| α-2 | 143 (125)b | |
| β | 54 (58) | |
| γ | 30 (25) | |
| δ | 24–27 (27)b | |
| K+ Channel | ||
| Drosophila Shaker A | — | 65–85 (70) |
| Rat drk1 | — | 130 (95) |
a First set of values is the apparent molecular weight determined biochemically (SDS-PAGE); weights in parentheses were obtained from cloned DNA sequences.
b The biochemically observed calcium channel α-2 and δ subunits are derived from proteolysis of a full-length α-2 translation product during biosynthesis.
For the skeletal muscle L-type Ca2+ channel, it was found that although a single large polypeptide (α1) was affinity labeled by the dihydropyridine (DHP) marker, at least four other polypeptides – α2, β, gamma (γ), delta (δ)–copurified with the DHP label. As with the Na+ channel, α1 seems to have the mechanisms required for ion channel operation. However, there is evidence that the other subunits are important accessory proteins that may be involved in the key role of this channel as the voltage sensor in excitation–contraction coupling, which is described in Chapter 45.
Also shown in Table 21.2 are data for voltage-gated K+ channels. The characterization of K+ channel subunits was achieved primarily through molecular genetic means, as discussed in Section IVF.
IIIE Reconstitution of Purified Proteins Confirms their Identity as Channels
An important criterion for purification was the reconstitution of the isolated material into artificial membranes and the demonstration that voltage-gated, sodium-selective, toxin-inhibitable channels were formed (see Miller, 1986). Thus, these experiments showed that purified toxin-binding material could be reinserted into small artificial lipid vesicles (liposomes) or planar lipid films (lipid bilayers) where they retained their ability to transport sodium selectively in response to voltage changes. These reconstituted channels had the same ability to respond to different pharmacological compounds, such as toxins and anesthetics, as the channels in natural tissue (reviewed in Catterall, 1992). Similar experiments have demonstrated that purified DHP receptors can form functional Ca2+ channels in bilayers (Catterall, 1988).
IIIF Limitations of Biochemical Characterization of Channels
The biochemical approach to channel purification thus gave important information about the size, chemical composition and polypeptide makeup of Na+ and L-type Ca2+ channels. Classically, the next steps would be to sequence the purified material and attempt to obtain a high-resolution structure from crystallographic approaches. However, although these techniques have worked well with a small number of soluble globular proteins, for several reasons they have been relatively unsuccessful with membrane proteins in general and voltage-gated channels in particular. First, sequencing a large polypeptide requires that it be enzymatically or chemically cleaved into overlapping smaller fragments amenable to Edman degradation techniques (which can only sequence from 25 to 50 residues at a time). In such an approach, each small fragment must be separately purified from the fragmented preparation. For the large polypeptides of Na+ and Ca2+ channels, the number of fragments generated by such cleavages is just too great for all of them to be purified separately. Furthermore, many of the interesting membrane-spanning segments do not fragment or isolate well because of their high degree of hydrophobicity (“fears water”). Finally, the amphipathic (i.e. polarized hydrophilic/hydrophobic) nature of membrane proteins prevents their crystallization; instead, in the absence of lipids or detergents, such purified preparations usually form amorphous aggregates or precipitates with little intrinsic order. Hence, the elaboration of detailed structure of biochemically purified channels has been hampered by the unfavorable physical properties of membrane proteins.
IV Channel Structure Investigation through Manipulation of DNA Sequences Encoding Channel Polypeptides
Fortunately, the limitations of classic biochemistry may be partly overcome by the application of techniques that allow one to identify and sequence DNA that encodes channel peptides. The next three subsections describe how the powerful methods of molecular biology are being applied to questions of interest to the cell physiologist. These approaches have given important insights into the structure of channels and how they work. This aspect of channel biology is considered in Sections IVA and IVB. However, molecular biology has also shown us that voltage-gated channels exist in a rich diversity of forms even within a single organism. This aspect of channel biology is described in Section IVC. First, a brief description of how recombinant DNA methods are used in such studies is provided for the novice reader (Fig. 21.2).
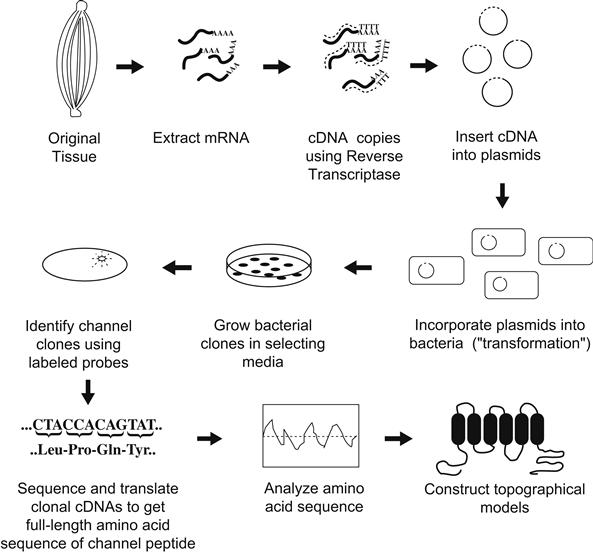
FIGURE 21.2 General strategy used to determine ion channel structure using recombinant DNA approaches.
IVA Primary Structure of Ion Channels Determined Using Recombinant DNA Technology
One starts with the same tissues shown to be enriched in channel protein because these are also probably enriched in messenger RNAs (mRNAs) encoding these channels. Using the retroviral enzyme reverse transcriptase, one may then make complementary DNA copies (cDNAs) of the mRNAs. Using enzymatic “scissors” (called restriction enzymes), one may then insert each cDNA obtained into a circular DNA plasmid. These plasmids have the ability to be replicated along with other genetic material in bacteria and also they encode an enzyme that will destroy certain antibiotics that would otherwise kill their bacterial hosts. The recombinant plasmids are next introduced into a bacterial host so that each bacterium will contain no more than a single plasmid with its unique cDNA insert. The bacteria are then diluted and spread over a plate of agar containing an antibiotic. Thus, bacteria without a plasmid are killed, whereas plasmid-containing bacteria can survive and replicate to form visible colony plaques. Since each such colony arises from a single original plasmid-transformed cell, they are known as clones. Each agar plate may contain many thousands of such clones, each clone having many cells with plasmids containing the identical cDNA insert.
The problem is to identify which of the many colonies has a particular cDNA that encodes for the channel protein of interest, among all of the cloned cDNAs that encode the myriad proteins of the original tissue. The severity of this problem can be appreciated from the fact that even highly enriched tissues such as eel electric organ may have at most only one message out of 10 000 or so that encodes a Na+ channel. Hence, there must be a rapid method for screening the colonies to identify the channel cDNA clones. This is where information from biochemical purification proves to be vital.
Despite the near impossibility of totally sequencing large Na+ or Ca2+ channel polypeptides, from a proteolytic digest one can readily purify a homogeneous preparation of a particular fragment. This fragment may then be partially sequenced using standard Edman degradation techniques. In practice, one only needs a few (perhaps six or so) residues of sequence to be reasonably sure that a unique segment of the protein is encoded. From this short amino acid segment, one can then synthesize (by automated means) a short DNA fragment, called an oligonucleotide, that encodes the segment. Further, this synthetic DNA may be radiolabeled and thus becomes a binding probe to identify cDNA inserts. Identification is done by placing a piece of special paper briefly on top of the agar plate and lifting off a few bacteria from each colony plaque onto the paper. The paper is then placed in a special solution that lyses the bacteria but binds the released DNA immediately on the surface. The radiolabeled synthetic probe may then be washed over the paper; it will bind by complementary interactions to the antisense strand of clonal plasmids encoding the channel protein containing the original sequence. After unbound probe is washed away, the paper is overlaid in the dark with x-ray film. Channel clones are thus identified as dark spots (usually only a few) on the film and may be traced back to the plaque on the original agar plate. This plaque is then lifted from the plate with a toothpick and grown in a nutrient broth to produce many bacteria. This technique provides enough cells from which the plasmids are purified by simple chemical means and the cDNA insert may be readily sequenced. Once the cDNA sequence is known, the genetic code is applied to derive the primary amino acid sequence of the channel protein.
This description of clonal sequencing omits many details. For example, the length of DNA that one may clone or sequence at one time is limited and complex strategies are often required to sequence the entire length of long channel polypeptides. In addition, ion channels that are heteromeric complexes will require that each subunit polypeptide be separately sequenced. Despite these apparent complexities, an organized group of researchers using automated equipment can screen hundreds of thousands of clones and sequence thousands of bases of interesting cDNA inserts in a relatively short time.
IVB Analysis of the Primary Structure of Large Polypeptide Voltage-Gated Channels
The electroplax Na+ channel was the first voltage-gated channel to be sequenced (Noda et al., 1984). As suggested by the biochemistry, it was found to be a long polypeptide of 1820 amino acids (Fig. 21.3). Subsequent cloning of mammalian channels showed them to be similar in size and sequence (see Numa and Noda, 1986). What can one learn about channel structure from such primary sequence data? Typically, one subjects the primary sequence data to several forms of analysis to derive a model of the appearance of the higher order structure. The first striking characteristic revealed in the Na+ channel sequence was the presence of four repeats (of about 200 amino acids each) that were highly (but not completely) homologous to one another. Furthermore, these internal repeats were found to have great potential for forming membrane-spanning domains of the channel. From such analyses, the model shown later in Fig. 21.6 was obtained.

FIGURE 21.3 Amino acid sequences of three Na+ channels obtained by cDNA cloning techniques. Rat I and II were obtained from rat brain cDNA libraries, while the eel Na+ channel is that expressed in the electric organ of Electrophorus electricus. Boxed residues show homology among the three channels, while dashed lines designate absent segments. Labeled brackets underneath the sequences refer to parts of the highly conserved putative transmembrane repeats (see text and Figs 21.6 and 21.7 for details). (From Numa and Noda, 1986.)
How are such analyses performed? Basically, the analyses occur in two or three separate steps. First, one analyzes the sequence of the peptide for regions of high hydrophobicity, on the expectation that membrane-spanning regions should have a high proportion of hydrophobic amino acids. To do this, hydropathy values that reflect its relative hydrophobicity are assigned to each amino acid residue. The assignment of such values is based largely on indirect physical measurements of the hydrophobicity, such as the ability of a given residue to partition from an aqueous solution to an oil phase. Such values may or may not reflect the chemical behavior of a residue in a long polypeptide chain, where chemical properties may be further influenced by neighboring residues. As a result, a number of such hydropathy tables have been developed by various investigators who have significant differences in the way they assign hydropathy weights to a given residue. One of the most popular tables is that of Kyte and Doolittle (1982) shown in Fig. 21.4. In most cases, the assignments are qualitatively obvious, especially for expected hydrophobic groups (e.g. the hydrocarbon chains or leucine or valine) or hydrophilic groups (e.g. the charged residues such as glutamate or lysine).
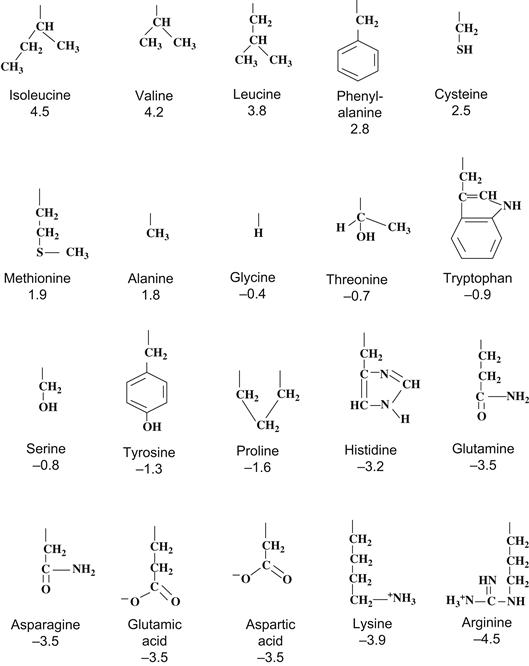
FIGURE 21.4 Structure and hydropathy values of amino acid residues. Arranged from most hydrophobic to most hydrophilic according to relative values assigned by Kyte and Doolittle (1982). Only side groups are shown with hydropathy values underneath.
Figure 21.5 shows hydropathy plots for the Na+ channel sequences given in Fig. 21.3. The plot is obtained by averaging the hydropathy values of a fixed segment (perhaps 7–15 residues). Starting with the first residues at the N-terminal end, this average is computed and the value is plotted versus the number of the residue in the middle of the segment. This averaging “frame” is then shifted by one residue toward the carboxyl end, the new hydropathy average is recomputed (i.e. the change due to the loss of the N-terminal side residue and the gain of the next C-terminal side residue in the sequence) and the value is plotted by incrementing the residue number by one. This frame-shift average is thus continued until the end of the protein is reached. The purpose of using the averaging frame is to smooth out sharp variations in hydropathy, so that a pattern may be more readily seen.
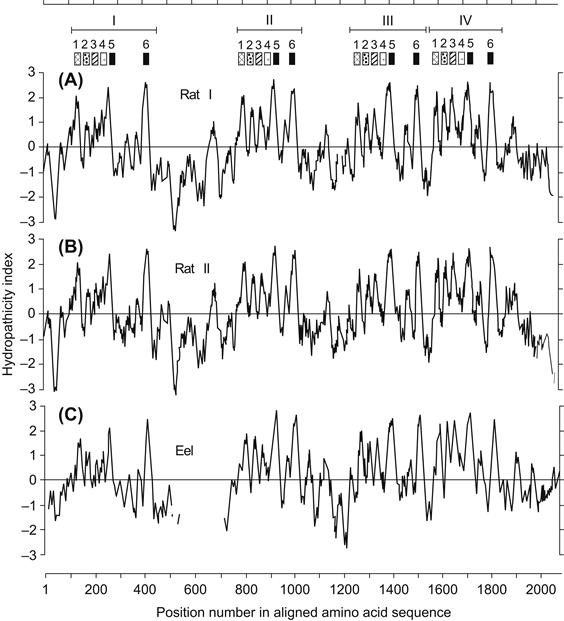
FIGURE 21.5 Hydropathy plots for the three Na+ channel sequences of Fig. 21.3. Brackets and numbered patterns at top refer to the locations of homologous repeats and transmembrane segments (see text and Fig. 21.7). (From Numa and Noda, 1986.)
As can be seen in Fig. 21.5, there is a repeated pattern of hydropathy for Na+ channels that reflects the conserved homology of the four internal repeat domains I–IV. Within each domain, there are five or six peaks of hydrophobicity that are each about 20 amino acids long. This is significant because such a length is considered optimal to span the hydrophobic interior of the lipid bilayer. However, hydrophobicity alone is not sufficient to predict a membrane-spanning region; instead, the residues must assume a favorable secondary structure, most commonly assumed to be an α-helix. In the helical conformation, the hydrophobic side chains project radially away from the axis of the helix and presumably into the surrounding lipid. Such a configuration prevents the very hydrophilic peptide backbone of the protein from exposure to the hydrophobic lipid environment and thus represents a favorable low free energy arrangement of the protein–lipid interface.
How are such secondary structures predicted from primary sequence? This too is a highly empirical process (Chou and Fasman, 1978). Basically, investigators have analyzed in detail a number of proteins for which there are high-resolution x-ray structures available at the amino acid level. By cataloging the frequencies with which given amino acids or short combinations of residues are found in helices, β-sheets, or turns, a set of empirical rules has been developed to predict the secondary structures that a given sequence of amino acids will most probably form (see Fig. 21.5). Although the general accuracy of such predictions has been debated, when combined with hydropathy information the method is apparently rather accurate for the prediction of membrane-spanning domains in membrane proteins. This is probably because of the severe thermodynamic constraints confronting polypeptides needing to cross a boundary of high hydrophobicity while interfacing with a highly hydrophilic aqueous phase on either side of the membrane and in the aqueous channel pore.
IVC How Is Channel Structure Formed?
In any case, such primary sequence analyses suggest that each internal domain has six α-helical membrane-spanning segments (see Numa and Noda, 1986). The question to consider next is how such an arrangement might form a transmembrane pore for the passage of ions. The answer is that the four transmembrane regions probably contribute equally to the pore walls in a “staves of a barrel” configuration (Fig. 21.6; see Guy and Seetharamulu, 1986). In fact, such a model was comforting to the original investigators, because a similar arrangement had previously been shown to exist among the separate subunits of the synaptic acetylcholine-gated channel, the first ion channel to be purified and sequenced via cDNA cloning.
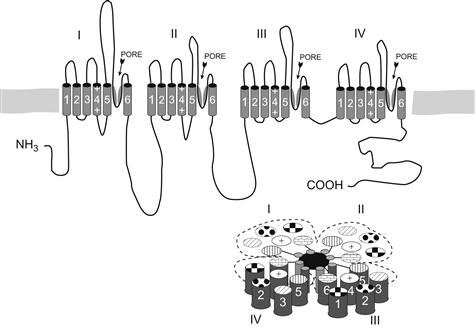
FIGURE 21.6 Model of Na+ channel structure based on analysis of primary sequence data. The top drawing shows linear placement of four homologous domains, loops and termini (outer surface of membrane is at the top). Possible pore and selectivity regions are labeled and shown as the angled segments between transmembrane segments 5 and 6. The drawing on the bottom right shows a hypothetical arrangement of homology repeats in the “staves of a barrel” configuration to form a pore (placement of transmembrane segments 1–6 is somewhat arbitrary).
IVD Sequence Homology among the Transmembrane Domains of Ion Channels
Analysis of the DHP-sensitive Ca2+ channel α-1 peptide revealed that it also has four internal homologous repeats (Fig. 21.7; see Tanabe et al., 1987). Furthermore, these repeats (but not the intervening loops) were found to be highly homologous to the Na+ channel repeats. The “staves of a barrel” configuration in which either homologous internal repeats (for Na+ and L-type Ca2+ channels) or highly homologous subunits (as in the case of transmitter-gated and K+ channel—see Section IVG) form a pore seems to be a widespread architecture among ion channels in general.
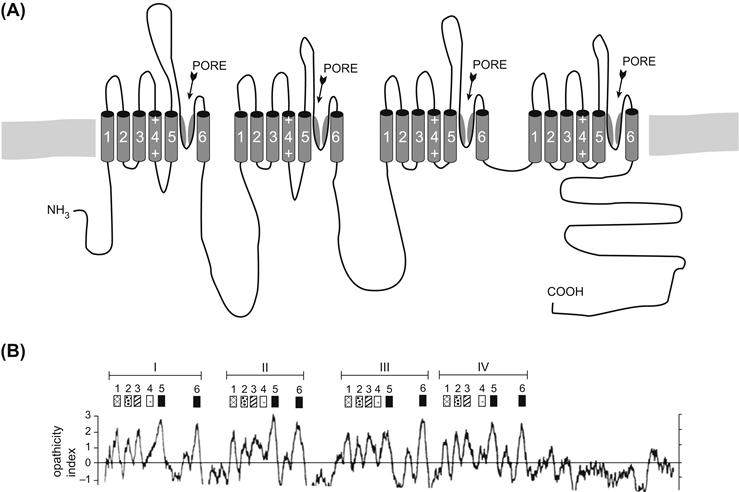
FIGURE 21.7 Model and hydropathy plot (bottom) of L-type skeletal muscle Ca2+ channel α-1 subunit. Compare to Figs. 21.5 and 21.6.
IVE How Are Topographical Predictions for Channel Structure Tested?
The modeling approach based on primary structure is highly uncertain and needs to be empirically confirmed for each channel. One of the most popular ways this has been done is through the use of site-directed antibodies raised to synthetic peptides encoding a short stretch (i.e. 10–20 residues) of the channel of which one wishes to know the topographical orientation. These antibodies may be applied to channels in their native membranes or intact cells and the relative sidedness of the epitope may be determined from the side of the membrane to which the antibody is bound. The approach has been used for Na+ channels in several laboratories and the topographical model shown in Fig. 21.6 has largely been confirmed (see Catterall, 1992). Similar approaches can be used in which impermeant group-reactive chemical reagents are applied to either intact cells or liposomes with oriented channels, followed by some method to identify the site of interaction on the protein (e.g. sequencing of proteolytic fragments). Alternatively, a synthetic, but highly antigenic, short epitope may be recombinantly inserted into the channel sequence and the mutant expressed in cultured cells. In this case, the same antibody may be used in all experiments to determine on which side of the membrane the epitope tag is located as a function of its position in the native channel sequence.
IVF The Genetic Approach Used to Identify Nucleic Acid Clones Coding for K+ Channels
K+ channels are extremely important in excitation and transduction phenomena. Partly because of the way these channels were cloned and partly because their structure is a simplified version of the long Na+ and Ca2+ channels, they are very valuable systems with which to study the general structures and mechanisms of the voltage-gated ion channel class. Such channels were not originally cloned via the usual biochemical purification–oligonucleotide probe approach described previously because there were few good chemical labels like tetrodotoxin or DHP.
A highly creative, yet complex, approach was taken independently by several laboratories that used behavioral mutants of the fruit fly Drosophila (e.g. see Papazian et al., 1987). In brief, mutant strains that had uncontrolled shaking when exposed to ether were identified. Electrophysiology then established that the muscles and certain neurons of such flies had missing or altered K+ currents of a specific type known as A-type currents that, like sodium currents, inactivate on depolarization. The locus of the genetic defect causing this behavior could be seen as an actual physical defect (translocation) in the polytene salivary chromosomes of these so-called Shaker mutants. Since extensive libraries of cloned identified chromosomal DNA fragments of Drosophila have long been available (called genomic clones), the investigators were able to obtain the DNA fragment of normal flies on which the mutant defect occurred. This allowed them to sequence such fragments and, by comparison with Shaker mutant sequences and by the identification of homologous cDNA fragments (derived as described earlier), the sequence of the A-type K+ channel could be inferred. Since the Shaker mutation allowed a completely genetic elucidation of the channel primary amino acid sequence, the A-current it manifests has become known as the Shaker-type K+ channel.
IVG K+ Channels are Homologous to a Single Internal Repeat of Na+ Channels
An initially surprising finding when the Shaker A-type channel sequence was analyzed was that the length of the sequence was much shorter than that of the other voltage-gated channels, but contained a single domain that was reminiscent of (but strictly speaking not very homologous to) the internal repeat domains of the Na+ or Ca2+ channels (Fig. 21.8; see Pongs, 1992). An obvious hypothesis for channel structure for the Shaker channel was that four molecules of this sequence (i.e. subunits) combined to form the “staves of a barrel” structure postulated for the long-channel polypeptides. This tetrameric assembly model has recently received strong support from several elegant experiments (MacKinnon, 1991).
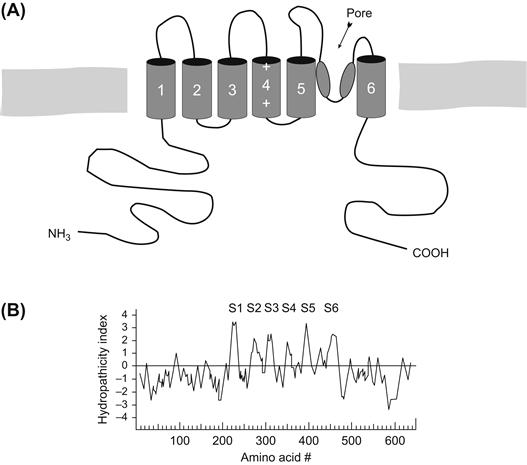
FIGURE 21.8 (A) Model and (B) hydropathy plot of Shaker B-type K+ channel. Compare to Figs. 21.5, 21.6 and 21.7.
V Molecular Mechanisms of Channel Function: How Does One Investigate Them?
We now discuss the methods that can be used to determine which parts of an elucidated channel sequence are involved in the basic channel properties of pore formation, ion selectivity and voltage-dependent gating. Here, too, the methods of recombinant DNA technology have proved to be of great use in that they allow the investigator to change the nucleic acid sequence encoding a channel in the manner desired. Using such mutagenesis techniques, the basic idea is to alter the amino acid sequence via deletion, addition, or change in the primary sequence in specific locations and then to test how such changes affect the function of the mutant channels. The hope is that by obtaining many such structure–function correlations, one might not only infer the part of the sequence that underlies, for example, channel activation, but also infer how this structure works as a mechanism.
VA Choosing Interesting Sites and Segments as Targets for Mutagenesis
How does one decide which of the many amino acids in a channel polypeptide to change? This is where the modeling approach described previously is so important, for it provides clues or testable hypotheses that focus mutagenesis attempts on reasonable guesses. For example, one might assume that a highly conserved sequence in channels from phylogenetically distant animals might reflect a functionally essential part of the molecule that cannot withstand evolutionary alteration without being deleterious. Another strategy might be to compare the regions of divergence in the sequence of the various voltage-gated channels in the hope of finding candidate regions involved in different functions among channels. For example, the difference in ion selectivity among Na+, K+ and Ca2+ channels must lie somewhere in the variations in their amino acid sequence. Both approaches have had a certain degree of success, although “homology” is a relative term and, in reality, large areas of channels are highly variable, even among isotypes of channels from the same organism (see Section VI). This makes it difficult to guess which of these many differences relates to specific functions.
Another approach is to take advantage of certain genetic disorders in which mutations in channel structure occur naturally with functional consequences. A number of human diseases and animal models have been shown to be caused by channel defects. The sequence of such channels can give important clues into structure–function relationships in channels, while directing artificial mutagenesis experiments.
VB Use of Expression Systems in Mutagenesis Studies
Another essential part of mutagenesis experimentation is the ability to introduce artificially altered genetic material into a cell that is capable of translating it into a protein molecule, processing the molecule appropriately (i.e. folding the polypeptide, assembling subunits, adding sugars or lipids) and inserting the protein into the cell surface. The protein then may be functionally characterized using the biophysical methods described in other chapters. The most popular such expression system is the oocyte of the South African frog Xenopus laevis (Fig. 21.9; Leonard and Snutch, 1991). In this system, one prepares mRNA from clones of the mutant channel and injects this message into the large (1–2 mm in diameter) oocytes with a micropipette. After waiting a few days for channel synthesis and membrane insertion to occur in sufficient numbers, one observes the expressed ionic currents via voltage-clamp methods, using large microelectrodes to impale the cell. Alternatively, one may also use a very large diameter electrode to patch-clamp a large area of membrane; such macropatch methods give better voltage control than the impaled electrode technique while allowing macroscopic current characterization. Finally, conventional single-channel analysis may be done in all the usual modes using standard patch pipettes. Thus, the oocyte allows the investigator to compare both the macroscopic and single-channel properties of the mutants with the normal wild-type channel.
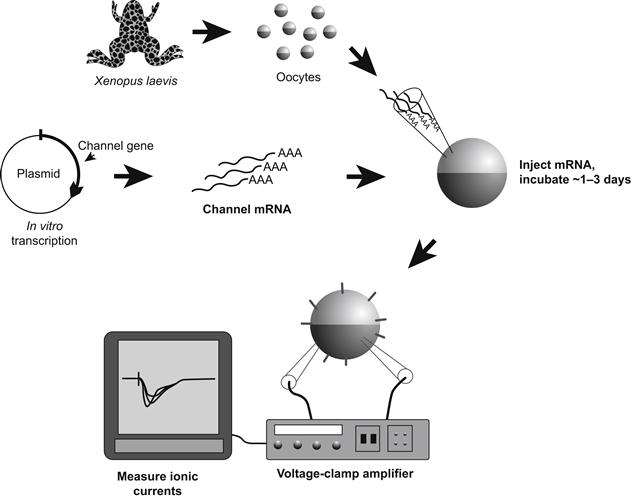
FIGURE 21.9 Xenopus oocyte expression system. For details, see text.
Although other expression systems in which the altered genetic material may be introduced into cells transiently via viruses or integrated permanently into the host genome (stable transfection) have been developed more recently, the use of such systems is beyond the scope of this chapter. Suffice it to say that the frog oocyte is currently the most popular and productive expression system with which to study structure–function relationships.
Because of its great potential in elucidating channel mechanisms, mutagenesis is being used by a large number of investigators. As a result, both new and altered concepts of channel function appear frequently in the scientific literature. In the brief account that follows, we can only summarize some of the fairly firm basics of gating, pore formation and ion selectivity.
VC Domains Involved in Voltage-Dependent Activation
The ability to respond to changes in transmembrane potential is a hallmark of voltage-gated channels. Thus investigators looked for common motifs in channel sequences suggestive of voltage sensor domains that might target mutagenesis among the large number of residues in cloned voltage-gated channels. In comparing Na+, DHP-sensitive Ca2+ and Shaker-type K+ channels, the fourth predicted helix in the repeat/subunit motif was seen to display a highly conserved sequence pattern (see Figs. 21.6, 21.7 and 21.8). This segment, known as S4, consists of a repeated triad of a positively charged residue (Arg or Lys), followed by two highly hydrophobic residues (such as Val, Leu, Ile; Fig. 21.10). While the number of such triads in the various S4 segments ranges from three to eight, its structure of both charged and hydrophobic residues suggests an element capable of responding to transmembrane voltage changes (see Catterall, 1992). Accordingly, a number of experiments have now been done in which both the charged and hydrophobic elements have been changed.
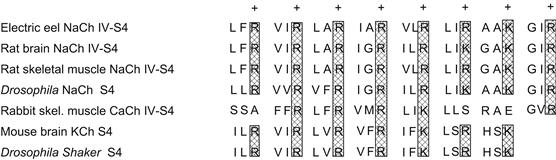
FIGURE 21.10 Comparison of S4 segments from different voltage-gated channels. Note the repeating triadic motif of positive charge (R, Arg; K, Lys) followed by two hydrophobic residues. Hatched areas show conservation of charge among channels; all amino acids designated by single letter codes.
In brief, S4 mutagenesis usually causes significant changes in the steady-state activation behavior of the affected channel (Fig. 21.11; Papazian et al., 1991; Lopez et al., 1991). Thus, shifts of the steady-state activation curve along the voltage axis occur with most changes to either charged or hydrophobic residues. In addition, changes in activation slope are frequently seen.
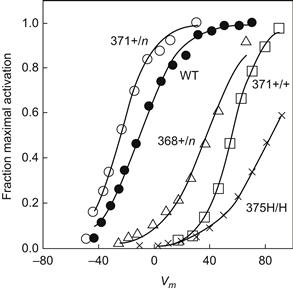
FIGURE 21.11 Effect of S4 mutations on Shaker K+ channel activation expressed in oocytes. Shown are steady-state activation curves for wild-type (WT) and various single amino acid changes. +/n, change from a positively charged residue to a neutral one; +/+, a change of positively charged residue for an other charged residue; H/H, replace one hydrophobic residue with another hydrophobic residue. (Data redrawn from Papazian et al., 1991 and Lopez et al., 1991.)
The question is whether the observed changes can be explained in terms of a mechanism by which S4 participates in gating (instead of the general hypothesis that S4 is some part of a gating structure). Unfortunately, to date the results obtained from mutagenesis do not discriminate very well among the various possible roles for S4 in gating, for example, as a gating sensor, a part of the transduction linkage to a gating element, or simply an unrelated part of channel structure. Thus, the magnitude or direction of changes in activation behavior does not systematically correlate with changes to S4 charge or sequence. In addition, others have questioned whether the charged groups in the S4 segments can completely account for all the gating charge that moves when channels activate (see Sigworth, 1995). In general, the mutagenesis approach is inherently limited in its ability to distinguish between alterations that directly affect a mechanistically important part of the molecule and those that indirectly cause functional effects through structural changes that propagate from mechanistically unrelated domains. This problem arises because protein structures are notoriously sensitive to long-range effects of such alterations, generically known as allosteric (“other site”) effects.
VD Limitations of Mutagenesis in the Study of Channel Mechanisms
The naive reader can understand the problem from a crude analogy in which many of us as children tried to understand the workings of a mechanical watch by a similar approach using simple tools. The child understands the function of the mechanism only by the movement of the hands. However, as we all know, a mechanical watch is quite a delicate machine. Thus, even the act of carelessly opening the case may cause the mechanism to cease working. Are we to conclude that the case is therefore part of the mechanism? Given access to the mechanism itself, the situation is highly complex, since the movement of many parts is required for the ultimate movement of the hands. For example, the result of stopping the second hand by interfering with a given gear tells one very little of the role that gear plays in how the watch operates.
For some time it has been known that the interactions of chemical substances or structural alterations with a site on one side of a protein can dramatically affect the function of another site at some distance, for example, on the other side of the protein. This concern and the potentially complex nature of channel-gating mechanisms should motivate the responsible practitioner of mutagenesis to display caution in interpreting results in mechanistic terms.
How, then, in the face of such limitations does one make progress in elucidating the mechanisms of channel function? In fact, the approach is philosophically identical to the way one attempts to prove any hypothesis, namely, to establish a circumstantial case by as many independent tests as possible. In the example of S4 and mutagenesis, for example, one might wish to demonstrate that mutations to any other part of the molecule have no effect on voltage-dependent activation as a sufficient (but not necessary) test. Unfortunately, mutations to other parts of channel sequence can have equally profound effects as S4 alterations (for example, to the pore-forming region described previously; see Yool and Schwarz, 1991). Thus, while these other mutations do not disprove a role for S4 as a sensor (or any other role), they do suggest, as in the mechanical watch, that a number of segments of the sequence are either directly involved in a complex mechanism of voltage gating or indirectly affect a gating process through allosteric/structural influences.
VE The Mechanism of Channel Inactivation
Genetic engineering studies of the inactivation process currently provide a better example of what one may learn mechanistically from the mutagenesis approach. Some of the most important such studies have involved the Shaker A-type channel. Here early workers found important clues to the segment involved in the inactivation process by comparing naturally occurring variants of the channel (isotypes; see Section VI). Thus, Fig. 21.12 shows the pattern of four such isotypes, called A, B, C and D (Timpe et al., 1988). Shaker B, C and D were identified from further testing of cDNA clones using probes constructed from the original Shaker A sequence as described earlier. When expressed in oocytes, these isotypes displayed very different kinetics of inactivation. Analysis of the regions of homology and differences among the isotypes shown in Fig. 21.12 indicates that fast inactivation properties correlate most strongly with the nature of the N-terminal segment (thought to be cytoplasmic; see Fig. 21.8).
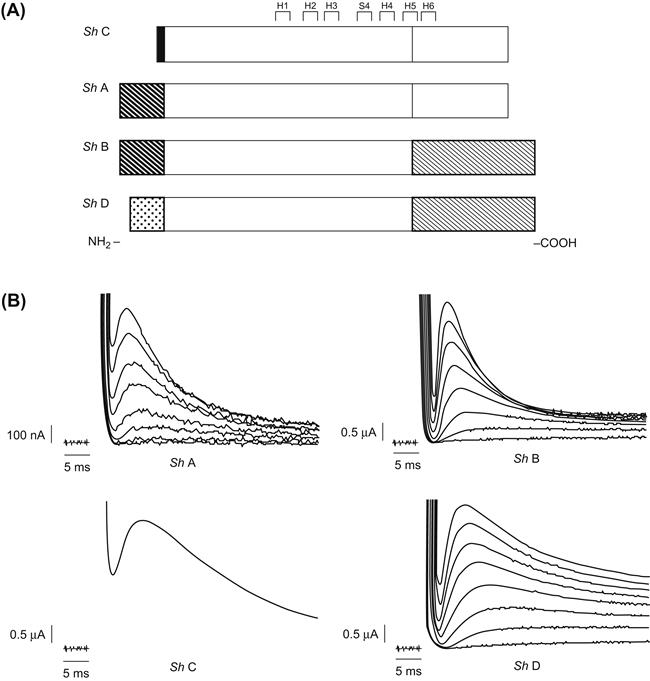
FIGURE 21.12 Gating kinetics of alternatively spliced Shaker isoforms expressed in oocytes. (A) Comparison of sequences, with patterned regions representing alternatively spliced domains that are different among isoforms (other unpatterned regions are identical in sequence). (B) Both current traces obtained from oocyte voltage-clamp show currents resulting from each isoform. (From Timpe et al., 1988. Copyright 1988 by Cell Press.)
Further evidence for the role of this segment in inactivation was obtained by constructing channel chimeras in which the Shaker A clone was modified by replacing its N-terminal segment with those from other isotypes. In each case, the mutant channel assumed the inactivation properties of the isotype donating the N-terminal segment, despite the fact that the rest of the molecule had the sequence of the fast-inactivating Shaker isotype (Aldrich et al., 1990).
Finally, investigators mutagenized specific regions of the fast-inactivating Shaker B segment to localize those residues involved in the inactivation process (Hoshi et al., 1990). Both by deleting small segments and by replacing others it was found that the crucial part of the molecule was a stretch of residues from the positions 6–83 (i.e. the N-terminal tail of the molecule seen in Fig. 21.8). In this segment there were distant effects of the alterations. Thus, mutations to the residues in the 23–83 residue region tended to produce changes in inactivation kinetics, whereas changes to those in the N-terminal side (6–22) of the segment tended to destroy inactivation completely (see Fig. 21.14).
What might be the mechanism of Shaker inactivation? Much earlier, Armstrong and coworkers had reported that internal perfusion of proteases in the squid destroyed the ability of Na+ channels to inactivate. On the basis of this and other experiments, Armstrong proposed that the inactivation mechanism is a cytoplasmic ball attached to a peptide tether that swings into an open channel to block it from the inside (Fig. 21.13; Armstrong and Bezanilla, 1977). In this hypothesis, the ball blocks the channel by binding to a receptor in the inside of the pore that is accessible only when the channel has opened (activated). The reader may recall the evidence that inactivation is coupled to activation and hence may be intrinsically voltage independent. Thus, the ball-and-chain model also explains these properties since the cytoplasmic ball would experience little of the transmembrane field but could swing into the channel only after it had activated and revealed its receptor (hence the coupling properties).
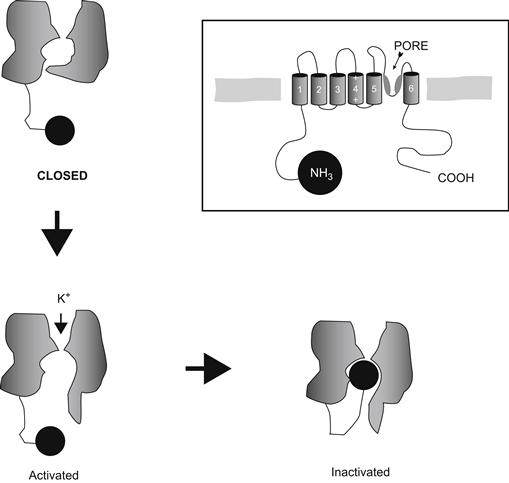
FIGURE 21.13 Ball-and-chain model of inactivation. Not shown are the three other balls that the other subunits of the channel will contribute.
In the case of the Shaker channel, Aldrich and colleagues proposed that the N-terminal segment that they identified was just such a mechanism, since changes to the length of the 23–83 segment affected kinetics (as might be expected from changing the length of the chain), while changes to the N-terminal segment (such as alterations of charge) that eliminated inactivation were postulated to prevent the ball from binding to its receptor in the activated channel.
How might such a model be further tested? Here the investigators performed a particularly elegant experiment (Fig. 21.14; Zagotta et al., 1990). First they constructed a mutant Shaker channel in which inactivation had been eliminated by a deletion of its own N-terminal segment. Next, a synthetic peptide that had the exact sequence of the deleted region was prepared. The mutant channel was then expressed in oocytes, where both excised macropatch and single-channel observations could be made. The investigators found that when the peptide was applied to the solution bathing the inside surface of the membrane, inactivation was restored to these mutant channels. Furthermore, the kinetics of the restored inactivation depended on the concentration of the peptide in the bath and the inactivation could be reversed by washing away the peptide-containing solution. These observations are thus consistent with a direct blocking interaction of the peptide with a site in the open channel and constitute strong evidence for the ball-and-chain mechanism of inactivation in A-type K+ channels.
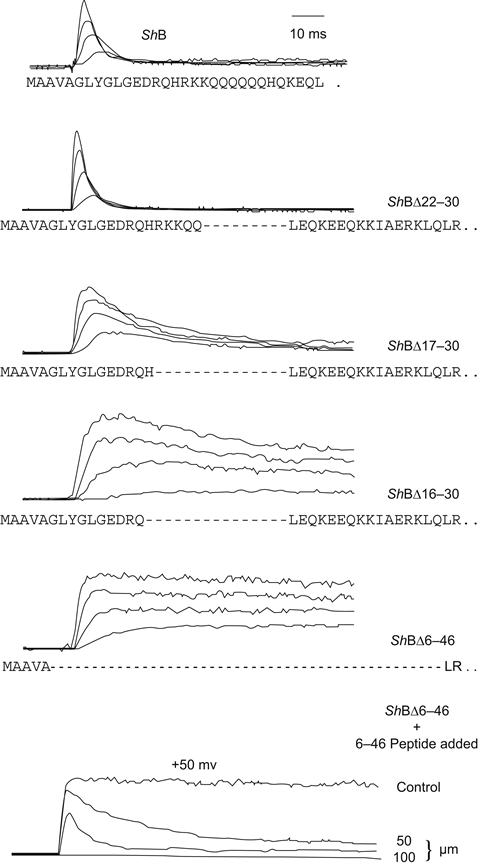
FIGURE 21.14 Identification of a putative ball and chain in Shaker B channels expressed in oocytes. Panels 2–5 from the top show the effect of deleting increasing lengths of a segment near the N terminus of the Shaker protein. Note that the deletion of residues 6–46 results in a non-inactivating K+ current. The bottom panel shows that inactivation properties of this latter mutant may be restored by application of the synthetic 6–46 peptide to the inner channel surface in a dose-dependent fashion. (Reorganized from Hoshi et al., 1990, and Zagotta et al., 1990.)
VF Other Gating-Related Domains in Voltage-Sensitive Channels
Analysis of the homology of the various cloned Na+ channels revealed that the longest segment of conservation lay in the postulated internal loop between repeats III and IV, even among animals of some phylogenetic distance (see Fig. 21.3). This high degree of conservation suggested a functionally important role for the segment. This idea was initially tested by constructing mutant Na+ channels in which the channel was expressed in oocytes in two pieces, with the genetic “cut” within this segment (Stuhmer et al., 1989). Interestingly, such artificial dimeric channels were assembled and expressed in the oocyte and displayed relatively normal activation and conductance; however, such channels did not inactivate. Further, site-directed antibodies raised to a synthetic peptide encoding this segment were found to slow or eliminate inactivation when they were applied to the cytoplasmic surface of cells expressing normally inactivating channels (Vassilev et al., 1989). Finally, mutagenesis directed to this segment produced channels with altered inactivation (Catterall, 1992).
However, the sequence of this segment has little resemblance to the N-terminal ball-and-chain Shaker domain and it is additionally tethered at both ends. Nonetheless, several lines of evidence now suggest that this domain operates analogously to the Shaker ball-and-chain in producing Na+ channel inactivation. Most notably, via mutagenesis to the III–IV loop, one can produce “inactivationless” sodium channels; in such mutants, inactivation can be restored with the internal application of the same synthetic Shaker peptide that restored inactivation to non-inactivating potassium channels in the experiments described above.
For DHP-sensitive Ca2+ channels, an isotype of the originally cloned skeletal muscle channel has been cloned from the heart. In the parent tissues, these channels share some characteristics, but differ greatly in the speed by which their calcium currents activate (heart channel currents being much more rapid in responding to voltage). Although the isotypes share considerable homology, there is still enough difference in the sequences to make it difficult to guess which residues might be responsible for activation kinetics. This problem was elegantly addressed by Beam, Tanabe and coworkers, who made chimeras of the two channels (Fig. 21.15; Tanabe et al., 1991). To focus the mutagenesis, it was reasoned that only the predicted transmembrane repeats should be involved in voltage-gated activation. Thus, the chimeras swapped the homologous repeats among constructs. As shown in Fig. 21.15, the kinetics of activation correlate almost completely with the kinetics of the donor of the first (I) repeat. This finding suggests that despite their approximate homology, the repeats in a given channel contribute differently to gating properties. This is perhaps expected from the observation that the S4 regions among these domains in different channels have somewhat different structures and numbers of charges (e.g. see Fig. 21.10). Having identified the domain responsible, these investigators have been able to narrow their search to shorter segments and, ultimately, will identify the residues involved in this activation phenomenon (Nakai et al., 1994).
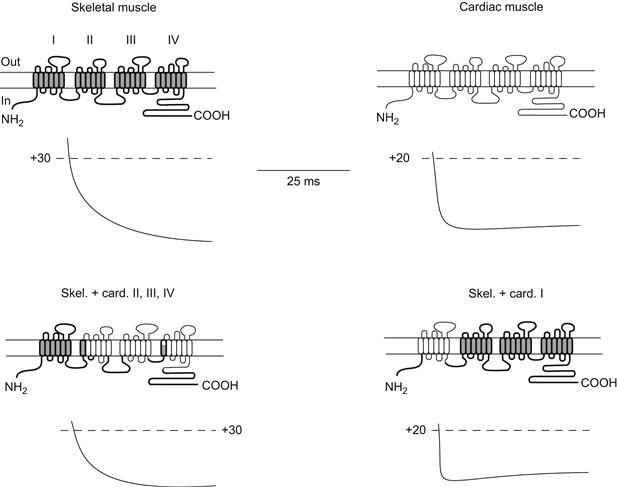
FIGURE 21.15 Identification of regions determining activation kinetics in L-type Ca2+ channels from skeletal muscle and heart. In these experiments, hybrids (chimeras) of skeletal muscle and cardiac channels were made in various combinations to localize the domains responsible for the differences in Ca2+ current activation kinetics in heart and skeletal muscle. cDNA encoding each construct was injected in cultured muscle cells (myotubes) from mutant mice unable to synthesize active L-type channels. Thus, these mutant cells would express only currents resulting from the genetically engineered DNA introduced into them. The top two panels show the current kinetics resulting from DNA encoding the wild-type skeletal muscle and cardiac channels. The bottom panels show two chimeras in which current kinetics correlate with the donor of repeat domain I. Most of the other possible combinations of channels were also made, with the same correlation of kinetics with domain I. (Redrawn from Tanabe et al., 1991.)
VG Pore Formation and Ion Selectivity
Pores are the defining structures of all ion channels. As such, they provide both an aqueous pathway for ions to traverse the membrane as well as a means for discriminating among ions for access to this path. Studies of the mechanisms of pores provide an especially vivid example of the integration of theoretical modeling, mutagenesis and molecular structural approaches.
Even before the advent of structural studies, Hille hypothesized that ion selectivity and pore formation were performed by the same parts of the channel structure. Experimental evidence also indicated that charged or polar oxygen groups lined the narrowest region of the pore, where cation recognition occurs (see Hille, 1992). This latter finding was a sensible one considering the chemistry of cations in solution. In aqueous solution, metal cations such K+, Na+ and Ca2+ are stabilized by electrostatic interaction with the electronegative oxygen atoms of water molecules. In the narrowest region of the pore, water molecules must be removed in order for the channel to recognize the preferred cation; lining this narrow region with oxygen-bearing groups provides a chemically favorable selectivity filter.
Based on this early work, investigators sought short protein segments that might line the aqueous pore with oxygen atoms. A candidate sequence was initially identified using sophisticated analysis of primary sequence information (Guy and Seetharamulu, 1986). This sequence is 20–25 amino acids long and forms part of the loop connecting the fifth and sixth predicted transmembrane helices in the voltage-gated cation channels (see Figs. 21.6, 21.7 and 21.8). Tests of this putative pore-lining loop’s (P-loop) role in pore formation were soon forthcoming.
One type of test was to determine whether site-directed mutations in the P-loop sequence affected the action of known pore-blocking agents on heterologously expressed mutant channels. Among the agents tested in this way were the K+ channel-blockers tetraethylammonium and charybdotoxin and the Na+ channel-blocker tetrodotoxin (MacKinnon and Yellen, 1990; MacKinnon et al., 1990; Terlau et al., 1991). Block by the externally effective agents tetrodotoxin or charybdotoxin was specifically affected by mutations at the extracellular end of the P-loop. Tetraethylammonium block of K+ channels, which occurs by blocker binding at either end of the narrow pore, was altered by mutations at both ends of the P-loop. Results such as these confirmed that the P-loop does indeed form at least part of the pore in voltage-gated ion channels. Does the P-loop also form the ion selectivity filter?
The answer to this question also comes from site-directed mutagenesis studies. In Shaker-type K+ channels, point mutations introduced into the P-loop profoundly altered the channel’s preference for K+ over Rb+ or NH4+ as shown in Fig. 21.16 (Yool and Schwarz, 1991). However, non-conducting channels were produced whenever mutations were introduced into one stretch of eight amino acids in K+ channel P-loops. This so-called K+ channel signature sequence is highly conserved in K+ channels found in bacteria to humans and it was therefore suspected of forming the narrow selectivity filter in K+ channels.
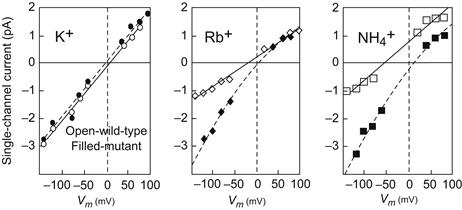
FIGURE 21.16 Alteration of Shaker B K+ channel ion selectivity caused by a mutation in the putative pore-forming region. In these experiments, Phe 433 was mutated to Ser. The oocyte system used to express both wild-type and mutant channels is described in the text. Single-channel currents were measured using patch-clamp methods in which the ion indicated in each panel was applied to the external surface of the channel with K+ on the inside in all cases. In these bi-ionic conditions, inward (negative) currents will be carried mainly by the test ion, while outward (positive) currents will be mostly K+ ion fluxes. As can be seen from the increased inward currents, permeability of the channel to Rb and NH4 ions was greatly enhanced in the mutants, while K+ was unaffected. (Data redrawn from Yool and Schwarz, 1991.)
Site-directed mutations in the P-loops of Na+ channels and Ca2+ channels also affect ion selectivity (Heinemann et al., 1992; Yang et al., 1993). In Na+ channels, a locus of four residues has been identified that confers selectivity for Na+ over Ca2+ (Fig. 21.17). Each of the four P-loops of a Na+ channel contributes one residue to the locus, which includes two carboxylate-bearing residues (Asp, Glu) and two non-carboxylate residues (Lys, Ala). Substitution of Glu residues for the Lys and Ala residues dramatically reduces Na+ selectivity and allows significant amounts of Ca2+ to permeate this mutant Na+ channel. The analogous locus in Ca2+ channels is comprised of four Glu residues. Functional analysis of the effects of mutations in this locus has shown that these four Glu residues form the core of the selectivity filter in Ca2+ channels (Ellinor et al., 1995).
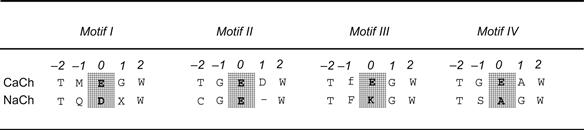
FIGURE 21.17 Alignment of Ca2+ and Na+ channel pore sequences. Position 0 in the alignments (shaded) marks the critical residues in the selectivity filter (EEEE locus in Ca2+ channels; DEKA locus in Na+ channels). High-voltage–activated calcium channels are identical to one another in these pore sequences, except at the ñ1 position in motif III: the “f” indicates that this position is occupied by either Phe (F) or Gly (G). In sodium channels, the residue at the +1 position in motif I (x) varies depending on channel isoform. Na+ channel sensitivity to tetrodotoxin depends partly on the residue present at this position.
As described above, oxygen atoms have long been thought to line the selectivity filter in voltage-gated, cation-selective channels. The presence of multiple carboxylate groups in the selectivity filters of Ca2+ and Na+ channels is consonant with the earlier work although, in principle, the carboxylates may either project into the pore or away from it. In fact, Ca2+ channel carboxylate groups apparently project into the aqueous pore and thereby provide the electronegative oxygen atoms needed to stabilize Ca2+ there. Evidence supporting this picture of selectivity filter structure derives from the fact that protonation of these carboxylate groups blocks current through Ca2+ channels; mutational replacement of the carboxylates by non-protonatable groups predictably alters proton block. Orientation of selectivity filter side chains has also been investigated using a technique called the “substituted cysteine accessibility method”. In this method, a sulfhydryl-containing Cys residue is substituted for the wild-type residue and a sulfhydryl-modifying agent is bath-applied to the mutant channel. If the side-chain of the substituted Cys projects into the pore, where it may be accessible to the sulfhydryl-modifying agent, then the modifying agent will attach itself to the Cys sulfhydryl and obstruct ion flow. Interruption of channel current thus reports that a particular side chain is oriented into the aqueous pore, whereas absence of block indicates that the side chain faces away from the pore. Application of this method to Na+ and Ca2+ channels indicates that the side chains in the carboxylate-containing loci of these channels all project into the pore (Chiamvimonvat et al., 1996). The extreme narrowness of K+ channel pores has prevented this technique from being effectively employed with these channels. However, the selectivity filter of K+ channels may be structurally distinct from that of Ca2+ channels and perhaps Na+ channels.
VH Crystal Structure of a Bacterial K+ Channel Pore Region
Results of these site-directed mutagenesis studies have been strikingly confirmed by the first x-ray crystallographic structure obtained for a member of the family of voltage-gated ion channels (Doyle et al., 1998). This structure was solved for a bacterial K+ channel, which though not in fact voltage-gated, is nevertheless a member of the family of voltage-gated ion channels: most significantly, it possesses the hallmark of all K+ selective channels, the K+ channel signature sequence. The crystal structure reveals that the selectivity filter is formed by part of the P-loop, with two K+ ions stabilized therein, probably by main-chain carbonyl oxygen atoms (Fig. 21.18). That uncharged carbonyl oxygens apparently line the selectivity filter in the bacterial K+ channel actually agrees with predictions, based on mutagenesis studies, for other K+ channels.
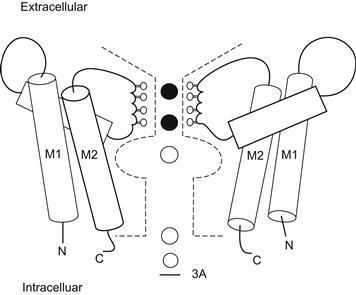
FIGURE 21.18 Cartoon illustration of the bacterial K+ channel structure. Only two of the four subunits of the channel are shown. Each subunit has a P-loop and two helices (M1, M2), similar to the structure of inwardly rectifying K+ channels. The segment of the P-loop that lines the ion permeation pathway projects carbonyl oxygen atoms into the pore. A pair of K+ ions is shown stabilized in the selectivity filter by the rings of carbonyl oxygens. (Adapted with permission from Choe, S. and Robinson, R. (1998). An ingenious filter: the structural basis for ion channel selectivity. Neuron. 20, 821–823. Copyright 1998 Cell Press.)
Why might K+ channels have evolved a selectivity structure that employs neutral, main-chain carbonyl oxygen atoms, whereas their evolutionary descendants, Ca2+ channels, apparently line their selectivity filter with negatively charged, side-chain carboxylate oxygens? Perhaps the negative charge of carboxylate oxygens is necessary in selecting for Ca2+, a charge-dense divalent cation, whereas uncharged carbonyl oxygen atoms are ideal in selecting for certain monovalent cations, such as K+. How then to account for Na+ channel selectivity structure? Na+ channels have evolved from Ca2+ channels and the Na+ channel selectivity filter apparently includes two side-chain carboxylate oxygens. K+ and Na+ have the same ionic charge but Na+ has a higher charge density owing to its smaller ionic radius. As in the case for Ca2+, the relatively high charge density of Na+ may require carboxylate oxygens for selectivity, though only two rather than the four present in the Ca2+ channel selectivity filter.
VI Isoforms of Voltage-Gated Channels as Part of a Large Superfamily
One of the most striking results of recombinant DNA analysis of channel clones has been the discovery of numerous channel isoforms expressed within the same organism and even within the same cell. Naturally, some degree of homology among channels of different organisms was expected and electrophysiological recordings showed that channels could be functionally diverse at the cellular level. Even a structural similarity among Na+, Ca2+ and K+ channels had been predicted by some investigators. However, the number and diversity of channels within a single organism revealed by molecular biology were rather unexpected. Channels of a functionally similar class (usually based on ion selectivity) are now called isotypes or isoforms by analogy to the phenomenon of isoenzymes.
We have previously discussed the Shaker isotypes. Shaker-derived sequences have also been used to construct probes that have discovered other families of K+ channels in flies, mammals and other species (Salkoff et al., 1992). These families are so extensive that they challenge investigators to develop appropriate nomenclature for their classification (Table 21.3; Chandy and Gutman, 1995). In any case, although they are all to some degree homologous with the original Shaker family, they have much higher degrees of sequence homology among members of their class. Functionally, they run the gamut from inactivating A-type channels to non-inactivating channels of the classic delayed rectifier sort. Interestingly, there is usually a much higher degree of sequence homology among channels of the same class from different animals than there is between channels of separate classes in the same animal. For example, Shaker channels from Drosophila are much more homologous to Shaker channels of mouse brain than they are to the Drosophila Shab-type channels.
TABLE 21.3. Properties of Major Potassium Channel Subfamilies in Fruit Flies and Mammals
| Drosophila Gene | Homologous Mammalian Genes (no.) | Properties |
| Shaker | Kv1.1–1.7 (7) | Extensively spliced in fly; inactivation fast to slow |
| Shab | Kv2.1, 2.2 (2) | Limited splicing in fly; intermediate inactivation kinetics |
| Shaw | Kv3.1–3.4 (4) | Limited splicing in fly and mammal; slow inactivation, delayed rectifier characteristics |
| Shal | Kv4.1–4.3 (3) | Limited splicing in fly; slow inactivation, delayed rectifier characteristics |
| eag | h-erg (3) | Limited splicing in fly and mammals; slow-activation delayed rectifier |
Recently, several other subfamilies of K+ channels have been discovered in mammalian and invertebrate genomes. Two of these, the erg and KQT subfamilies, are important in mammalian cardiac function. Mutants of these channels have been associated with serious inherited heart and nervous system disorders in humans, in particular the long QT syndrome discussed in other chapters in this book. In any case, in mammals the number of K+ channel isoforms is already quite large: currently there are 12 known potassium channel subfamilies totaling over 100 isoforms, with more being discovered at frequent intervals (see Coetzee et al., 1999).
Considerable diversity is also found among Na+ and Ca2+ channels (Agnew and Trimmer, 1989; Hofmann et al., 1994). For the main channel-forming α subunits (see Figs. 21.6 and 21.7), there are thought to be at least 10 Na+ channel and nine Ca2+ channel genes in the mammalian genome. Some of these isoforms are expressed very selectively in certain tissues, such as brain, or in only certain cells types or at very specific stages of development.
The term “isoform” is rather non-specific in that it does not convey a sense of relative relatedness among channels. An evolving system of classification refers to the set of voltage-gated channels as a superfamily, while the selectivity types (i.e. Na+, Ca2+ or K+ channels) form families. Because of the impressive degree of diversity found for K+ channels (see Table 21.3), investigators have formally proposed a uniform system of nomenclature in which the closely related isoforms such as Shaker are referred to as subfamilies. A separate nomenclature for isoforms of Ca2+ channel subunits has also been developed. Further consideration of channel classification will undoubtedly be useful in understanding the process of channel evolution.
VIA How Do Isoforms Arise?
In general, two separate mechanisms that create such diversity have been identified. First, there is the expected situation in which distinctly separate genes encode different channels in the same beast. Such is largely the situation for mammalian Na+ channel and Ca2+ channel isoforms. However, a second mechanism has been found, called alternative splicing, in which a given segment of a channel polypeptide may be encoded in several different stretches of genomic DNA known as exons. This is the case for Shaker in Drosophila; thus, in Fig. 21.12, the various Shaker isoforms are generated by mixing and matching the transcripts of different exons. During transcription, some sort of regulatory mechanism exists that decides which one of the alternative coding RNAs for a given segment will be incorporated in the final mRNA. The other undesired homologous segments are then excised from the transcript and the desired RNA segments are spliced together. Currently, there is evidence for the operation of both mechanisms in vertebrates and invertebrates. However, most mammalian systems studied to date appear to rely more heavily on separate genes than alternative splicing whereas, in the fruit fly, both mechanisms appear common.
Finally, for all multisubunit channels there is the possibility of diversification through various combinations of each set of subunit isotypes. The best-studied examples are the heteromeric formation of channels by different K+ channel subunit isotypes (Salkoff et al., 1992). For example, if the same cell synthesized Shaker A and Shaker B channel subunits, one might find heteromeric complexes of A and B types as well as the homomeric types. In fact, experiments with the oocyte expression system into which both types of mRNA have been injected show that such heteromeric assembly can occur, yielding functional channels with characteristics different from homomultimers of either channel type. Thus, it is theoretically possible that distinct channel properties may be generated by heteromeric assembly of K+ channel subunits. Whether this occurs in nature is currently being investigated.
On the other hand, oocyte experiments show that heteromeric association among subunits from different families (e.g. Shaker and Shal) does not occur. Apparently different families have unique structural domains that allow only homomeric assembly among members of their family. This discovery has led to a recent set of chimeric experiments in which domains among a fly Shaker channel and a distantly related mammalian delayed rectifier channel (drkl) were exchanged in an attempt to identify the segments specifying assembly specificity (Li et al., 1992). Such a segment was found on the N-terminal part of the peptide. Thus, substitution of the Shaker segment into the drkl cDNA produced a channel drkl subunit that was able to co-assemble with native Shaker.
VIB Why Are Channels So Diverse in a Given Organism?
One obvious reason for the presence of so many channel isotypes in the same animal is that each isotype has different functional properties that are appropriate to the function of its host cell. For example, high-frequency, repetitive firing requires short-duration action potentials that could be created by rapidly gating Na+ channels and fast-inactivating A-type K+ channels. In addition, isotypes may be differentially sensitive to intracellular modulators that allow cellular mechanisms to alter the properties of specific channels selectively in response to physiological effectors. On the other hand, many of the discovered isotypes have no significant functional differences among them. In this case, it is possible that it is not the differences in the channels themselves that are important, but rather the way the isotypes are expressed. Thus, in a number of cases it has been shown that expression of one isotype over another occurs developmentally or in response to a physiological stimulus (Ribera and Spitzer, 1992). Such specific expression is thought to be controlled by genetic elements outside the coding region for the channel, known as regulatory elements. These stretches of DNA respond specifically to one of many possible soluble factors by increasing (in the case of promoters and enhancers) or inhibiting (silencers) transcription of the channel gene (see Maue et al., 1990). Separate genes, then, potentially allow the cell to express selectively a given channel in response to a given stimulus or at a specific time during development. Finally, there is the possibility that a given isotype sequence encodes information used by the cellular machinery to localize or cluster it in very discrete locations, such as the nodes of Ranvier in myelinated nerve fibers (Dugandzija-Novakovic et al., 1995), neuronal cell bodies, or dendrites (Maletic-Savatic et al., 1994). The study of the diversity of channels and their biological purpose will continue to be a highly active area of cell biology for some time.
VII Future Directions
The current molecular biological approaches will no doubt be productive for some time. However, it is likely that better understanding of channel mechanism will require more direct and precise resolution of the higher order structure of channel molecules. The problems inherent in a crystallographic approach have been discussed. However, the problem is currently being solved by recombinantly synthesizing smaller segments of channel proteins that may be amenable to crystal formation. To be useful, these small segments must retain a significant part of the structure they have in the original protein (a serious concern). Alternatively, small segments may be structurally studied using advanced spectroscopic techniques, such as magnetic resonance. Finally, some important information has been obtained by direct imaging of purified channels using electron microscopy and modern instruments that promise greatly improved resolution at the molecular level are being developed. Finally, the study of the genetic regulation of channel expression will be expanded through the further study of regulatory domains in the DNA. This will undoubtedly increase our appreciation of ion channels as important elements in the metabolism of the entire organism.
BIBLIOGRAPHY
1. Agnew WS, Trimmer J. Molecular diversity of voltage-sensitive sodium channels. Annu Rev Physiol. 1989;51:401–418.
2. Aldrich RW, Hoshi T, Zagotta WN. Differences in gating among amino terminal variants of Shaker potassium channels. Cold Spring Harbor Symp Quant Biol. 1990;55:19–27.
3. Armstrong CM, Bezanilla F. Inactivation of the sodium channel II Gating current experiments. J Gen Physiol. 1977;70:567–590.
4. Catterall WA. Molecular properties of dihydropyridine sensitive calcium channels in skeletal muscle. J Biol Chem. 1988;263:3535–3538.
5. Catterall WA. Cellular and molecular biology of voltage-gated sodium channels. Physiol Rev 1992;(Suppl. 72):S15–S48.
6. Chandy KG, Gutman GA. Voltage-gated K+ channel genes. In: North RA, ed. CRC Handbook of Receptors and Channels. Boca Raton: CRC Press; 1995;:1–71.
7. Chiamvimonvat N, Perez-Garcia MT, Ranjan R, Marban E, Tomaselli GF. Depth asymmetries of the pore-lining segments of the Na+ channel revealed by cysteine mutagenesis. Neuron. 1996;16:1037–1047.
8. Chou PY, Fasman GD. Empirical predictions of protein conformation. Annu Rev Biochem. 1978;47:251–276.
9. Coetzee WA, Amarillo Y, Chiu J, et al. Molecular diversity of K+ channels. Ann NY Acad Sci. 1999;868:233–285.
10. Doyle DA, Cabral JM, Pfuetzner RA, et al. The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science. 1998;280:69–77.
11. Dugandzija-Novakovic S, Koszowski AG, Levinson SR, Shrager P. Clustering of K+ channels and node of Ranvier formation in remyelinating axons. J Neurosci. 1995;15:492–503.
12. Ellinor PT, Yang J, Sather WA, Zhang J-F, Tsien R. Ca2+ channel selectivity at a single locus for high affinity Ca2+ interactions. Neuron. 1995;15:1121–1132.
13. Guy HR, Seetharamulu P. Molecular model of the action potential sodium channel. Proc Natl Acad Sci, USA. 1986;83:508–512.
14. Heinemann SH, Terlau H, Stuhmer W, Imoto K, Numa S. Calcium channel characteristics conferred on the sodium channel by single mutations. Nature. 1992;356:441–443.
15. Hille B. Ionic Channels of Excitable Membranes. Sunderland: Sinauer Associates; 1992.
16. Hofmann F, Biel M, Flockerzi V. Molecular basis for Ca2+ channel diversity. Ann Rev Neurosci. 1994;17:399–418.
17. Hoshi T, Zagotta WN, Aldrich RW. Biophysical and molecular mechanisms of Shaker potassium channel inactivation. Science. 1990;250:533–538.
18. Isom LL, De Jongh KS, Patton DE, et al. Primary structure and functional expression of the β1 subunit of the rat brain sodium channel. Science. 1992;256:839–842.
19. Kyte J, Doolittle RF. A simple method for displaying the hydropathic character of a protein. J Mol Biol. 1982;157:105–132.
20. Leonard J, Snutch TP. The expression of neurotransmitter receptors and ion channels in Xenopus oocytes. In: Glover DM, Hanes BD, eds. Molecular Neurobiology: a Practical Approach. New York: Oxford University Press; 1991;:161–182.
21. Li M, Jan YN, Jan LY. Specification of subunit assembly by the hydrophilic amino-terminal domain of the Shaker potassium channel. Science. 1992;257:1225–1230.
22. Lopez GA, Jan YN, Jan LY. Hydrophobic substitution mutations in the S4 sequence alter voltage-dependent gating in Shaker K+ channels. Neuron. 1991;7:327–336.
23. MacKinnon R, Heginbotham L, Abramson T. Mapping the receptor site for charybdotoxin, a pore-blocking potassium channel inhibitor. Neuron. 1990;5:767–771.
24. MacKinnon R, Yellen G. Mutations affecting TEA blockade and ion permeation in voltage-activated K+ channels. Science. 1990;250:276–279.
25. MacKinnon R. Determination of the subunit stoichiometry of a voltage-activated potassium channel. Nature, (London). 1991;350:232–235.
26. Maletic-Savatic M, Lenn NJ, Trimmer JS. Differential spatiotemporal expression of K+ channel polypeptides in rat hippocampal neurons developing in situ and in vitro. J Neurosci. 1994;15:3840–3851.
27. Maue RA, Kraner SD, Goodman RH, Mandel G. Neuron-specific expression of the rat brain type II sodium channel gene is directed by upstream regulatory elements. Neuron. 1990;4:22–231.
28. Miller C, ed. Ion Channel Reconstitution. New York: Plenum; 1986.
29. Miller JA, Agnew WS, Levinson SR. Principal glycopeptide of the tetrodotoxin/saxitoxin binding protein from Electrophorus electricus: isolation and partial physical and chemical characterization. Biochemistry. 1983;22:462–470.
30. Nakai J, Adams BA, Imoto K, Beam KG. Critical roles of the S3-segment and S3–S4 linker of repeat I in activation of L-type calcium channels. Proc Natl Acad Sci, USA. 1994;91:1014–1018.
31. Noda M, Shimizu S, Tanabe T, et al. Primary structure of Electrophorus electricus sodium channel deduced from cDNA sequence. Nature. 1984;312:121–127.
32. Numa S, Noda M. Molecular structure of sodium channels. Ann NY Acad Sci. 1986;479:338–355.
33. Papazian DM, Timpe LC, Jan YN, Jan LY. Cloning of genomic and complementary DNA from Shaker, a putative potassium channel gene from Drosophila. Science. 1987;237:749–753.
34. Papazian DM, Timpe LC, Jan YN, Jan LY. Alteration of voltage- dependence of Shaker potassium channel by mutations in the S4 sequence. Nature. 1991;349:305–310.
35. Pongs O. Molecular biology of voltage-dependent potassium channels. Physiol Revs 1992;(Suppl. 72):S69–S88.
36. Ribera AB, Spitzer NC. Developmental regulation of potassium channels and the impact on neuronal differentiation. In: Narahashi T, ed. Ion Channels, Vol 3. New York: Plenum Press; 1992;:1–38.
37. Salkoff L, Baker K, Butler A, Covarrubias M, Pak MD, Wei A. An essential set of K+ channels conserved in flies, mice, and humans. TINS. 1992;15:161–166.
38. Sharp AH, Imagawa T, Leung AT, Campbell KP. Identification and characterization of the dihydropyridine-binding subunit of the skeletal muscle dihydropyridine receptor. J Biol Chem. 1987;262:12 309–12 315.
39. Sigworth FJ. Charge movement in the sodium channel. J Gen Physiol. 1995;106:1047–1051.
40. Stuhmer W, Conti F, Suzuki H, et al. Structural parts involved in the activation and inactivation of sodium channels. Nature. 1989;339:597–603.
41. Tanabe T, Adams BA, Numa S, Beam KG. Repeat I of the dihydropyridine receptor is critical in determining sodium channel activation kinetics. Nature. 1991;352:800–803.
42. Tanabe T, Takeshima H, Mikami A, et al. Primary structure of the receptor for calcium channel blockers from skeletal muscle. Nature. 1987;328:313–318.
43. Terlau H, Heinemann SH, Stuhmer W, et al. Mapping the site of block by tetrodotoxin and saxitoxin of sodium channel II. FEBS Lett. 1991;293:93–96.
44. Timpe LC, Jan YN, Jan LY. Four cDNA clones from the Shaker locus of Drosophila induce kinetically distinct A-type potassium currents in Xenopus oocytes. Neuron. 1988;1:659–667.
45. Vassilev P, Scheuer T, Catterall WA. Inhibition of inactivation of single sodium channels by a site-directed antibody. Proc Natl Acad Sci, USA. 1989;86:8147–8151.
46. Yang J, Ellinor PT, Sather WA, Zhang J-F, Tsien RW. Molecular determinants of Ca2+ selectivity and ion permeation in L-type Ca2+ channels. Nature. 1993;366:158–161.
47. Yool AJ, Schwarz TL. Alteration of ionic selectivity of a K+ channel by mutation of the H5 region. Nature. 1991;349:700–704.
48. Zagotta WN, Hoshi T, Aldrich RW. Restoration of inactivation in mutants of Shaker potassium channels by a peptide derived from ShB [see comments]. Science. 1990;250:568–571.