Chapter 31
Ligand-Gated Ion Channels
Chapter Outline
III. Classes of Ligand-Gated Ion Channels
IV. Basic Physiological Features
VI. Neuronal Acetylcholine Receptor Channels
VII. γ-Aminobutyric Acid and Glycine Receptor Channels
I Summary
Ligand-gated ion channels are membrane proteins that are fundamental signaling molecules in neurons. These molecules are localized in the plasmalemma and on intracellular organelles and can be gated by both intracellular and extracellular ligands. The neurotransmitter-gated ion channels discussed in this chapter mediate fast excitation and inhibition in the nervous system and have now been well characterized by physiological and molecular studies. Studies using the technique of voltage and patch-clamp recording have examined the three basic features of an ion channel: gating, conductance and selective permeability. In general, ligand-gated ion channels can be described by kinetic models involving binding of two or more ligand molecules that induce a conformational change in the protein. As a result, a central, water-filled pore opens and conducts ions at very high rates of up to 107 ions per second. Channel activity is terminated when the channel closes or when it enters a non-conducting (desensitized) state. In vertebrates, acetylcholine (Ach) and glutamate receptors are cation channels, whereas γ-aminobutyric acid (GABA) and glycine receptors are anion channels. The ACh, GABA and glycine receptors constitute the Cys-loop receptor superfamily that presumably evolved from a common ancestral gene. Glutamate receptors comprise a separate protein family. The ligand-binding domain of glutamate channels shares homology with bacterial amino acid binding proteins, whereas the pore shares homology with the pore of voltage-dependent ion channels.
Each of the neurotransmitter-gated channels appears to be comprised of a hetero-oligomeric complex of closely related subunits surrounding a central ion pore. For each receptor type there are a number of possible subunit combinations that can form functional channels. AChR subunits have four transmembrane domains with the second transmembrane domain lining the pore. Charged amino acid residues near the mouth of the pore are important in determining the selectivity and conductance of the individual channel types. The glutamate subunits have three transmembrane domains and a re-entrant loop domain that forms a portion of the pore. The recent availability of crystal structures of a homomeric Cys-loop receptor and a glutamate receptor will allow atomic level resolution of the conformational movements involved in binding and gating of these channels.
II Introduction
Ion channels are fundamental signaling molecules in virtually all cells. Studies of ion channel in the plasmalemma of excitable cells have provided the foundation for our knowledge of these important molecules. In the nervous system, voltage-gated ion channels mediate action potentials (APs) and trigger neurotransmitter release, whereas ligand-gated channels are responsible for chemical signaling mediated by classical fast-acting transmitters. Neurotransmitters also trigger slower synaptic responses that are mediated by G-protein-coupled receptors.
Until the 1980s, the molecular properties of ion channel proteins were largely inferred from physiological and biophysical studies (Hille, 2001). In retrospect, many of the predictions from early biophysical studies have been confirmed and extended following the elucidation of the amino acid sequence of voltage- and ligand-gated ion channels by molecular cloning. However, the increasing knowledge of the three-dimensional molecular structure of ion channels and their homologs in lower organisms now permit sophisticated correlations of protein structure with function (Sine and Engel, 2004; Mayer, 2005). These advances have dramatically increased our understanding of the molecular operation of these prototypic membrane proteins. These molecules have captured the interest of many outstanding scientists whose studies of ion channel function and structure have led to several Nobel Prizes (Neher, 1992; Sakmann, 1992; McKinnon, 2004). Neurological disorders can also be caused by mutations in ion channels, the so-called “channelopathies” (Kullmann, 2010) and by autoimmune mechanisms that target ligand-gated ion channel proteins (Graus et al., 2010).
A biophysical approach to ion channels was pioneered by studies of voltage-gated ion channels in squid axon. These studies established that a conductance change was associated with the AP and that this conductance change could be attributed to selective increases in the membrane permeability to Na+ and K+ ions, with the energy for the process derived from the transmembrane ion gradients created by the Na+, K+-ATPase. These findings suggested that there were discrete pores in the membrane that accounted for the Na+ and K+ conductance. However, it was not until the 1970s that the existence of ion channels as discrete membrane proteins was confirmed, first by measurements of the statistical properties of currents through populations of ion channels, a technique called fluctuation or noise analysis. Later, this conclusion was refined by the introduction of patch-clamp recording, which allowed measurement of the current through single ion channels (Neher, 1992; Sakmann, 1992). Patch-clamp recording revolutionized the study of ion channels because it allowed detailed biophysical studies of the electrical activity of single molecules. Protein purification and molecular cloning revealed that ion channels are a large and diverse group of membrane proteins. Although ligand-gated ion channels vary in structural details, they share a common basic structure in that they are composed of multiple subunits arranged around a central water-filled pore. Each subunit is a polypeptide encoded by a separate gene. Generally, there are a large number of possible subunit combinations; thus, the particular subunits expressed in a certain class of neurons can result in channels with distinct functional characteristics.
This chapter is divided into three topic areas. In the first, the categories of ligand-gated ion channels are briefly reviewed. The basic physiological properties and general molecular structure of ligand-gated channels are then discussed using the muscle nicotinic acetylcholine receptor (AChR) and the N-methyl-D-aspartate (NMDA)-type glutamate receptor as the prototypes. In the third section, the features of neurotransmitter-gated channels that are responsible for fast synaptic transmission are discussed with an emphasis on their distinctive characteristics. Channels gated by acetylcholine (ACh), γ-aminobutyric acid (GABA), glycine and glutamate are included as representative examples. Related topics on ion channels are discussed in other chapters in Sections III through V of this book. The reader is also referred to the excellent monographs and reviews listed in the references for further details.
III Classes of Ligand-Gated Ion Channels
Ligands that activate ion channels in nerve cell membranes can be roughly divided into two major categories, neurotransmitters and intracellular ligands (Table 31.1). The neurotransmitter-gated ion channels involved in fast chemical synaptic transmission have been extensively studied. These include ACh, which is the transmitter at the vertebrate neuromuscular junction and in autonomic ganglia and the amino acids, L-glutamate and GABA, which mediate the majority of fast excitatory and inhibitory synaptic transmission, respectively, in the vertebrate central nervous system. Each of these ligands also activates receptors that are coupled to second messengers via GTP-binding (G) proteins. G-protein-coupled receptors generally mediate slower and neuromodulatory transmembrane signaling. Other neurotransmitters that activate ligand-gated ion channels in various cell types include serotonin, glycine, histamine and adenosine triphosphate (ATP). For example, the 5HT3 receptor activated by serotonin is a ligand-gated ion channel (Julius, 1991). ATP is released from nerve terminals in several pathways and activates the P2X family of channels (Burnstock, 2007; Browne et al., 2010). The transient receptor potential (TRP) channels provide a particularly large and diverse group that are activated not by classical neurotransmitters, but rather by diverse chemical as well as physical stimuli (Wu et al., 2010).
TABLE 31.1. Ligand-Gated Ion Channels
| Ligand | Receptor | Ion Selectivitya |
| Neurotransmitters | ||
| Acetylcholine | Muscle, neuronal AChRs | NS |
| Glutamate | AMPA, kainate, NMDA | NS |
| GABA | GABAA | Cl |
| Glycine | GlyR | Cl |
| Serotonin | 5HT3 receptor | NS |
| ATP | P2X receptor | NS |
| Intracellular Ligands | ||
| Calcium | Calcium-dependent channels | K, Cl, NS |
| Cyclic nucleotides | cGMP and cAMP receptors | NS |
| ATP | ATP-dependent channel | K |
| IP3 | Calcium release channel | Ca |
a Abbreviations for ion selectivity are: NS: non-selective cation permeability, some with calcium permeability; Cl: permeable to chloride; K: permeable to potassium; and Ca: permeable to calcium.
In addition to neurotransmitter-gated ion channels, increasing attention has focused on ligand-gated ion channels that are activated by intracellular ligands. These ligands include Ca2+, cyclic nucleotides, ATP and inositol trisphosphate (IP3). Intracellular ligands can increase (Ca2+, cAMP, IP3) or decrease (cGMP, ATP) activity of the associated channel. These channels play important roles in cell function. For example, Ca2+-dependent potassium, chloride and non-specific cation channels modify the excitability of the nerve cell membrane and thus can alter the AP and release of transmitter from nerve terminals. The cyclic nucleotide (cAMP, cGMP)-gated channels mediate sensory transduction in the visual and olfactory system. Light results in the closing of cGMP channels in vertebrate photoreceptors whereas odors trigger the opening of cAMP-gated channels in olfactory receptor neurons. (See Chapters 35 and 39 for more discussion of channels gated by cyclic nucleotides.)
IV Basic Physiological Features
Patch-clamp techniques allow investigators to define the three fundamental properties of any ion channel – conductance, selective permeability and gating – in molecular terms. In addition to these fundamental properties, modulation, channel block and desensitization are processes that alter the activity of ligand-gated ion channels and are important in shaping the impact of channel activity on membrane excitability. The biophysical basis of these properties will be discussed first and then the structural features of ligand-gated channels that control these properties will be considered.
Conductance reflects the flux of charged ions through the channel and is measured in picoSiemens (pS). Most ligand-gated ion channels have a conductance of 5–50 pS, which corresponds to the movement of more than 1×106 ions per second through the channel pore. These high flux rates initially suggested that ligand-gated channels must contain a water-filled pore, because this rate is much higher than that predicted for other transport mechanisms, such as pumps or exchangers. The size of the single-channel conductance (γ) is characteristic for a given channel. Several openings of a glutamate-activated channel in a hippocampal neuron are shown in Fig. 31.1. The conductance of the open channel is usually near 50 pS, but drops occasionally to subconductance states of 8, 35, 40 and 45 pS. The conductance of a channel increases as the permeant ion concentration is raised, but eventually saturates at concentrations well above physiological levels. This behavior can be described by Michaelis–Menten kinetics, suggesting that ions bind to sites within the pore rather than simply obeying the laws of free diffusion. Most permeant ions have a low Km of approximately 100 mM (Km is the concentration at which the conductance is half-maximal, as defined by the Michaelis–Menten equation); thus the ions bind for only a microsecond or so before continuing through the pore.
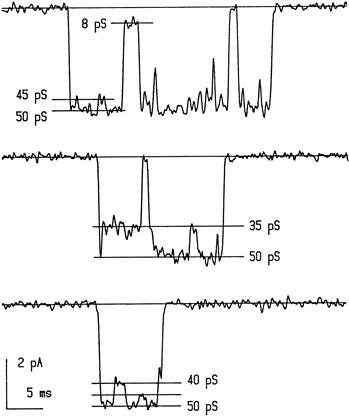
FIGURE 31.1 Activity of single ligand-gated channels as measured by patch-clamp recording. Examples of NMDA-type glutamate channel recording in an outside-out membrane patch obtained from a cultured hippocampal neuron. Downward deflections indicate opening of the channel in the presence of ligand (in this case, 20 μM NMDA). The channel usually opens to the 50 pS level, but may switch to lower levels of 8, 35, 40 or 45 pS before closing to the baseline level. (Modified with permission from Jahr and Stevens (1987). Nature. 325, 522–525. Copyright 1987 by Macmillan Magazines Limited.)
The current carried by the opening of a single-channel is measured in picoamperes (pA) with the single-channel current given by:
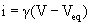 (31.1)
(31.1)
where V is the membrane potential and Veq is the equilibrium potential, at which no net current is measured. This equation is simply Ohm’s law, where (V−Veq) is the electrochemical driving force for ions that can pass through the channel. For example, if the extracellular and cytoplasmic concentrations of permeant ions are equal, then Veq is 0 mV. Thus, as the membrane potential changes, the size of the single-channel current also changes; a plot of this relationship is called a current–voltage or I-V plot. For channels, such as the muscle AChR, that are permeable to Na+ and K+, the reversal potential is 0 mV and the I-V curve is nearly linear. However, some channels deviate from linear behavior and show inward or outward rectification (i.e. the channel passes current in one direction better than in the other), analogous to the behavior of some voltage-gated K+ channels.
Channels are not equally permeable to all ions, i.e. they exhibit selective permeability. As listed in Table 31.1, most ligand-gated ion channels are permeable to either monovalent cations or anions. The selective permeability of channels is partly due to the physical size of the pore. For example, the muscle AChR is permeable to cations with diameters up to about 6.5 Å. In general, cationic ligand-gated ion channels are less selective than voltage-gated ion channels, which are highly selective for Na+, K+ or Ca2+, but more selective than gap junction channels, which are permeable to small molecular weight molecules such as cyclic nucleotides as well as to ions. Because anions and cations are approximately the same size, it is obvious that the pore dimensions cannot totally account for selective permeability. As discussed later, positively- and negatively-charged amino acid residues at the entrances to the channel and within the pore provide a means for this discrimination. Selective permeability is extremely important in considering the function of an ion channel. For example, channels that are permeable to Na+ and K+ do not significantly alter the ion concentrations on either side of the membrane but, instead, provide an electrical signal that depolarizes the neuron and brings the membrane potential closer to the threshold for AP generation. However, channels with significant Ca2+ permeability can transiently increase the cytoplasmic Ca2+ concentration and thus act as a biochemical signal. The relative Ca2+ permeability of ligand-gated channels, such as the NMDA receptor and some nicotinic receptors, is an important aspect of their role in neuronal function. The reader is referred to the monograph by Hille (2001) for a full discussion of channel permeability and its measurement.
Channel gating refers to the conformational change in the ion channel protein that is triggered by ligand binding. The conformational change results in a rapid switch between conducting (open) and non-conducting (closed) states of the channel. The gating behavior of channels has many parallels with the allosteric behavior of enzymes, with the binding of ligand providing the free energy necessary to maintain the channel in the open conformation. Gating between the open and closed configuration is extremely rapid (approximately 10 μs) and thus is beyond the resolution of standard recording methods. Each channel opening usually lasts a few milliseconds, but can, on some occasions, last up to several hundred milliseconds. Binding of more than one agonist molecule is usually necessary to open a channel. The binding of multiple agonist molecules creates a sigmoidal dose–response relationship, characteristic of the cooperative binding of substrates to enzymes. The steep activation created by sigmoidal kinetics prevents the channel from opening in the presence of a low concentration of agonist, which could be quite important in preventing desensitization of synaptic receptors. In general, the higher the binding affinity of the agonist, the longer the channel will remain open. This is because the channel can remain open (or reopen) until the agonist dissociates.
The gating of ligand-gated ion channels can be described by multistate kinetic diagrams as shown in Fig. 31.2A. Such state diagrams have been developed for many ligand-gated ion channels, based on the analysis of open and closed time distributions obtained in single channel recording. For example, the presence of two exponentials in an open time histogram implies the existence of two open states. The minimal kinetic model for a ligand-gated channel usually has at least four states, including closed and unbound, closed and bound, open and bound and non-conducting (desensitized). For the case shown in Fig. 31.2A and B, note that the channel opens only when two agonist molecules are bound. For fast-acting neurotransmitters, the concentration of transmitter in the synaptic cleft is typically very high for a brief period. At high concentrations of agonist the receptors quickly reach the fully liganded but closed state because transition to this state is determined by the concentration of agonist as well as the binding rate, rb. Thus, the rising and falling phase of the synaptic response largely reflects the unbinding rate, ru, the channel opening and closing rates (β and α) and, in some cases, the rates in and out of the desensitized state, Rd. Classical pharmacological terms can be interpreted in terms of these kinetic schemes (Colquhoun, 1998). Agonists open the channel, whereas antagonists bind, but apparently do not cause a sufficient conformational change in the channel protein to open the channel. The fraction of time the bound channel spends in the open state is called the open probability and may differ between agonists – an agonist that activates the channel with higher probability therefore has a higher efficacy in the nomenclature of classical receptor pharmacology.
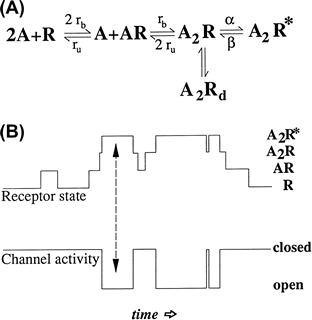
FIGURE 31.2 Kinetics of ligand-gated channels. (A) Example of a four-state kinetic scheme used to describe the behavior of ligand-gated ion channels. In this example, two agonist molecules (A) must bind before the channel can open (A2R∗) or enter a non-conducting desensitized state (A2Rd). The rate constants for agonist binding and unbinding are designated rb and ru, and the rate constants for channel opening and closing are designated as β and α. (B) Relationship of the state of the receptor to the open and closing of the channel. The channel opens only when the receptor is in the A2R∗ state as indicated by the arrows.
Earlier studies of the activity of ligand-gated channels were generally limited to equilibrium measurements because of the limits in the speed of application of agonists to cells or cell-free patches. Such equilibrium measurements are difficult to interpret in terms of state diagrams such as that shown in Fig. 31.2. In addition, the duration of fast-acting transmitters such as glutamate is generally brief, suggesting that the synapse itself operates under non-equilibrium conditions. However, the development of rapid solution exchange techniques allowed agonists to be applied within several hundred microseconds to membrane patches, usually in the “out-side-out” configuration to allow agonist in the bath to access the extracellular face of the receptor. Such approaches appear to more closely resemble conditions at synapses and are particularly useful in evaluating the role of channel modulation, channel block and desensitization on synaptic responses. An example of the response of a membrane patch from a hippocampal neuron to a brief application of glutamate is shown in Fig. 31.3.
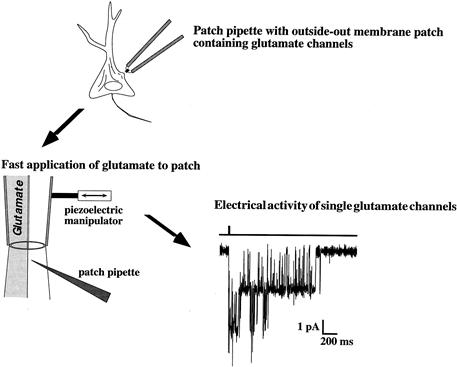
FIGURE 31.3 Use of rapid application methods to study kinetics of ligand-gated channels. The patch-clamp technique allows small membrane patches to be removed from a cell, such as a hippocampal neuron, with the extracellular side of the membrane facing the bath (outside-out configuration). The tip of the pipette is then placed in the interface of two rapidly moving streams of solution. A piezoelectric manipulator moves the solutions to change the solution flow over the patch in less than 1 ms, which mimics what occurs at glutamate release sites. The response, recorded as the opening of glutamate channels of the NMDA type, greatly outlasts the brief application of agonist. The bar above the channel activity indicates the duration of the glutamate application.
In general, the conductance and selective permeability of ligand-gated channels are not subject to regulation. However, a number of allosteric control mechanisms can profoundly affect gating (Changeux and Edelstein, 1998). These mechanisms can be manifest as desensitization, channel block and allosteric modulation. Desensitization refers to the loss of the response during the continued presence of agonist and was first noted in studies of the muscle AChR. At the single-channel level, desensitization reflects a non-conducting state of the channel. This phenomenon has now been seen for most ligand-gated channels and can be described by including an extra bound, but non-conducting, state in the kinetic scheme (see Fig. 31.2A). Depending on the channel, the non-conducting state may be accessible from either the closed state (A2R) or the open state (A2R∗). Desensitization represents a distinct conformation of the receptor as has been demonstrated by structural studies of AMPA receptors (see below). Very rapid desensitization may play a role in terminating the neuronal response to neurotransmitters at some synapses, whereas slower desensitization may actually prolong synaptic responses as channels re-enter open states after transiting through desensitized states (Jones and Westbrook, 1996).
Ligand-gated channels can be plugged by ions or drugs, a phenomenon referred to as channel block. If the blocking particle is charged, such as a large ion, the block will be influenced by the voltage across the membrane. Thus, the reduction in channel activity will be more pronounced at some membrane potentials. The voltage-dependent block of NMDA-type glutamate channels by extracellular Mg2+ is an example of this phenomenon (Ascher and Nowak, 1987). At the single channel level, channel block is usually seen as rapid interruptions (“flickers”) of the open state as illustrated in Fig. 31.4A and B. A characteristic feature of most forms of channel block is that the blocker is only effective when the channel is open. Thus, the binding site for the blocker resides within the pore of the channel, or at least at a site that is only available in the open conformation of the channel. Channel block is the mechanism of action for a number of drugs, such as the local anesthetic lidocaine and psychoactive compounds, such as phencyclidine (PCP).
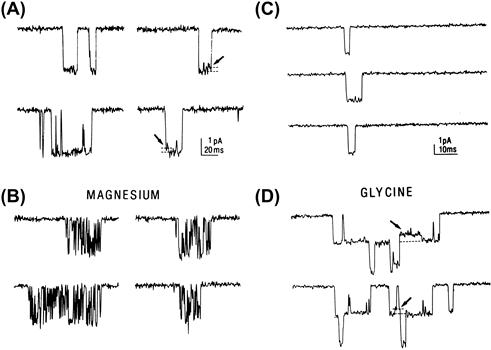
FIGURE 31.4 NMDA-type glutamate receptors illustrate two common mechanisms of channel regulation: channel block and allosteric modulation. (A and B). Channels activated by NMDA in a membrane patch from a hippocampal neuron in the absence of extracellular Mg2+ remain open (downward deflection) for several milliseconds before closing (A). However, in the presence of Mg2+ (B), the openings are interrupted by brief closures due to plugging of the pore by Mg2+ ions. (C and D) The frequency of NMDA channel opening is increased in the presence of extracellular glycine (D) compared to glycine-free solutions (C). The stepwise increase in current in part D indicates that at least two channels were present in the membrane patch. Because glycine is required for NMDA channel opening, the occasional openings in part C probably reflect contamination of the solution with low levels of glycine. (Modified with permission from Ascher, R. and Nowak, L. (1987). Trends Neurosci. 10, 284–288.)
Channel gating is also subject to modulation by either covalent modification, such as phosphorylation (Swope et al., 1999), or via the non-covalent binding of modulators to the channel. Most ligand-gated channels undergo phosphorylation of intracellular portions of the channel protein; however, the resulting effect on channel function is dependent on the specific channel and the type of kinase involved. Phosphate groups are highly charged and thus can modify interactions in local regions. For example, phosphorylation of the AChR enhances desensitization and may also be important in clustering of AChR molecules at sites of innervation. The list of non-covalent modulators is long and includes Ca2+, toxins, drugs and some endogenous substances such as protons and steroid hormones. These modulators usually act in a non-competitive manner, i.e. they do not directly interfere with ligand binding. Most of these reagents affect gating by altering the rate of channel opening or the time spent in the open state. For example, glycine binds to the NMDA channel and results in an increased probability of opening (see Figs. 31.4C and D). This is a dramatic example of allosteric regulation because the channel will not open unless both glycine and the transmitter (glutamate) are bound. A less profound, but equally important, example is the upregulation of GABAA receptor activity by benzodiazepines (Olsen and Sieghart, 2009).
V Molecular Structure
The ligand-gated ion channels can be divided into three main structural classes: the Cys-loop receptors, the glutamate receptors and the P2X receptors. Channels in these three families are formed as combinations 5, 4 and 3 subunits, respectively (Fig. 31.5). We discuss here structural information on the pentameric Cys-loop receptor superfamily that includes the nicotinic, GABAA, glycine and 5-HT3 receptors, and the tetrameric glutamate receptor family that includes the AMPA, kainate and NMDA receptors. For the Cys-loop and glutamate receptor families, structural studies are providing a more and more detailed understanding of the main functional features of ligand-gated ion channels: binding of ligand, the gating and ion selectivity of the channel and the coupling of ligand binding to channel gating. For information on the unique structure of the trimeric P2X receptors activated by ATP, see Browne et al. (2010).
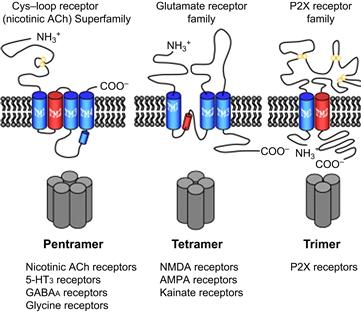
FIGURE 31.5 The topology of the three families of ligand-gated ion channel subunits. The general topologies of ligand-gated receptor subunits for each of the main receptor families are shown in the upper row. The stoichiometry of subunits in an assembled receptor are shown in the lower row. The Cys-loop family of receptors are pentameric, whereas the glutamate receptors are tetrameric and P2X receptors are trimeric. The three protein families also differ in the general structure of extracellular, intramembrane and intracellular domains. The red cylinders indicate the intramembrane pore region through which ions flow, M2 (TM2) in the case of Cys-loop and P2X receptors and the P loop for glutamate receptors. (Modified with permission from Collingridge, G.L., Olsen, R.W., Peters, J., Spedding, M. (2009). Neuropharmacology. 56, 2–5.)
The AChR led the way in determining the molecular structure of ligand-gated channels, beginning with biochemical studies of the muscle-type receptor (Karlin, 1991). This analysis took advantage of the high density of AChRs in the electric organ of the ray (Torpedo californica) and the high-affinity snake toxin, α-bungarotoxin, that binds to muscle-type AChRs. Protein purification revealed an approximately 250-kDa complex with two α-bungarotoxin binding sites per complex and separation on denaturing gels revealed polypeptides of 40, 50, 60 and 65 kDa designated as the α, β, γ and δ subunits. Because of the molecular weight of the complex and the binding of toxin molecules, a pentameric structure with two α subunits and single copies of β, γ and δ was proposed. Using the partial amino acid sequence of the purified subunits, the cDNAs encoding AChRs in the Torpedo electric organ were cloned and sequenced in the early 1980s. Incorporation of the purified protein complex in lipid bilayers or expression of AChR mRNA in Xenopus oocytes demonstrated channels activated by acetylcholine, confirming the identity of the molecule. Consistent with the proposed combination of subunits in intact receptors, the expression of AChRs in oocytes was much greater when all four subunit mRNAs were included. The α subunit was essential in order to obtain responses to acetylcholine.
Based on the hydrophobicity of amino acid residues in the primary sequence, the proposed transmembrane topology included four putative transmembrane domains (M1–M4), large N-terminal domain and short C-terminal extracellular domains and two cytoplasmic loops – a short one between M1 and M2 and a longer loop containing phosphorylation sites between M3 and M4. The early evidence that M2 lines the pore came from amino acid substitutions within that domain. In addition, a 23-amino-acid peptide fragment containing the M2 region of the Torpedo δ AChR subunit formed a cation channel when incorporated into lipid bilayers. The analysis of the primary amino acid sequence gives limited information about three-dimensional protein structure. Subsequently, two-dimensional crystals of AChRs from the Torpedo electric organ were analyzed with the electron microscope (Miyazawa et al., 1999), providing a good first estimate of the overall shape and structural features of the AChR.
Yet it was identification and high-resolution crystal structure of an acetylcholine-binding protein (AChBP) from the freshwater snail (Lymnaea stagnalis) that provided the next level of resolution (Smit et al., 2001; Brejc et al., 2001). Thc AChBP is a pentamer of identical subunits that is homologous to the extracellular ACh-binding portion of the AChR and thus provided improved molecular resolution of ligand-binding domain. Combining AChBP with two-dimensional arrays of Torpedo channels began to reveal the relationship of the extracellular binding domains and the structural movements that lead to opening of the intramembrane channel (Unwin, 2005). Crystal structures of two full length homomeric channels related to the Cys-loop family now allow the first glimpse of the structure of an entire protein of this class. Interestingly, these structures are not those of well-studied members of the Cys-loop family, but rather include a pentameric ligand-gated channel from bacteria (Hilf and Dutzler, 2009) and a glutamate-gated chloride channel from C. elegans (Hibbs and Gouaux, 2011).
Each of the two binding pockets in the Cys-loop receptors is composed of hydrophobic and aromatic amino acid residues contributed by discontinuous segments of an α and a non-α subunit. The Cys-loop receptors get their name from an N-terminal 13 amino acid sequence flanked by cysteines that form a disulfide bond and make a closed loop in the extracellular domain. The binding pocket is capped by the “C-loop” contributed by the alpha subunit, which must be open for agonist access and is locked in place when the agonist is bound and thus fully enveloped. The structure also suggests a mechanism by which agonist binding can result in channel opening through linkers that connect to the first transmembrane domain. The new Cys-loop receptor crystal structures promise a much clearer picture of the conformational movements that underlie the open, closed and desensitized states.
As with the Cys-loop receptors, the proposed quarternary structure of glutamate channels involves multisubunit complexes surrounding a central pore (Fig. 31.6). However, cloning of the glutamate channels revealed that they differed substantially from the Cys-loop receptors in their primary amino acid sequence (Nakanishi, 1992; Hollmann and Heinemann, 1994). Notable differences include the much longer N-terminal domain (approximately 500 residues). In addition, the second hydrophobic domain of the glutamate subunits is a re-entrant loop that enters and exits the membrane from the cytoplasmic side without fully traversing the membrane. This “P” loop shows homology to the P loop of voltage-gated channels that lines the channel pore and controls ion selectivity and conductance. A prokaryotic glutamate receptor may provide an evolutionary link between the voltage-gated potassium channels and glutamate receptors (Chen et al., 1999). Secondly, the region between the third and fourth hydrophobic domains is extracellular in glutamate subunits whereas it is cytoplasmic in the AChR subunits. Finally, the C-terminus of the glutamate subunits is cytoplasmic. These unique features provided important clues to the location of the transmitter binding site(s), the behavior of the channel pore and sites of interaction with proteins that anchor and/or localize glutamate receptor subunits at synapses.
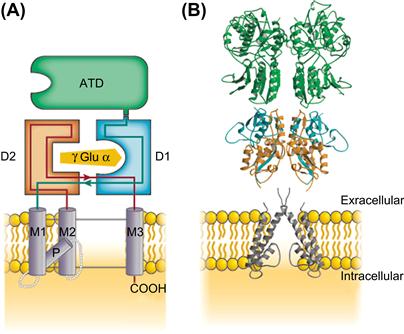
FIGURE 31.6 The domain organization of glutamate receptor subunits. (A) As shown in this schematic diagram, the extracellular region contains two domains, the ATD shown in green and the agonist-binding domain show in orange and blue surrounding an agonist molecule. The hydrophobic transmembrane domains M1, M2 and M3 as well as the P loop of the pore are shown in lavender. The intracellular domain is truncated in this schematic (-COOH). (B) A glutamate receptor assembles as a dimer of dimers. One representative dimer is shown here based on the crystal structures. The color-coded domains illustrate the secondary structure of α-helices and β-sheets that contribute to each domain. (Modified with permission from Mayer, M.L. (2006). Nature. 440, 456-462.)
The extracellular component of glutamate receptors constitutes two separate domains (see Fig. 31.6). The amino terminal domain (ATD) of approximately 380 amino acids is involved in subunit assembly. The agonist-binding domain is discontinuous, being formed from the first extracellular region (D1) and the extracellular loop between the second and third transmembrane regions (D2). Both of these domains are homologous to bacterial proteins that bind amino acids. This homology led to a proposed model for the glutamate binding site based on the crystal structure of the bacterial periplasmic binding proteins and subsequently confirmed based on chimeric receptors with domains from two glutamate subunits with distinct pharmacology (Stern-Bach et al., 1994). In GluN1, glycine occupies the agonist-binding domain instead of a glutamate, which binds to the GluN2 subunit. Selectivity for glycine over glutamate is conferred by the volume of the agonist-binding cavity resulting from steric constraints from residues present only in GluN1 (Furukawa and Gouaux, 2003).
Unlike AMPA or kainate receptors, which can form homomeric channels, functional NMDA channels are heteromeric, requiring two GluN1 and two GluN2 subunits. The general tetrameric organization of glutamate receptors was confirmed with the solving of a homomeric AMPA receptor structure (Sobolevsky et al., 2009). Unlike Cys-loop receptors, agonist binding in glutamate receptors occurs in a subunit-autonomous manner, requiring no inter-subunit interactions. The ligand binding regions of NMDA receptors are arranged as dimeric pairs (see Fig. 31.6B). Within a subunit, ligand binding occurs at the D1/D2 interface, which results in the closure of this structure in a clamshell fashion. These domain movements are thought to be associated with channel gating behavior, specifically, opening, closing and desensitization.
VI Neuronal Acetylcholine Receptor Channels
Studies of acetylcholine receptors began with the skeletal muscle isoforms (Steinbach, 1989), but AChRs are also expressed in neurons. The embryonic muscle AChR consists of an α2 βγδ pentamer, but later in development the γ subunit is replaced by an ε subunit. A much greater heterogeneity of receptor subunits is involved in the expression of AChRs at central synapses. (Dani and Bertrand, 2007). Neuronal AChRs mediate excitatory synaptic transmission in autonomic ganglia and at the recurrent collateral synapse of spinal motoneurons onto Renshaw cells (inhibitory interneurons). The functional role of nAChRs in the central nervous system was more difficult to establish despite the well-known action of nicotine (via cigarette smoke) on human behavior. nAChRs differ in several respects from muscle AChRs. First, α-bungarotoxin blocks muscle AChRs, but not most nAChRs. However, α-bungarotoxin binding sites represent a distinct subtype of nAChRs that have faster kinetics and a higher calcium permeability than other nAChR subtypes. The Ca2+ permeability of some nAChRs and changes in channel properties during synaptogenesis suggest that various nAChR subtypes play a role in synaptic plasticity and development. The activity of nAChRs are also modulated by phosphorylation. The kinase activation can be due to the actions of neuropeptides, such as vasoactive intestinal peptide and substance P, that are co-released with acetylcholine at some synapses.
Perhaps even more than for other ligand-gated channels, the cloning and characterization of subunit genes greatly accelerated studies of the structure and function of nAChRs (Role, 1992). Unlike the muscle AChRs, the nAChR pentamer is composed of only two classes of subunits, aptly named α and β. Subunits with cysteines at residues 192 and 193 are designated as α; subunits lacking these cysteines are called β. The genes for nine α (α2–10) and three β (β2–4) subunits have been cloned from rat and chick brain cDNA libraries. The stoichiometry of each channel molecule can be either homo-oligomeric (five alpha subunits) or heteromeric (usually with two α and three β subunits). Expression of single subunits and combination of subunits in Xenopus oocytes narrowed the possible combinations of subunits in functional channels. For example, α2–6 and β2–4 form heteromeric channels, whereas α7, α8 and α9 do not. Combinations of subunits differ in their single-channel properties, ligand affinity and sensitivity to antagonists such as α-bungarotoxin. The α4 β2 receptor is a common heteromer in the CNS. It has a relatively high affinity for nicotinic agonists and thus is a principal target of the nicotine in tobacco smoke. Homo-oligomeric α7 channels have a relatively low affinity for ACh and rapidly desensitize. This subunit has the closest homology to the snail AChBP and has been used to probe the structural substrates of channel properties such as desensitization, ion flux and selectivity. Homo-oligomeric α7, α8 and α9 channels are sensitive to α-bungarotoxin, but α7 is the predominant subunit of these three in brain. Presynaptic homomeric α7 receptors with their high calcium permeability can enhance the release of glutamate and other transmitters. The α9 subunit is unique due to its limited expression in the outer hair cells of the cochlea as well as its mixed muscarinic/nicotinic pharmacology.
Many central neurons have responses to exogenous application of nicotinic agonists and there is a wide distribution of nAChR subunit genes as revealed by in situ hybridization. However, there is a reasonable correlation between pathways with functional nAChRs (e.g. the cholinergic input to the hippocampus and medial habenula from the medial septum and diagonal band), the presence of nicotinic binding sites and the expression of nAChR subunits. Studies of nAChRs in different neuronal cell types have revealed a spectrum of channel properties and pharmacological characteristics, suggesting that individual neurons express several combinations of nAChR subunits. For example, in situ hybridization studies of neurons in the medial habenular nucleus have revealed two α and three β subunit genes. As for many classes of ligand-gated channels, the role of specific nAChR subunits has been investigated using genetically-engineered knockout and knock-in mice to investigate subunit-specific functions. Interest in the nAChRs has been further fueled by studies indicating that mutations in α4 or β2 subunits cause a genetic form of human epilepsy.
VII γ-Aminobutyric Acid and Glycine Receptor Channels
Synaptic inhibition in the central nervous system (CNS) is mediated largely by GABAA and glycine receptors. These ligand-gated receptor channels are selectively permeable to anions, principally Cl− under physiological conditions. GABA-gated Cl− channels are designated GABAA receptors to distinguish them from the G-protein-coupled GABAB receptor (Padgett and Slesinger, 2010). GABAA and glycine receptors are members of the Cys-loop receptor family. Unlike other mammalian Cys-loop receptors that are non-selective cation channels, GABAA and glycine channels are selectively permeable to anions.
Virtually all CNS neurons have GABAA receptors, whereas the anatomical distribution of glycine receptors is generally restricted to the brainstem and spinal cord. GABAA receptors are often localized on proximal dendrites of central neurons, but also are expressed on axon initial segments and distal dendrites. Because the Cl− equilibrium potential in many neurons is more negative than the resting potential, the opening of GABAA or glycine channels hyperpolarizes the cell membrane potential and reduces excitability. In addition to hyperpolarizing the membrane potential, the opening of large numbers of these channels lowers the membrane electrical resistance. Thus GABAA channels at proximal dendrites effectively “shunt” excitation traveling down the dendrite from excitatory synapses on more distal dendritic branches. In some neurons, particularly during early development, the Cl− equilibrium is more positive than the resting potential, resulting in depolarizing GABAA or glycine responses. Depolarizing GABAA responses occurring in axons can increase excitability and neurotransmitter release. Finally, some inhibitory synapses in the spinal cord and brainstem contain both GABAA and glycine receptors. Analysis of unitary release events at these sites indicates that single synaptic vesicles contain both GABA and glycine and that a subpopulation of postsynaptic sites contains both receptor types (Jonas et al., 1998). As with other ligand-gated neurotransmitter receptors, molecular studies have revealed anchoring and regulatory proteins that interact with glycine and GABAA receptors, such as gephyrin (Fritschy et al., 2008) and GABA receptor-associated protein (GABARAP; Mohrluder et al., 2009). Gephyrin was identified as a cytoplasmic protein that interacts directly with glycine receptors. Gephyrin also interacts with tubulin and the actin-binding protein profilin and thus acts as a bridge between glycine receptors and the cytoskeleton. Gephyrin is also co-localized with GABAA receptors at postsynaptic sites but, unlike glycine receptors, has not been shown to bind to GABAA receptors. GABARAP interacts with many GABAA receptor subtypes, as well as binding to gephyrin and tubulin. Interaction with these cytoplasmic factors may alter the localization and trafficking of GABAA and glycine receptors as well as create zones of localized signal transduction.
The behavior of single GABAA and glycine channels can be described by a kinetic scheme similar to that of the nAChR with the binding of two agonist molecules required for channel opening (Macdonald and Twyman, 1992). Analysis of the openings and closings of single GABAA channels suggests that the channel may open briefly following the binding of a single GABAA molecule and into two longer-lived open states from the doubly liganded configuration. Comparison of the total open duration of singly- and doubly-liganded receptors demonstrates that occupancy of both agonist sites results in many more channel openings. Channels may close and re-enter longer-lived open states before the agonist dissociates from the receptor. These so-called bursts are composed of short closings interrupting a series of openings and they can last tens of milliseconds. Desensitization of GABAA channels results in long closed intervals that are grouped with bursts into clusters lasting up to several hundred milliseconds. These clusters are important in determining the duration of inhibitory postsynaptic potentials at some synapses (Jones and Westbrook, 1996).
The drugs that act on GABAA and glycine channels comprise a fascinatingly rich assortment of clinically important compounds (Olsen et al., 1991). Because these channels underlie synaptic inhibition in the CNS, enhancement or reduction in their activity can lead to profound changes in brain function, including amnesia (increased GABAA activity) or seizures (decreased GABAA activity). Antagonists for these receptors include strychnine, which inhibits glycine receptors; bicuculline, which inhibits GABAA receptors; and picrotoxin, which inhibits both receptor types. The GABAA receptor is also the target of sedative-hypnotic drugs, such as the benzodiazepines and barbiturates. Benzodiazepines (BDZ) increase the probability of channel opening, whereas barbiturates appear to act by prolonging long channel openings (bursts). The pharmacology of benzodiazepine modulation of the GABAA receptor is particularly interesting, because compounds either can enhance channel opening (BDZ agonists), reduce channel opening (BDZ inverse agonists) or block the effects of BDZ agonists (BDZ antagonists). GABAA receptor activity is also modulated by alcohol, volatile anesthetics, such as isoflurane, and some steroid anesthetics (or their endogenous equivalents, the neurosteroids).
Using benzodiazepines and strychnine as selective ligands, GABAA and glycine receptors were purified as multimeric protein complexes, each with molecular weights of approximately 50–60 kDa. The solubilized receptor complex had a molecular weight of approximately 250 kDa, suggesting that, as for the AChR, five subunits constitute a receptor. Subsequent molecular cloning identified a series of receptor subunits for both receptors. The glycine subunits include the strychnine-binding subunit (α) of which four have been cloned and a single β subunit, with a stoichiometry of (α)2(β)3 for receptors from mature animals. Interestingly, the immature form of the glycine receptor contains only α subunits. Gephyrin binds to the β subunit, thus the interaction between gephyrin and glycine receptors is limited to the adult form. Nineteen GABAA subunits have been identified and grouped according to sequence similarity. These include six α, three β, three γ, three ρ subunits and single δ, ε, π, and Θ subtypes (Wisden and Seeburg, 1992; Olsen and Sieghart, 2009). In heterologous systems, expression of single GABAA or glycine receptor subunits can result in functional homomeric receptors. However, given the broad co-expression patterns of many GABAA and glycine receptor subunits and the functional heterogeneity of native receptors, homomeric receptors probably occur rarely. The large number of GABAA receptor subunits provides a formidable challenge in determining which combinations form functional receptors in neurons. GABAA and glycine receptor subunit expression also varies during development and with neuronal cell type. Based on pharmacology, expression, biochemistry and subcellular localization, at least 26 different types of native GABAA receptors have been identified in CNS neurons (Olsen and Sieghart, 2009).
Subunit composition can have a strong influence on the biophysical and pharmacological properties of GABAA and glycine receptors. The GABA and benzodiazepine binding sites reside at the interface between an α subunit and a β or γ subunit (usually γ2), respectively. The γ2 subunit is broadly and highly expressed in the CNS and genetic deletion greatly reduces BDZ binding sites in the brain. Interestingly, the α6 subunit has a low affinity for BDZ agonists, but still can bind BDZ inverse agonists or antagonists, which may explain benzodiazepine-insensitive GABAA receptors in some neurons. Homomeric receptors composed of the GABAA receptor ρ subunit are bicuculine-insensitive, weakly antagonized by picrotoxin and insensitive to BDZs, barbiturates and neurosteroids. These channels also show distinct gating properties and conductances compared to other GABAA receptors. They were initially referred to as GABAC receptors. However, due to their sequence similarity and proposed structure, they are currently thought of as a subtype of GABAA receptors. The three ρ subunits (ρ1, ρ2 and ρ3) are expressed throughout the CNS but expression is predominantly in several cell types in the retina.
VIII Glutamate Receptor Channels
Although it was known from the early 1950s that L-glutamate was a neuroexcitant, it was not until the 1980s that the role of glutamate-gated ion channels in central synaptic transmission was widely accepted (Collingridge and Lester, 1989; Westbrook and Jahr, 1989). It is now clear that glutamate receptor channels mediate a substantial portion of fast excitatory transmission in the brain and spinal cord through the simultaneous activation of two types of ion channels co-localized at excitatory synapses. Characterization of this family of ligand-gated ion channels was initially based on selective activation by the exogenous amino acid ligands: NMDA, kainate and quisqualate. AMPA (α-amino-3-hydroxy-5-methyl-4-isoxazole proprionic acid) replaced quisqualate as one of the prototypic ligands because quisqualate also activates a G-protein-coupled receptor. It soon became apparent that NMDA receptors were distinct from kainate and AMPA receptors, but for some time it was debated whether kainate and AMPA (i.e. non-NMDA) responses arose from the same or different receptors. It is now clear that AMPA and kainate receptors are distinct molecular entities, although kainate can activate both classes of receptors.
Initial studies of native AMPA/kainate receptor channels demonstrated many of the same features as AChRs. In hippocampal pyramidal neurons, AMPA receptor activation leads to brief openings (1–5 ms) of monovalent cation channels that show little or no voltage dependence. However, in some neurons, particularly interneurons, AMPA receptors have a higher calcium permeability and are inwardly rectifying. AMPA receptors rapidly desensitize when gated by glutamate and agonist dissociation (unbinding) and desensitization (agonist still bound but the channel is non-conducting) can contribute to the decay of the synaptic current. The behavior of AMPA channels seems well suited to the rapid relay of information that characterizes the fast component of excitatory postsynaptic potentials in the brain.
The NMDA channel provides perhaps the best example of the linkage between fundamental properties of ion channels and the electrical activity of single cells and complex behavioral phenomena (Bekkers and Stevens, 1990; McBain and Mayer, 1994). This linkage is primarily based on three features of NMDA channels: (1) voltage-dependent block of the open channel by extracellular Mg2+; (2) a relatively high permeability to Ca2+; and (3) slow channel kinetics resulting in long-lasting channel activity. Because Mg2+ ions are positively charged and thus sense the membrane electric field, they are drawn into the open NMDA receptor channel at negative membrane potentials. Mg2+ binds in the channel pore and impedes the flow of permeant ions, even though the agonists (glutamate and glycine) remains bound. However, during synaptic activity as the cell membrane depolarizes, Mg2+ falls out of the channel and permeant cations flow through the channel. Thus, ion flux through the channel is voltage dependent. This mechanism differs from other voltage-dependent ion channels, such as the sodium or calcium channel, where voltage dependence results from an intrinsic conformational change in the channel protein. However, the end result is the same; current flows through the NMDA receptor channel more effectively with depolarization and thus acts as a positive feedback on synaptic activation.
Unlike many AMPA channels, Ca2+ contributes a substantial fraction of the flux through NMDA channels. However, the rate of flux of Ca2+ through the NMDA channel is slower than Na+ because Ca2+ transiently binds with higher affinity to sites within the channel (energy wells). Thus most of the current through open NMDA channels is due to Na+, which is present in 50-fold excess in the extracellular space compared to calcium. Nonetheless, the 5–10% of the current that is carried by Ca2+ is sufficient to act as a biochemical signal for such processes as the induction of long-term potentiation in the hippocampus, an experimental model of associative learning. Excessive activation of glutamate receptors may also cause neuronal damage, presumably through elevations of intracellular Ca2+ concentration and subsequent activation of proteases and free radical formation. This mechanism, called excitotoxicity, has been implicated in brain damage due to prolonged seizures, following strokes and may also contribute to loss of neurons in several degenerative neurological diseases.
Because no high-affinity ligands were available for purification of glutamate receptors, molecular biologists were forced to use the somewhat laborious task of expression cloning to isolate cDNA clones for glutamate receptor subunits. The first AMPA receptor subunit (GluA1) was isolated by this technique in 1989 and the first NMDA receptor clone (GluN1) was also isolated by expression cloning in 1991. Using these initial AMPA and NMDA clones to screen for homologous sequences, 16 related glutamate receptor subunits were isolated from rat and mouse cDNA libraries. On the basis of glutamate receptor sequence homology and the characteristics of the expressed receptors, the subunits can be grouped into three categories: four AMPA subunits (GluA1-4, formerly called GluR1–4 or A–D), five high-affinity kainate subunits (GluK1–5, formerly GluR5–7, KA-1, KA-2) and seven NMDA subunits (GluN1, GluN2A–D, GluN3A–B, formerly NR1, NR2A–D, NR3A–B). GluD1 and GluD2 have significant homology to glutamate receptor subunits yet they do not bind glutamate agonists. They are referred to as orphan subunits and their function remains unclear. The GluD2 subunit is particularly intriguing because a mutation in the gene encoding this protein is responsible for the neurological phenotype of the Lurcher mouse. Relatively little is known about GluN3 subunits and it is unclear whether neurons make NMDA receptors that contain NR3 subunits. Curiously, when GluN1 and GluN3A or GluN3B are co-expressed in heterologous cells, they form excitatory channels that are gated by glycine rather than glutamate.
Some AMPA receptor subunits can combine to form functional homo-oligomeric receptors and this has revealed some interesting and curious phenomena. For example, the genes for GluA2, GluK1 and GluK2 code for a glutamine in the P loop region. However, the mRNA undergoes editing such that the expressed protein has an arginine residue at this site. This switch has a profound effect on the behavior of the channel. The current evoked by homo-oligomeric expression of unedited subunits shows marked inward rectification and an increased permeability to Ca2+. The edited versions have a linear current–voltage relationship and are permeable only to monovalent cations (Na+ and K+). Because the synaptic response mediated by AMPA receptors in hippocampal pyramidal cells has a linear current–voltage relationship, most AMPA receptors in these neurons contain one or more edited copies of GluA2. However, in interneurons in cortex and hippocampus as well as in dorsal horn neurons of the spinal cord, AMPA receptors lacking edited GluA2 subunits show inward rectification and increased permeability to Ca2+.
Glutamate channels, particularly NMDA channels, are regulated by a variety of allosteric mechanisms and by phosphorylation. The most dramatic are the actions of glycine as a co-agonist that is required for the opening of NMDA channels and extracellular Mg2+ that blocks NMDA channels. Binding of two molecules of glutamate and two molecules of glycine are required to activate an NMDA channel. Because glutamate channels are tetramers, it is thought that each subunit contributes to agonist binding. The glycine concentration in cerebrospinal fluid exceeds the EC50 for the glycine site, thus it is likely that the glycine site is tonically saturated. Recent evidence indicates that endogenous D-serine may also bind to the glycine site, with approximately the same affinity and efficacy as glycine. Although AMPA receptor channels do not require a co-agonist, given their homology with NMDA receptor channels, four molecules of glutamate are probably necessary to activate AMPA channels (Rosenmund et al., 1998). Other regulatory factors that affect NMDA channel activity include extracellular protons, Zn2+, polyamines, redox potential and intracellular Ca2+. The ability of these factors to affect NMDA receptors can depend on subunit composition. For example, the GluN1/GluN2A receptors are almost 100-fold more sensitive to Zn2+ than GluN1/GluN2B receptors. There are numerous phosphorylation sites on the C-terminal domains of AMPA and NMDA receptor subunits that can be regulated by protein kinase A and C, tyrosine kinases and Ca2+/calmodulin-dependent kinase (Chen and Roche, 2007). Kinase activation can alter any of a number of channel properties including gating and membrane trafficking. Phosphorylation of AMPA and NMDA receptors can also affect receptor trafficking that can then govern the ability of synapses to respond to stimuli that trigger long-term changes in synaptic efficacy. Activation of the NMDA receptor itself and the subsequent Ca2+ influx can result in PKC activation, which can then alter AMPA receptor trafficking. The impact of these regulatory mechanisms on synaptic function is a continuing area of investigation (Lee, 2006).
As with other neurotransmitter receptors, glutamate receptors are clustered in the postsynaptic membrane at synaptic sites. Recent studies have revealed a number of proteins that are involved in the clustering and localization of glutamate receptor channels. Glutamate channels are imbedded in the postsynaptic density (PSD) that contains receptors, regulatory proteins and cytoskeletal proteins. Biochemical studies suggest that calmodulin and cytoskeletal proteins can interact with C-terminal domains of several NMDA receptor subunits at domains that are involved in the regulation of NMDA receptor desensitization, suggesting an intriguing link between dynamic regulation of channel activity (e.g. by compartmentalized regulatory proteins) and structural features, such as channel anchoring or localization at synapses. A family of proteins with a PDZ domain are involved in interactions with AMPA and NMDA subunits (Ehlers et al., 1996; Sheng and Pak, 2002). TARPs (transmembrane AMPA receptor regulatory proteins) are a family of proteins that share homology with auxiliary subunits of voltage-gated calcium channels. TARPs alter the gating and pharmacological sensitivity of AMPA receptors (Milstein and Nicoll, 2008) and thus are considered auxiliary AMPA receptor subunits (Mayer, 2005). Other recently identified proteins, like CKAMP44, may have similar regulatory actions on AMPA receptors. The interactions between glutamate receptors and their associated proteins is important in receptor lateral mobility and trafficking, synapse development and the dynamic regulation of activity of ligand-gated channels in a broad range of brain functions including learning and memory.
BIBLIOGRAPHY
1. Ascher P, Nowak L. Electrophysiological studies of NMDA receptors. Trends Neurosci. 1987;10:284–288.
2. Bekkers JM, Stevens CF. Computational implications of NMDA receptor channels. Cold Spring Harbor Symp Quant Biol. 1990;55:131–135.
3. Brejc K, van Dijk WJ, Klaassen RV, et al. Crystal structure of Ach-binding protein reveals the ligand-binding domain of nicotinic receptors. Nature. 2001;411:269–276.
4. Browne LE, Jiang LH, North RA. New structure enlivens interest in P2X receptors. Trends Pharmacol Sci. 2010;31:229–237.
5. Burnstock G. Physiology and pathophysiology of purinergic neurotransmission. Physiol Rev. 2007;87:659–797.
6. Changeux J-P, Edelstein SJ. Allosteric receptors after 30 years. Neuron. 1998;21:959–980.
7. Chen BS, Roche KW. Regulation of NMDA receptors by phosphorylation. Neuropharmacology. 2007;53:362–368.
8. Chen GQ, Cui C, Mayer ML, Gouaux E. Functional characterization of a potassium-selective prokaryotic glutamate receptor. Nature. 1999;402:817–821.
9. Collingridge GL, Lester RA. Excitatory amino acid receptors in the vertebrate central nervous system. Pharmacol Rev. 1989;41:143–210.
10. Collingridge GL, Olsen R, Peters J, Spedding M. A nomenclature for ligand- gated ion channels. Neuropharmacology. 2009;56:2–5.
11. Colquhoun D. Binding, gating, affinity and efficacy: the interpretation of structure-activity relationships for agonists and of the effects of mutating receptors. Brit J Pharmacol. 1998;125:923–947.
12. Dani JA, Bertrand D. Nicotinic acetylcholine receptors and nicotinic cholinergic mechanisms of the central nervous system. Annu Rev Pharamacol Toxicol. 2007;47:699–729.
13. Ehlers MD, Mammen AL, Lau LF, Huganir RL. Synaptic targeting of glutamate receptors. Curr Opin Cell Biol. 1996;8:484–489.
14. Fritschy JM, Harvey RJ, Schwarz G. Gephyrin: where do we stand, where do we go?. Trends Neurosci. 2008;31:257–264.
15. Furukawa H, Gouaux E. Mechanisms of activation, inhibition and specificity: crystal structures of the NMDA receptor NR1 ligand-binding core. EMBO J. 2003;22:2873–2885.
16. Graus F, Saiz A, Dalmau J. Antibodies and neuronal autoimmune disorders of the CNS. J Neurol. 2010;257:509–517.
17. Hibbs RE, Gouaux E. Principles of activation and permeation in an anion-selective Cys-loop receptor. Nature 2011; May 15 [Epub ahead of print].
18. Hilf RJ, Dutzler R. A procaryotic perspective on pentameric ligand-gated ion channel structure. Curr Opin Struct Biol. 2009;19:418–424.
19. Hille B. Ionic Channels of Excitable Membranes. 3rd ed. Sunderland, MA: Sinauer Associates Inc.; 2001.
20. Hollmann M, Heinemann S. Cloned glutamate receptors. Annu Rev Neurosci. 1994;17:31–108.
21. Jahr CE, Stevens CF. Glutamate activates multiple single channel conductances in hippocampal neurons. Nature. 1987;325:522–525.
22. Jonas P, Bischofberger J, Sandkühler J. Corelease of two fast neurotransmitter at a central synapse. Science. 1998;281:419–424.
23. Jones MV, Westbrook GL. The impact of receptor desensitization on fast synaptic transmission. Trends Neurosci. 1996;19:96–101.
24. Julius D. Molecular biology of serotonin receptors. Annu Rev Neurosci. 1991;14:335–360.
25. Karlin A. Explorations of the nicotinic acetylcholine receptor. Harvey Lectures. 1991;85:71–107.
26. Kullmann DM. Neurological channelopathies. Annu Rev Neurosci. 2010;33:151–172.
27. Lee H-K. Synaptic plasticity and phosphorylation. Pharmacol Therapeut. 2006;112:810–832.
28. Macdonald RL, Twyman RE. Kinetic properties and regulation of GABAA receptor channels. Ion Channels. 1992;3:315–343.
29. MacKinnon R. Nobel lecture. Potassium channels and the atomic basis of selective ion conduction Biosci Rep. 2004;24:75–100.
30. Mayer ML. Glutamate receptor ion channels. Curr Opin Neurobiol. 2005;15:282–288.
31. Mayer ML. Glutamate receptors at atomic resolution. Nature. 2006;440:456–462.
32. McBain C, Mayer M. N-methyl-D-aspartic acid receptor structure and function. Physiol Rev. 1994;74:723–759.
33. Milstein AD, Nicoll RA. Regulation of AMPA receptor gating and pharmacology by TARP auxiliary subunits. Trends Pharmacol Sci. 2008;29:333–339.
34. Miyazawa A, Fujiyoshi Y, Stowell M, Unwin N. Nicotinic acetylcholine receptor at 4.6 A resolution: transverse tunnels in the channel wall. J Mol Biol. 1999;288:765–786.
35. Mohrluder J, Schwarten M, Willbold D. Structure and potential function of gamma-aminobutyrate type A receptor-associated protein. FEBS J. 2009;279:4989–5005.
36. Nakanishi S. Molecular diversity of glutamate receptors and implications for brain function. Science. 1992;258:597–603.
37. Neher E. Ion channels for communication between and within cells. Science. 1992;256:498–502.
38. Olsen RW, Sapp DM, Bureau MH, Turner DM, Kokka N. Allosteric actions of central nervous system depressants including anesthetics on subtypes of the inhibitory gamma-aminobutyric acidA receptor-chloride channel complex. Ann NY Acad Sci. 1991;625:145–154.
39. Olsen RW, Sieghart W. GABAA receptors: subtypes provide diversity of function and pharmacology. Neuropharmacology. 2009;56:141–148.
40. Padgett CL, Slesinger PA. GABAB receptor coupling to G-proteins and ion channels. Adv Pharmacol. 2010;58:123–147.
41. Role LW. Diversity in primary structure and function of neuronal nicotinic acetylcholine receptor channels. Curr Opin Neurobiol. 1992;2:254–262.
42. Rosenmund C, Stern-Bach Y, Stevens CF. The tetrameric structure of a glutamate receptor channel. Science. 1998;280:1596–1599.
43. Sakmann B. Nobel lecture Elementary steps in synaptic transmission revealed by currents through single ion channels. Neuron. 1992;8:613–629.
44. Sheng M, Pak DT. Ligand-gated ion channel interactions with cytoskeletal and signaling proteins. Annu Rev Physiol. 2002;62:755–778.
45. Sine SM, Engel AG. Recent advances in Cys-loop receptor structure and function. Nature. 2004;440:448–455.
46. Smit AB, Syed LI, Schaap D, et al. A glial-derived acetylcholine-binding-protein that modulates synaptic transmission. Nature. 2001;411:261–268.
47. Sobolevsky AI, Rosconi MP, Gouaux E. X-ray structure, symmetry and mechanism of an AMPA-subtype glutamate receptor. Nature. 2009;462:745–756.
48. Steinbach JH. Structural and functional diversity in vertebrate skeletal muscle nicotinic acetylcholine receptors. Annu Rev Physiol. 1989;51:353–365.
49. Stern-Bach Y, Bettler B, Hartley M, Sheppard PO, O’Hara PJ, Heineman SF. Agonist selectivity of glutamate receptors is specified by two domains structurally related to bacterial amino acid-binding proteins. Neuron. 1994;13:1345–1357.
50. Swope SL, Moss SJ, Raymond LA, Huganir RL. Regulation of ligand-gated ion channels by protein phosphorylation. Adv Second Messenger Phosphoprotein Res. 1999;33:49–78.
51. Unwin N. Refined structure of the nicotinic acetylcholine receptor at 4 A resolution. J Mol Biol. 2005;346:967–989.
52. Westbrook GL, Jahr CE. Glutamate receptors in excitatory neurotransmission. Sem Neurosci. 1989;1:103–114.
53. Wisden W, Seeburg PH. GABAA receptor channels: from subunits to functional entities. Curr Opin Neurobiol. 1992;2:263–269.
54. Wu LJ, Sweet TB, Clapham DE. International Union of Basic and Clinical Pharmacology LXXVI Current progress in the mammalian TRP ion channel family. Pharmacol Rev. 2010;262:381–404.