Chapter 33
Excitation—Secretion Coupling
Chapter Outline
III. Cellular Components Involved in Excitation–Secretion Coupling
IIIA. Interaction of Cytoskeletal Structures with Transmembrane Signaling Molecules
IIIB. Actin-Binding Proteins Important in Signaling
IIIC. Interaction between Microtubules and Microtubule-Associated Proteins in Signaling
IIID. Actin-Binding, Docking and Fusion Proteins of the Secretory Granules
IV. Cellular and Molecular Events in Chromaffin, Mast Cells and Neuronal Synaptic Vesicles
IVA. Dynamic Changes in the Cytoskeletal Networks are Required for Exocytosis
IVB. Physical Events Associated with the Fusion of Vesicles to Plasma Membrane
I Summary
The first step in the secretory process for peptide hormones and neurotransmitters involves synthesis, modification and sorting of the molecules to be secreted. These secretory molecules are packaged in secretory granules or vesicles, which are then transported to the cell periphery, where they are released in the extracellular space by fusion with the plasma membrane. This complex process is named exocytosis. Exocytosis is an all-or-none phenomenon in which Ca2+ plays a pivotal role.
Interaction of cytoskeletal structures with transmembrane signaling is an important feature of excitation–secretion coupling. Actin-binding proteins are in large part responsible for cytoskeleton–receptor interactions. In resting cells, more than 95% of the phosphatidylinositol bisphosphate (PtdInsP2) may be complexed with profilin. This interaction promotes actin polymerization. Following receptor activation, phospholipase C activity increases to a level where profilin protection can be overcome, resulting in PtdInsP2 hydrolysis. In resting cells, the bulk of gelsolin is cytosolic. When the cell is activated, there is a transient rise in [Ca2+]i due to InsP3 action and/or Ca2+ influx, which activates gelsolin, causing a 200-fold increase in its affinity for actin. This results in rapid filament side-binding, followed by severing and capping, inducing a dramatic disruption of existing actin network structure in the vicinity of [Ca2+]i elevation.
Dynamic changes in the cytoskeletal network are required for exocytosis. The subplasmalemmal area of secretory cells is characterized by the presence of a highly organized cytoskeletal network where F-actin, together with specific actin-binding proteins, forms a dense viscoelastic gel. Some actin-associated proteins, such as fodrin, caldesmon, gelsolin and scinderin, exist on secretory granule membranes, linking actin microfilaments to secretory granules. Caldesmon and synapsin I are other proteins that are associated with synaptic vesicles. Synapsin I binds to spectrin and actin microfilaments and may serve as an anchor between synaptic vesicles and the cytoskeleton. Synapsin I phosphorylation results in the release of synaptic vesicles from their anchoring site on the cytoskeleton, allowing the vesicles to move to the active synaptic zones.
Secretion is a process that requires (1) the movement of secretory vesicles toward the plasma membrane, (2) the fusion of vesicles with the plasma membrane and (3) subsequent release of secretory contents into the cell exterior. The process of secretion is mediated by contractile elements either associated with secretory vesicles or present elsewhere in the cell. As microfilaments (F-actin) are preferentially localized in the cortical surface of the chromaffin cell, F-actin may act as a barrier to the secretory granules, impeding their contact with the plasma membrane. Upon stimulation, fodrin rearranges into patches beneath the plasma membrane. Such a redistribution could be related to the clearing of exocytotic sites at the level of the plasma membrane. Scinderin is a cytosolic protein that shortens actin filament length when Ca2+ is present in the medium. Stimulation induces both redistribution of scinderin from the cytosol to the cell cortex and F-actin disassembly, which precedes exocytosis. Docking of granules on the plasma membrane is an important step in exocytosis. Several proteins are involved in this process: synaptobrevin, syntaxin and SNAP-25, which form the SNAP receptor (SNARE). Priming of the granules is also a pivotal step in exocytosis. N-ethylmalimide-sensitive fusion protein (NSF) primes the granule through ATP hydrolysis; once primed, an increase in [Ca2+]i triggers the release. All these processes require the presence of Ca2+ in the extracellular medium. Therefore, only secretagogues that induce Ca2+ entry are able to produce these effects.
In regulated exocytosis, fusion of secretory granules with the plasma membrane is triggered by an appropriate signal. Every time a secretory granule fuses with the plasma membrane, the total capacitance of the cell increases by a value proportional to the surface of the new membrane added to the existing cell membrane. Capacitance step values generally range from 1 to 30 fF, corresponding to granule diameters of 0.2 to 1 pm. Freeze-fracture images of exocytosis reveal the presence of narrow pores formed between the granules and the plasma membrane called fusion pores. The size of the pore increases as fusion progresses, allowing for the release of vesicle contents.
Control of exocytosis occurs not only in electrically excitable cells, but also in non-excitable systems in response to receptor activation. The regulatory pathways that couple stimulation and secretion vary widely among cell types. In many cells, Ca2+ is the key signal for triggering exocytosis. Ca2+ influx from the external medium is crucial for secretion, but internal Ca2+ release from internal stores also plays a pivotal role, depending on cell type. In some cases, Ca2+ alone is not able to trigger secretion, but the presence of a guanine nucleotide together with Ca2+ is necessary and sufficient for exocytosis.
II Introduction
The process of excitation–secretion coupling is completely different depending on the peptide/amine or lipid nature of the secretory products. The secretory process for peptide and amine molecules begins with the synthesis, modification and sorting of the molecules to be secreted. Synthesis occurs in the rough endoplasmic reticulum (RER) and sorting occurs in the Golgi complex. The secretory molecules are packaged in secretory granules or vesicles, which are then transported to the cell periphery before they are released in the extracellular space by fusion with the plasma membrane. This complex process is named exocytosis or reverse pinocytosis and can be operated either by a constitutive or a regulated mechanism. Constitutive secretion is unregulated and closely follows the rate of synthesis of the secretory products. This form of secretion occurs in many cell types, including lymphocytes, hepatocytes and pancreatic β cells. In regulated secretion, fusion of the secretory granules with the plasma membrane is initiated by a specific signal (ligand-receptor coupling), triggered by an increase in cytosolic calcium concentration ([Ca2+]i). In steroid-secreting cells (adrenal cortex, ovary, testis), the process of synthesis begins with cholesterol stored in lipid droplets followed by subsequent steps occurring in mitochondria and smooth endoplasmic reticulum. It is generally assumed that steroids are free to diffuse throughout the aqueous cytoplasm and lipid phase of the plasma membrane. Secretory vesicles are not present and secretion and/or release of steroids is tightly coupled to steroid synthesis.
Although the process of synthesis differs, stimulation of secretion of peptide hormones, neurotransmitters and steroids involves similar cascades of molecular events. After binding to their specific receptors, the stimuli activate second messenger production, several cascades of phosphorylation/dephosphorylation of intracellular proteins, cytoskeleton reorganization, synthesis of new products and release of secretory products. Cytoskeleton and Ca2+ ion are certainly the most important players involved in this excitation–secretion coupling. However, while disruption of the actin network is necessary to trigger fusion of secretory vesicles with the cell membrane, a well-preserved organization seems important for steroid release.
III Cellular Components Involved in Excitation–Secretion Coupling
IIIA Interaction of Cytoskeletal Structures with Transmembrane Signaling Molecules
The cytoskeletal elements are described in Chapter 5. Therefore, the brief descriptions given here on microfilaments and microtubules are aimed at understanding the mechanisms involved in excitation–secretion coupling (Schmidt and Hall, 1998). In the living cell, actin filaments, F-actin (consisting of two staggered, parallel rows of monomers, G-actin, non-covalently bound and twisted into a helix), interact with several proteins, such as vinculin, α-actinin, villin and fodrin, which cross-link and bundle microfilaments into a well-organized three-dimensional network. They can also cross-link myosin, forming a contractile network, or form a very dense network at the cell periphery, the cell cortex (Figs. 33.1A and 33.2A). This dynamic organization of microfilaments depends on a large and diverse group of actin-binding proteins, including profilin and caldesmon (which bind G-actin) and gelsolin and scinderin (which cap and sever F-actin). On the other hand, polymerization, stabilization and modulation of microtubules depend on several microtubule-associated proteins (MAPs) that adorn the tubulin-containing core of the tubules. The complete cell cytoskeletal network includes interaction between microfilaments, microtubules and intermediate filaments (see Figs. 33.1 and 33.2 and Chapter 5).
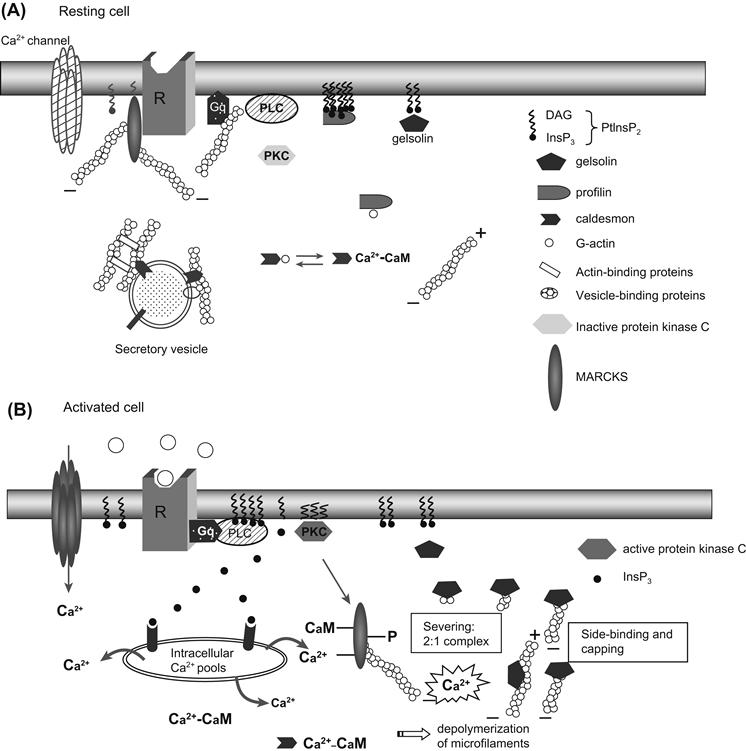
FIGURE 33.1 Interaction of cytoskeletal structures with transmembrane signaling. In resting cells (A), receptors, G proteins, phospholipase C (or adenylyl cyclase) and phosphoinositides are not linked together, but are associated with microfilaments or actin-associated proteins. For example, profilin interacts with high affinity with eight molecules of PtdInsP2, protecting them from hydrolysis by phospholipase C; gelsolin is also associated with PtInsP2; the protein caldesmon is associated with both actin monomers and calmodulin; MARCKS (myristoylated, alanine-rich C kinase substrate) is a specific protein kinase C substrate associated with the cytoplasmic face of the membrane under resting conditions. In its non-phosphorylated form, MARCKS cross-links actin, favoring a rigid actin meshwork at the membrane level. In activated cells (B), ligand binding to its receptor activates phospholipase C, which hydrolyzes PtdInsP2 and leads to InsP3 and diacylglycerol (DAG). The rise in [Ca2+] (due to the InsP3 binding to intracellular pools of Ca2+) induces F-actin depolymerization. Moreover, calmodulin–caldesmon complex binds to Ca2+, gelsolin binds, severs and caps actin filaments into 2:1 complexes to the barbed ends. Activated protein kinase C (now at the membrane) phosphorylates MARCKS, which is released from the membrane. All these modifications decrease cell rigidity, making the actin network more plastic.
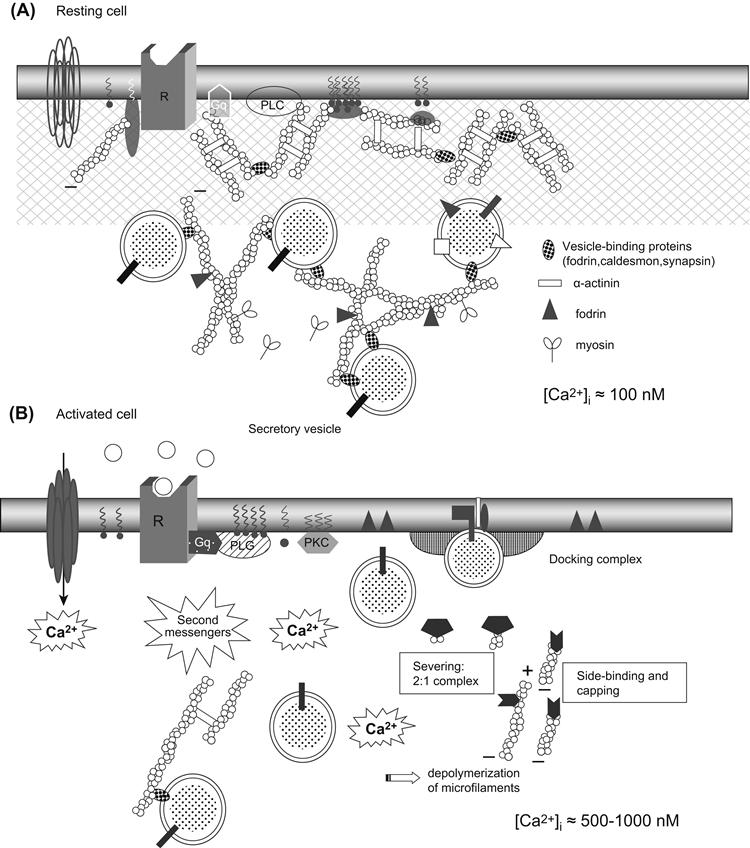
FIGURE 33.2 Dynamic changes in cytoskeleton during exocytosis. In resting cells ([Ca2+]i ≈100 nM) (A), microfilaments interact with several proteins (including fodrin and α-actinin) that cross-link and bundle microfilaments into a well-organized three-dimensional network. In vesicle-secretory cells, microfilaments can also cross-link myosin, forming a contractile network, or form a very dense network at the cell periphery, the cell cortex. Activation of cells by appropriate stimulation (B) is associated with a reduction in the cell cortex rigidity, due to characteristic changes in cytoskeletal properties from “gel” (solid) to “sol” (liquid) states. These changes are primarily under the control of Ca2+, which cooperates with phospholipid messengers to induce microfilament disruption. An increase in intracellular calcium ([Ca2+]i (≈0.5–1 μM) induces several cellular modifications. In addition to capping and severing of the actin microfilaments (see Fig. 33.1), there is dissociation of actin from actin-associated proteins, such as fodrin, patching of fodrin along the plane of the plasma membrane (docking site) and dissociation of caldesmon from secretory vesicles. These events result in a decrease in viscosity, which favors movement of granules toward the plasma membrane releasing sites. Actin–myosin interactions could facilitate granule displacement in cytosol.
Association of cell surface molecules with the cytoskeleton is widely believed to be one of the earliest consequences of cellular activation for many systems. These cell surface molecules include not only receptors, but also integrins, the receptors of the components of extracellular matrix. Appropriate stimulation of receptors and integrins has immediate and profound effects on the organization and activity of the microfilaments (Aplin et al., 1998). Several studies have shown that microfilament disruption (with cytochalasins) or microtubule disruption (with colchicine or vinblastine) increased exocytosis (i.e. peptide or amine secretion), although steroid hormone secretion is decreased or abolished. In other words, these observations indicate that cytoskeleton is a barrier in the former, but a requirement for the latter. In both cases, microfilaments are active players in the process of excitation–secretion coupling. Several studies indicate that receptors (either G-protein-coupled or tyrosine kinase types), α subunits of heterotrimeric G proteins (αi, αs, αq), monomeric G proteins (Rho, Rac, Cdc42), second-messenger-activating enzymes (adenylyl cyclase, phospholipase C [PLC]), Ca2+, K+ or Cl− channels (see Chapter 31) and proteins up or downstream from second messenger production (phosphatases, protein kinase C [PKC], mitogenic associated protein kinase [MAPK]) are associated with microfilaments. For example, anchoring of PKC-e to F-actin is required for glutamate release in nerve endings of neuronal cells (Janmey, 1998). In general, these associations occur mainly via acting-binding proteins, which bind effectors or phosphoinositides through specific domains, called plecktrin homology domains, (PH domains). PH domains are essential for the membrane recruitment of several proteins that contain them and are frequently accompanied by other motifs (Src homology domains SH2 and SH3 and proline-rich, Dbl homology [DH], or GTP exchange domains), indicating that the recruitment of the PH domain protein would nucleate sites on the membrane for protein complex assembly (Inglese et al., 1995; Janmey, 1998; Martin, 1998). In addition, close functional association between G proteins and microtubules has also been extensively described (Popova et al., 1997). Such observations indicate that the cytoskeleton operates as a matrix improving the efficiency of the signal transduction cascade and that actin-binding proteins are in large part responsible for these cytoskeleton–membrane receptor interactions.
IIIB Actin-Binding Proteins Important in Signaling
The physical properties of actin networks depend on the length of microfilaments and the architecture of the three-dimensional network formed by interaction between actin and several actin-binding proteins. The aim of this section is to point attention to the F-actin cross-linking proteins which may be regulated by Ca2+ and thus involved in the process of excitation–secretion coupling (Schmidt and Hall, 1998).
Profilin was the first actin-binding protein described in signaling. Profilin also contains a PtdInsP2 binding site and binding of PtdInsP2 to this site triggers the dissociation of profilin from actin, promoting actin polymerization (see Fig. 33.1A). PtdInsP2 molecules bind to profilin with a stoichiometry of about 8:1. This 8:1 association of PtdInsP2:profilin complexes protects PtdInsP2 from cleavage by phospholipase Cγ (PLCγ). In a similar fashion, also in resting conditions, PLCγ may also bind profilin. However, when PLCγ becomes tyrosine phosphorylated (by appropriate ligand activation), its affinity for PtdInsP2 increases to a level where profilin protection can be overcome, resulting in PtdInsP2 hydrolysis. Hydrolysis of one or two of the eight PtdInsP2 molecules bound to each profilin leads to a rapid decrease in PtdInsP2 affinity and release of the remaining profilin-bound PtdInsP2 molecules (see Fig. 33.1B). Thus, the binding of profilin to PtdInsP2 not only liberates polymerization-competent G-actin, but also affects hydrolysis of PtdInsP2 by PLCγ (Forscher, 1989; Janmey, 1998).
Gelsolin is a Ca2+-dependent F-actin severing molecule which, along with villin, fragmin, adseverin and scinderin, is able to sever actin filaments. These proteins bind to actin filaments and bend and cleave them in a Ca2+-dependent manner and afterwards cap the barbed filament ends. Severing actin filaments promotes cell cortex transition from a “gel” state to a “sol” state upon addition of Ca2+. Gelsolin contains two spatially separate binding sites: a G-actin Ca2+-sensitive site in the C-terminal domain and a PtdIns-sensitive site closer to the N-terminal portion. In resting cells ([Ca2+]i ≈100 nM), the bulk of gelsolin is cytoplasmic and a small proportion is PtdInsP2-associated. Gelsolin has little affinity for actin under these conditions and is in an actin-free state (see Fig. 33.1A). When the cell is activated, there is a transient rise in [Ca2+] due to InsP3 action or Ca2+ influx that then activates gelsolin, causing a 200-fold increase in its affinity for F-actin. This results in rapid filament side-binding, followed by severing and capping of any free barbed filament ends, inducing a dramatic disruption of existing actin network structure in the vicinity of Ca2+ elevation (see Fig. 33.1B). The activities of these severing proteins are inhibited by binding to PtdInsP2.
In the absence of PtdIns turnover, much of the PtdInsP2 is likely to be tightly associated with profilin and thus unavailable to gelsolin. If gelsolin is activated under these conditions (e.g. by an increase in [Ca2+]i, independent of the PtdIns turnover), severing of actin networks is observed, but without subsequent actin reassembly. This leads to two possible modes of gelsolin activation: (1) calcium influx produced by activation of PtdInsP-independent pathways (i.e. voltage- or agonist-gated Ca2+ channels) leads to actin severing and capping only; (2) in contrast, activation of these same Ca2+ channels concomitant with PtdInsP turnover results in severing and capping followed by actin polymerization, i.e. actin remodeling (Forscher, 1989; Janmey, 1998).
In summary, association of actin-binding proteins with phosphoinositides causes opposite effects to those observed upon association with Ca2+. In the former, such associations (profilin: PtInsP2) promotes actin polymerization, favoring the cortical actin “gel” near the plasma membrane, while increased Ca2+ concentration favors association of actin-binding proteins (profilin, gelsolin) with Ca2+, inducing depolymerization and thus “sol” state of actin filaments.
Caldesmon is a calmodulin-dependent actin-binding protein that, at low Ca2+ concentrations (100 nM), binds and cross-links actin monomers, inhibiting actin polymerization. Under these conditions, caldesmon interacts reversibly with secretory granules. At greater concentrations of Ca2+ (μM ranges), Ca2+-calmodulin complex binds to caldesmon and reverses this inhibition. The flip-flop regulation of caldesmon may be important for secretory vesicle function during the changes in intracellular Ca2+ levels observed upon stimulation (see Figs. 33.1 and 33.2).
MARCKS (myristoylated, alanine-rich C kinase substrate) is a specific protein kinase C substrate that is targeted to the membrane by its amino-terminal binding domain. In resting cells, MARCKS associates with the cytoplasmic face of the membrane. In its non-phosphorylated form, MARCKS cross-links actin, favoring a rigid actin meshwork at the membrane level (see Fig. 33.1A). Activated protein kinase C phosphorylates MARCKS, which remains associated with actin filaments, but can no longer cross-link actin fibers, making the actin network more plastic (see Fig. 33.1B). In addition, an increase in intracellular Ca2+ concentration promotes binding of calmodulin to MAR-CKS, inhibiting its actin cross-linking activity, again resulting in a less rigid actin meshwork. Thus, PKC induces a local destabilization of the actin skeleton through the phosphorylation of MARCKS. MARCKS is phosphorylated when synaptosomes are depolarized, suggesting a role in secretion (Arbuzova et al., 1998).
Several newly identified proteins, such as the focal adhesion molecule (p125FAK), paxillin and the small GTP-binding protein Rho are also closely implicated in actin polymerization after hormonal stimulation. Tyrosine phosphorylation of p125FAK and paxillin and their association with cytoskeleton and βγ subunits of G proteins have been recently identified as early events in the action of several growth factors and G-protein-coupled receptors (such as angiotensin II and vasopressin). However, to date, their role has been ascribed to regulating cell adhesion, motility or proliferation rather than secretion (Inglese et al., 1995; Tapon and Hall, 1997; Hall, 1998).
IIIC Interaction between Microtubules and Microtubule-Associated Proteins in Signaling
As mentioned earlier, polymerization, stabilization and plasticity of microtubules depend on several microtubule-associated proteins (MAPs), which differentially cross-link microtubules (Matus, 1988). Recent studies have shown that some MAPs could be implicated in the secretory process (Gundersen and Cook, 1999). MAPs are substrates for several protein kinases, including a Ca2+-calmodulin-dependent kinase, a cAMP-dependent kinase, tyrosine kinases and protein kinase C. Both cAMP (via protein kinase A) and Ca2+ (via Ca2+-calmodulin kinase) lead to phosphorylation of MAP-2. Moreover, MAP-1 and MAP-2 are responsible for binding secretory granules to microtubules, either in cells from the anterior pituitary gland, the β cells of the endocrine pancreas or in synaptic vesicles of neurons. These results strengthen the probable role of MAPs in secretory processes.
IIID Actin-Binding, Docking and Fusion Proteins of the Secretory Granules
Granule vesicles for peptides or amine products not only allow secretory tissues to store large amounts of secretory products in a relatively small volume, but also protect this material from intracellular degradation while providing a very efficient means for transporting and releasing fixed quantities of secretory material.
IIID1 Actin-binding Proteins Associated with Secretory Granules
Induction of exocytosis is associated with a loss in the cell cortex rigidity, due to characteristic changes in cytoskeletal properties from “gel” to “sol” states. These changes are primarily under the control of Ca2+, which cooperates with phospholipid messengers to induce microfilament disruption (see Figs. 33.1B and 33.2B) (Tchakarov et al., 1998). Moreover, several pieces of evidence indicate that microfilaments, rather than microtubules or intermediate filaments, are responsible for the mechanical changes initiated by Ca2+. Chromaffin cells of the adrenal medulla and mast cells from the immune system synthesize and, along with neuronal synaptic vesicles, store and secrete large amounts of neurotransmitters or neuropeptides. These cells contain numerous electron-dense secretory granules which discharge their contents into the extracellular space by exocytosis. The subplasmalemmal area is characterized by the presence of a highly organized cytoskeletal network. F-actin seems to be exclusively localized in this area and, together with specific actin-binding proteins, forms a dense viscoelastic gel.
Fodrin, vinculin, α-actinin and caldesmon, four actin-binding proteins, as well as gelsolin and scinderin, two actin-severing proteins, are found in the plasmalemmal region. Moreover, fodrin, caldesmon and α-actinin binding sites also exist on secretory granule membranes, indicating that actin filaments can also link to secretory granules (see Fig. 33.2A). Chromaffin granules can be entrapped in this subplasmalemmal lattice and thus the cytoskeleton acts as a barrier preventing exocytosis (Burgoyne, 1995; Burgoyne and Morgan, 1998a).
Synapsin I, a phosphoprotein and a substrate for protein kinase A and calmodulin-dependent protein kinase II (CaM kinase II), is associated with synaptic vesicles. Synapsin I also binds to spectrin and actin microfilaments and may serve as an anchor between synaptic vesicles and the cytoskeleton. The affinity of synapsin I for synaptic vesicles is decreased by phosphorylation and neurotransmitter release is preceded by a reversible phosphorylation of synapsin I. Therefore, synapsin I phosphorylation results in the release of synaptic vesicles from their anchorage sites on the cytoskeleton, thus allowing the vesicles to move to the active exocytosis zones.
IIID2 Docking and Fusion Proteins
In addition to the actin-binding proteins, several proteins from the secretory vesicle membrane, the plasma membrane and the bulk cytosol interact together to form the core complex (or a docking complex) with docking, priming and fusioning properties (or budding properties). The most important of these properties are described next (Südhof, 1995; Fernandez-Chacon and Südhof, 1999).
The vesicular proteins synaptobrevins (or vesicle-associated membrane proteins, VAMPS) and the plasma membrane proteins syntaxin and synaptosome-associated protein (SNAP-25, Mr 25 kDa) form the three complex membrane proteins (called the core complex) essential to the regulated exocytosis machinery in neurons and neuroendocrine and endocrine cell types. Moreover, the potential involvement of the cytosolic proteins, N-ethylmalimide-sensitive fusion protein (NSF) and the soluble NSF attachment proteins (SNAPs) (α, β, γ isoforms), was demonstrated by the discovery that all these proteins interact to form a complex named SNARE (for SNAP receptor) (Fig. 33.3). Based on in vitro studies, a model for neurotransmitter release was proposed in which synaptic vesicles become docked at the plasma membrane by a specific pairing of VAMP (the vesicle membrane, v-SNARE) with syntaxin 1 and SNAP-25 (the target membrane, t-SNARE). The proposed sequence for docking and priming is described hereafter (Südhof, 1995; Aroeti et al., 1998; Burgoyne and Morgan, 1998a). Before and/or during docking, syntaxin is bound to Munc 18 and synaptophysin to synaptobrevin. These two complexes, syntaxin–Munc18 and synaptophysin–synaptobrevin, must dissociate in order for the core complex to be formed. Formation of this complex is considered to be the first step of vesicle priming. Syntaxin and SNAP-25 (t-SNARE) can then bind tightly together to form a high-affinity site for synaptobrevin (v-SNARE) located on the vesicle membrane; the core complex has a stoichiometry of 1:1:1. The trimeric core complex serves as a receptor (SNARE) for the soluble SNAPs (not related to SNAP-25). NSF will only interact with SNAPs (α, β) bounded on the trimeric complex. The NSF-SNAP receptor forms a multisubunit particle that sediments at 20S; it may form the core of a generalized apparatus catalyzing bilayer fusion (Söllner et al., 1993). NSF is a trimeric protein that cross-links multiple core complexes into a network. The core complex is then disrupted by enzymatic activity of NSF under ATP hydrolysis. Botulinum A and tetanus toxins are toxin proteases able to digest synaptobrevin, SNAP-25 and syntaxin. When entering in the nerve terminal, they irreversibly inhibit exocytosis. However, the number of docked granules is not decreased by the toxins, indicating that the primary function of the core complex is fusion and not docking. Hydrolysis of ATP by NSF is followed by ATP-independent steps (see later) sensitive to temperature, H+ and Ca2+. The last step is Ca2+-sensitive and likely involves a Ca2+ sensor at the site of exocytosis.
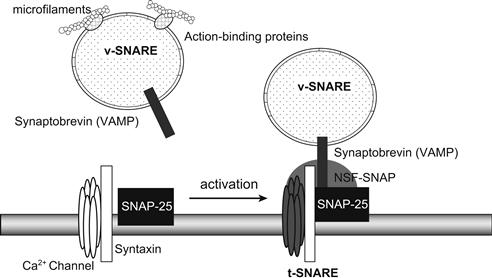
FIGURE 33.3 SNARE, the receptor involved in docking and fusion. The 20S particle that forms the core complex contains several interacting proteins. v-SNARE, related to synaptobrevin (VAMP) and located on the vesicle, binds to t-SNARE, related to syntaxin and SNAP-25 and located on the plasma membrane, to form the SNARE or SNAP receptor. SNAPs and NSF can then bind to the receptor to complete the core complex. Numerous SNARE-related proteins, each specific for a single kind of vesicle or target membrane, ensure vesicle-to-target specificity. Binding of synaptobrevin relies on the inhibition of the Ca2+ channel.
Synaptotagmins (Syt) are membrane glycoproteins found in brain secretory vesicles of which eight forms have been cloned. One of these, synaptotagmin I (Syt I), plays a pivotal role in the Ca2+-triggered neurotransmitter release as a Ca2+ sensor (Südhof and Rizo, 1996; Goda and Südhof, 1997; Burgoyne and Morgan, 1998b). The functional implication of multiple synaptotagmins is unknown. Syt I binds Ca2+ cooperatively and undergoes a Ca2+-dependent conformational change; the coefficient of cooperativity (4) is similar to that observed for Ca2+-triggered release. Syt I also binds phospholipids as a function of Ca2+ with high affinity (half maximal binding, 5–6 μM). Syntaxin, one of the proteins of the core complex, in addition to its role in the docking and fusion process of the vesicle, controls the entry of Ca2+ by the voltage-dependent Ca2+ channels (L- and N-type). Indeed, binding of syntaxin to the Ca2+ channel prevents the opening of the channel by membrane depolarization. When the vesicle docks to the membrane by binding to syntaxin and SNAP-25, the inhibition of syntaxin on the channel is relieved, allowing voltage-dependent opening of the channel (see Fig. 33.3) (Geppert and Südhof, 1998).
In addition to phospholipids and syntaxin, Syt I binds to neuroxins, a family of neuronal cell surface proteins, and to AP-2, a protein complex involved in synaptic vesicle endocytosis. In addition to a Ca2+ sensitivity similar to Ca2+-triggered release, the role of Syt I as a Ca2+ sensor has been illustrated in several ways. Knockout mice for Syt I have impaired Ca2+-triggered transmitter release, but release can still be obtained by Ca2+-independent agents, such as hypertonic sucrose or the excitatory neurotoxin α-latrotoxin (the receptor for α-latrotoxin belongs to the neuroxin family). Synaptotagmin Drosophila mutants show a severe but incomplete block of neurotransmission with an altered Ca2+-dependence in some mutants. Injection of synaptotagmin peptide in squid nerve terminal inhibits release and vesicles accumulate possibly by competing for a common effector. Synaptotagmins are also able to interact with non-neuronal syntaxin, indicating that they can play a role in a variety of cell types from endocrine and immune systems.
Synaptophysin and the related protein synaptogyrin are major integral membrane proteins of small presynaptic vesicles. The presence of phosphorylation sites for tyrosine kinase of the Src family could indicate that phosphorylation modulates protein activity. The primary structure was deduced from the cDNA sequence, leading to the proposition that synaptophysin and synaptogyrin span the membrane four times with N- and C-terminals located in the cytoplasm. Ubiquitous isoforms of these two proteins are co-expressed in all cells and could represent invariant components of trafficking organelles.
The annexin family includes several Ca2+- and phospholipid-binding proteins with conserved structure. Two of these are thought to be involved in the mechanism of exocytosis. Synexin (annexin VII) is a calcium-binding protein (47 kDa) with four transmembrane domains. This protein demonstrates a voltage-dependent Ca2+ channel activity. Synexin is able to induce the aggregation of chromaffin vesicles in the presence of Ca2+. A model for the synexin-driven Ca2+-dependent membrane fusion has been proposed in which synexin monomers polymerize as the concentration of Ca2+ increases. The polymerized synexin forms a hydrophobic bridge between the two membranes. Annexin II (calpactin) is located on the cytoplasmic side of the plasma membrane in chromaffin cells. Sites of phosphorylation for protein kinase C and camp- and calmodulin-dependent protein kinases have been localized within the N-terminal domain; phosphorylation of these sites could inactivate the protein. A possible role for annexin II could be to link the granule with the plasma membrane (docking) and/or to induce fusion (Burgoyne, 1991).
Exocytotic fusion mechanisms also involve PtdIns transfer protein, PtdIns 4-kinase (PI4K), and PI5K to promote formation of PtIns4,5P2 on the vesicle membrane (Martin, 1998).
IV Cellular and Molecular Events in Chromaffin, Mast Cells and Neuronal Synaptic Vesicles
Exocytosis is an all-or-none phenomenon in which Ca2+ plays a pivotal role. Ca2+ is required for second messenger activity, for the control of cytoskeletal dynamics and for the vesicle–plasma membrane fusion process. In neurons, neuroendocrine cells and some endocrine cells, the electrical activity of the cell, the action potential, leads to the opening of voltage-dependent Ca2+ channels with a subsequent increase in cytosolic Ca2+. Neurotransmitter release at synaptic and neuromuscular junctions, peptide hormone secretion and catecholamine release by the adrenal medulla all belong to this class. In non-excitable cells, the triggering Ca2+ signal is provided by release of Ca2+ from intracellular stores after appropriate stimulation. Depletion in Ca2+ from these intracellular pools activates an influx of Ca2+, which is responsible for the sustained increase in [Ca2+]i observed in many cell types. Moreover, this Ca2+ influx provides Ca2+ ions for the replenishment of internal stores.
IVA Dynamic Changes in the Cytoskeletal Networks are Required for Exocytosis
Secretion is a process that requires (1) the movement of secretory vesicles toward the plasma membrane, (2) the fusion of vesicles with the plasma membrane, and (3) subsequent release of secretory contents in the cell exterior.
As explained before, in resting conditions, actin filaments (F-actin) are preferentially localized in the cell surface and act as a barrier to the secretory granules, impeding their contact with the plasma membrane (Trifaró et al., 1992). Stimulation of chromaffin or mast cells as well as neuronal synaptic vesicles produces disassembly of the actin network and removal of the barrier (Tchakarov et al., 1998) (see Fig. 33.2). Several observations support this concept: (1) direct evidence for an actin barrier has come from the use of drugs that affect actin assembly and disassembly. Cytochalasin B and DNase I prevent actin assembly and drive the system toward net disassembly and increased secretion in permeabilized chromaffin cells. (2) Studies using fluorescent rhodamine-labeled phalloidin (a drug that stabilizes actin filaments in vitro and stops actin disassembly on stimulation) and actin antibodies have shown, in resting cells, a strong cortical fluorescent ring of filamentous actin. Cholinergic receptor stimulation produces a fragmentation of the fluorescent ring, leaving cell cortical areas devoid of fluorescence. These changes are accompanied by a decrease in F-actin associated with a concomitant increase in G-actin (Tchakarov et al., 1998) (Fig. 33.4). (3) Results from the use of toxins also support the concept of a cortical actin barrier. Botulinum C2 toxin, which ADP-ribosylates actin and inhibits actin polymerization, enhances secretion in PC12 cells. In contrast, tetanus and botulinum A toxins, which block actin disassembly, inhibit exocytosis upon cholinergic stimulation.
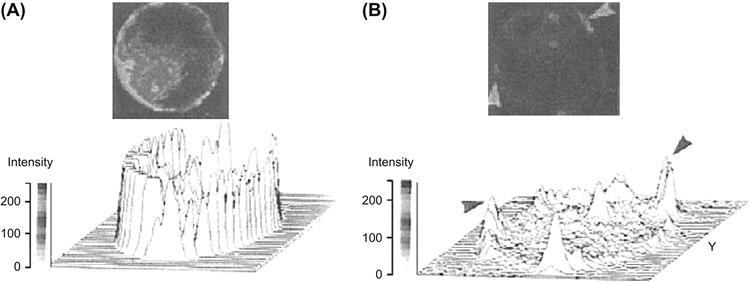
FIGURE 33.4 Rhodamine–phalloidin fluorescence of chromaffin cell cytoskeleton. Cultured chromaffin cells were incubated for 40 s in Locke’s solution in the absence (A) or in the presence (B) of 10 μM nicotine. Cells were fixed and processed for fluorescence studies. Resting chromaffin cells showed a bright and continuous cortical fluorescent ring (A), while nicotinic-receptor-stimulated cells showed disruption in the cortical fluorescent ring. Some fluorescent patches are shown by arrowheads (B). Three-dimensional image analysis of the same patterns is shown below each cell. In control cells, there is a uniform cortical fluorescence intensity pattern, whereas in stimulated cells, the cortical fluorescent intensity pattern shows irregularities such as valleys and peaks. These peaks correspond to the patches observed in cells. (Adapted with permission from Tchakarov et al., 1998.)
Several actin-binding proteins present in the cell cortex undergo changes upon stimulation. In a resting cell, fodrin is localized in the cell cortex. On stimulation, it rearranges into patches beneath the plasma membrane. This redistribution could be related to the clearing of exocytotic sites at the plasma membrane. Scinderin is a cytosolic protein that shortens actin filament length when Ca2+ is present in the medium. Stimulation induces both redistribution of scinderin from cytosol to cell cortex and F-actin disassembly, which precedes exocytosis. Thus, stimulation-induced redistribution of scinderin and F-actin disassembly produce subplasmalemmal areas of decreased cytoplasmic viscosity and high secretory vesicle mobility. All these processes require the presence of Ca2+ in the extracellular medium. Therefore, only secretagogues that induce Ca2+ entry are able to produce these effects (see Fig. 33.2B).
In isolated chromaffin cells, stimulation with nicotinic agonists can result in secretion of about 30% of the total catecholamine. Electron microscopic observations show that a small number of granules lie within the exclusion zone of the cell cortex. This demonstrates the importance of changes in cortical actin to allow movement of the bulk of the granules involved in a full secretory response. Nevertheless, low levels of exocytosis, due to these granules in the cortical exclusion zone, could occur without generalized changes in cortical actin. Thus, in a physiological situation, where relatively few exocytotic events occur per stimulus, changes in cortical actin may not be necessary for the initial wave of exocytosis, but are required for the movement, into the cortical exclusion zone, of granules ready for the next stimulus. A similar picture has emerged from studies on the nerve terminal cytoskeletal phosphoprotein, synapsin I, which is believed to cross-link synaptic vesicles and release them following depolarization and phosphorylation of the synapsin I.
IVB Physical Events Associated with the Fusion of Vesicles to Plasma Membrane
In regulated exocytosis, fusion of the secretory granules with the plasma membrane is triggered by an appropriate signal. The fusion process, following the triggering signal, can be very fast, such as in mammalian nerve terminals, with delays of less than 0.2 ms between the action potential and exocytosis. In some cells, however, delays of 0.2 s (chromaffin cells) to 50 s (mast cells) are observed. This delay is thought to be caused by the time required for production of second messengers possibly involved in exocytosis and removal of the cytoskeletal barrier that immobilizes the vesicles. However, the physical interactions between the granule membrane and the plasma membrane remain similar whether the exocytotic delay is fast or slow. Accordingly, the following fusion events will be described along general lines based on a sequence proposed by Almers (1990).
IVB1 Capacitance Jump
Each time a secretory granule fuses with the plasma membrane, the total capacitance of the cell increases by a value proportional to the surface area of the new membrane added to the existing cell membrane. Assuming that biological membranes have a constant specific capacitance of about 1 μF/cm2 allows a simple calculation of the granule size. Upon fusion of a single vesicle, the capacitance value increases abruptly to a new stable value as the membrane of the vesicle and the plasma membrane become continuous and the vesicle lumen opens into the extracellular space. Figure 33.5 illustrates the equivalent circuitry of a resting cell (Fig. 33.5A) and that of a cell undergoing exocytosis (Fig. 33.5B). During degranulation, several granules fuse with the plasma membrane, which produces a typical staircase recording (Fig. 33.5C). Degranulation in three different cell types are presented in Fig. 33.6: human neutrophil (Fig. 33.6A), guinea pig eosinophil (Fig. 33.6B) and horse eosinophil (Fig. 33.6C). Note that the amplitude of the individual step capacitance is lower in human neutrophil than in horse eosinophil, reflecting the different sizes of the vesicles. A capacitance step amplitude histogram is built by measuring the step height of a large number of individual events. An example of this is illustrated in Fig. 33.6D for neutrophils. The capacitance step amplitudes range from 1 to 6 fF (1 to 6×10−15 F), with a greater number of events having an amplitude of 2 fF. Assuming a spherical shape, the diameter of the granule can be calculated from the step change in capacitance, δCm in fF, by the relation:
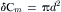 (33.1)
(33.1)
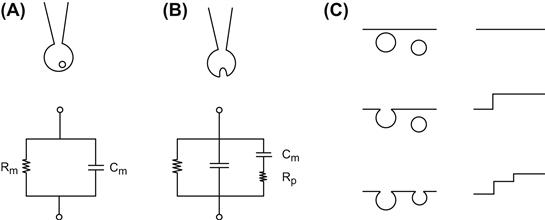
FIGURE 33.5 Capacitance measurement and equivalent electrical circuit. (A) The cell is patch-clamped in the whole-cell configuration. The cell is at rest and an unfused granule is shown near the plasma membrane. The equivalent circuit is represented by membrane resistance R in parallel with membrane capacitance Cm. The series resistance due mainly to the micropipette has been omitted for clarity. (B) Once the granule has fused with the plasma membrane, its capacitance Cv, proportional to the surface of membrane added, is added to the total capacitance of the cell. Rp is the fusion pore resistance, which will rapidly decrease as the pore dilates. (C) The fusion of a granule with the plasma membrane increases the capacitance by step. Granules of identical sizes induce the same increases in capacitance.
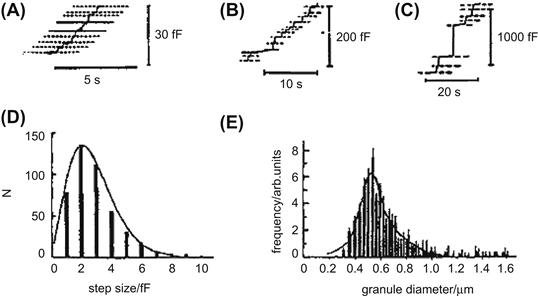
FIGURE 33.6 Analysis of step capacitance. Capacitance recordings obtained from different cells: (A) human neutrophils, (B) guinea pig eosinophils, (C) horse eosinophils. Note the staircase appearance of the recordings, which reflects the sequential fusion of individual granules and the size of the step capacitances, which are proportional to the size of the granules. (D) Frequency distribution of step sizes for human neutrophils. (E) Distribution of granule size for guinea pig eosinophils derived from capacitance measurement. (Adapted with permission from Lindau and Gomperts, 1991.)
The frequency distribution of the sizes of the granules obtained from the capacitance step amplitude histogram is represented in Fig. 33.6E for guinea pig eosinophils. The distribution is fitted (smooth line) by the sum of two Gaussian curves with means of 520 and 590 nm (Lindau and Gomperts, 1991). This size distribution fits in well with the morphometric data obtained by direct microscopic observation of the secretory vesicles. Capacitance step measurements have been achieved in a variety of cell types, including adrenal chromaffin cells, mast cells, pancreatic acinar cells, neutrophils, eosinophils, basophils, pituitary lactotrophs and nerve terminals derived from the posterior pituitary. Capacitance step values generally range between 1 and 30 fF, corresponding to granule diameters of 0.2 to 1 μm.
Giant vesicles with diameters ranging from 1 to 5 μm are found in mast cells from a strain of genetically defective beige mice (strain C57BL/6J-bgj/bgj). In these mice, mast cells and other granulocytes are unable to limit the size of their secretory vesicles; mast cells contain 10 to 40 giant vesicles that can easily be observed under photonic microscopy. These cells thus provide an ideal material for exocytosis studies and, for this reason, have been extensively used. The capacitance method offers the possibility to study degranulation in real time online; time analysis of the secretory process reveals that the granules fuse sequentially, one by one, with the plasma membrane. However, in mast cells, capacitance step analysis demonstrates the presence of step values greater than 60 fF, which could not be produced by the fusion of a single vesicle. A detailed analysis of the capacitance step histogram reveals a multimodal distribution of granule size, which indicates that the larger granules could be formed by the fusion of two to five single granules with each other.
IVB2 Capacitance Flickering
The pattern of the staircase increase in capacitance during degranulation demonstrates that each step builds upon the previous one, indicating that the fusion event is irreversible. However, closer observation of capacitance jumps in mast cells reveals the existence of “on” and “off” steps. The “on” step is produced by the opening of a small-diameter pore, called a fusion pore, which adds the surface of the vesicle to that of the cell. Once opened, the fusion pore can close quickly and reopen (Fig. 33.7). Rapid oscillation between open and closed states gives the appearance of a flickering of the capacitance (Almers, 1990). Size distribution of capacitance steps during the flickering period shows that large steps are absent and that all steps remain in the range of values expected for single vesicle fusion events. The size of the fusion pore is proportional to its conductance. An unexpected result is that the reclosing of the pore occurs, not only in small-diameter pores (low conductance), but also in larger-sized pores having a conductance of several nS. Capacitance flickering is rather frequent in mast cells, but very few have been observed in eosinophils. This raises the question of the undetectable presence of flickering during the fusion of all vesicles before the irreversible fused state is attained. Indeed, the initial phase of fusion is a very fast process which cannot be faithfully recorded by the speed of the recording techniques used. The earliest steps as well as short-lived flickering are certainly missed.
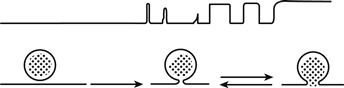
FIGURE 33.7 Flickering of the membrane capacitance. Fusion of the granule is reversible and can oscillate between an unfused state and a fused state. Each time the granule fuses with the plasma membrane, the capacitance of the cell increases by a step. If the fusion pore closes, the capacitance recovers its initial value; incomplete recovery indicates very brief closures exceeding the speed of the recording system. Eventually, a stable fused state can be reached.
IVB3 Fusion Pore
Freeze-fracture images of exocytosis in mast cells and neutrophils reveal the presence of narrow pores formed between the granules and the plasma membrane. The size of the pore increases as fusion progresses, allowing the release of vesicle contents. The presence of pores joining two secretory vesicles is also observed (Lindau and Gompertz, 1991). Electrically, the first opening of the fusion pore generates a brief transient current from which several parameters can be deduced.
Size of Fusion Pore
The conductance of the fusion pore can be calculated from the current transient produced by movement of the charges between two differently charged membranes, i.e. the plasma membrane and vesicle membrane. The initial value of the current, I0, is related to the initial pore conductance, g0, by the relationship:
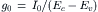 (33.2)
(33.2)
where Ec is the potential across the plasma membrane and Ev the potential across the vesicle membrane.
The initial conductance of the fusion pore in mast cells was found to be 230 pS. The corresponding pore diameter was calculated assuming a resistivity of 100 Ω-cm and a pore length of 15 nm. The abrupt increase in conductance corresponds to the all-or-none opening of a pore having an inner diameter of less than 2 nm. For comparison purposes, gap junction channels have conductances that vary from 80 to 240 pS and a diameter of approximately 2 nm. It can thus be proposed that the fusion pore is a large protein spanning across two membrane thicknesses and having a structure and function resembling that of a channel. Data obtained from electron microscopic observations reveal that the smallest pores that can be observed have diameters between 30 and 50 nm, values far higher than the values reported from conductance measurements. A rapid dilatation of the pore soon after its formation might explain this discrepancy. Once the pore has been formed, the conductance increases abruptly to a value near 250 pS. Thereafter, the pore begins to dilate, possibly by infiltration of lipid molecules between the subunits of the protein structure, followed by an increase in conductance. The pore conductance increases from 500 pS to 3000 pS in about 25 ms; a plateau is reached after 150 ms, corresponding to a pore diameter of more than 16 nm.
Does the Fusion Pore Leak?
Once established, the diameter of the fusion pore is similar to that of the gap junction, between 1.5 and 2 nm. Since gap junctions allow the intercellular passage of molecules weighing up to 1.9 kDa, the question can be asked as to whether granule contents can leak through the fusion pore. In guinea pig eosinophils, the irreversible fusion of the granule with the plasma membrane is occasionally preceded by a long-lived fusion pore having a conductance of 70–250 pS. When granules were loaded with the fluorescent dye quinacrine, no release of the dye in the extracellular medium could be observed during the life of the fusion pore. Only after the fusion pore had completely dilated and the vesicle had reached its irreversible state of fusion was the dye released. These results indicate that the fusion pore is too narrow for the release of granule contents.
However, computations based on the diffusion of a small molecule such as histamine predict that a granule with a diameter of 0.8 mm should release its contents rapidly through the opening of the fusion pore. Experimental proof was provided by Neher and collaborators using bovine chromaffin cells (Chow et al., 1992). Secretion was measured by voltametry, while cells were studied by voltage-clamp. The cells were stimulated by depolarizing the membrane from a holding potential of −60 mV to a step potential of +10 mV for 25 ms to activate the Ca2+ channels. The amperometric signals, which represent the detection of the released catecholamine molecules by the carbon electrode (potential of 800 mV), were transient with a fast or slow rising phase and variable amplitudes. A histogram of integrals of current transient amplitude obtained on several cells showed that the mean charge transfer had a value of 0.76 pC, which is equivalent to the release of 2.36 × 106 molecules of catecholamine. Sometimes, larger events were detected, presumably corresponding to multigranular exocytosis. One interesting and surprising feature was that the majority of the fast-rising events were preceded by a small “foot” or “pedestal,” as illustrated in Fig. 33.8A. The mean duration of the foot was 8.26 ms and the mean charge 34 fC, equivalent to 1.05 × 105 molecules (Fig. 33.8B). The foot was interpreted as reflecting a slow leakage of catecholamine molecules through the fusion pore formed during the early step of the fusion process. A second important result of this study was the discovery and quantification of a long latency period between the end of the stimulus and catecholamine release. The majority of the secretory events occurred 5 to 100 ms after the end of the electrical stimulus, which is rather long when compared to nerve endings, where the delay is about 1 ms. A complex cascade of intracellular events triggered by the increase of cytosolic Ca2+ concentration could be responsible for this long latency.
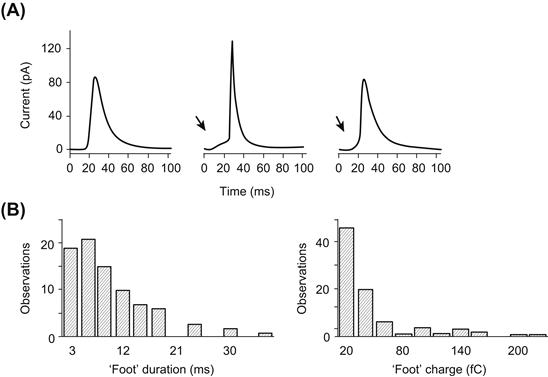
FIGURE 33.8 Amperometric current recorded in chromaffin cells. (A) The amperometric signal recorded with the voltametry method shows a fast rising phase followed by a slower decrease. Occasionally, the rising phase is preceded by a pedestal or foot. The foot signal is thought to be due to the leak of catecholamine by the fusion pore before its complete dilatation. (B) Histogram of the foot signal duration (left panel) and histogram of the charge of the foot signal (right panel), with a mean of 34 fC corresponding to 1.05×105 molecules. (Adapted with permission from Chow et al. (1992). Nature. 356, 60–63. Copyright 1992 Macmillan Magazines Limited.)
IVB4 Membrane Tension as a Driving Force for Fusion
As previously described, flickering is characterized by the opening and closing of the fusion pore. In some cases, it has been shown that after a period of flickering, the capacitance of the plasma membrane declines to a value lower than its initial value (Monck et al., 1990). Figure 33.9A shows that this decrease in capacitance is paralleled by an increase in the conductance of the fusion pore (Fig. 33.9B), thus establishing a relationship between the dilatation of the pore and decreased plasma membrane capacitance. Once the flickering stops, the conductance recovers its initial value but the whole-cell capacitance remains lower. These results are interpreted as reflecting a decrease of the plasma membrane surface due to a net transfer of material to the granule membrane. The difference between the “on” and “off” steps (found in one-half of the transient fusions with values between −2 to −4 fF) is proportional to the duration of contact between the plasma membrane and the secretory vesicle (Fig. 33.9C). The rate of cell surface area reduction is 0.16 μm2·s−1. The transfer of membrane is facilitated by the fact that the membrane of the secretory vesicle is under tension. Upon fusion, a movement of phospholipid molecules occurs from the plasma membrane to the granule membrane. A possible mechanism for generating tension in the granule membrane is osmotic swelling. However, fusion can proceed in isotonic, hypotonic or hypertonic solutions with no change in the kinetics of capacitance increase.
FIGURE 33.9 Decrease of total membrane capacitance after fusion. Capacitance (A) and conductance (B) measurements during transient fusion of a giant secretory granule from beige mouse mast cell. Capacitance and conductance increase on fusion. The fusion pore closes twice with a decrease in conductance and capacitance. Note that the conductance returns to its initial level but that the capacitance of the cell is lower than its initial value. This indicates a reduction of the surface of the plasma membrane due to a leak of lipid molecules toward the granule membrane. (C) Histogram showing the size distribution of the capacitance difference measured after transient fusion in mast cells. (Reproduced with permission from Monck et al. 1990.)
IVB5 Fusion Steps
Several lines of evidence favor the hypothesis that a pore-forming protein could be involved in the fusion process. The abrupt opening of the fusion pore with an initial conductance of 250 pS, similar to the conductance of the gap junction channel and the occurrence of rapid flickering are the strongest arguments. Prior to their fusion, secretory granules are docked to the plasma membrane. In synaptic nerve endings, docking is localized to a restricted region and, upon stimulation, yields localized secretions. Docked granules have been observed in a variety of systems, including chromaffin cells. In these cells, localized secretion was also reported, depending on the applied stimulus.
IVC Control of Exocytosis
Exocytosis occurs in a variety of electrically excitable and non-excitable systems in response to receptor activation. The regulatory pathways that couple stimulation and secretion vary widely among cell types. In the following section, analysis of the factors controlling exocytosis mainly focuses on two well-studied systems: the chromaffin cell from the adrenal medulla, which belongs to the class of excitable cells, and the mast cell from the immune system, a non-excitable cell.
IVC1 Effectors of Exocytosis
Calcium Signaling and Sources of Calcium
In many cells, Ca2+ is the key signal for triggering exocytosis. In chromaffin cells, the resting Ca2+ concentration has a value ranging between 50 and 100 nM. Chromaffin cells can be stimulated by two different classes of acetylcholine receptors. The nicotinic receptor has a channel-like structure consisting of five transmembrane subunits. The binding of two acetylcholine molecules opens the channel, which leads to a large net influx of Na+ ions. This influx causes a membrane depolarization which can activate voltage-dependent Ca2+ channels. The nicotinic receptor channel is also permeable to K+ ions and, to a lesser extent, Ca2+ ions. The muscarinic receptor belongs to the family of the seven-span transmembrane domain receptor proteins, which utilizes the G protein cascade pathway as its signal-transducing mechanism. Stimulation of chromaffin cells with the cholinergic agonist nicotine induces a rapid rise in [Ca2+]i (measured with Ca2+-sensitive fluorescent dye) up to 1 μM; this [Ca2+]i increase is followed by F-actin disassembly (see Fig. 33.4) and exocytosis. Muscarinic stimulation of chromaffin cells also increases [Ca2+]i but does not induce secretion.
In a Ca2+-free external medium, secretion is abolished regardless of the stimulus. This observation reinforces the fact that Ca2+ influx from the external medium is crucial for secretion in chromaffin cells. Several voltage-dependent Ca2+ channels have been described in chromaffin cells, namely: (1) the L-type dihydropyridine-sensitive channels; (2) the N-type ω-conotoxin-sensitive dihydropyridine-insensitive Ca2+ channels; and (3) the dihydropyridine-sensitive facilitation Ca2+ channels. Electrical depolarization, K+ depolarization and nicotinic stimulation open the Ca2+ channels, allowing an immediate influx of Ca2+ ions from the external medium. The video-imaging technique allows the recording of [Ca2+]i with good spatial definition. As shown in Fig. 33.10 (right panel), the increase in [Ca2+]i is restricted to the immediate vicinity of the plasma membrane after activation of Ca2+ channels. The requirement of high Ca2+ concentration for exocytosis implies that secretory granules are stored near the Ca2+ channels. Recent experimental evidence confirms that when Ca2+ channels are activated, the concentration of Ca2+ at the secretory sites could range from 10 to 100 pM, creating a Ca2+ microdomain (Neher, 1998).
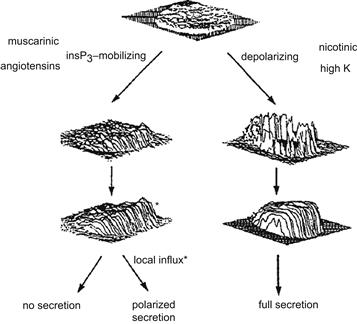
FIGURE 33.10 Ca2+ signal in a chromaffin cell in response to various agonists. The distribution of [Ca2+]i is obtained by video-imaging. The figure illustrates the fact that level and distribution of [Ca2+]i are important for full secretion. The right portion of the figure shows that a depolarization of the plasma membrane, induced by high [K+]o or nicotinic agonist, opens voltage-activated Ca2+ channels with an immediate and high [Ca2+]i increase at the periphery of the cell. Afterwards, [Ca2+]i increases in the entire cell due to release of Ca2+ from internal stores; a full secretion is obtained. The left portion of the figure shows that release of Ca2+ from the IP3-sensitive pool is not sufficient to induce secretion, although some localized secretion can be obtained in the region of the cell where [Ca2+]i increase was the highest. (Adapted with permission from Burgoyne, 1991.)
In chromaffin cells, internal Ca2+ release is provided by two different pools: the InsP3-sensitive pool and the Ca2+-induced Ca2+ release pool (CICR). The activation of the PtdIns-specific phospholipase C by the Gq-protein-coupled receptor or by cytosolic Ca2+ elevation induces the hydrolysis of PtdInsP2, thus generating two messengers: InsP3 and diacylglycerol (DAG) (see Figs. 33.1 and 33.2). The binding of InsP3 to specific sites on the endoplasmic reticulum induces Ca2+ release. InsP3 receptors were also found in secretory granules of endocrine and neuroendocrine cells. Their activation by InsP3 induces the release of Ca2+ ions from the granules and provides a localized increase of [Ca2+]i. Ca2+ ion alone or caffeine can trigger the release of Ca2+ from the second pool (CICR). The muscarinic receptor induces the release of Ca2+ from the InsP3-sensitive store without Ca2+ channel activation. The observed Ca2+ increase is low and sometimes localized to one pole of the cell (Fig. 33.10, left panel). Despite this [Ca2+]i increase, there is little or no stimulation of secretion at the site where [Ca2+]i has increased. A more uniform [Ca2+]i increase can be generated by release from the Ca2+-induced Ca2+ release pool by caffeine or from the InsP3 pool by introduction of GTP-γS into the cell. However exocytosis still remains unstimulated. These results conclusively demonstrate that, in chromaffin cells, only a considerable rise in Ca2+ concentration in the proximity of the plasma membrane is able to trigger exocytosis. This highly localized [Ca2+]i increase can only be achieved via the Ca2+ channels and not by a small increase due to release from internal stores.
Mast cells are able to synthesize, store and secrete histamine. However, Ca2+ alone is not able to trigger histamine secretion. There is a requirement for a guanine nucleotide together with Ca2+ for exocytosis. Mast cells are stimulated by antigenic binding on cell surface IgE receptors or by compound 48/80. A sudden rise in Ca2+ concentration (released from InsP3-sensitive pool) is observed, reaching a level of up to several micromolar. The signal is transient and [Ca2+]i declines within several seconds to its original baseline value. Degranulation begins soon after this transient response. Two important features should be noted concerning [Ca2+]i and secretion in mast cells: (1) the [Ca2+]i transient is not dependent on the presence of Ca2+ in the external medium; and (2) simultaneous recording of capacitance and membrane conductance reveals that conductance is constant throughout the duration of exocytosis. These results indicate that Ca2+ entry through Ca2+ channels is not a required signal for exocytosis, but rather its release from internal stores. A Ca2+ current, called ICRAC (calcium-release activated calcium), was described in mast cells (Hoth and Penner, 1992); ICRAC is activated by the depletion of intracellular Ca2+ pools. The role of ICRAC in non-excitable cells could be to maintain a high [Ca2+]i and to replenish empty Ca2+ stores after stimulation. A possible direct role in exocytosis has not yet been shown.
One conclusion is that Ca2+ release from internal stores is sufficient for exocytosis in mast cells. However, intracellular application of InsP3 induces a transient [Ca2+]i increase but does not trigger secretion, emphasizing that the signal is more complex. Application of the non-hydrolyzable GTP analog GTP-γS (100 μM) inside a mast cell (via a patch pipette) induces a transient [Ca2+]i increase, after a delay of 10–20 s, followed by an increase in capacitance. GTP- γS activates G proteins, including the Gq protein coupled to PLC, with a resulting production of InsP3 and DAG. Ca2+ is released from the InsP3-sensitive pool by the same mechanism as with the direct application of InsP3 described above. However, in this case, exocytosis is stimulated, leading to the conclusion that GTP- γS activates a G protein that plays a crucial role in exocytosis.
Guanine Nucleotides
As seen earlier, Ca2+ and a guanine nucleotide are necessary and sufficient effectors to ensure secretion in mast cells. The stable analog GTP- γS is the commonly used guanine nucleotide, although any ligand that binds to G protein is able to stimulate secretion. In chromaffin cells, two types of observations have been recorded: (1) GTP- γS increases the Ca2+ sensitivity of secretion in permeabilized cells; and (2) GTP analogs stimulate secretion in a Ca2+-independent manner (Morgan and Burgoyne, 1990). GTP analogs do not enhance the secretion induced by high Ca2+ concentration (10 μM), which indicates that the two stimuli (GTP and Ca2+) act on the same exocytotic pathway.
Evidence for the involvement of a GTP-binding protein in secretion has been obtained from a variety of cells: neutrophils; platelets; parathyroid; pituitary lactotroph, gonadotroph and melanotroph; insulin-secreting cells; and pancreatic acinar cells. Various effects have also been observed in these cells: GTP or GTP analogs behave as effectors able to trigger a Ca2+-independent secretion, or as modulators that increase Ca2+ sensitivity, or more surprisingly as inhibitors that block the exocytotic pathway at a late stage. The exact nature of the G proteins is not yet known, although some analogy can be made with GTP proteins involved in vesicular traffic (Rothman and Orci, 1992).
The small Ras-like GTPases are involved in the formation, transport and fusion of vesicles. In yeast, the small Ras-related GTP-binding protein SEC4 is required for targeting and/or fusion of vesicles to the plasma membrane. Rab3A from the rab family of proteins has been located in the membrane of neurosecretory vesicles. More than 30 Rabs have been identified. They have a regulatory role in secretion. In its GTP-bound form, Rab3A inhibits secretion, possibly by stabilizing a fusion-incompatible conformation. Under an appropriate signal, GTP is hydrolyzed to GDP with the help of GAP (a GTPase-activating protein); this exchange of GTP for GDP releases the inhibition. The fusion can then proceed to subsequent steps. Two accessory proteins are involved in the cycle: rabphilin-3A, which binds to the GTP-Rab3A form, and GDI (a GDP-dissociation inhibitor), which binds to the GDP-Rab3A form. Rabphilin is phosphorylated by many kinases and contains binding sites for Ca2+ and phospholipids. In endocrine cells, Nac2 binds GTP-Rab3A; this protein has a high similarity to the Rab3A-binding domain of rabphilin, but lacks the Ca2+ binding sites and phospholipids. GDI binds to Rab3A in its GDP form and removes it from the membrane. After dissociation of the GDI-GDP-Rab3A complex, GTP-Rab3A can bind again to another vesicle. Thus, rabphilin-3A and GDI control the Rab3A cycle but do not directly participate in the fusion (Südhof, 1995; Geppert and Südhof, 1998).
IVC2 Modulators of Exocytosis
ATP
Permeabilized cells rapidly lose their secretory response to agonists unless ATP is present in the medium. However, the sequence of exocytosis can be separated in an early phase that requires MgATP to proceed and a late phase that is MgATP-independent. ATP could act at various levels of the secretory response, acting as a substrate for protein phosphorylation or modulating various kinases involved in secretion. The last ATP-requiring steps in exocytosis have been identified; they involve ATPase, NSF and the formation of PtdInsP2. NSF forms a large (20S) complex with the attachment proteins (SNAPs) and SNAP receptors (SNAREs), which has been proposed to be the fusion particle (see Fig. 33.3). The hydrolysis of ATP by NSF is thought to produce energy for the fusion (priming step). The formation of PtdInsP2 by phosphatidylinositol 4-phosphatase-5-kinase and a phosphatidylinositol transfer protein requires the presence of ATP. PtdInsP2 is thought to be involved in the interaction between cytoskeleton and secretory granules (docking step). Docking and priming of secretory granules are thus ATP-dependent. During vesicle transport through the Golgi, ATP hydrolysis by NSF occurs after docking, but in granule secretion no such evidence exists.
Cyclic Nucleotides
In chromaffin cells, cAMP concentration increases after cholinergic stimulation, leading to the hypothesis that cAMP could have a role in exocytosis. In pancreatic β cells, the cAMP-dependent protein kinase A enhances secretion. However, contradictory results have been reported on the effects of high and low cAMP concentrations on secretion modulation, i.e. inhibition and potentiation, respectively. Nicotinic-induced secretion in chromaffin cells is inhibited by high cGMP concentration, whereas low concentrations potentiate the secretory response.
Calmodulin
A calmodulin-binding protein of 65 kDa, called 65-CMBP or p65, has been found in several secretory vesicles, such as synaptic, neurohypophyseal, chromaffin, platelets and pancreatic islet granules. This protein also binds to phospholipids with high affinity. A possible role of calmodulin in exocytosis was investigated by blocking its action. The calmodulin antagonist calmidazolium inhibits secretion from intact chromaffin cells. Antibodies raised against calmodulin inhibit the Ca2+-dependent binding of calmodulin to vesicle membrane by acting on the docking and/or fusion steps of exocytosis. However, some reports indicate that the less specific calmodulin inhibitor trifluoperazine (TFP) has no effect on permeabilized cells. At present, it appears that calmodulin is not essential for Ca2+-dependent exocytosis, although interaction between calmodulin and cytoskeletal proteins should nevertheless be considered.
Protein Kinase C
A role for PKC in secretion could be inferred from the fact that any agonist that stimulates secretion also induces activation of PKC. PKC has a modulating role in Ca2+ affinity, by decreasing Ca2+ requirements for exocytosis. Inhibition of PKC by staurosporine or through downregulation partially inhibits secretion. However, in gonadotrophs, PKC-stimulated luteinizing hormone exocytosis is independent of Ca2+. Obviously, the role of PKC in exocytosis is not yet completely understood. It is possible that PKC is not essential for Ca2+-dependent exocytosis (modulator), but that a second Ca2+-independent pathway could coexist in which PKC acts as an effector of secretion. Indeed, it has been recently shown in chromaffin cells that PKC acts at a late stage in exocytosis before the final Ca2+-sensitive step. The role of PKC would be to increase the size of the readily releasable pool (RRP) of secretory granules by speeding their maturation after they dock with the plasma membrane. Moreover, direct stimulation of PKC by the phorbol ester PMA (phorbol 1,2-myristate-1,3-acetate) leads to a disruption of the actin network near the plasma membrane, which increases the number of docked granules. Several proteins involved in the regulation of the cytoskeleton are substrates for PKC: annexin I, annexin II and MARCKS (see Fig. 33.1).
Phospholipase A2 and Arachidonic Acid
A possible fusogen role of arachidonic acid was inferred from the fact that, in vitro, granules aggregated by synexin and Ca2+ fuse together if arachidonic acid is added to the medium. As previously mentioned for PKC activation, arachidonic acid is produced each time secretion is stimulated. Inhibition of phospholipase A2 and arachidonic acid metabolism inhibits secretion of chromaffin cells due to a blockage of Ca2+ entry.
IVC3 Secretory Granule Pools
In bovine chromaffin cells, increase in [Ca2+]i triggers secretion at various rates: ultrafast secretion (time constant <0.5 s), fast secretion (time constant 3 s) and slow secretion (time constant 10 to 30 s). Nevertheless there is a weak correlation between the rate of secretion and [Ca2+]i levels, the three types of responses are mainly observed for 10 to 50 μM, above 80 μM and around 170 μM [Ca2+]i, respectively. The time constant of the increase in membrane capacitance can be considered as a measure of the hormone released from a particular store. This indicates that the secretory granules are in various states of releasability. The ultrafast response comes from vesicles docked to the plasma membrane and immediately available for release by an increase in [Ca2+]i; they belong to the immediately releasable pool (IRP). A second pool of granules are located near the membrane in a nearly releasable pool (NRP) and can be released within seconds of a rise in [Ca2+]i; the fast response originates from this pool. Finally, the slow response originates from vesicles from a depot store in the bulk cytoplasm (Neher and Zucker, 1993). Movement of granules from the NRP to the IRP is Ca2+-dependent and could involve calpactin in the docking process. However, not all the docked granules are available for the last steps, opening of the granule induced by Ca2+. The fastest response, the exocytotic burst, mobilizes only one-tenth of the docked granules, which indicates that, after docking, granules undergo a maturation process in the IRP. Figure 33.11 describes a possible sequence for exocytosis in endocrine cells. PKC and ATP hydrolysis are involved in the docking and priming steps. Thereafter, three steps have been proposed based on their sensitivity to temperature, blockage by acidification and Ca2+ ion (three or four ions) triggering; PKC is thought to play a role in this sequence. The pivotal role of ATP in secretion has been outlined, however, a large pool of granules can be released in the absence of ATP by an increase in [Ca2+]i. This indicates first, that ATP hydrolysis by NSF is an early step in the priming process and, second, that if the energy of hydrolysis powers fusion, it remains stored in the core complex until [Ca2+]i increases (Parsons et al., 1995; Gillis et al., 1996).
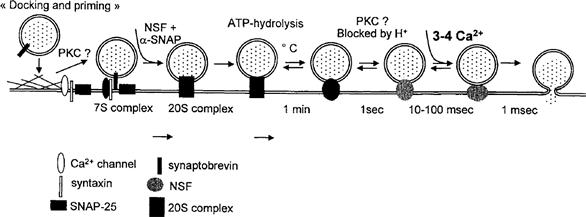
FIGURE 33.11 Docking and fusion of secretory granules in endocrine cells. Docking of vesicles involves the proteins synaptobrevin and syntaxin/SNAP-25. In the first step of docking, protein kinase C (PKC) could have a role in the disruption of the actin barrier. ATP hydrolyzed by NSF is thought to produce energy for fusion. Several steps have been identified after the ATP-dependent step based on their temperature, H+ and Ca2+ dependence. PKC could be involved before the final Ca2+-dependent step. (Adapted with permission from Parsons et al., 1995 and Gillis et al., 1996.)
V Hormone Release in Endocrine Cells
VA Polypeptide and Thyroid Hormones
Most of the events described previously could be applied for most of the endocrine secretory cells.
VA1 Secretion of Insulin
Insulin secretion by the pancreatic β cells provides an excellent example of a cellular activity that requires direction. Insulin is packaged in secretory vesicles which have to migrate to the plasma membrane and fuse with it to release the entrapped insulin. Both microscopic and biochemical studies have shown that secretory granules are linked to microtubules which direct attached vesicles to the cell surface. However, a cortical band of fine microfilaments is consistently observed in β cells. Alteration of this cell web by cytochalasin B is associated with an enhancement of glucose-induced secretion of insulin by isolated islets. This microfilamentous web plays an important role in the exocytosis of insulin secretory granules by controlling access to the cell membrane via a mechanism probably similar to that previously described for chromaffin cells. Ca2+ appears to initiate the cascade of events by which microtubules facilitate the displacement of granules toward the cell membrane. Glucose metabolism increases intracellular concentration of ATP, which closes the ATP-sensitive K+ channels, consequently inducing cell depolarization and Ca2+ influx, while cAMP modifies the intracellular distribution of Ca2+ by increasing the cytosolic pool at the expense of Ca2+ bound to intracellular organelles. Protein kinase C also appears to be involved in the secretion of insulin.
VA2 Secretion of Pituitary Hormones
Anterior pituitary cells, in their diversity and heterogeneity, provide a rich source of models for secretory function. Secretion of the pituitary hormones is controlled by both specific hypothalamic-releasing peptides and neurotransmitters. Upon binding to their receptors, these agonists activate both Ca2+ influx by different types of Ca2+ channels (voltage-activated channels, ligand-activated channels and second messenger-activated channels) and second messenger production, thus resulting in hormone secretion. As a second messenger, cAMP appears to be as potent as InsP3 and PKC in interacting with Ca2+ to trigger secretion. As for insulin secretion, cytoskeletal structures are tightly associated with hormone release. Purified secretory granule membranes co-sediment with microtubules, MAPs being involved in this association. This suggests that microtubules facilitate the movement of secretory granules from the Golgi apparatus to the plasma membrane, by providing tracks along which the granules can move. The granule membrane can then dissociate from the microtubules and fuse with the cell membrane, followed by exocytosis and release of the hormone into the circulation. Moreover, actin and microtubules are cross-linked by MAPs, forming three-dimensional networks. This cross-linking activity can be inhibited if MAPs are heavily phosphorylated. These observations suggest that MAPs might play an important role in the binding of secretory granules to tubulin and actin. Binding of actin to secretory granules suggests a role for actin in the final steps of exocytosis, as described previously.
VB Stimulation of Steroid Synthesis and Secretion
While disruption of the actin network is necessary to trigger fusion of secretory vesicles with the cell membrane, a well-preserved organization seems important for steroid release. Several studies have shown that microfilament disruption (with cytochalasins) or microtubule disruption (with colchicine or vinblastine) decreases or blocks second messenger production and steroid secretion, either from the adrenal cortex or gonads. In contrast with peptide hormones, steroids are not packaged in vesicles. Steroid hormones are synthesized from cholesterol contained in lipid droplets. The process of steroidogenesis begins in the mitochondria, where cholesterol is converted to pregnenolone, from which others steroids are synthesized. Stimuli induce rapid increase in the production and secretion of steroids, without cytoplasmic storage. More recent biochemical analysis and fluorescence studies indicate that ligand-receptor interaction induces a rapid polymerization of actin, demonstrated by a rise in the proportion of F-actin over G-actin and by an increased interaction of F-actin with the membrane (Fig. 33.12A and B versus C and D). Moreover, Gq/11 protein localization overlaps F-actin distribution and cell activation is accompanied by a rapid translocation of Gq/11 and F-actin from the cytosol to the membrane, an association essential in promoting phospholipase C activation (Côté et al., 1997).
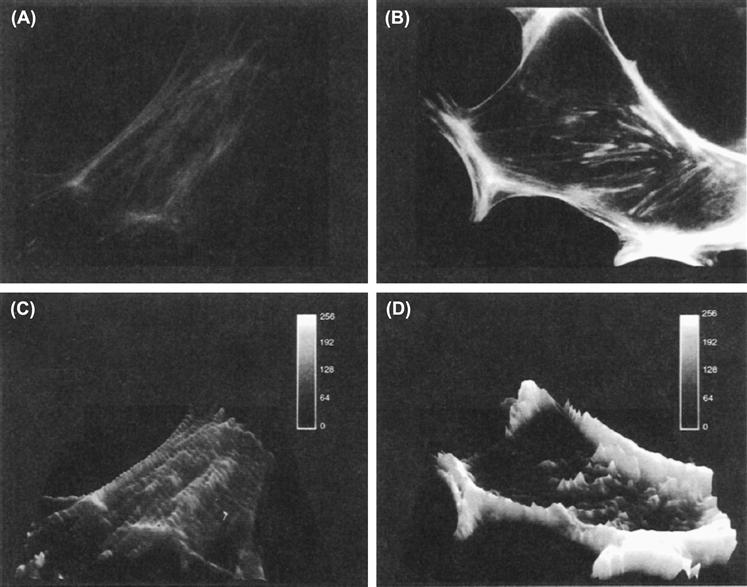
FIGURE 33.12 Effect of Ang II on immunofluorescence labeling of actin in rat glomerulosa cells. Rat glomerulosa cells were cultured for 3 days on plastic coverslips and then incubated without (A) or with (B) 100 nM angiotensin II for 1 min. After formaldehyde fixation and permeabilization with 0.1% Triton X-100, cells were processed for immunofluorescence labeling of actin using rhodamine-phalloidin. (C) and (D) represent the same images as (A) and (B) after computer analysis. Bars represent 13 μm. (Adapted from Côté et al., 1997.)
It has been shown that microfilaments are strictly required for both spontaneous and stimuli-induced corticosteroid secretions, including those utilizing the cAMP-dependent pathways (ACTH, serotonin and vasoactive intestinal peptide), as well as those utilizing the phosphoinositide pathway (angiotensin II and acetylcholine). They are involved in a common and probably late step of steroidogenesis (translocation of the DOC from RER to mitochondria). In contrast, microtubules seem involved in an early step in the mechanism of hormone action, probably at the level of their interaction with the α subunit of the G protein. Microtubules and microfilaments are closely associated with the plasma membrane. That colchicine and vinblastine stimulate basal steroidogenesis may be explained by the fact that most of the tubulin is linked to cholesterol, in lipid droplets. Thus, by acting on tubulin, antimicrotubular drugs could release cholesterol, causing an increase in basal steroid secretion (Feuillolley and Vaudry, 1996). Moreover, in adrenocortical cells, findings suggest that intermediate filaments could facilitate or increase the transport of cholesterol to mitochondria in response to ACTH and cAMP. Figure 33.13 summarizes the role of the three types of cytoskeletal fibers in the steroidogenic effects of the main stimuli of corticosterone and aldosterone secretions (Feuillolley and Vaudry, 1996).
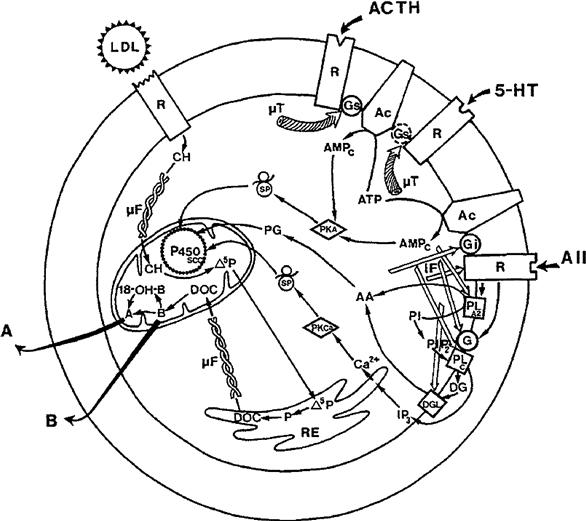
FIGURE 33.13 Cytoskeleton implication in adrenal corticosteroidogenesis. Microtubules (μT) are implicated in the action of ACTH and serotonin (5-HT), probably at a level of Gs protein. Microfilaments (μF) control the transfer of steroid precursors (cholesterol from lipid droplets or membrane low-density lipoproteins to mitochondria and from mitochondria to endoplasmic reticulum). Intermediate filaments (IF) appear to be implicated in the action of angiotensin II (AII), between hormone-receptor coupling and second messenger production. A, aldosterone; AA, arachidonic acid; AC, adenylyl cyclase; cAMP, cyclic adenosine monophosphate; ATP, adenosine triphosphate; B, corticosterone; DAG, diacylglycerol; DOC, 11-deoxycorticosterone; 18-OH-B, 18-hydroxycorticosterone; P, progesterone; P450scc, P450scc cytochrome; Δ5P, pregnenolone; PG, prostaglandins; PI, PtdIns; PIP2, PtdInsP2; PKA, protein kinase A; PKCa, protein kinase Ca2+-dependent; PLA2, phospholipase A2; PLC, phospholipase C; PS, protein synthesis. (Reproduced with permission from Feuilloley et al., 1989.)
Acknowledgments
The authors thank Dr Mylène Côté for critical review of the revised version and fruitful discussions and Dusan Chorvat, Jr, from the International Laser Center, Bratislava (Slovakia), for image analysis.
BIBLIOGRAPHY
1. Almers W. Exocytosis. Annu Rev Physiol. 1990;52:607–624.
2. Aplin A, Howe A, Alahari S, Juliano R. Signal transduction and signal modulation by cell adhesion receptors: the role of integrins, cadherins, immunoglobulin-cell adhesion molecules, and selectins. Pharmacol Rev. 1998;50:197–263.
3. Arbuzova A, Murray D, McLaughlin S. MARCKS, membranes, and calmodulin: kinetics of their interaction. Biochim Biophys Acta. 1998;1376:369–379.
4. Aroeti B, Okhrimenko H, Reich V, Orzech E. Polarized trafficking of plasma membrane proteins: emerging roles for coats, SNAREs, GTPases and their link to the cytoskeleton. Biochim Biophys Acta. 1998;1376:57–90.
5. Burgoyne RD. Control of exocytosis in adrenal chromaffin cells. Biochim Biophys Acta. 1991;1071:174–202.
6. Burgoyne R. Mechanisms of catecholamine secretion from adrenal chromaffin cells. J Physiol Pharmacol. 1995;46:273–283.
7. Burgoyne R, Morgan A. Analysis of regulated exocytosis in adrenal chromaffin cells: insights into NSF/SNAP/SNARE function. Bioessays. 1998a;20:328–335.
8. Burgoyne R, Morgan A. Calcium sensors in regulated exocytosis. Cell Calcium. 1998b;24:367–376.
9. Chow RH, Von Rüden L, Neher E. Delay in vesicle fusion revealed by electrochemical monitoring of single secretory events in adrenal chromaffin cells. Nature. 1992;356:60–63.
10. Côté M, Payet M-D, Dufour M-N, Guillon G, Gallo-Payet N. Association of the G protein αq/α11-subunit with cytoskeleton in adrenal glomerulosa cells: role in receptor-effector coupling. Endocrinology. 1997;138:3299–3307.
11. Fernandez-Chacon R, Südhof T. Genetics of synaptic vesicle function: toward the complete functional anatomy of an organelle. Annu Rev Physiol. 1999;61:753–776.
12. Feuillolley M, Vaudry H. Role of the cytoskeleton in adrenocortical cells. Endocr Rev. 1996;17:269–288.
13. Forscher P. Calcium and polyphosphoinositide control of cytoskeletal dynamics. Trends Neurosci. 1989;12:468–474.
14. Geppert M, Südhof T. RAB3 and synaptotagmin: the yin and yang of synaptic membrane fusion. Annu Rev Neurosci. 1998;21:75–95.
15. Gillis KD, Mössner R, Neher E. Protein kinase C enhances exocytosis from chromaffin cells by increasing the size of the readily releasable pool of secretory granules. Neuron. 1996;16:1209–1220.
16. Goda Y, Südhof T. Calcium regulation of neurotransmitter release: reliably unreliable?. Curr Opin Cell Biol. 1997;9:513–518.
17. Gundersen G, Cook T. Microtubules and signal transduction. Curr Opin Cell Biol. 1999;11:81–94.
18. Hall A. Rho GTPases and the actin cytoskeleton. Science. 1998;279:509–514.
19. Hoth M, Penner R. Depletion of intracellular calcium stores activates a calcium current in mast cells. Nature. 1992;355:353–356.
20. Inglese J, Koch W, Touhara K, Lefkowitz R. G beta gamma interactions with PH domains and Ras-MAPK signaling pathways. Trends Biochem Sci. 1995;20:151–156.
21. Janmey P. The cytoskeleton and cell signaling: component localization and mechanical coupling. Physiol Rev. 1998;78:763–781.
22. Lindau M, Gomperts BD. Techniques and concepts in exocytosis: focus on mast cells. Biochim Biophys Acta. 1991;1071:429–471.
23. Martin T. Phosphoinositides as signaling molecules. Annu Rev Cell Dev Biol. 1998;14:231–264.
24. Matus A. Microtubule-associated proteins: their potential role in determining neuronal morphology. Annu Rev Neurosci. 1988;11:29–44.
25. Monck JR, Alvarez de Toledo G, Fernandez JM. Tension in secretory granule membranes causes extensive membrane transfer through the exocytotic fusion pore. Proc Natl Acad Sci USA. 1990;87:7804–7808.
26. Morgan D, Burgoyne RD. Stimulation of Ca2+-inde-pendent catecholamine secretion from digitonin-permeabilized bovine adrenal chromaffin cells by guanine nucleotide analogues Relationship to arachidonate release. Biochem J. 1990;269:521–526.
27. Neher E. Vesicle pools and Ca2+ microdomains: new tools for understanding their roles in neurotransmitter release. Neuron. 1998;20:389–399.
28. Neher E, Zucker RS. Multiple calcium-dependent processes related to secretion in bovine chromaffin cells. Neuron. 1993;10:21–30.
29. Parsons TD, Coorssen JR, Horstmann H, Almers W. Docked granules, the exocytic burst, and the need for ATP hydrolysis in endocrine cells. Neuron. 1995;15:1085–1096.
30. Popova JS, Garrison JC, Rhee SG, Rasenik MM. Tubulin, Gq, and phosphatidylinositol 4,5-bisphosphate interact to regulate phospholipase Cβ1 signalling. J Biol Chem. 1997;272:6760–6765.
31. Rothman JE, Orci L. Molecular dissection of the secretory pathway. Nature. 1992;355:409–415.
32. Schmidt A, Hall M. Signaling to the actin cytoskeleton. Annu Rev Cell Dev Biol. 1998;14:305–338.
33. Söllner T, Whiteheart SW, Brunner M, et al. SNAP receptors implicated in vesicle targeting and fusion. Nature. 1993;362:318–323.
34. Südhof TC. The synaptic vesicle cycle: a cascade of protein-protein interactions. Nature. 1995;375:645–653.
35. Südhof T, Rizo J. Synaptotagmins: C2-domain proteins that regulate membrane traffic. Neuron. 1996;17:379–388.
36. Tapon N, Hall A. Rho, Rac and Cdc42 GTPases regulate the organization of the actin cytoskeleton. Curr Opin Cell Biol. 1997;9:86–92.
37. Tchakarov L, Zhang L, Rose S, Tang R, Trifaro J. Light and electron microscopic study of changes in the organization of the cortical actin cytoskeleton during chromaffin cell secretion. J Histochem Cytochem. 1998;46:193–203.
38. Trifaró J-M, Vitale ML, Rodriguez Del Castillo A. Cytoskeleton and molecular mechanisms in neurotransmitter release by neurosecretory cells. Eur J Pharmacol. 1992;225:83–104.