Die Daten sind nun vorverarbeitet, und wir überlegen, was wir eigentlich damit machen wollen.
Mit Supervised Learning (überwachtem Lernen) bezeichnet man ein vom Menschen gesteuertes Lernen. Dabei wird ein System mit bekannten Daten trainiert, und dieses Wissen wird dann auf bisher nicht bekannte Daten angewendet. Insbesondere soll eine mathematische Funktion gelernt werden, die Features als Eingabe bekommt und die entsprechenden Labels als Ausgabe haben soll. Die Parameter dieser Funktion sollen so optimiert werden, dass die bekannten Daten möglichst gut wiedergegeben werden, dass aber auch gleichzeitig die Labels von neuen Daten vorhergesagt werden können.
In diesem zentralen Teil des Buchs fangen wir mit Grundlagen an und arbeiten uns dann durch die einzelnen Ansätze des überwachten Lernens.
Der Code und die erzeugten Grafiken für dieses Kapitel sind wieder unter der URL https://github.com/DJCordhose/buch-machine-learning-notebooks (https://bit.ly/ml-kug) als Notebook kap4.ipynb verfügbar.
Als erstes Beispiel nehmen wir den bekannten Boston-Housing-Datensatz (https://archive.ics.uci.edu/ml/machine-learning-databases/housing/), der die Immobilienpreise verschiedener Vororte Bostons auflistet. Der Datensatz enthält neben den Preisen 13 weitere Attribute der Häuser bzw. der Nachbarschaft, in der sich diese befinden. In unserem Beispiel wollen wir die Relation der Immobilienpreise (medianer Wert pro Bezirk) zu der durchschnittlichen Zimmeranzahl untersuchen.
Zunächst lädst du die Daten aus der Scikit-Learn-Bibliothek. Die Labels (Preise) sind in dem Datenobjekt im Attribut target und die 13 Features im Attribut data gespeichert:
import sklearn.datasets as data
boston = data.load_boston()
Die Form der Daten und des Targets bekommst du über die shape-Abfrage:
boston.data.shape, boston.target.shape
Anhand der Ausgabe kannst du sehen, dass 506 Datensätze mit jeweils 13 Features vorhanden sind:
((506, 13), (506,))
Die sechste Spalte in boston.data gibt die durchschnittliche Zimmeranzahl an. Wir speichern Features und Labels in gewohnter Weise in den Variablen X und y:
X = boston.data[:, 5:6]
y = boston.target
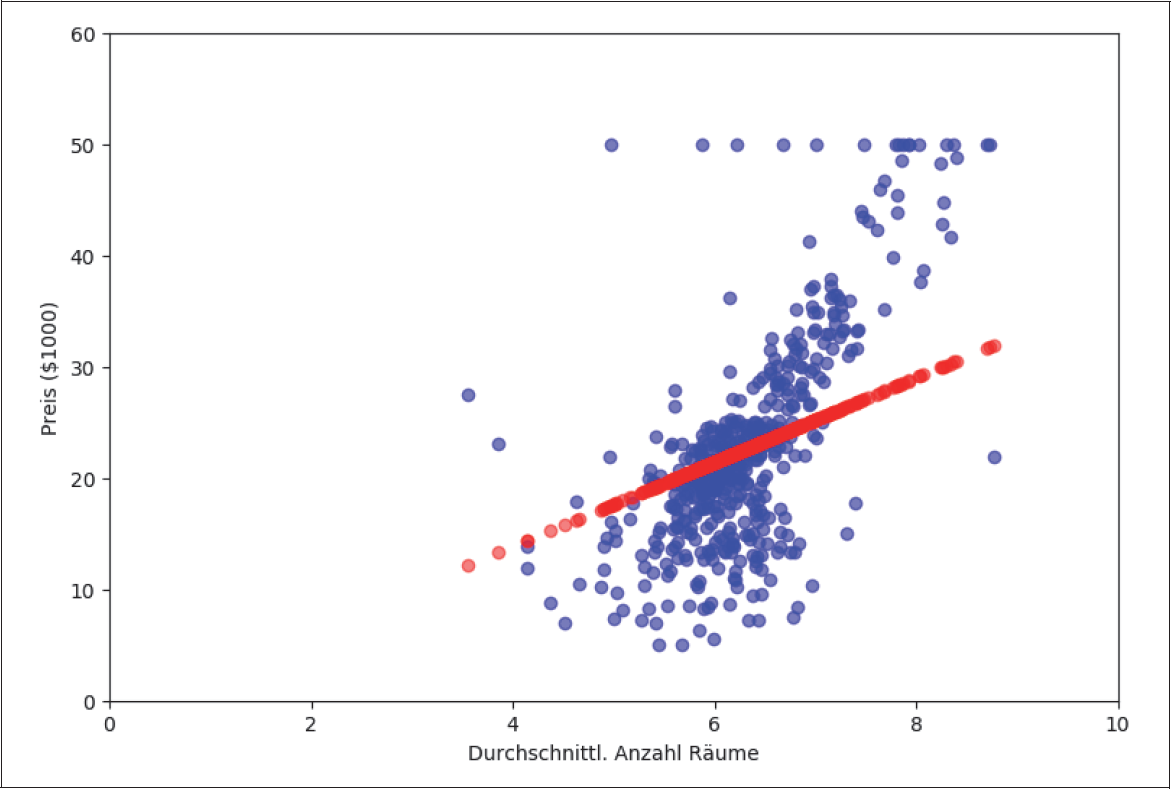
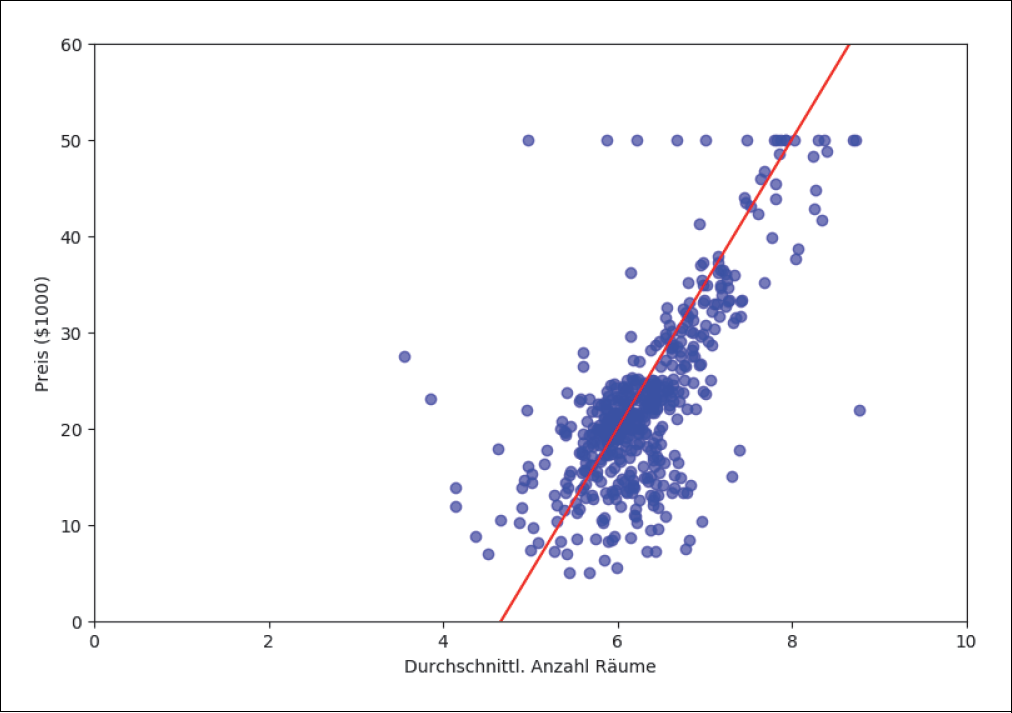
Wenn wir den Preis (target) gegen die Zimmeranzahl auftragen, können wir eine gewisse Tendenz erkennen. Wie erwartet, steigen die Preise mit der Zimmeranzahl (Abbildung 4-1). Wir hegen den Verdacht, dass die Relation mehr oder weniger linear ist. Mit dem Auge können wir sogar ganz grob eine Linie erraten. Die Gerade y = 15 · X – 70 (rote Linie) in Abbildung 4-1 scheint eine gute erste Schätzung zu sein.
Ist das aber die »beste« Gerade? Oder gibt es bestimmte Methoden, wie man die »passendste« Gerade finden kann? Genau darum geht es beim Machine Learning: Wie findest du möglichst automatisch bzw. mithilfe eines selbstlernenden Algorithmus die optimale Lösung zu einem Problem? Und da lineare Regression ein schon lange bekanntes und sehr gut verstandenes Problem ist, wollen wir dich Schritt für Schritt durch diese erste Aufgabe führen.
Abbildung 4-1: Boston-Housing-Datensatz
Eine Gerade in der x-y-Ebene kann mathematisch folgendermaßen formuliert werden:
y = a ⋅ x + b
wobei y in unserem Beispiel den Preis darstellt, der in einer bestimmten Relation zu x, der Zimmeranzahl, steht. In der Machine-Learning-Sprache sind die y-Werte die Labels und die x-Werte die Features. a und b sind die Parameter des linearen Regressionsmodells. In unserem Fall kann man a als die Steigung und b als den y-Schnittpunkt grafisch interpretieren.
Es gibt verschiedene Möglichkeiten, wie du dem Lernalgorithmus mitteilst, welche die »beste« Kurve (in diesem Fall eine Gerade) sein soll. Dieser Algorithmus soll die optimalen Parameter selbst herausfinden, d.h. »erlernen«. Zu dem Zweck definieren wir eine sogenannte Loss-Funktion (manchmal auch Cost- oder Error-Funktion genannt). Wir zeigen dir erst einmal so eine Funktion L (für Loss) und erläutern sie dann Schritt für Schritt:

Wir bilden die Summe aller Abweichungen des tatsächlichen y-Werts (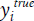 ) von dem y-Wert (
) von dem y-Wert (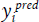 ), den unsere Geradenfunktion vorhersagt. Die Vorhersage können wir durch die Geradenfunktion von oben ersetzen, damit wir außer a und b keine weiteren Unbekannten in unserer Formel haben. Den wahren Wert kennen wir aus dem target-Feld unseres Datensatzes.
), den unsere Geradenfunktion vorhersagt. Die Vorhersage können wir durch die Geradenfunktion von oben ersetzen, damit wir außer a und b keine weiteren Unbekannten in unserer Formel haben. Den wahren Wert kennen wir aus dem target-Feld unseres Datensatzes.
Mit dieser Funktion sucht der Lernalgorithmus der linearen Regression diejenigen Parameter a und b, die die Summe aller Abweichungen minimiert. Das Quadrat benutzen wir, weil wir die Abweichungen immer positiv zählen, außerdem werden Punkte, die weit weg von der Geraden liegen, besonders stark gewichtet. Das heißt, größere Abweichungen tragen im erhöhten Maß zum Gesamtfehler bei.
Der einfachste Lernalgorithmus, der mithilfe einer solchen Loss-Funktion die optimalen Parameter a und b findet, ist die Methode Ordinary Least Square (OLS) bzw. die Methode der kleinsten Quadrate. Sie ist die klassische schon seit Jahrhunderten bekannte Methode und steht praktisch als Synonym für lineare Regression. Details zu OLS findest du z.B. in Wikipedia: https://en.wikipedia.org/wiki/Ordinary_least_squares oder https://de.wikipedia.org/wiki/Methode_der_kleinsten_Quadrate.
Wir machen es uns etwas einfacher und erhalten die Lösung mithilfe der Scikit-Learn-Bibliothek: Wir laden das Modul LinearRe gression, erzeugen eine Instanz des Regressors und trainieren ihn mit unseren Daten. Dazu rufen wir die fit()-Methode mit der Zimmeranzahl X auf und geben ihr auch die tatsächlichen Preise y mit:
import sklearn.linear_model as lm
lr = lm.LinearRegression() # Regressorinstanz
lr.fit(X, y) # Training des Regressors mit den Daten
Nur zur Erinnerung: Wir beschäftigen uns hier mit Supervised Learning, d.h., wir trainieren mit uns bekannten Daten und erhoffen uns im ersten Schritt, ein Modell zu erhalten, das zumindest diese Daten reproduzieren kann. Unser Modell ist dabei durch unsere Geradenfunktion und die beiden Parameter a und b definiert. Dies kann bereits interessant sein, weil wir eine Idee von der Struktur der Daten bekommen. Typischerweise legt man es beim Training des Modells aber auf Generalisierung an. Das bedeutet, wir wollen nicht nur die bereits bekannten Werte vorhersagen, sondern auch Werte, die wir bisher nicht gesehen haben.
Erst einmal sehen wir uns aber an, wie gut das Training funktioniert hat, und zwar über den Aufruf der score()-Methode:
lr.score(X, y) # Validierung
In unseren Fall ist das:
0.48352545599133429
Die voreingestellte Metrik für die score()-Methode für lineare Regression ist der sogenannte R2-Score. Es ist ein Maß für die Güte dieses Modells. Für uns reicht es hier erst einmal, zu wissen, dass die Güte des Modells desto besser ist, je näher der Score an 1 liegt. Da die Daten sehr stark gestreut sind, ist der Score weit von 1 entfernt.
Mehr über die Metrik und eine ausführliche Beschreibung der Güte eines Modells findest du in Kapitel 6, Modellvalidierung. Darin werden wir auch darauf eingehen, warum es für die Validierung des Modells günstig ist, nicht die Werte zu überprüfen, die wir bereits für das Training benutzt haben. In diesem Kapitel konzentrieren wir uns aber auf die Funktionsweise der verschiedenen Methoden und validieren erst einmal auf den Trainingsdaten.
Die Vorhersage des trainierten Modells für bestimmte Features erhält man über die predict()-Methode. Hier gibst du wieder die komplette Liste der Zimmeranzahl hinein und erhältst die vorhergesagten Preise:
y_pred = lr.predict(X)
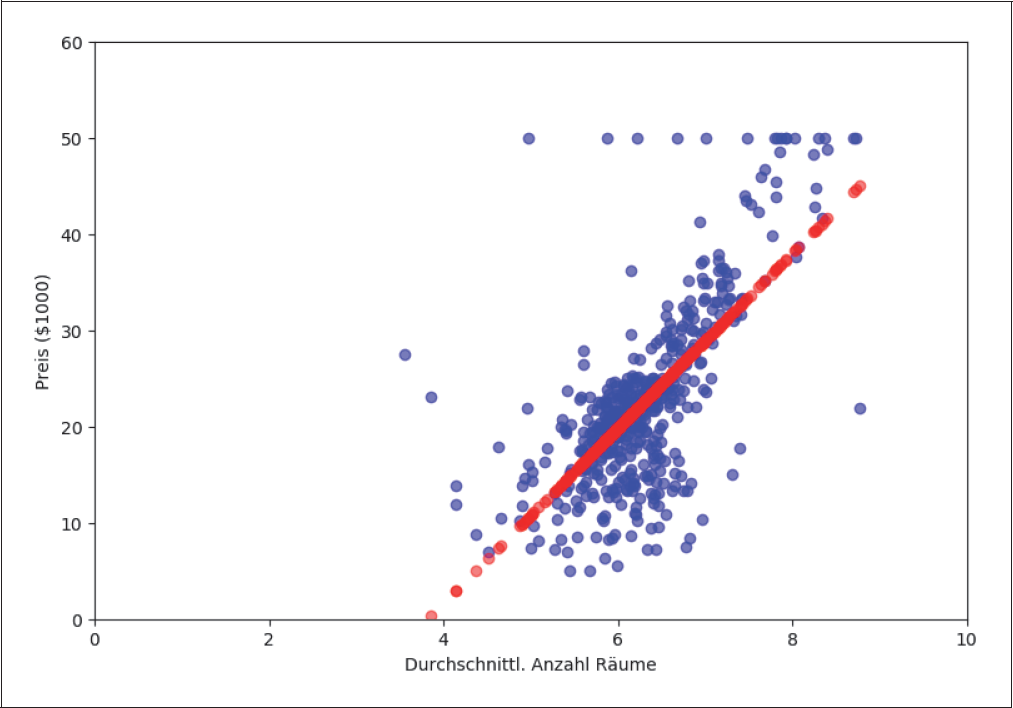
In Abbildung 4-2 werden die wahren Werte (blau) mit der Vorhersage aus der predict()-Methode (rot) für alle Trainingsdaten verglichen.
Abbildung 4-2: Ordinary Least Square Regression
Das Praktische an der Scikit-Learn-Bibliothek ist, dass alle Regressor- und Klassifikatorklassen im Großen und Ganzen die gleiche API besitzen. Das wird im Kasten unten noch mal zusammengefasst:
Grundlegende API von Scikit-Learn (Sklearn)
Zunächst erzeugst du eine Instanz einer Klassifikator- oder Regressionsklasse, z.B.:
import sklearn.linear_model as lm
lr = lm.LinearRegression()
Das Training der Daten X mit den Labels y wird immer über die Methode fit(), eventuell mit zusätzlichen Optionen, aufgerufen:
lr.fit(X, y)
Die Güte des Modells kann man mit der Methode score() berechnen:
lr.score(X, y)
Dabei solltest du beachten, dass bestimmte Score-Methoden voreingestellt sind. In der Regel liefern sie einen Wert zwischen 0 und 1, wobei ein großer Wert für hohe Güte steht. Für manche Probleme und Daten sind eventuell andere Score-Definitionen (auch Metriken genannt) aussagekräftiger.
Die Voraussage des trainierten Modells für neue Daten rufst du mit der Methode predict() auf:
lr.predict(X)
Stochastic Gradient Descent (SGD) heißt ein Algorithmus, der mithilfe einer vorher definierten Loss-Funktion die optimalen Parameter a und b finden soll. Dieser Optimierungsalgorithmus wird heutzutage sehr häufig in Lernalgorithmen angewendet. Mit SGD kann man sehr gut die Art und Weise veranschaulichen, wie viele moderne Machine-Learning-Algorithmen funktionieren.
Für unser Boston-Housing-Problem starten wir mit irgendwelchen zufällig ausgewählten Parametern a und b. Im nächsten Schritt hangeln wir uns an der durch die Loss-Funktion beschriebenen Kurve zum Minimum hinunter (siehe Abbildung 4-3), indem wir die Steigung der Kurve durch die Ableitung der Funktion an den aktuellen Parameterwerten berechnen. Wenn etwas Negatives herauskommt, gehen wir nach rechts. Kommt etwas Positives raus, gehen wir nach links.
Die Ableitung oder das Differential einer Funktion beschreibt so etwas wie die Veränderung oder die Tendenz der Funktion bzw. der Kurve. Ist die Ableitung an einem bestimmten Punkt positiv, wird die Funktion für größere x größer und für kleinere x kleiner. Umgekehrt, bei einem negativen Ableitungswert, wird die Funktion für größere x kleiner und für kleinere x größer. Die Ableitung kann man grafisch auch als Steigung der Tangente an dem jeweiligen Punkt der Kurve darstellen. Tangentenhälften einer Loss-Funktion für einen gegebenen Datensatz, die in Richtung niedrigerer Werte zeigen, sind in Abbildung 4-3 dargestellt.
Abbildung 4-3: Ableitung der Loss-Funktion
Sollte die Ableitung 0 sein, haben wir unser Ziel erreicht, wir sind am Minimum unserer Loss-Funktion angelangt. Genauer gesagt, könnte es auch nur ein sogenanntes lokales Minimum sein oder sogar ein (lokales) Maximum (Abbildung 4-4).
Lokale Minima und Maxima kannst du durch unterschiedliche Strategien vermeiden. Eine Maßnahme ist z.B., die Schrittweite (die Learning Rate), mit der wir nach links oder rechts gehen, geschickt dynamisch anzupassen (siehe unten).
Bei der Stochastic-Gradient-Descent-Methode wird diese iterative Minimierung möglichst effizient ausgeführt. Sie wird sehr häufig auch bei anderen Lernalgorithmen, z.B. bei neuronalen Netzen, die wir in Kapitel 7, Neuronale Netze und Deep Learning, beschreiben, benutzt.
Der Begriff Gradient steht für eine mehrdimensionale Ableitung in eine bestimmte Richtung. In einer Dimension gibt es nur »rechts« oder »links«. Bei drei Dimensionen gäbe es z.B. auch »vorne«, »hinten«, »oben«, »unten« oder sogar eine bestimmte Kombination daraus. Descent ist der »Abstieg«. Beides zusammen bedeutet in unserem Fall also die Richtung, in die man gehen muss, um zum Minimum der Funktion zu gelangen.
Abbildung 4-4: Maxima und Minima
Nun müssen wir uns überlegen, wie wir mit dieser Information unsere beiden Parameter a und b schrittweise in Richtung des Minimums anpassen. Den oben erläuterten Gradienten, also die mehrdimensionale Ableitung von L, beschreiben wir wie in der Mathematik üblich mit ∇L. Diesen multiplizieren wir mit der ebenfalls bereits erwähnten Learning Rate η, also dem Faktor, der bestimmt, wie stark die Änderung pro Iteration sein soll. Wir erhalten dann pro Schritt eine Korrektur von η ⋅ ∇L, die wir von den bisherigen Werten abziehen müssen.
Wenn wir alle Parameter, auch Gewichte genannt, in einen Vektor w zusammenfassen, lautet die Update-Regel für einen Iterationsschritt somit:
wi+1 = wi − η ⋅ ∇L
In unserem eindimensionalen Regressionsfall ist w = (a, b), da wir bei unserer Geradengleichung ja die Parameter für die Steigung und die Verschiebung herausfinden wollen. Hier die Geradengleichung noch einmal zur Erinnerung:
y = a ⋅ x + b
Bei jeder Iteration werden a und b nach dieser Vorschrift verändert, bis die Änderung null oder nur noch sehr gering ist.
Die Learning Rate ist ein sehr wichtiger Parameter des Lernalgorithmus. Stellt man ihn zu klein ein, wird es unter Umständen sehr lange dauern, bis wir auch nur in die Nähe des Minimums gelangen (dabei spricht man oft auch von »konvergieren«). Stellt man die Learning Rate zu groß ein, besteht die Gefahr, dass der Algorithmus immer wieder über das Minimum hinwegspringt und wir nie eine zufriedenstellende Lösung finden.
Die Learning Rate richtig zu tunen, ist schwierig. Eine mögliche Strategie ist es, mit einer relativ großen Learning Rate zu starten und sie dann stetig immer kleiner werden zu lassen. Weitere sehr clevere Algorithmen (z.B. RmsProp oder Adam) berücksichtigen die Gradienten voriger Iterationsschritte und ermitteln daraus eine Richtung, die schneller konvergiert. Wir werden die Methoden in diesem Buch zwar nicht besprechen, aber du kannst dir mehr Informationen z.B. hier beschaffen: http://sebastianruder.com/optimizing-gradient-descent/.
Wenn du viele Daten hast und viele Features auswerten möchtest, kann die Berechnung des Gradienten sehr lange dauern oder sehr viel Speicherplatz beanspruchen, weil dafür alle Daten mit allen Feature-Dimensionen im Speicher gehalten werden müssen. Deshalb ist es eine gute Strategie, immer nur einen Datensatz auszuwerten und dementsprechend die Parameter a und b zu aktualisieren. Wenn man das auf diese Weise Schritt für Schritt für jeden einzelnen Datensatz macht, kommt man am Ende auf ein sehr ähnliches Ergebnis, als hätte man alle Daten auf einmal berechnet. Dies ist eine sogenannte stochastische Annäherung, die manchmal schneller konvergiert. Auf jeden Fall beansprucht sie weniger Speicherressourcen (für jede Berechnungsiteration muss nur ein Datensatz zum selben Zeitpunkt im Speicher liegen). Es gibt noch eine Mittellösung, die sogenannte Mini-Batch-Gradient-Descent-Methode, bei der man das Update immer nur für z.B. 10 oder 100 Datensätze durchführt.
In Sklearn wenden wir SGD auf unser Problem folgendermaßen an:
Der Score kann je nach zufälliger Initialisierung etwas schwanken …
lr.score(X, y)
… er ist aber in jedem Fall schlechter als bei OLS. In unserem Experiment ist es dieser:
0.3732024974814111
Wir visualisieren den Unterschied in Abbildung 4-5.
Abbildung 4-5: Stochastic Gradient Descent, erster Versuch
Woran liegt das? Der SGD-Algorithmus beendet die Optimierung, wenn sich der Fehler für eine Reihe von Durchläufen nicht nennenswert ändert. Dieser Wert ist auf 1e-3 voreingestellt und für uns zu hoch. Wir setzen diesen Wert außer Kraft – damit durchlaufen wir die voreingestellten 1.000 Durchläufe und bekommen einen Score nahe an 0,5:
lr = lm.SGDRegressor(tol=None)
lr.fit(X, y)
lr.score(X, y)
> 0.48274229859866685
Schon ziemlich nah an dem optimalen Ergebnis, oder nicht? Das können wir auch noch einmal an der Visualisierung in Abbildung 4-6 sehen.
Abbildung 4-6: Stochastic Gradient Descent
Das war schon unser erstes Machine-Learning-Beispiel, genauer gesagt, das erste Regressionsmodell. Als Nächstes wollen wir mit dir ein Klassifikationsmodell erstellen.
Die logistische Regression ist eine der fundamentalen Techniken für Klassifikationen. Viele Klassifikationsmethoden lassen sich auf diese zurückführen. Deshalb möchten wir die grundlegende Methodik der logistischen Regression im Folgenden detaillierter beschreiben mit dem Ziel, dass du eine gewisse Intuition oder Anschauung dafür erlangst.
Während die lineare Regression für die meisten recht anschaulich ist, besteht bei der logistischen Regression nicht unbedingt eine direkte bildliche Beziehung zu den Formeln. Ohne eine mathematisch saubere Herleitung der logistischen Regression zu geben, möchten wir dir im Folgenden eine anschauliche Begründung ihrer Anwendung skizzieren.
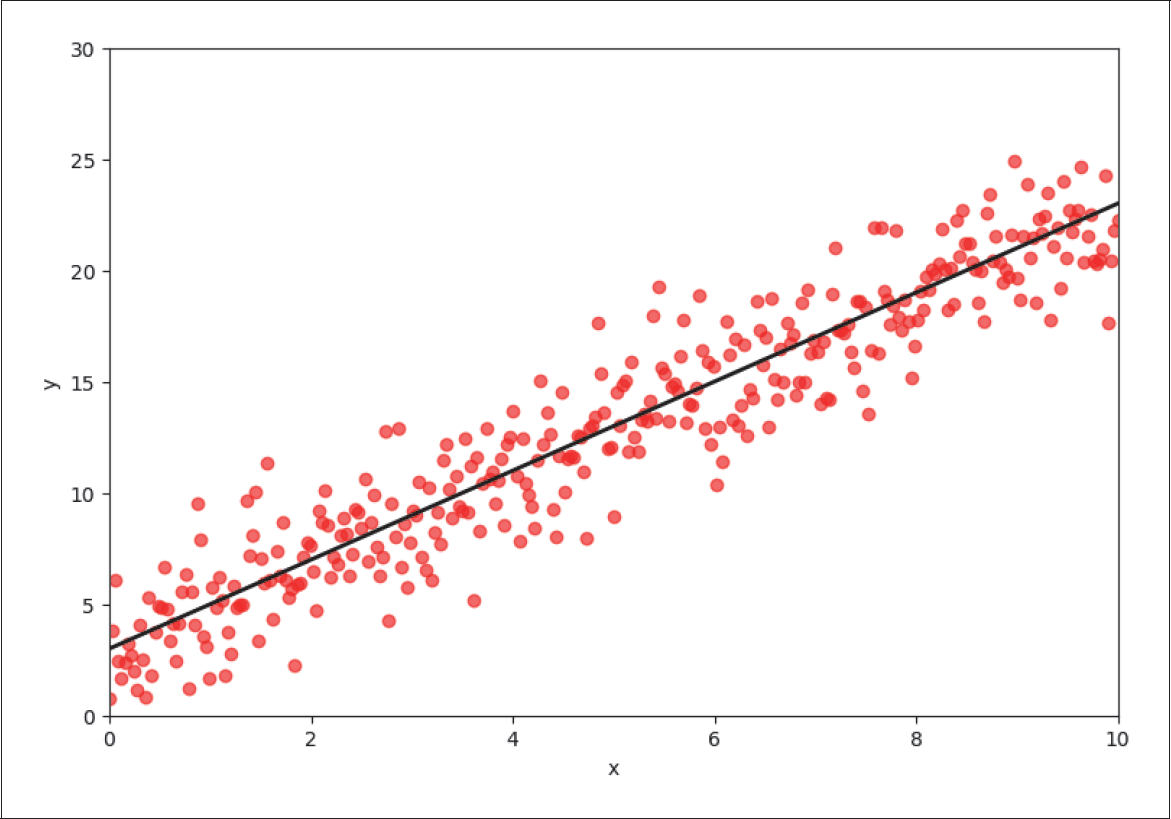
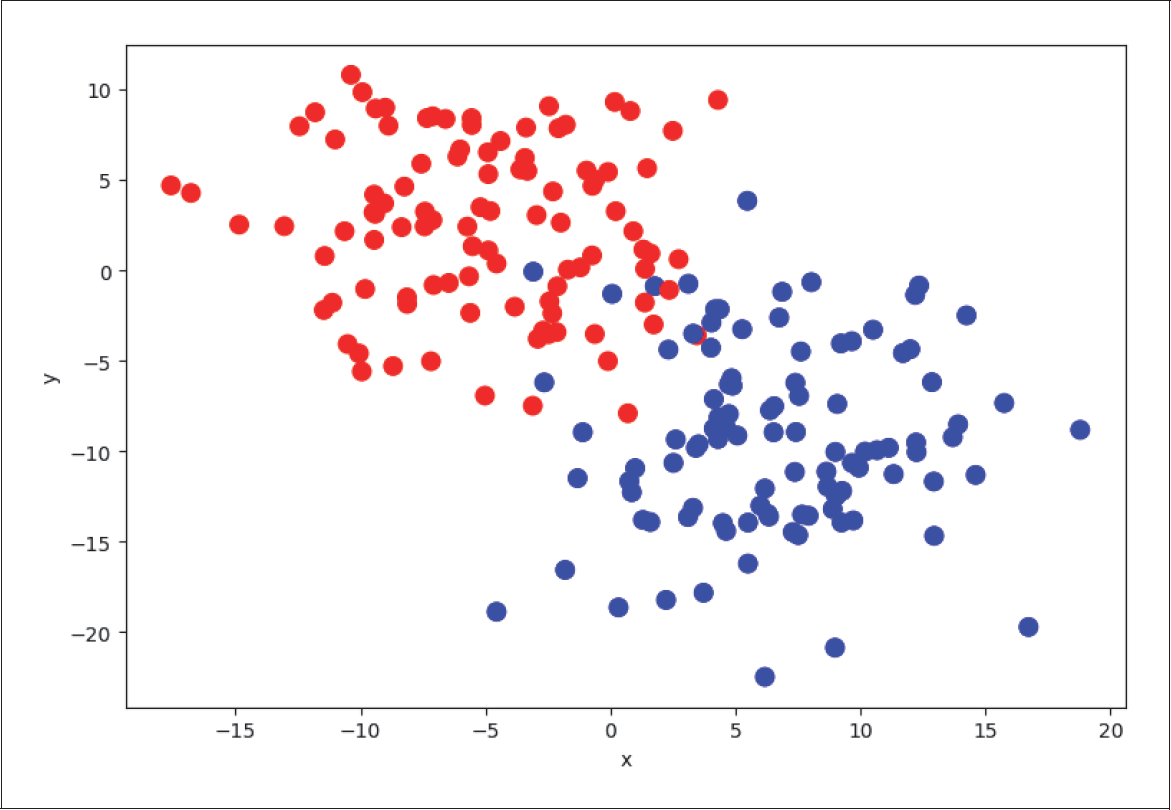
Regression und Klassifikation scheinen auf den ersten Blick ganz unterschiedliche Problemstellungen zu sein. Auf den zweiten Blick gibt es aber einige Gemeinsamkeiten, die wir uns zunutze machen können. Um die Beziehung zwischen logistischer und linearer Regression zu veranschaulichen, erzeugen wir zunächst einen neuen Regressionsdatensatz mit 300 zufällig angeordneten Datenpunkten.
In Abbildung 4-7 ist das Label y gegen das Feature x aufgetragen, und die berechnete Regressionsgerade ist in Schwarz eingezeichnet.
Abbildung 4-7: Lineare Regression
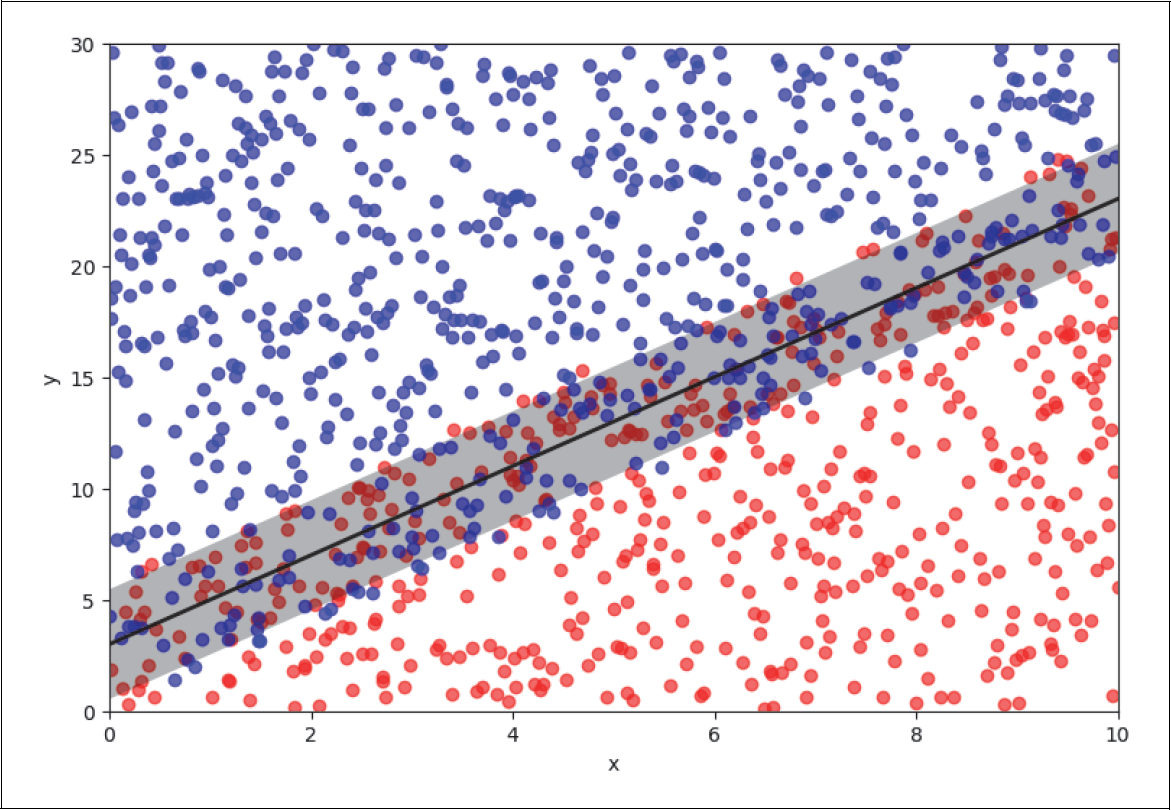
In Abbildung 4-8 betrachten wir nun ein Klassifikationsproblem. Es sind zwei verschiedene Klassen in Rot und Blau aufgetragen, die man durch eine Linie (in Schwarz) mehr oder weniger gut trennen kann. Diese beiden Klassen haben wir absichtlich so generiert, dass die trennende Linie identisch mit der Regressionsgeraden in Abbildung 4-7 ist.
Abbildung 4-8: Klassifikationsproblem
Das ursprüngliche Regressionsproblem können wir nun in ein Klassifikationsproblem umformulieren: Wir suchen jetzt nicht die beste Linie, die die Daten wiedergibt, sondern diejenige, die die beiden Klassen am besten trennt. Da es sich um ein binäres Klassifikationsproblem handelt, ordnen wir der roten Klasse das Label 1 und der blauen das Label 0 zu.
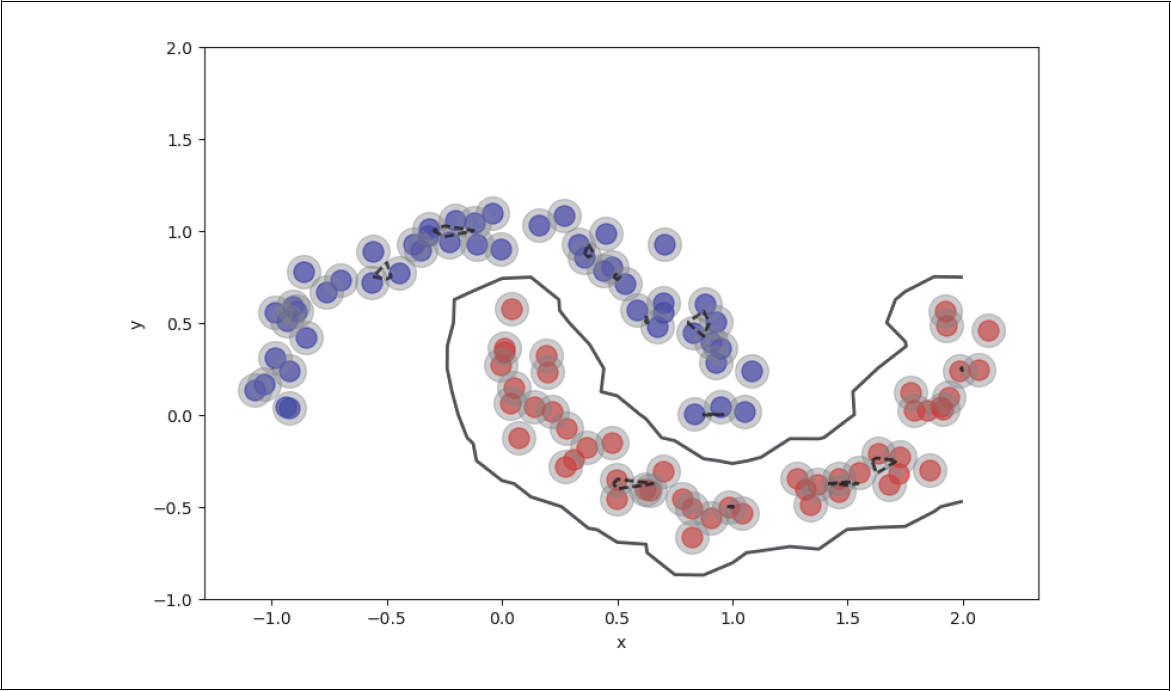
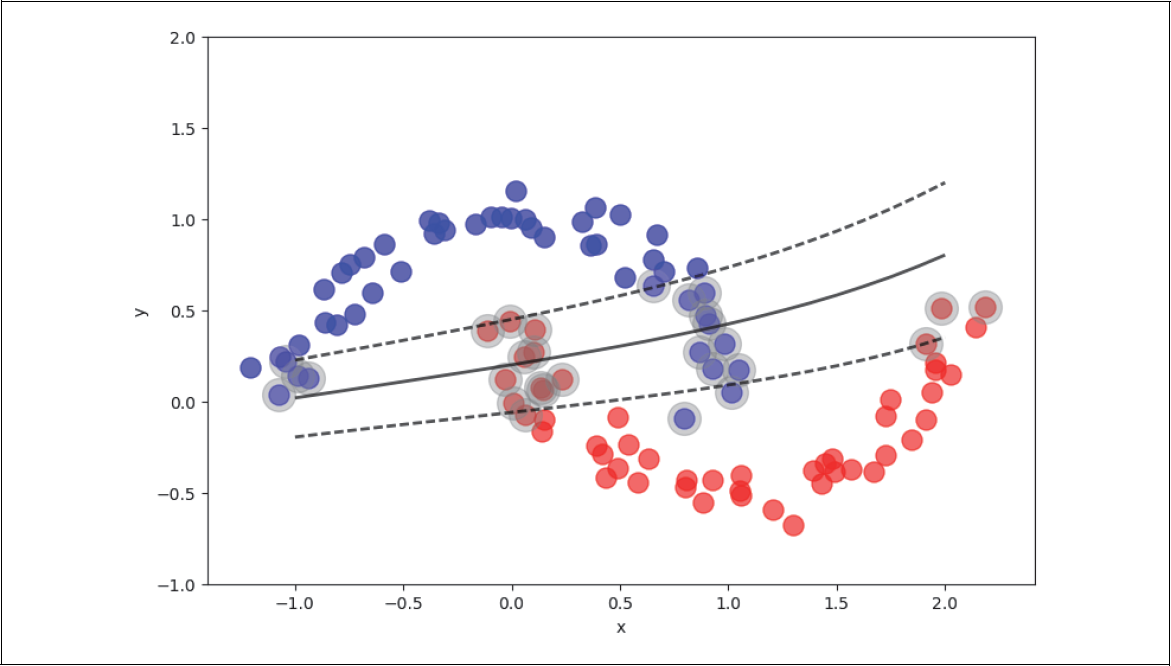
Wir bewegen uns nun entlang der ursprünglichen schwarzen Regressionsgerade und betrachten das Verhältnis von roten zu blauen Punkten als Funktion des Abstands von der Geraden. In Abbildung 4-9 haben wir das einmal für den Abstand von zwei Einheiten von der Geraden (graues Band) angedeutet.
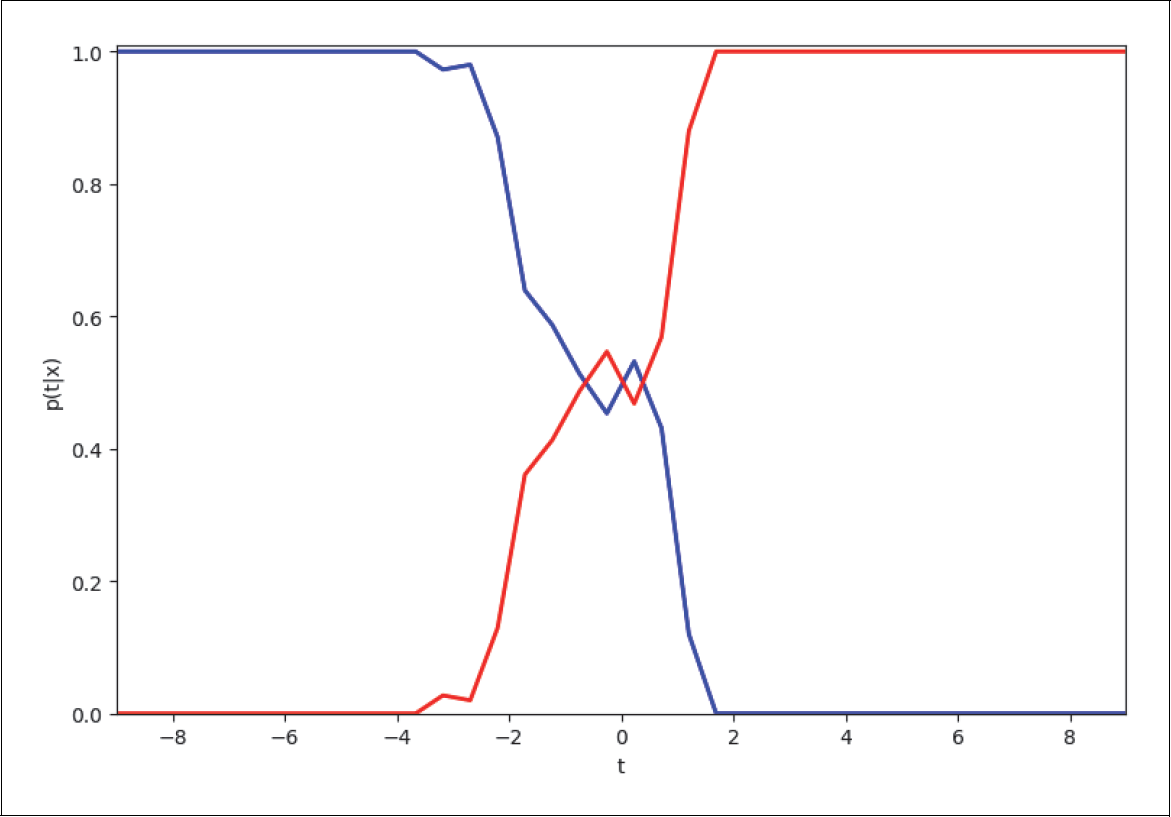
Nun stellen wir in Abbildung 4-10 dieses Verhältnis von roten zu blauen Punkten als Funktion des Abstands t zur Geraden p(t|x) dar. In der Mitte ist die Wahrscheinlichkeit etwa 50%, entweder blaue oder rote Punkte zu finden. Je weiter man nach rechts geht, desto größer ist die Wahrscheinlichkeit, dass man mehr rote als blaue sieht. Ganz weit rechts werden wir wahrscheinlich nur noch rote sehen. Das Gleiche gilt umgekehrt, wenn wir links der Geraden schauen.
Abbildung 4-9: Trainingsbeispiele entlang der Geraden
Abbildung 4-10: Anzahl Beispiele als Funktion des Abstands zur Geraden
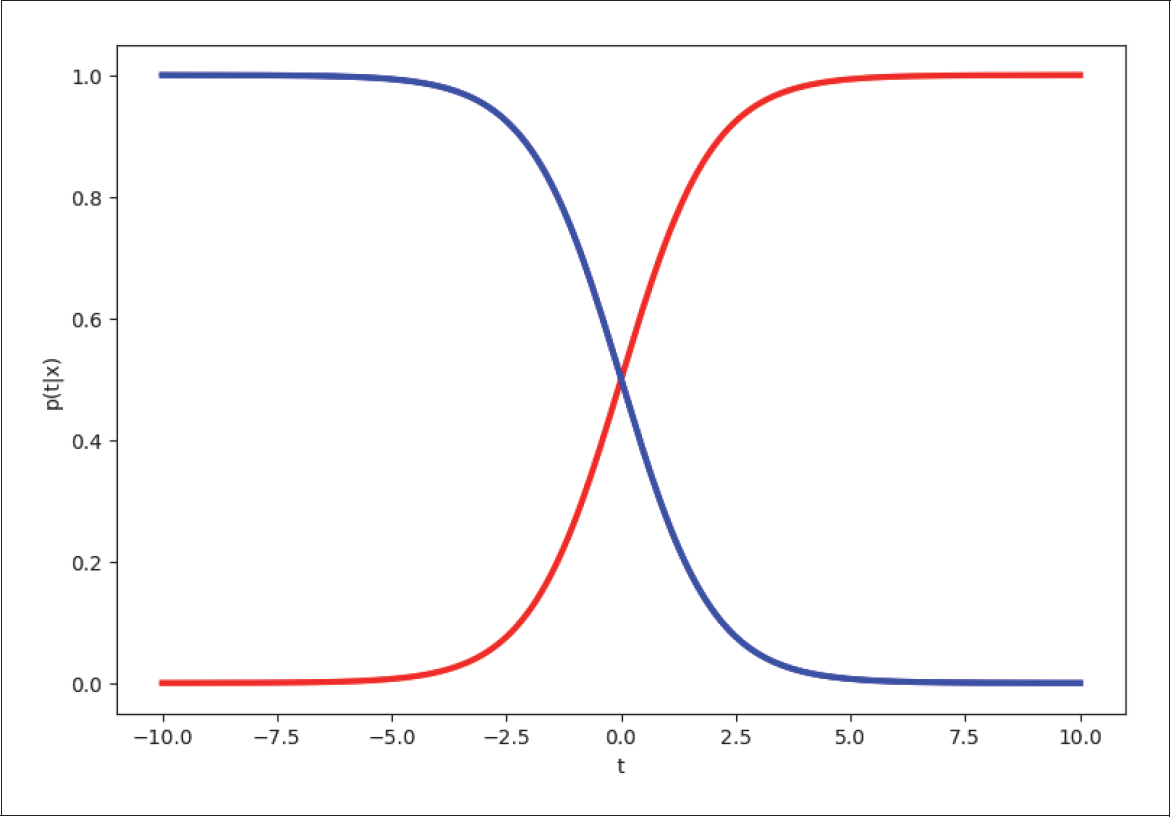
In vielen Fällen können wir die Kurve in Abbildung 4-10 mit der Kurve in Abbildung 4-11 modellieren.
Abbildung 4-11: Verteilungsmodell
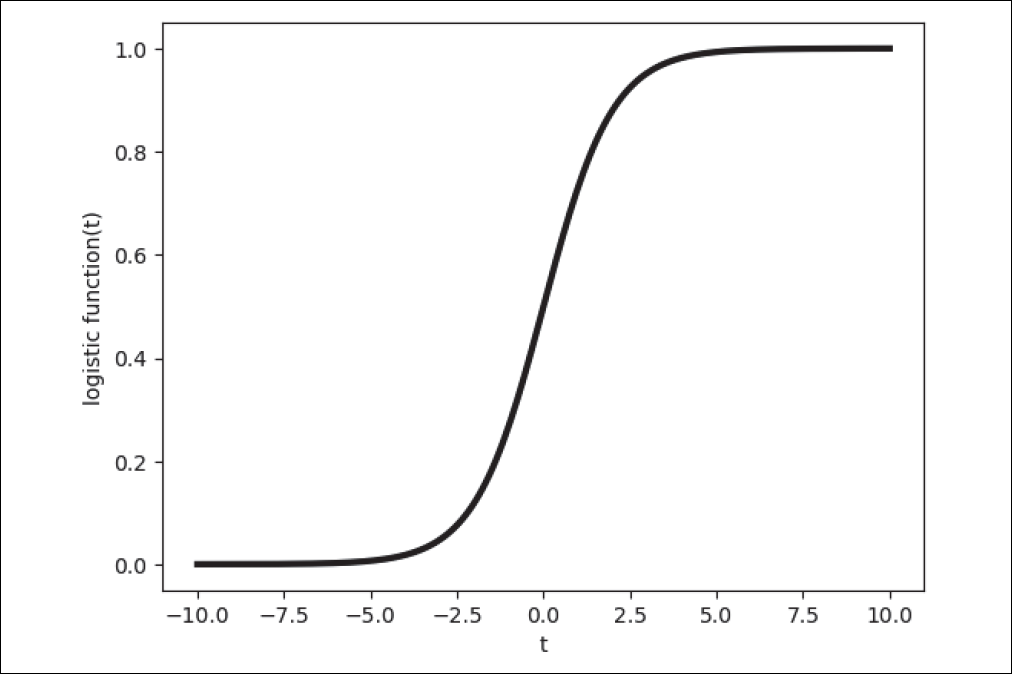
Dieses Modell macht die Annahme, dass man die Wahrscheinlichkeiten mit der logistischen Funktion beschreiben kann. Die Definition der logistischen Funktion s, wegen ihrer Form auch Sigmoid-Funktion genannt (siehe Abbildung 4-12), lautet:
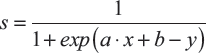
Die logistische Funktion für unser Beispiel in zwei Dimensionen ist in Abbildung 4-13 dargestellt.
Abbildung 4-12: Logistische Funktion (Sigmoid-Funktion)
Abbildung 4-13: Logistische Funktion zweidimensional
Nach dieser anschaulichen Einführung wollen wir im nächsten Abschnitt eine Klassifizierung unserer Irisdaten mithilfe der logistischen Regression durchführen.
In Sklearn kannst du den Klassifkator LogisticRegression() aus der Bibliothek linear_model aufrufen:
import sklearn.linear_model as lm
logr = lm.LogisticRegression()
Sklearn erkennt an den Shapes der Inputs X und y automatisch, wie viele Features für den Datensatz berücksichtigt werden sollen. Im obigen Beispiel hatten wir die zwei Features X und y. Der Irisdatensatz enthält drei Klassen und vier Features. Wir können nun mit nur wenigen Zeilen die etwas komplexeren Irisdaten mit logistischer Regression auswerten:
from sklearn.datasets import load_iris
iris = load_iris()
X = iris.data y = iris.target
X.shape, y.shape
> (150, 4) (150,)
logr.fit(X,y)
logr.score(X,y)
> 0.95999999999999996
Dabei werden im vierdimensionalen Feature-Raum die linearen Hyperflächen ermittelt. Eine Linie kann man mit einem Punkt in zwei Bereiche aufteilen, bei einem zweidimensionalen Raum geht das mit einer Linie. Bei höheren Dimensionen spricht man von Hyperebenen oder -flächen, die den n-dimensionalen Feature-Raum aufteilen. Die Hyperfläche hat also immer eine Dimension weniger als der Feature-Raum. In diesem vierdimensionalen Beispiel ist die Hyperdimension also drei.
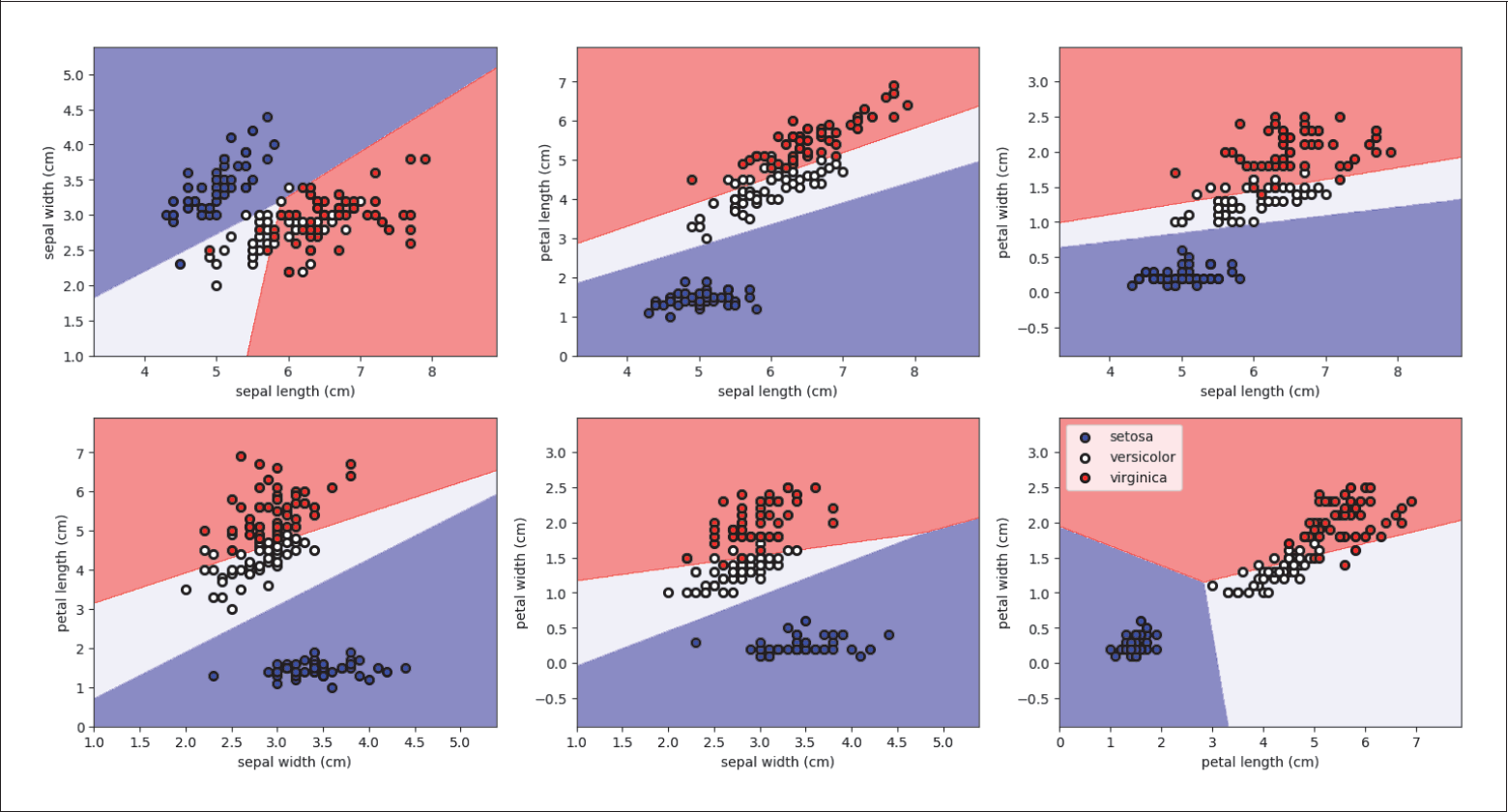
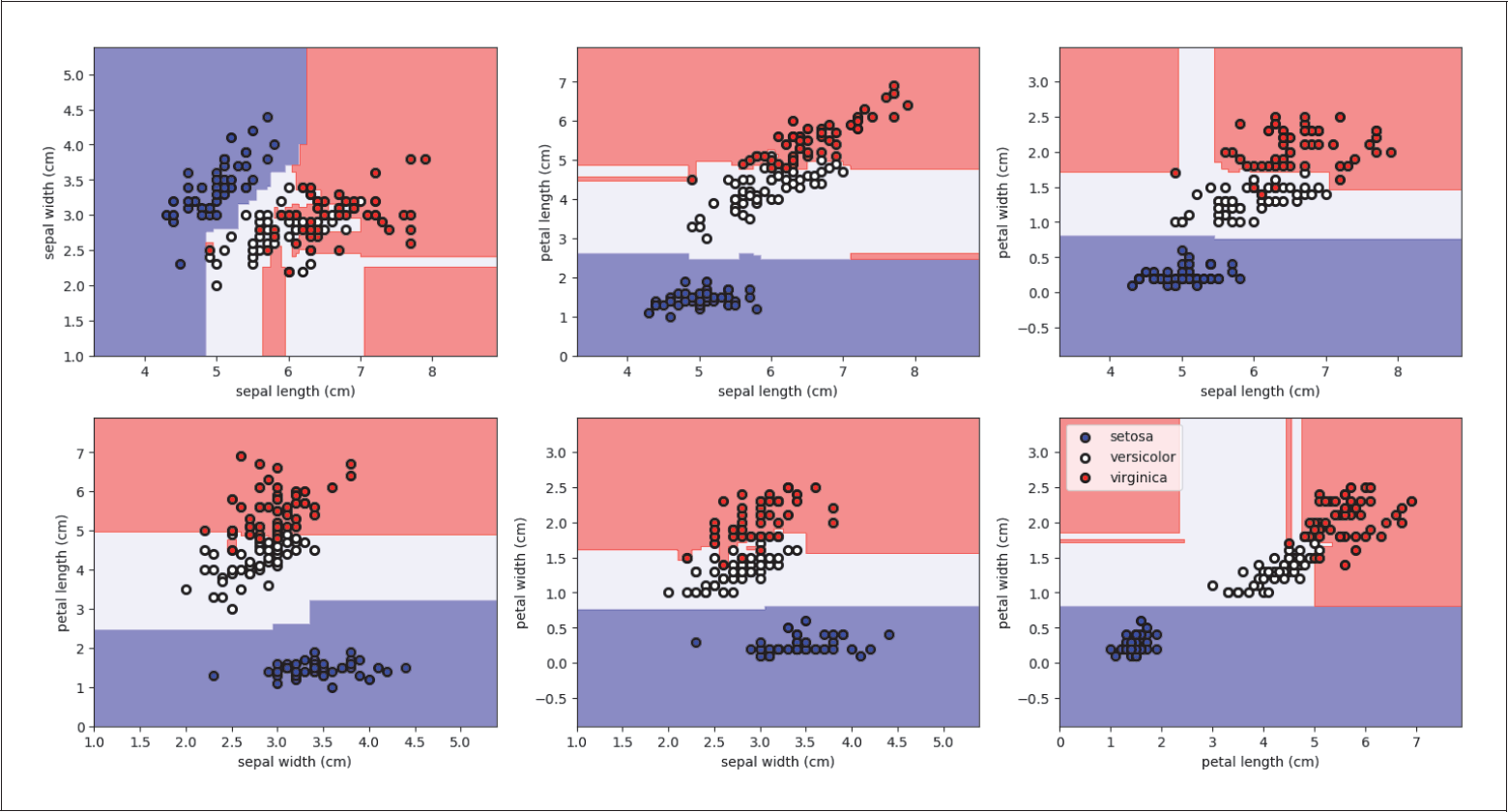
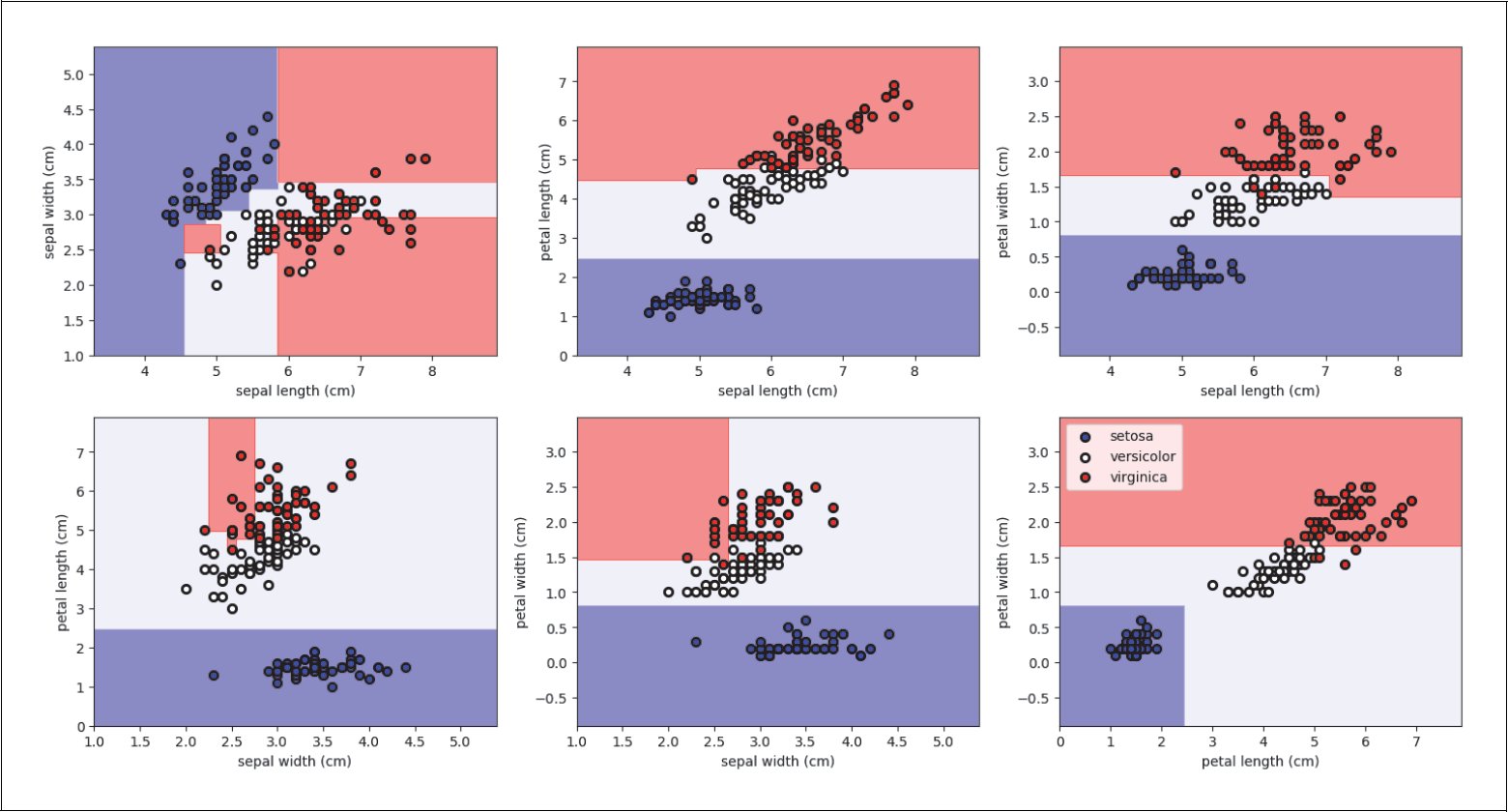
Auf diese Weise berücksichtigt man die Korrelationen zwischen allen vier Features. Wenn man sich z.B. nur paarweise zwei Features anguckt und dort jeweils die beste Gerade bestimmt (siehe Abbildung 4-14), würde man jeweils nur die paarweisen Korrelationen berücksichtigen und nicht alle vier gleichzeitig. Wichtige Informationen hätte man nicht genutzt. Im folgenden Code geben wir die Scores für jede mögliche Gerade aus:
for pair in [[0, 1], [0, 2], [0, 3], [1, 2], [1, 3], [2, 3]]:
X = iris.data[:, pair]
y = iris.target
logrfit = logr.fit(X, y)
print(logr.score(X, y))
Wie du anhand dieser Ausgaben sehen kannst, sind alle Scores geringer als der Score, den wir mit allen vier Features erreicht haben:
> 0.766666666667
> 0.926666666667
> 0.906666666667
> 0.926666666667
> 0.946666666667
> 0.873333333333
Abbildung 4-14: Paarweise Klassifikation des Irisdatensatzes
Als Nächstes wollen wir eine weitere sehr effektive Methode mit einem etwas anderen Ansatz betrachten, die sogenannte Support Vector Machine (SVM). In Sklearn wird diese Klassifikationsmethode auch Support Vector Classifier genannt (SVC). Ihr Vorteil gegenüber der logistischen Regression ist, dass sie bei kleineren Datensätzen oft bessere Ergebnisse liefert. Dies hängt mit dem Problem der hohen Dimensionen zusammen, das wir in Kapitel 5, Feature-Auswahl, diskutieren.
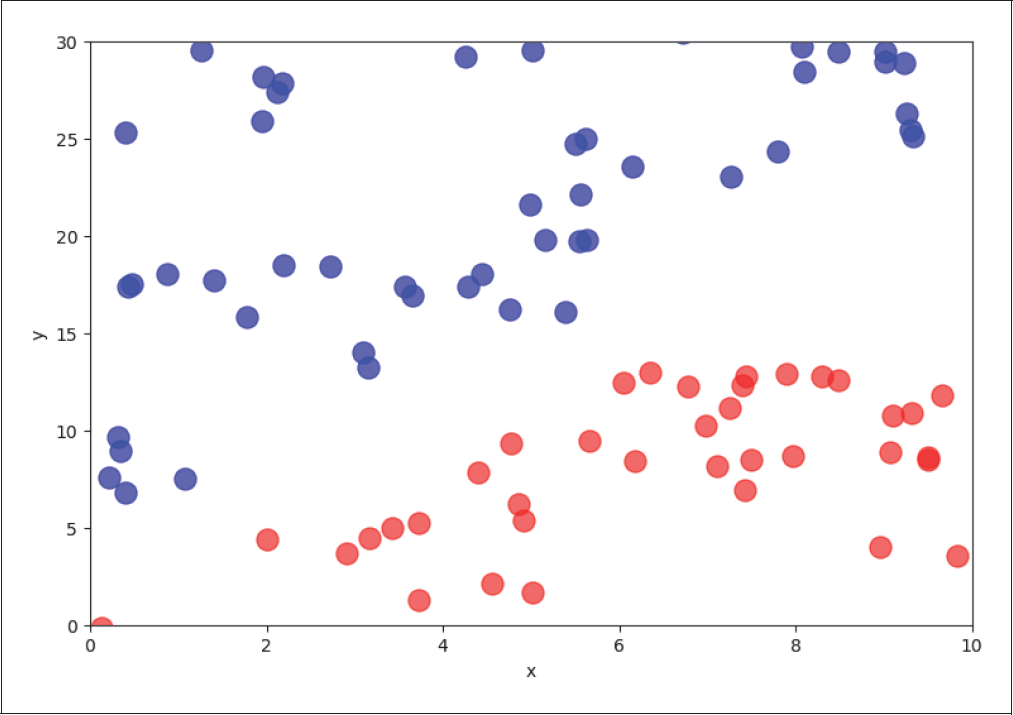
Wir wollen zunächst ein einfaches Klassifikationsproblem betrachten, bei dem die beiden Klassen in der Feature-Ebene (x-y) linear separierbar sind, was bedeutet, dass es keine Überlappungen zwischen zwei Klassen gibt. Dies kannst du gut in Abbildung 4-15 erkennen.
Abbildung 4-15: Zwei separable Klassen
Bei der logistischen Regression haben wir nach einer Geraden gesucht, die die beiden Klassen in der x-y-Ebene am besten trennt. Die logistische Loss-Funktion diente dabei als Kriterium. Die Minimierung der logistischen Loss-Funktion kann man interpretieren als Minimierung der Klassifizierungsunsicherheit oder als Minimierung der Missklassifizierungswahrscheinlichkeit.
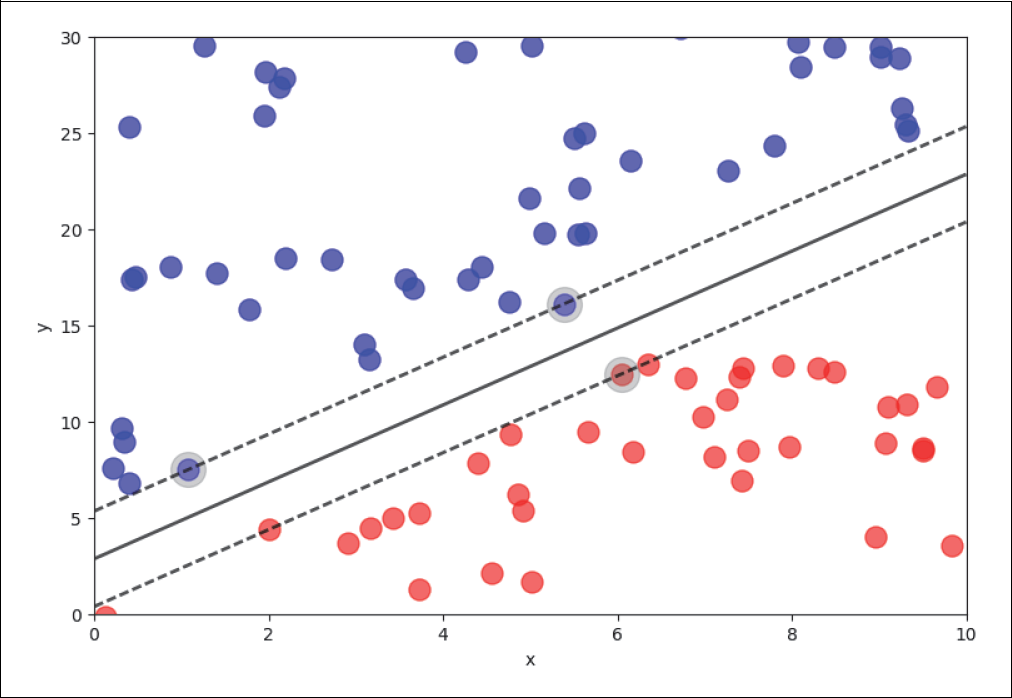
Die lineare Support-Vector-Machine-Methode sucht nach der Geraden (der durchgezogenen Linie in Abbildung 4-16), die den größtmöglichen Abstand zu den jeweils nächstgelegenen Punkten (Support Vectors) der verschiedenen Klassen besitzt. Wie schon im vorigen Abschnitt erwähnt, nennt man diese Trennungsgerade in höheren Dimensionen auch Hyperfläche. Die Region zwischen den beiden gestrichelten Linien kann als Trennungsband (Margin) interpretiert werden. Im Fall von separierbaren Problemen, wie in unserem Beispiel hier, kann man eine Region finden, die die beiden Klassen perfekt trennt (Hard-Margin):
from sklearn.svm import SVC
model1 = SVC(kernel='linear', C=1)
model1.fit(X, y)
model1.score(X, y)
Wir erhalten den erwarteten perfekten Score:
> 1.0
Abbildung 4-16: Support Vector Machine – Hard Margin
Diese Hyperfläche, in unserem Fall also die Trennungsgerade, findet man, indem man die sogenannte Hinge-Loss-Funktion minimiert:
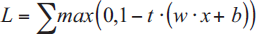
Dabei bezeichnet t das Label (–1, 1: blau, rot), und w·x + b bezeichnet die Geradenfunktion in Vektorschreibweise. Wenn w also der Normalenvektor der Geraden ist und x der Vektor eines Punkts, kann man w·x, also das skalare Vektorprodukt, auch als ein Maß für den Abstand des Punkts x zu der Geraden interpretieren. Wir wollen w·x + b maximieren, also 1 –(w·x + b) minimieren.
Für die Berechnung der Lösung ist es wichtig, dass nur wenige Punkte diese Hyperfläche bestimmen. Im separierbaren Fall sind es lediglich die Punkte, die auf den Grenzen der Margin liegen. Diese heißen Support Vectors (eingegraute Punkte in Abbildung 4-16) und geben dieser Methode auch ihren Namen. Die Punkte, die auf der falschen Seite der Linie liegen, wollen wir mit einem negativen Faktor belegen, also 1 – t·(w·x + b).
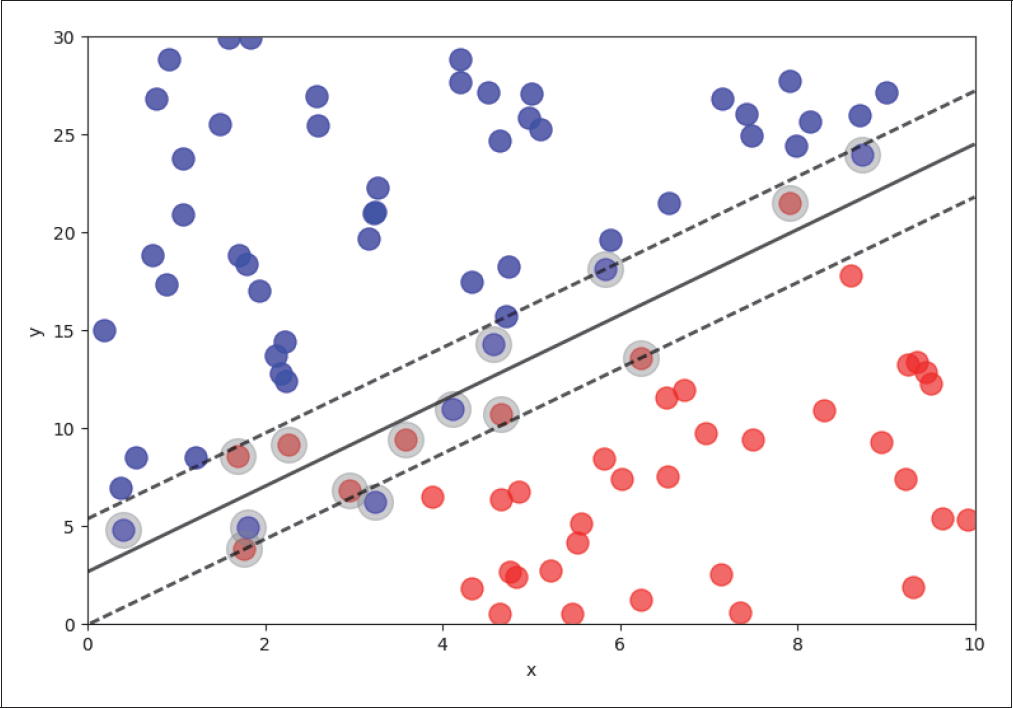
Für die Betrachtung von nicht linear separierbaren Klassen haben wir, wie in Abbildung 4-17 zu sehen, zwei Klassen mit Überlappungen generiert.
Abbildung 4-17: Linear nicht separable Klassen
Bei linear nicht separablen Klassen ist es sinnvoll, der Loss-Funktion einen weiteren Term hinzuzufügen, der die Toleranz von Punkten auf der falschen Seite kontrolliert:

Je größer λ gewählt wird, desto stärker erhöht man die Toleranz dieser missklassifizierten Punkte an der Grenzlinie.
Sklearn benutzt die externe Bibliothek libsvm (https://www.csie.ntu.edu.tw/~cjlin/libsvm/), die den Parameter C zur Regulierung verwendet. C ist umgekehrt proportional zu λ: C ~ 1/λ. Je kleiner C gewählt wird, desto stärker erhöht man die Toleranz dieser missklassifizierten Punkte an der Grenzlinie. Man weicht die Region regelrecht auf, deshalb heißt sie auch Soft-Margin (siehe Abbildung 4-18).
Abbildung 4-18: Soft-Margin
Für einen C-Wert von 100 erhalten wir beispielsweise einen Score von über 0,95:
Wenn wir C von 100 auf 0.01 ändern, weichen wir die Margin auf (siehe Abbildung 4-18):
model3 = SVC(kernel='linear', C=1.E-2)
model3.fit(X, y)
print(model3.score(X, y))
> 0.946666666667
Der optimale Wert für C hängt vom Problem und den jeweiligen Daten ab. Meist findet man ihn nur durch Ausprobieren. Die Anpassung der Parameter zur Vermeidung von Overfitting vor und während der Trainingsphase nennt man Regularisierung. Methoden zur Optimierung und Regularisierung von Parametern besprechen ausführlicher in Kapitel 5, Feature-Auswahl.
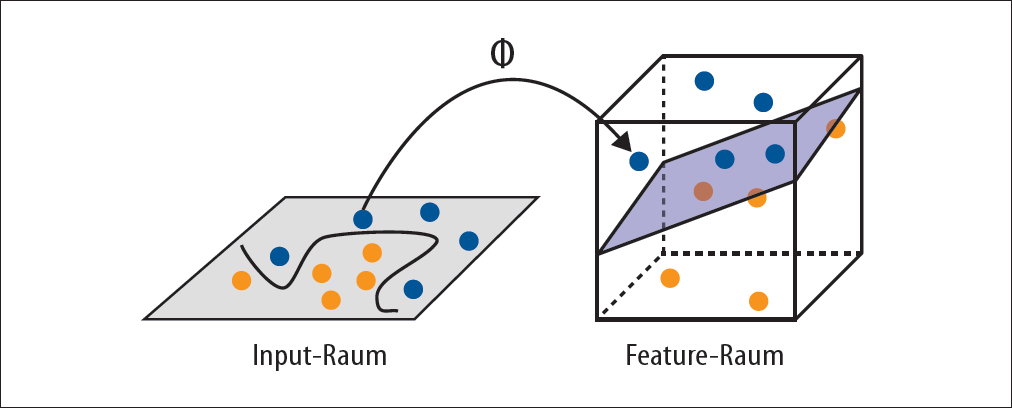
Um nicht-lineare Verteilungen zu trennen, bedient man sich eines Tricks. Dabei werden die ursprünglichen Koordinaten, die durch unsere Input-Variablen vorgegeben sind, in einem neuen Feature-Koordinatensystem so transformiert, dass die Klassen wieder durch eine Gerade trennbar sind. Grundsätzlich können fast alle Funktionen durch eine Summe von verschiedenen Basisfunktionen approximiert werden, z.B. durch Polynome oder Gaußfunktionen. Letztere ist ein Spezialfall einer sogenannten Radial-Basis-Funktion (RBF), die sehr häufig benutzt wird.
SVM bedienen sich eines weiteren Tricks: Statt die Koordinatentransformationen für alle Trainingsbeispiele durchzuführen und dann die Distanzberechnung in dem Feature-Raum vorzunehmen, werden Kernelfunktionen eingesetzt. Mit Kernelfunktionen kann man die Distanz in dem Feature-Raum berechnen, ohne die Koordinatentransformation explizit auszuführen. Das ist der sogenannte Kerneltrick, der bei hohen Dimensionen viel Rechenaufwand sparen kann.
Diese Vorgehensweise ist äquivalent zu einem Mapping der Input-Variablen vom Input-Raum in den Feature-Raum, bei der die Hyperfläche linear ist (Abbildung 4-19).
Abbildung 4-19: Input-Raum-Transformation
Ein Beispiel für die Anwendung von RBF-Kerneln in SVM ist in Abbildung 4-20 dargestellt.
Abbildung 4-20: SVM mit RBF-Kernel
Eine perfekte Trennung erhält man durch die Anwendung einer SVM mit Radial-Basis-Funktionen (RBF) als Kernel:
model4 = SVC(kernel='rbf', C=1E6, gamma=1.)
model4.fit(X, y)
print(model4.score(X,y))
> 1.0
Zur Gruppe der Radial-Basis-Funktionen gehören alle Funktionen, deren Wert nur abhängig von der Distanz zu einem bestimmten Mittelpunkt ist. Gaußfunktionen gehören damit z.B. auch zu den RBF. Der Parameter Gamma für die RBF bestimmt dabei, wie schnell oder wie langsam der Wert der Funktion in Abhängigkeit zur Distanz vom Mittelpunkt fällt. Je größer der Gammawert, desto schneller wird der Funktionswert kleiner.
Somit sind RBF sehr gut geeignet, um beliebige Kurven anzupassen oder zu glätten. Jede Kurve kann durch eine Summe von RBF angenähert werden. Den Einfluss jeder einzelnen Funktion kann man über den jeweiligen Gammawert kontrollieren. Die einzelne Funktion hat jeweils einen großen oder geringen lokalen Einfluss auf die Gesamtfunktion. Man kontrolliert somit die Granularität der Approximation.
Genau so kann man auch die Granularität des SVM-Klassifikators kontrollieren. Im obigen Beispiel wurde ein Gammawert von 1 benutzt. Die Bedeutung der Granularität im Fall von Klassifikatoren können wir illustrieren, indem wir Gamma auf 100 erhöhen. Wir sehen, dass der Klassifikator überfittet, weil er jedem einzelnen Trainingsbeispiel zu viel Wert einräumt, das Modell ist zu komplex (siehe Abbildung 4-21).
Verringern wir hingegen den Gammawert auf 0,001, sehen wir Underfitting. Die Granulation ist zu gering, um die Struktur abzubilden, das Model ist zu einfach (siehe auch Abbildung 4-22):
model5 = SVC(kernel='rbf', C=1E6, gamma=1.)
model5.fit(X, y)
print(model5.score(X,y))
> 1.0
Abbildung 4-21: SVM-Overfitting
Abbildung 4-22: SVM-Underfitting
Wenden wir die SVM mit linearem Kernel auf den Irisdatensatz an, erhalten wir schon bessere Ergebnisse als mit linearer logistischer Regression:
iris = load_iris()
X = iris.data
y = iris.target
model6 = SVC(kernel='linear', C=1, gamma=1)
model6.fit(X, y)
print(model6.score(X,y))
> 0.98
Mit nicht-linearem RBF-Kernel und etwas Tunen erreichen wir sogar 100%. Wie oben bereits gezeigt, ist hier ein Overfitting wahrscheinlich. Dieses Thema werden wir für diesen Fall in Kapitel 6, Modellvalidierung, aber noch näher betrachten.
model7 = SVC(kernel='rbf', C=1, gamma=1)
model7.fit(X, y)
print(model7.score(X,y))
> 0.98
model8 = SVC(kernel='rbf', C=1000, gamma=1)
model8.fit(X, y)
print(model8.score(X,y))
> 1.0
Die nicht-linearen dreidimensionalen Hyperflächen, die von der SVM ermittelt werden, lassen sich sehr schwer im vierdimensionalen Feature-Raum darstellen. Deshalb verzichten wir hier wieder darauf. Einen gewissen – aber dennoch eingeschränkten – Eindruck erhältst du, wenn wir wieder paarweise die zweidimensionalen Entscheidungsflächen darstellen (siehe Abbildung 4-23).
Abbildung 4-23: Paarweise RBF-SVM-Klassifikation der Irisdaten
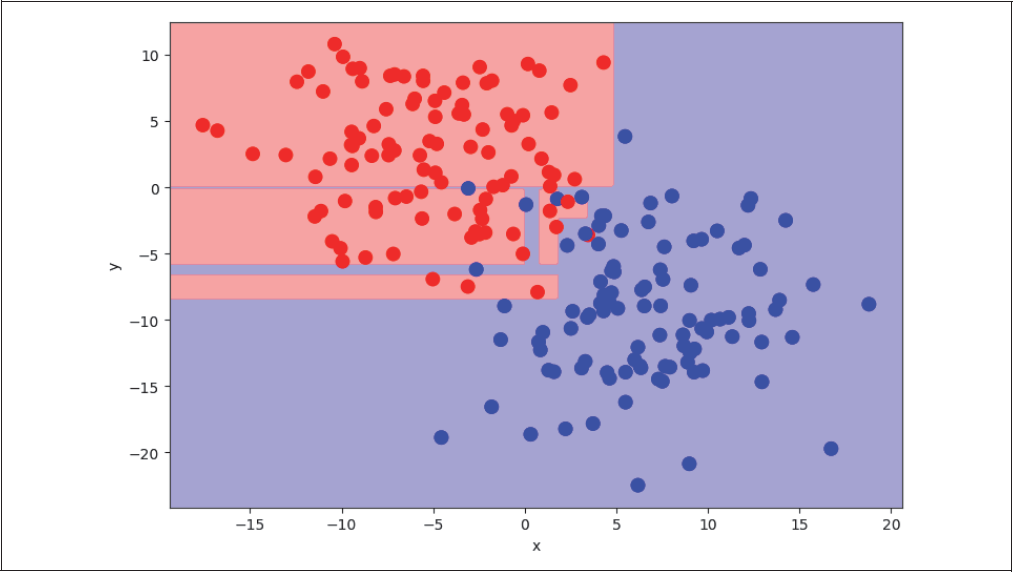
Decision Trees haben einige Vorteile gegenüber den beiden schon beschriebenen Klassifikationsmethoden. Man benötigt in der Regel keine so aufwendige Vorverarbeitung (Preprocessing), weil kategorische Input-Variablen nicht als Zahlen codiert werden müssen. Du kannst sogar numerische und kategorische Features simultan benutzen. Zudem kann eine Rangfolge der relevantesten Features nach der Trainingsphase relativ leicht extrahiert werden. Dies ist vor allem wichtig, wenn du die Entscheidungskriterien des Modells nachvollziehen möchtest. Decision Trees tendieren aber auch zu Overfitting. Es gibt einige Techniken zur Regularisierung, aber diese sind in der Praxis oft recht aufwendig. Häufig sind Ergebnisse nicht reproduzierbar, weil kleine Änderungen in den Daten manchmal schon zu gänzlich unterschiedlichen Baumstrukturen führen können.
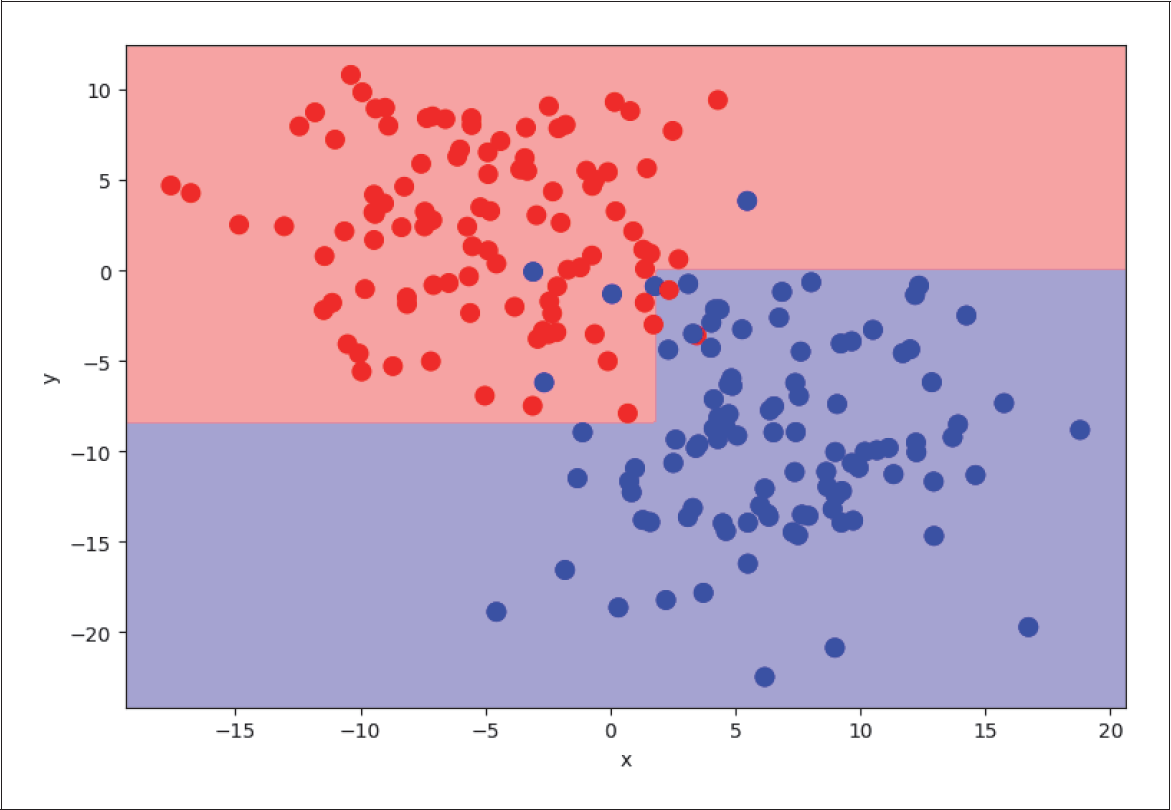
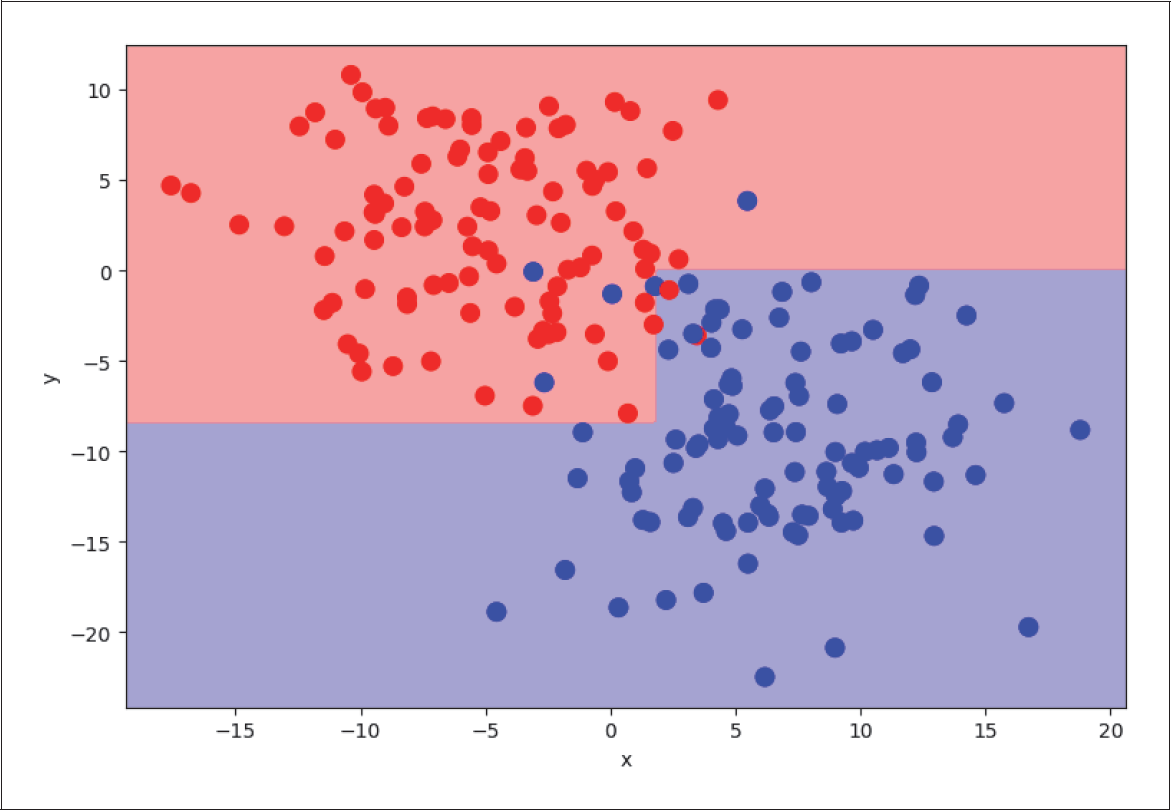
Als einfaches Beispiel erzeugen und betrachten wir wieder zwei verschiedene Klassen (rot und blau dargestellt in Abbildung 4-24) mit den Features x und y.
Abbildung 4-24: Decision Tree mit zwei Klassen
Wenn wir den DecisionTreeClassifier von Sklearn auf dieses Problem anwenden, können wir die Entscheidung des Klassifikators wieder mithilfe farbiger Entscheidungsflächen visualisieren (siehe Abbildung 4-25):
from sklearn.tree import DecisionTreeClassifier
tree = DecisionTreeClassifier()
tree.fit(X, y)
print(tree.score(X,y))
> 1.0
Abbildung 4-25: Entscheidung eines Decision-Tree-Klassifikators
Eine schematische Darstellung des Entscheidungsbaums könnte in etwa aussehen wie in Abbildung 4-26 (in der Abbildung wurden aus Platzgründen die Verzweigungswerte auf ganze Zahlen gerundet).
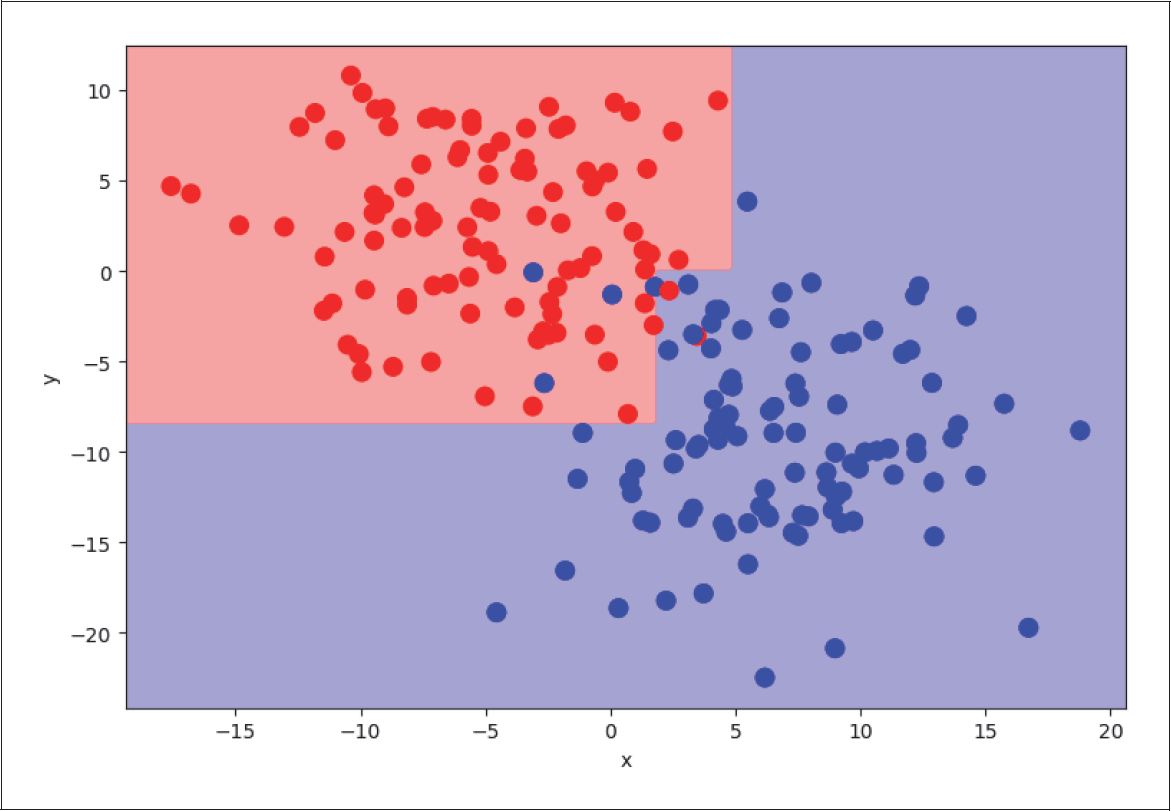
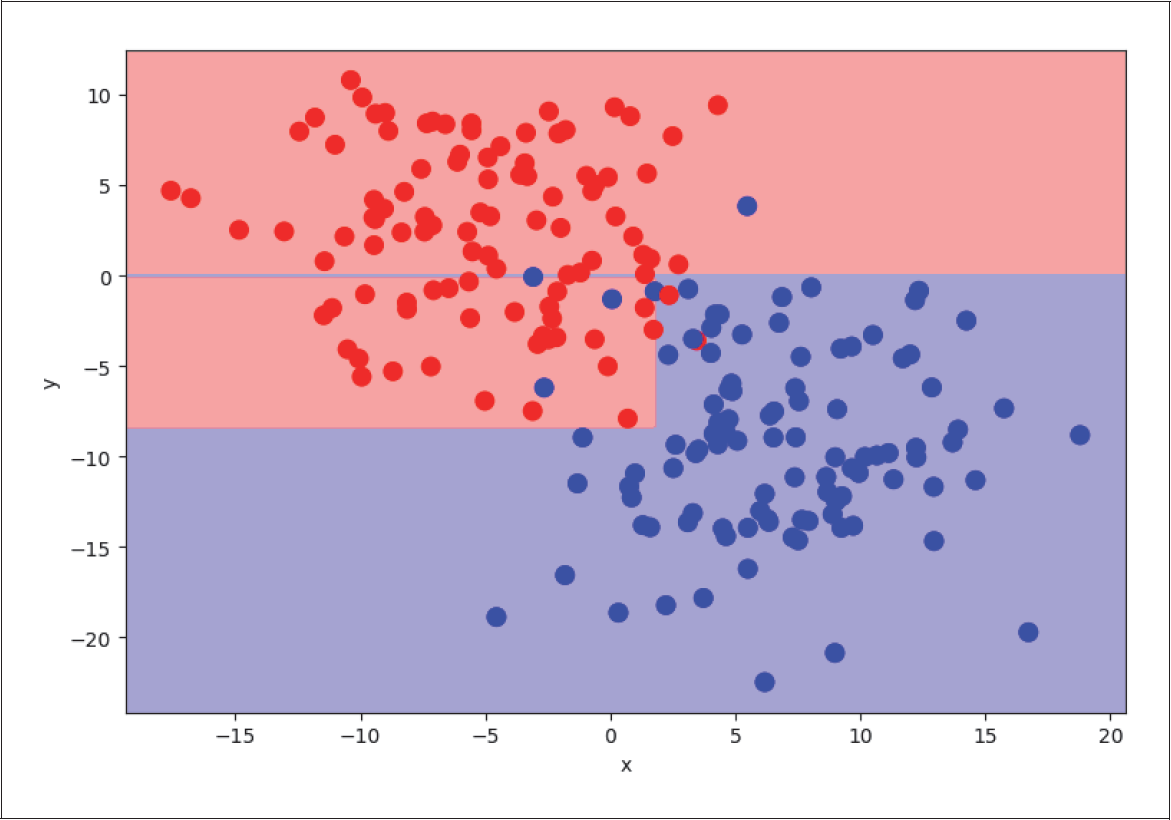
Auf der ersten Ebene teilt der Algorithmus alle Trainingsbeispiele in zwei Zweige: in die Beispiele mit dem x-Wert kleiner als 1,75 (erster Zweig) bzw. größer als 1,75 (zweiter Zweig, siehe Abbildung 4-27). Selbst mit dieser sehr groben Einteilung kommen wir bereits auf einen erstaunlich guten Score. Wir setzen dazu max_depth auf 1, um nur die erste Ebene zu nutzen:
tree1 = DecisionTreeClassifier(max_depth=1)
tree1.fit(X, y)
print(tree1.score(X,y))
> 0.91
Abbildung 4-26: Schematischer Entscheidungsbaum
Abbildung 4-27: Level 1 Decision Tree
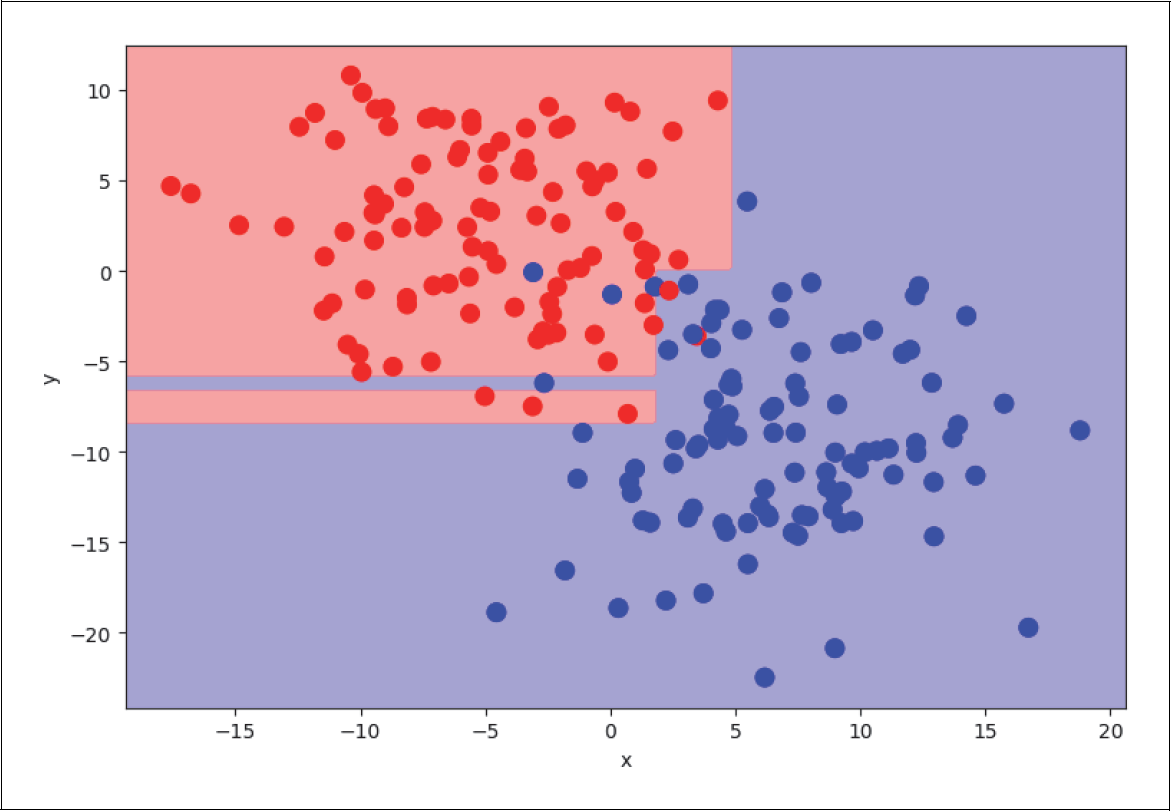
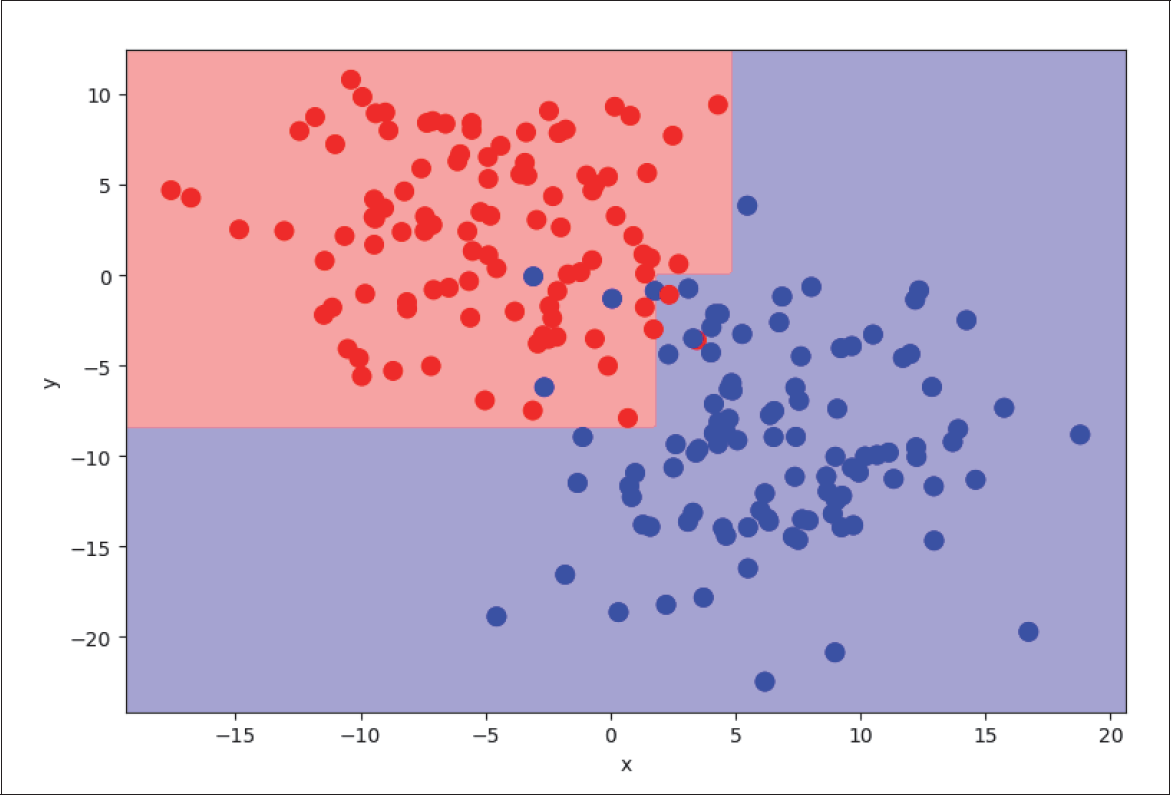
Auf der zweiten Ebene werden die jeweiligen Zweige wiederum in je zwei weitere Zweige geteilt. Der erste Zweig (x <= 1,75) wird geteilt in die Beispiele mit y < –8,4 und y > –8,4, während der zweite Zweig (x > 1,75) bei etwa y = 0 geteilt wird (siehe Abbildung 4-28):
tree2 = DecisionTreeClassifier(max_depth=2)
tree2.fit(X, y)
print(tree2.score(X,y))
> 0.97
Abbildung 4-28: Level 2 Decision Tree
Auf die gleiche Weise geht es weiter auf der dritten und vierten Ebene etc. (siehe Abbildungen 4-29 bis 4-31), und zwar so lange, bis nur noch Beispiele von lediglich einer Klasse in einem Zweig (Branch) übrig sind. Die Klassifikation für den Zweig ist dann abgeschlossen, und der Zweig wird als Blatt (Leaf) bezeichnet:
tree3 = DecisionTreeClassifier(max_depth=3)
tree3.fit(X, y)
> 0.975
tree4 = DecisionTreeClassifier(max_depth=4)
tree4.fit(X, y)
print(tree4.score(X,y))
> 0.985
tree5 = DecisionTreeClassifier(max_depth=5)
tree5.fit(X, y)
print(tree5.score(X,y))
> 0.99
Auf der vierten und fünften Ebene (siehe Abbildungen 4-30 und 4-31) kannst du aber ein Problem erkennen: Overfitting. Der Algorithmus konzentriert sich zu sehr auf Einzelbeispiele und verliert dadurch an Generalisierungsfähigkeiten. Wir erkennen das intuitiv daran, dass kleine Entscheidungsinseln oder -raster entstehen, die nur durch einzelne wenige Trainingsbeispiele verursacht werden.
Abbildung 4-29: Level 3 Decision Tree
Abbildung 4-30: Level 4 Decision Tree
Abbildung 4-31: Level 5 Decision Tree
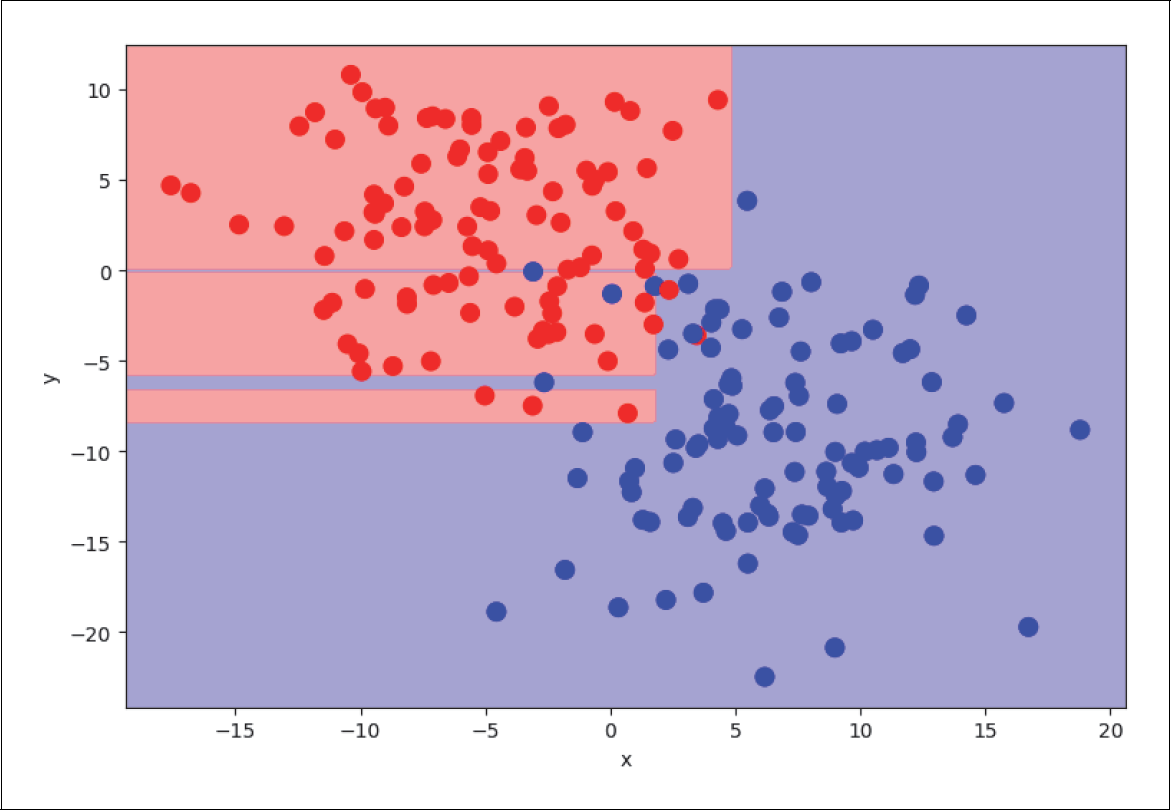
Wir können Overfitting unterdrücken, wenn wir einige der Voreinstellungen ändern. Eine Möglichkeit ist, die maximale Anzahl der Ebenen anzugeben. Wenn wir in unserem Beispiel diese maximale Anzahl auf 2 festlegen, sehen die Entscheidungsflächen folgendermaßen aus (siehe Abbildung 4-32):
tree = DecisionTreeClassifier(max_depth=2)
tree.fit(X, y)
print(tree.score(X,y))
> 0.97
Abbildung 4-32: Regularisierung: maximale Anzahl an Levels
Eine weitere Möglichkeit besteht darin, die Mindestanzahl von 10 Beispielen pro Verzweigung anzugeben (siehe Abbildung 4-33):
tree = DecisionTreeClassifier(max_depth=2,
min_samples_split=10)
tree.fit(X, y)
print(tree.score(X,y))
> 0.98
Abbildung 4-33: Regularisierung: Mindestanzahl von Beispielen pro Branch-Splitting
Die Festlegung der maximalen Anzahl von Blättern auf 8 führt zu folgendem Ergebnis (siehe Abbildung 4-34):
tree = DecisionTreeClassifier(max_depth=2,
min_samples_leaf=1,
max_leaf_nodes=8)
tree.fit(X, y)
print(tree.score(X,y))
> 0.98
Zusammenfassend können wir nun den Decision-Tree-Algorithmus mit allen besprochenen Regularisierungen auf den Irisdatensatz anwenden und erhalten folgende Ergebnisse:
X = iris.data
y = iris.target
from sklearn.tree import DecisionTreeClassifier
tree = DecisionTreeClassifier(max_depth=2,
min_samples_leaf=1,
min_samples_split=2,
max_leaf_nodes=8)
print(tree.score(X,y))
> 0.99333333333
Abbildung 4-34: Regularisierung: maximale Anzahl von Leaf-Knoten
Das Resultat ist eine dreidimensionale Hyperebene im vierdimensionalen Feature-Raum. Diese können wir natürlich wieder nicht darstellen, deshalb zeigen wir dir die paarweisen zweidimensionalen Entscheidungsflächen in Abbildung 4-35. In diesem Fall gibt es sechs solcher Flächen. Sie stellen die Projektionen der dreidimensionalen Hyperfläche auf die jeweilige zweidimensionale Ebene dar. So bekommst du zumindest einen gewissen Eindruck der resultierenden Hyperebene.
Wie schon in der Einleitung angedeutet, können Decision Trees sehr effektiv sein, aber sie tendieren leicht zu Overfitting. Eine Regularisierung mithilfe der vielen Parameter ist möglich, in der Praxis aber oft zu aufwendig. Weiter unten werden wir dir im Rahmen der Ensemble-Methoden auch die Random-Forest-Methode vorstellen, die einige dieser Probleme löst.
Abbildung 4-35: Paarweise Entscheidungsflächen des Decision-Tree-Klassifikators für die Irisdaten
Vorher aber sehen wir uns für ein besseres Verständnis in einem kleinen Exkurs an, wie auf jeder Ebene die Entscheidung getroffen wird, auf welche Weise die Zweige aufgespaltet werden. Als Voreinstellung wird das Gini-Kriterium angewendet. Dieses Kriterium ist ein Maß für die Klasseninhomogenität (engl. Impurity) der Beispiele, die in einen Zweig fallen:
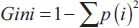
Dabei bezeichnen i die Klasse und p die Wahrscheinlichkeit einer Klasse (approximiert durch den relativen Anteil der Beispiele in Klasse i). Je kleiner der Gini-Wert, desto größer ist die Klassenhomogenität des Splits. Teilt der Split die Beispiele für einen Zweig nur in eine Klasse i auf, wird die Wahrscheinlichkeit für Klasse i 1 und 0 für alle anderen. Die Gini Impurity ist dann null. Sind aber gleich viele Beispiele für alle Klassen i in dem Zweig, werden alle Wahrscheinlichkeiten klein und die Gini Impurity maximal.
Alternativ kannst du Entropie als Kriterium anwenden. Die Entropy ist definiert als
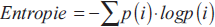
Sie ist ebenfalls ein Maß für die Homogenität der Klassen. Sie hat ähnliche Eigenschaften wie die Gini Impurity, und man erhält in der Praxis sehr ähnliche Ergebnisse mit beiden. Das Gini-Kriterium ist bei der Berechnung etwas schneller, da kein Logarithmus angewendet wird, und wird deshalb etwas häufiger benutzt. Details dazu findest du wieder im Notebook zu diesem Kapitel.
Ein großer Nachteil von Decision-Tree-Klassifikatoren ist das Overfitting. Wie wir im vorigen Abschnitt gesehen haben, kann man Overfitting unterdrücken, indem man verschiedene Regularisierungsparameter manuell einstellt. Die optimalen Parameter hängen aber nicht nur von den Features ab, sondern auch von der Anzahl der Trainingsbeispiele. Die Suche nach den optimalen Parametern oder eine Automatisierung ist in der Praxis häufig sehr umständlich und aufwendig.
Die Bagging-Methode des Random Forest Classifier ist ein Beispiel für eine Ensemble-Methode, die Overfitting reduzieren kann. Beim Random-Forest-Klassifikator wird der Datensatz zunächst in kleinere zufällige Subsamples aufgeteilt (wobei Duplikate ausdrücklich erlaubt sind). Für jedes dieser Subsamples wird wiederum ein Decision Tree Classifier mit einem zufälligen Subsample der Features generiert. Die jeweiligen Ergebnisse werden am Ende mit gleicher Gewichtung zusammengefasst. Es wird bei Regression also der Durchschnitt aller Einzelentscheidungen für die Gesamtentscheidung genommen bzw. bei der Klassifikation die Mehrheit.
In der Voreinstellung in Sklearn werden zehn zufällige Klassifikatoren (n_estimators) generiert. Zusätzlich kann man auch noch die Parameter der einzelnen Decision Trees einstellen (max_depth etc.).
from sklearn.ensemble import RandomForestClassifier
rf = RandomForestClassifier()
> RandomForestClassifier(bootstrap=True,
class_weight=None, criterion='gini',
max_depth=None, max_features='auto',
max_leaf_nodes=None,
min_samples_leaf=1, min_samples_split=2,
min_weight_fraction_leaf=0.0, n_estimators=10,
n_jobs=1,
oob_score=False, random_state=None, verbose=0,
warm_start=False)
Wenden wir den so initiierten Random Forest Classifier auf unsere Irisdaten an, erhalten wir folgendes Ergebnis:
rf.fit(X, y)
print(rf.score(X, y))
> 0.995
In Abbildung 4-36 wird der Klassifikator wieder als paarweise Entscheidungsflächen visualisiert.
Abbildung 4-36: Paarweise Entscheidungsflächen des Random-Forest-Klassifikators für die Irisdaten
Random Forests haben in der Regel eine bessere Performance als einzelne Decision Trees. Sie neigen auch weniger zu Overfitting. Ansonsten besitzen sie weiterhin alle Vorteile von Decision Trees, z.B. die Information über die Relevanz der einzelnen Features während des Trainings, und aufwendiges Preprocessing ist ebenfalls nicht notwendig.
Bei der Bagging-Methode oben werden die Ergebnisse von möglichst guten Einzelklassifikatoren zusammengefasst, um eine bessere Generalisierung zu erreichen. Im Gegensatz dazu versucht man mit der Boosting-Methode, viele verschiedene schwache Einzelklassifikatoren zu trainieren. Dabei geht man in der Regel iterativ vor. In jedem Iterationsschritt wird ein zufälliger schwacher Klassifikator hinzugenommen, und zusätzlich bekommen die missklassifizierten Trainingsbeispiele höhere Gewichte und die korrekt klassifizierten geringere. Sukzessive werden dadurch viele spezialisierte Klassifikatoren generiert, die kombiniert werden zu einem Gesamtklassifikator. Die Idee von Boosting ist es, Overfitting zu reduzieren, indem man viele schwache (nicht komplexe) Klassifikatoren, die jeder für sich overfitten, kombiniert.
Es gibt verschiedene Boosting-Algorithmen. Einer der bekanntesten ist der AdaBoost-Algorithmus, der auch in Sklearn benutzt werden kann:
from sklearn.ensemble import AdaBoostClassifier
bdt = AdaBoostClassifier()
bdt.fit(X,y)
print(bdt.score(X,y))
> 0.96
Die Ergebnisse des Boosted Decision Tree, angewendet auf den Irisdatensatz, sind in Abbildung 4-37 visualisiert.
Boosted Decision Trees können bessere Ergebnisse liefern als Random Forests, wenn man die Parameter richtig anpasst. Das Tunen der vielen Parameter ist wesentlich aufwendiger als bei Random Forests, und bessere Ergebnisse sind nicht garantiert. Ein Overtunen von Boosted Decision Trees führt leichter zu Overfitting.
Abbildung 4-37: Paarweise Boosted-Decision-Tree-Klassifikation der Irisdaten