TABLE 6-1. EL ESCORIAL CRITERIA—MODIFIED13,15
TABLE 6-1. EL ESCORIAL CRITERIA—MODIFIED13,15The motor neuron diseases (MNDs) are categorized by their pathological affinity for the voluntary motor system including anterior horn cells, certain motor cranial nerve nuclei, and corticospinal/bulbar tracts. Amyotrophic lateral sclerosis (ALS), also known as Lou Gehrig disease, is the most notorious of these disorders. The boundaries of what is and what is not ALS, particularly in the context of early diagnosis of individual patients, remain imprecise. In this Chapter, in an attempt to distinguish ALS from other MNDs, we consider ALS to be a disorder that has the following characteristics: (1) the clinical manifestations are dominated by signs attributable to voluntary motor system dysfunction, (2) the disease progresses rapidly both within and between different body regions, (3) that life expectancy is <5 years from clinical onset in the vast majority of cases, (4) and that no other etiology can be identified.
Despite its characterization as a motor system degeneration, ALS is best conceptualized as a multisystem disorder.1 This perspective is reinforced by both a clinical and pathological overlap between ALS and frontotemporal lobar degeneration (FTLD) (pathological) and frontotemporal dementia (clinical).2 Consequently, ALS is more correctly considered as a disorder in which dysfunction of the voluntary motor system involvement is the dominant source of morbidity (in the majority of cases) but in which involvement of other neurological systems at times clinically, and more commonly pathologically, develops.
The uncertain etiology of sporadic ALS (sALS) and the increasingly complex biology of ALS contribute to a lack of coherence in ALS nosology. This confusion applies to both historical and contemporary perspectives. In 1849 and 1850, respectively, Duchenne and Aran described progressive muscular atrophy (PMA), a disorder they believed to be of muscular origin. PMA has been long recognized however, to result from anterior horn cell degeneration.3 In 1860, Duchenne first described a syndrome of progressive dysphagia and dysarthria and coined the term progressive bulbar palsy (PBP).3 In 1874, Charcot and Cruveilhier recognized that corticospinal tracts and anterior horn cells were often affected concomitantly. Their description serves as the basis for our current construct of ALS.3 In the next year, Erb described primary lateral sclerosis (PLS), a progressive disorder of corticospinal tracts, without (at least initially) evidence of muscle atrophy, fasciculation, or weakness.3 ALS, PMA, PBP, and PLS are accepted by most, but not all neurologists as interrelated entities. PMA and PLS are clinically defined by the type of motor neuron affected. PBP on the other hand, is defined by site of disease onset, regardless of the type of motor neuron involved. Although survival in PMA and particularly PLS will on average exceed that of ALS, there is considerable overlap.4–10 Survival in ALS does not differ significantly between different disease categories as described by the El Escorial criteria (EEC) (see below) including the original EEC-suspected category that is synonymous with PMA, that is an exclusively lower motor neuron (LMN) presentation.9 Many patients with these initially limited MND (PBP, PMA, and PLS) phenotypes evolve into ALS. Unfortunately, phenotypic classification does not provide a mechanism by which to predict the natural history of disease in an individual patient. Patients with prolonged (>5 years) survival have a similar distribution of phenotypes as do patients with typical natural histories.11 Individual patients fulfilling ALS criteria may have indolent courses whereas patients with PMA may progress rapidly with approximately a third of PMA patients dying within 3 years of symptom onset.8,12
Recognizing the pragmatic limitations imposed by the imprecise MND boundaries, Lord Brain, in his text of 1962, proposed the “lumped” concept of MND, presumably to acknowledge and circumvent uncertainties of the split classification. To this day, MND serves as a synonym for ALS and other forms of MND within the United Kingdom.3 Arguably, it represents the most intellectually honest means to classify these disorders until a biological basis to justify lumping or splitting becomes available.
There have been three international consensus conferences that have attempted to provide an ALS classification scheme that is both accurate and clinically pragmatic. The first of these met in El Escorial, Spain in 1990. As there were no recognized effective treatments at that time, the primary goal was to define ALS with a high degree of sensitivity and specificity for research purposes. The proceedings of this meeting led to the subsequent publication of the EEC (Table 6-1).13 Shortly thereafter, Riluzole was reported to alter the natural history of the disease, the first (and to this date only) drug treatment shown to do so.14 In response to this, with promise of other effective treatments, and in recognition that the stringency of the EEC would hamper enrollment into clinical trials, a subsequent meeting was held in Airlie House, Virginia in 1998.15 The purpose of this convocation was to modify the EEC in order to allow earlier diagnosis without reducing diagnostic specificity thus facilitating for earlier and expedited clinical trial enrollment.
TABLE 6-1. EL ESCORIAL CRITERIA—MODIFIED13,15

Unfortunately, the Airlie House revision of the EEC may not have resulted in earlier participation in clinical trials.9 Despite the virtual universal acceptance of EEC and its Airlie House modifications as the “gold standard” for ALS diagnosis, they may continue to sacrifice sensitivity for specificity.16 Patients with early MNDs are frequently restricted from participation in clinical trials at a time in their disease when they are presumably most likely to be treatment responsive. Studies suggest that only 56% of patients clinically thought to have ALS will fulfill definite or probable Airlie House diagnostic criteria at the time of diagnosis.9 In addition, it has been recognized that the EEC classification at diagnosis, for example, definite, probable, possible, or suspected, does not correlate with clinical course and survival.9 Furthermore, up to 10% of a clinically defined ALS population will die without achieving either of these levels of diagnostic certainty.9 In support of this, postmortem examination of patients who would not fulfill definite or probable ALS diagnostic criteria because of phenotypes restricted to upper motor neuron (UMN) or LMN findings will have pathological confirmation of ALS.17,18 The diagnosis of probable or definite ALS via EEC is dependent upon either clinical or lab demonstration of both UMN and LMN findings in two or three body regions (cranial, cervical, thoracic, lumbosacral) respectively. The reliable demonstration of UMN findings can be difficult and subjective, particularly in the thoracic and cranial regions. In addition, there is no reliable surrogate marker for UMN involvement as there is for lower motor disease (EMG). For these reasons, the early diagnosis of ALS via EEC/Airlie House criteria remains problematic.
In an attempt to improve sensitivity without sacrificing specificity in ALS diagnosis, a third consensus conference of experts convened in Awaji Island, Japan in 2006.19,20 The premise of conference participants was that fasciculation potentials, particularly when “complex or unstable,” represented an adequate surrogate for fibrillation potentials and positive waves as a marker of ongoing denervation. In an attempt to maintain adequate disease specificity, the authors dictated that these unstable and complex fasciculation potentials had to occur in the context of two additional features: (1) the patient had clinical features suggesting ALS, and (2) fasciculation potentials had to occur in muscles concomitantly with chronic motor unit action potential (MUAP) changes. Subsequent studies supported the Awaji hypothesis by suggesting that diagnostic sensitivity increased from 28% to 60% in comparison to EEC while maintaining the same specificity of 96%.21,22
Although marketed as an evidence-based document, none of the citations supporting the widespread application of the Awaji criteria have offered convincing evidence that patients achieving an early diagnosis of ALS utilizing these criteria evolve into definite ALS as determined by existing gold standards of either a typical clinical course or postmortem confirmation. In addition, the proponents imply that the demonstration of “unstable” fasciculation potentials provides a reliable means to distinguish ALS from less malignant causes of fasciculation potentials.19,20 In a separate manuscript however, one of the authors of the Awaji manuscript refute the contention that “benign” and “malignant” fasciculation potentials can be reliably distinguished from one another.23 We are in agreement with all three sets of criteria that consider both a clinically weak muscle and a strong muscle that electrodiagnostically displays both ongoing and chronic changes of denervation to be equivalent in defining the anatomical extent of the disease. We, like others, remain unconvinced that fasciculation potentials, unstable or otherwise, should be accepted as a surrogate marker of ongoing denervation in the absence of other EDX abnormalities.24
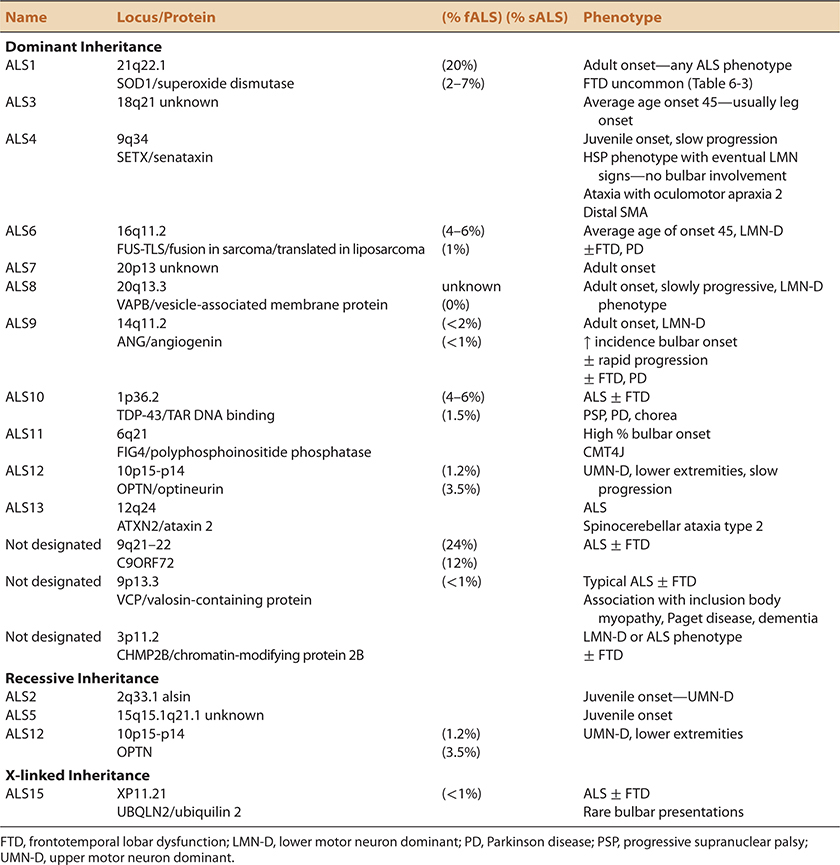
Aran first reported the occurrence of ALS in multiple family members in 1848. Nonetheless, the concept of familial ALS (fALS) was dismissed by Charcot and largely ignored until the discovery of the first ALS gene mutation in the early 1990s (Table 6-2). At least 5–10% of ALS cases associated with 13 currently recognized gene mutations occur as a result of a single gene (Mendelian) mutation.25 In addition, it seems likely that genetic influence confers disease susceptibility in some portion of sporadic cases. It has been reported that there is an approximate eightfold increased lifetime risk of developing ALS in siblings or progeny of apparent sALS patients.26
TABLE 6-2. FAMILIAL ALS25,27,30,42,43
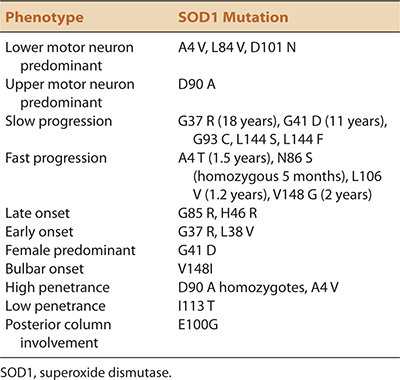
fALS, like sALS displays phenotypic heterogeneity. Other than for an earlier average age of onset, there are no distinguishing features that allow clinical distinction between inherited and sporadic cases. The full spectrum of phenotypic heterogeneity of ALS is evident even within different point mutations of the same fALS (SOD1) gene (Table 6-3). fALS may occur with juvenile or adult onset, slow or fast progression, limb or bulbar onset, UMN or LMN predominance, and in the presence or absence of frontotemporal dysfunction.27–30 Not only do all of these different phenotypic variations occur in both fALS and sALS, they seem to occur with similar prevalence rates. As in sALS, fALS patients may never fulfill the clinical EEC requirements for probable or definite ALS during the patient’s lifetime.
TABLE 6-3. PHENOTYPIC VARIABILITY OF SOD MUTATIONS IN FALS25,27,28,66
As alluded to previously, the historical conceptualization of ALS, either inherited or sporadic, is that of a degenerative disorder of anterior horn cells and pyramidal tracts. This construct is confounded by the recognition that both the clinical manifestations and pathology of ALS may affect extrapyramidal, cerebellar, and particularly, cognitive systems.1,2 It has been estimated that anywhere from 10% to 75% of ALS patients will have subtle or overt abnormalities in executive function, behavior, and/or language if carefully sought for, implicating frontotemporal lobar dysfunction (FTD).2,27,31,32 Estimates of overt dementia range from 15% to 40%.2 Conversely, if patients with FTD are carefully examined, it has been suggested that 14% will have signs of definite ALS and additional 36% signs of possible disease.33 MND may precede, occur concurrently, or follow the onset of FTD.34 In this chapter, we will use the acronym FTD to represent frontotemporal dysfunction without necessarily implicating overt blown dementia. The intent of this departure from consensus criteria nomenclature is that it allows us to consider the full spectrum of frontotemporal deficits, not only those fulfilling the Neary diagnostic criteria for dementia.2,35,36
Estimates of FTD prevalence however, originate in large part from Western cultures where prolonged survival and the use of tracheostomy-assisted mechanical ventilation (TAMV) occur infrequently. Experiences in cultures where long-term ALS survivors are more prevalent, related to increased utilization of TAMV, suggest that the occurrence of dementia increases over time in ALS patients. This further supports the concept of ALS as a multisystem disorder.37,38 ALS with dementia or involvement of other neurological systems is designated as ALS plus by the EEC.13,39 Frontotemporal dysfunction in fALS is increasingly recognized but has not been as well described as in sALS. This may reflect ascertainment bias as FTD has been estimated to occur less in those with SOD1 mutations, the most historically prevalent genotype of fALS prior to the recognition of the C9ORF72 mutation.2,40
Although the focus of this chapter is ALS, the strong association with FTD and FTLD justifies a few comments relevant to the epidemiology of the latter disorder. In addition to ALS, FTLD may occur in association with cortical basal ganglionic degeneration, progressive supranuclear palsy, or other neurodegenerative conditions. In approximately 10% of cases, it is associated with an apparent autosomal dominant mode of transmission.41 The genes most commonly related to heritable FTD that have been identified to date include microtubule-associated protein tau (MAPT) and progranulin (PGRN). MND occurs uncommonly with these mutations. Like fALS however, the locus for many fFTD cases remains unidentified.41
FTD co-associates with a number of fALS genotypes. Recently, hexanucleotide repeat mutations of the C9ORF72 gene have been identified as the most common cause of fALS and/or frontotemporal dementia, representing 12% of fFTD, 3% of apparent sFTD, 24% of fALS, and 4% of apparent sALS in North America.42 In Finland, the impact of this mutation is even greater with 46% of fALS and 21% of apparent sALS cases resulting from chromosome 9 open reading frame 72 (C9ORF72) gene expansion.43 Other mutations that are less frequent causes of FTD, but more common causes of ALS with or without FTD occur in the chromatin-modifying protein 2B (CHMP2B), fused in sarcoma (FUS), TAR DNA-binding protein (TARDBP), polyphosphoinositide phosphatase (FIG4), ubiquilin 2, and valosin-containing protein (VCP) genes.25,44 As mentioned, FTD is recognized infrequently with SOD1.40
Within the general population, MNDs are uncommon. The incidence of ALS averages 1.8/100,000 across all studies. This incidence appears to be increasing both within and outside the boundaries of an aging population.1 The average age of onset is 56 years for sporadic disease and 46 years for most dominantly inherited forms of the disease.25,27 Teenagers and the elderly may be afflicted. Although it is widely believed that environmental factors must play at least some role in ALS pathogenesis, epidemiologic studies have failed to identify any reproducible risks other than age, gender, increased body mass index, and potentially cigarette smoking.45–49 The historical inability to identify environmental risks may be related in part to methodology.48,49,51 Although ALS has been reproducibly shown to occur 1.5 times more frequently in men, this ratio diminishes with advancing age and may not be true for bulbar-onset disease which seems particularly prevalent in older women.49–52 There have been nests of apparent increased incidence. The ALS–Parkinsons–Dementia complex formerly endemic in Guam and other Western Pacific regions is the most notable example.53 The neuronal inclusions in this disorder contain tau however, not the ubiquitin characteristic of typical (non-SOD1) ALS, suggesting that Guamanian ALS may be a different disorder. Otherwise, neither ethnicity, geography, nor occupation has been reproducibly demonstrated to alter risk.
The majority of ALS patients succumb within 2 to 5 years from symptom onset without ventilatory and nutritional support.9,52,54 The range extends however from less than 1 year to more than 10 years without TAMV. It is estimated that a quarter of individuals will survive more than 5 years.12,52 A vital capacity of less than 50% of predicted is associated with the need for hospice, death, or need for mechanical ventilation within 6 months.55 Progression seems to follow a linear course, although the abrupt loss of a critical function may provide the appearance of stepwise deterioration.54 Patients with PBP are said to have a shorter average life expectancy, although many lead protracted existences if aspiration risk is minimized and ventilation preserved or supported.54 Young males seem to live longer on average.54 Rilutek, participation in a multidisciplinary clinic, noninvasive positive pressure breathing, and possibly percutaneous gastrostomy are interventions that have modest benefits in prolonging life expectancy.56
 CLINICAL FEATURES
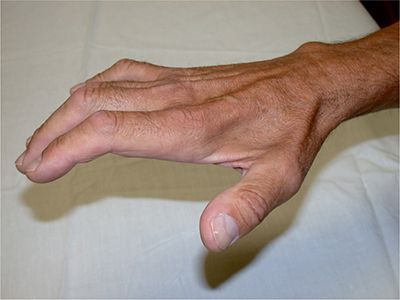
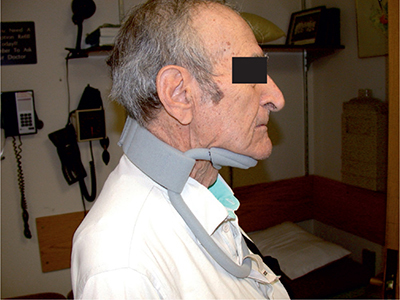
CLINICAL FEATURESALS is characterized by painless, progressive muscle weakness and atrophy (Fig. 6-1). The site of onset, the relative predominance of UMN or LMN signs, and the rate of progression are variable. Patients typically present when functional difficulties cause them to acknowledge their deficit. These initial deficits are frequently asymmetric and sometimes monomelic depending on patient vigilance. In instances where the patients do not seek early medical attention, or their physicians do not recognize the significance of the problem, the patients may not be diagnosed until their disorder is fairly advanced, months or even a year or more after onset. The initial deficits may be initially limited in distribution but should affect more than one nerve and nerve root distribution in limb-onset cases. Less commonly, the initial symptoms may be cramps, dysarthria, dysphonia or dysphagia, or related to impaired ventilation. Occasionally, the initial clinical manifestations may be head drop or bent spine syndromes related to paraspinal muscle weakness (Fig. 6-2). In this latter circumstance, the patient may actually present with back pain due to the disordered posture caused by inadequate spine support.
Figure 6-1. Hand atrophy in amyotrophic lateral sclerosis.
Figure 6-2. Head drop with cervical collar in amyotrophic lateral sclerosis.
Fasciculations are more commonly recognized by the examining physician than by patients. On occasion, they may be the initial manifestation of MND, particularly in those who have a pre-existing awareness of their potential significance. Patients presenting with a chief complaint of fasciculations without weakness, atrophy, or abnormal EMGs rarely have or evolve into ALS.57 In our experience, benign fasciculations are described more than seen, tend to occur most frequently in the calf muscles, and when observed are typically repetitive in the same location in any given muscle at any specific point in time. Conversely, fasciculations that are continuous and multifocal both within and between muscles, even in the absence of weakness, are ominous. This uncommon pattern of visible frequent and multifocal fasciculations without weakness is rarely seen in other circumstances other than MND, anticholinesterase overmedication being one notable exception. Lastly, the absence of fasciculations in patients with painless weakness does not preclude the diagnosis of ALS, particularly in those with excessive subcutaneous tissue.58 An increased frequency of muscle cramping is also common in MND. In our experience, provocation of cramps in muscles (with the exception of the calves) during manual muscle testing is seen with some frequency in ALS patients is uncommon in other disorders.
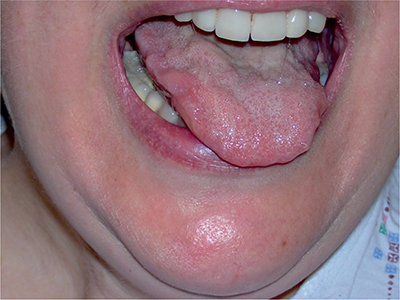
As previously alluded to, the definite clinical diagnosis of ALS is dependent on the demonstration of both LMN and UMN signs, which progress both within and between different body regions. Frequently ALS may be dominated by LMN, or less frequently UMN signs. This may occur at onset or in some patients throughout their entire disease course. Signs of LMN involvement include muscle weakness, atrophy out of proportion to disuse, attenuation or loss of deep tendon reflexes, cramps, and fasciculations.59 When LMN weakness impairs coordination, it does so to an extent proportionate to the degree of weakness.59 LMN features in cranial musculature in ALS are most frequently and convincingly manifest in the tongue. Atrophy is noted by the crenated, as opposed to the normal rounded edges, and fasciculations best noted with the tongue lying quietly on the floor of the mouth (Fig. 6-3). Tongue strength is best tested by pushing against the bulge in the cheek created by the patient “pocketing” their tongue on either side. Weakness of neck flexion and extension are common in ALS. Weakness of facial (e.g., eye closure) and jaw opening and closing muscles occur but are typically less evident. Ptosis and ophthalmoparesis are notable for their absence.
Figure 6-3. Tongue atrophy in amyotrophic lateral sclerosis.
UMN manifestations are more diverse and, at times, more subjective than their LMN counterparts. The elicitation of Babinski signs, sustained clonus, pathologically hyperactive deep tendon reflexes, and spasticity are objective and universally accepted manifestations of UMN pathology. Unfortunately, they are not overt in a significant percentage of ALS cases, thus confounding and delaying the clinical diagnosis. Other presumptive signs of corticospinal tract pathology include reflex spread (e.g., finger flexion with percussion of the brachioradialis tendon, hip flexion with percussion of the Achilles tendon), synkinesis (coactivation of muscles not required to accomplish a requested movement), Hoffman signs, and preservation of reflexes in a wasted and weak extremity. The latter is arguably the most prevalent of the subjective UMN sign demonstrable in ALS patients.52 In cranial innervated muscles, unequivocal UMN signs may be difficult to demonstrate. Increased emotional lability (pseudobulbar affect) and spastic dysarthria are perhaps the most frequently occurring UMN signs in this region but are more subjective than objective. Forced yawning is nonspecific. Exaggerated gag or jaw reflexes are more objective but do not occur with particularly great frequency. Arguably, slowness of attempted rapid blinking or tongue movements, in the absence of weakness, implicates central nervous system (CNS) pathology and provides support for UMN involvement in someone whose presentation is otherwise dominated by LMN signs. The same may be said for synkinesis, for example, the concomitant movement of the jaw with requested, rapid side-to-side tongue movements.
Coordination is impaired early with UMN involvement in a manner disproportionate to the degree of weakness.59 There is frequently a “UMN stickiness” resulting in delayed activation of requested movements associated with the normal or near-normal strength. Often, muscles not required for an attempted motion are inappropriately (synkinetically) activated. For example, contralateral foot tapping may occur with requested unilateral foot-tapping movements. With UMN disease, foot dorsiflexion strength may be normal but delayed in initiation and preceded by great toe dorsiflexion. UMN signs may be transient in ALS, as they may develop and then disappear as LMN-induced weakness evolves and trumps UMN manifestations. For example, a Babinski sign may be lost as the extensor hallucis muscle weakens.
Recognizing the value of the EEC and the Airlie House revision, we find it helpful to categorize our patients both by onset site and phenotype. Onset sites include bulbar, upper limb, lower limb, or rarely truncal or ventilatory locations. Phenotypic categories include PMA, lower motor neuron dominant (LMN-D) ALS, ALS, upper motor neuron dominant (UMN-D) ALS, and PLS. We define PMA as muscle weakness and atrophy associated with hypo- or areflexia in involved segments. LMN-D is applied to individuals with dominant LMN features associated with suggestive, but not definite UMN signs as listed above. Typically, these are individuals mentioned above whose deep tendon reflexes are either preserved or mildly increased in the involved body regions. UMN-D disease is defined by the absence of LMN signs clinically but with unequivocal signs of denervation on EMG that are not readily explained by an alternative mechanism. PLS is defined as a progressive UMN syndrome occurring without an alternative explanation without either clinical or electrodiagnostic evidence of LMN disease.
In most series, ALS is the most common presentation of MND, although even in these patients, the morbidity appears to stem primarily from LMN disease.9,59–62 In probability, most classifications consider LMN-D to represent ALS.52 In approximately two-thirds of cases, the initial site of involvement is in a limb, typically distally and asymmetrically located in a hand or foot.8,9,52,58,59 Initial weakness may occur in proximal muscles as well. A definite diagnosis cannot be made until combined UMN and LMN signs spread over a period of months, both within and outside the initially affected body region. A diagnosis of ALS meeting EEC definite or probable criteria, allowing clinical trial eligibility, is obtained in only 31–56% of cases at the time of the initial examination.9 Despite diagnostic limitations imposed by EEC, the combination of UMN and LMN findings confined to multiple segments in one limb is sinister when unassociated with pain or sensory symptoms.
It is estimated that PMA phenotype represents anywhere between 2% and 10% of MND patients.7,8,22,52,61 This variation is undoubtedly based on whether deep tendon reflex preservation in a weak limb is or is not considered to lie within the boundaries of PMA.7 A LMN-D presentation is estimated to occur in 7%, 26%, 29%, and 18% respectively in those whose disease begins in the bulbar, cervical, thoracic, and lumbosacral regions.9 These statistics may be biased however, by the ease or difficulty in identifying UMN or LMN signs in any given region. For example, UMN findings in the thoracic region may be underrepresented due to the insensitive clinical means of detection. Of those who do not manifest UMN signs at onset, 22% will develop them and 90% will evolve into EEC probable or definite disease.9 Even in the patients who fail to develop UMN features during life, the pathological features of ALS will be identified on postmortem examination.17,18,63–65
Other observations, supporting the concept that PMA and ALS exist as a continuum include the documentation that these disorders have overlapping natural histories. Although PMA and LMN-D patients live longer on average than patients classified as having ALS, individual patients in any of these categories may have malignant courses.7,8,12 On average, symmetric presentations and individuals who continue to have monomelic involvement after prolong periods of observation tend to have the more indolent courses.7,8 As would be expected, those whose measurements of ventilatory or limb strength decline precipitously have life expectancies that parallel ALS despite an absence or paucity of UMN signs.7,8 Additional arguments in support of PMA and LMN-D as part of the ALS spectrum include the recognition that PMA phenotypes occur in at least five different SOD1 fALS genotypes. The A4 V SOD1 mutation, a rapidly progressive PMA phenotype, represents the most common SOD mutation in North America and represents the most dramatic example of this phenomenon.62,66 FTD prevalence is estimated at 17% in PMA patients, providing further support for a common biology in the two syndromes.67
At times, slowly progressive forms of LMN-predominant ALS may remain confined to both upper extremities over protracted periods, producing a syndrome that has been described as flail arm, bibrachial amyotrophic diplegia (BAD), or “man-in-the-barrel” syndrome.68,69 This disorder more commonly affects the shoulder girdles initially in comparison to the more common LMN-D forms of ALS which are more likely to begin distally and progress more rapidly. A similar more indolent syndrome, referred to a lower extremity amyotrophic diplegia (LAD) may affect both lower extremities, rendering the individual paraparetic for protracted periods before spreading to other regions.69 In our opinion, both BAD and LAD are best conceptualized as PMA variants.
Between 25% and 40% of individuals present with “bulbar-onset” disease, that is, dysarthria or less commonly dysphagia.9,70 As in limb-onset ALS, PBP may be dominated by UMN characteristics, LMN characteristics, or both. As with limb-onset cases, unequivocal UMN and LMN signs occurring concomitantly in cranial innervated muscles, even in the absence of limb involvement, are ominous. As in PMA and PLS, fALS may have a PBP presentation. This PBP presentation more commonly occurs in women than in men. Life expectancy in PBP has been repeatedly demonstrated to be on average less than in limb-onset disease particularly if there is an associated language-dominant FTD.71 This prognosis does not necessarily apply to individual patients as mortality may be related more to the importance of the functions jeopardized early in the course (breathing and swallowing) than a reflection of the biology of the disease.72 In some individuals with sporadic PBP, signs and symptoms may remain confined to the “bulbar” musculature for considerable time, affecting the physicians’ confidence in the accuracy of their diagnosis, particularly with a UMN-D presentation. Some reports suggest both the prevalence and severity of FTD are increased in bulbar as opposed to limbonset cases whereas others do not. FTD has been reported to occur in as many as 48% of PBP cases if carefully sought for.73–76 One report suggests that there is an increased incidence of language-dominant FTD in patients with bulbar-onset disease.71
ALS beginning as an UMN exclusive (PLS) or dominant (UMN-D) process is less common than phenotypes dominated by LMN or bulbar dysfunction.77–81 Approximately 2–5% of ALS cases begin with a PLS phenotype.78 The average age of onset in virtually every series is about 50 years, approximately 10 years younger than typical ALS.78 Three-quarters of PLS cases involve the legs, initially creating an inability to effectively run or hop. In most cases, the onset is asymmetric and at times may be hemiparetic, referred to as the Mills variant.78 In approximately 15% of cases, PLS affects the bulbar muscles initially. In 10% of cases, the upper extremities are the first region to become symptomatic.5,6 It is commonly held that ALS spares the anterior horn of the sacral segments. As a result, it is commonly held that genitourinary symptoms do not occur in this disease. In our experience, PLS is an exception to this rule as urinary urgency and urgency incontinence may occur. Presumably this results from detrusor–sphincter dyssynergia. To further cement the biological relationship between PLS and ALS, PLS patients also appear susceptible to frontotemporal dysfunction and FTLD.81,82
The distinction between PLS from UMN-D ALS is based on whether LMN signs are absent (PLS) or present.4,5 In the aforementioned reports, the authors defined the threshold for LMN involvement by EMG as abnormalities in more than two muscles (minimal number of muscles studied not defined). These abnormalities could include fibrillation potentials/positive waves, fasciculation potentials, or evidence of mild MUAP enlargement consistent with denervation and reinnervation. Presumably, these limits were defined so as to not exclude patients with minor EMG abnormalities secondary to a separate, unrelated, neurogenic injury. It has been suggested that focal weakness, bulbar symptoms at onset, or later development of weight loss and declining ventilatory function predict transition to ALS.5
The natural history of PLS is more favorable than PMA or ALS. In nine reported series, mean disease duration ranged between 7 and 14 years.80 The majority (80%) of individuals with PLS who evolve into ALS do so within the first 4 years of their disorder.5 Conversely, development of LMN features, either clinically or electrodiagnostically, may not develop until 20 or more years after the initial symptoms at which time the distinction between ALS and PLS becomes moot. The natural history of patients who do not have clinical or electrodiagnostic evidence of LMN involvement during the first 4 years of their illness is statistically superior to those who do. ALS may not be initially considered in the differential diagnosis when the initial manifestations are dominated by UMN features. Presumably, this results from the relative rarity of this condition in comparison to the other more common causes of progressive UMN disease. Typically, PLS is considered, and appropriately diagnosed, only after imaging, CSF and other investigations fail to provide an alternative explanation for a patient’s worsening spasticity.
In up to 2.7% of cases, ALS may initially present with symptoms attributable to ventilatory muscle weakness.83,84 These may escape initial detection due to their sometimes protean clinical manifestations including disordered sleep, early morning headache, fatigue, or altered sensorium. Involvement of ventilatory muscles may not be recognized until the more classic manifestations of dyspnea on exertion or orthopnea occur. It is not rare for patients to notice dyspnea for the first time after meals or while bending over to tie their shoes which restrict movement of weak diaphragms. On occasion, ALS may be first recognized in an individual who cannot be weaned from the ventilator following elective intubation. Paradoxical abdominal movements or a drop in vital capacity of more than 10% in the supine position indicates diaphragmatic weakness in patients with a suspected neuromuscular cause of ventilatory symptoms.
ALS and frontotemporal dysfunction represent overlapping disorders which exist in a continuum.2,31–34 This association is important for a number of reasons including insight into a potential common biology. In addition, a reduced life expectancy in ALS patients with concomitant frontotemporal dysfunction has been suggested.2,71,85 Alternative management strategies may be required when ALS and FTD coexist.2 Both ALS and FTD may occur on either a sporadic or hereditary basis. Although it has been previously suggested that FTD may be more prevalent in patients with familial disease, others feel that the prevalence of cognitive impairment is similar in both sALS and fALS.86 Both ALS and FTD may exist as individual disorders or develop collectively, either on a clinically evident or strictly a pathological basis. In the latter circumstance, the second disorder may or may not become clinically manifest during the lifetime of the patient.
It is estimated that 15% of patients who present with apparent sporadic FTD will have ALS and another 30% will have features suggestive of MND.33 Conversely, 30–50% of ALS patients in most series and up to 75% in some who undergo careful testing will be identified as having some alteration in behavior, executive function, or language.2,31,72,74,85 Estimates of dementia fulfilling Neary criteria are estimated between 15% and 41% in ALS patients.2,29,76,87 ALS and FTD may precede the development of the other, or both may present concomitantly.33 On occasion, patients with FTD and ALS will be found to have concomitant Parkinsonism as well.33 The concurrence of ALS, a movement disorder, and dementia should raise the consideration of one of the known genetic causes of this triad (see above) (Table 6-2).
Minor cognitive and behavioral changes in MND patients may be overlooked for a number of reasons. They may be subtle and therefore escape detection unless appropriate screening instruments are utilized. The detection of these changes may also be obscured by the patient’s writing and speaking difficulties. Behavioral changes, when recognized, may be misattributed to known consequences of ALS such as hypercapnia, depression, or pseudobulbar affect.
A nomenclature for frontotemporal dysfunction, with or without ALS, has been established and continues to evolve.2 Patients may have a behavioral syndrome characterized by apathy, altered social and interpersonal conduct, emotional blunting, and loss of insight.87 Alternatively, the primary deficit may be one of expressive language with speech that is dysgrammatical, associated with paraphasic errors and word-finding difficulties. Frontotemporal dysfunction may also manifest as a semantic, predominantly receptive language disorder in which the significance of words and objects lose meaning. Executive dysfunction, which is the loss of the ability to plan and organize tasks by maintenance of attention or the ability to shift sets to accomplish a goal-directed task, represents a significant component of frontotemporal dysfunction as well. Executive dysfunction has been estimated to occur in anywhere between 22% and 35% of ALS patients who do not fulfill the criteria for overt dementia.2,34,88
Tests of verbal fluency provide a sensitive screening method for FTD patients with cognitive impairment.2,74 Normal values are 8 and 13 respectively for number of words generated in 1 minute beginning with the letter D and names of animals.2,74 Another strategy that may be even more sensitive for detecting set-shifting difficulties is to ask the patient to provide a word beginning with a specific letter alternating with a different category, for example, men’s first names. Normative values for this task are seven or more pairs in 1 minute. Perseveration may be readily detected if two consecutive responses belonging to the same category are provided. A potentially useful screening test in individuals with unintelligible speech is “antisaccade” testing (the ability to look in the opposite direction in response to a lateralized visual stimulus). A patient should make no more than two errors in 10 attempts to be considered normal. There are numerous standardized tests that can be efficiently administered during a routine clinic visit. It should be pointed out that the mini-mental state examination is not a particularly sensitive test for detection of early frontotemporal dysfunction. The authors prefer the Montréal cognitive assessment (MOCA) as their standardized screening instrument of choice due to its availability, ease of use, and ability to assess frontotemporal function.89 Behavioral abnormalities are best assessed by specific behavioral inventories.
DIAGNOSIS AND DIFFERENTIAL DIAGNOSISsALS remains a clinical diagnosis supported by the exclusion of potentially mimicking disorders for which testing exists. In cases where the clinical diagnosis of ALS is indisputable, it can be argued that the predominant goal of testing is to validate the credibility of the diagnosis in the eyes of the patient and their family. There are a number of suggested algorithms for the evaluation of the ALS suspect based on differential diagnostic considerations.59,100,132 It is our practice to perform limited “routine” testing in ALS suspects. It is our opinion that testing should be done on a case-by-case basis as the differential diagnostic emphasis differs depending on whether it is a UMN-D, LMN-D, or bulbar-onset phenotype (Table 6-4). Disorders that are dominated by LMN features provide the largest number of differential diagnostic considerations.
TABLE 6-4. ANCILLARY TESTING IN SUSPECTED ALS PATIENTS

Multifocal motor neuropathy (MMN) is the most common LMN-D ALS mimic in most series (discussed in detail in Chapter 14).133–138 It is distinguished from ALS by clinical, EDX, and serological means, and in some cases by response to diagnostic trials of IVIG. Potentially distinguishing clinical features include a slower rate of progression, motor deficits occurring in nerve rather than segmental (myotomal) distribution, and absence of cranial nerve and overt UMN signs.137 Nonetheless, preservation or slight exaggeration of deep tendon reflexes in an affected limb may serve as a confounding variable.137 The most characteristic laboratory feature is motor nerve conduction block but this can be elusive if located in very proximal or distal nerve segments. Antibodies directed at the GM1 ganglioside have been found in high titer in between 30% and 80% of these patients.138,139 In adults with sporadic multifocal or diffuse LMN syndromes, we routinely do more extensive motor conductions looking for conduction block and order GM1 antibody screens. In addition, we consider MR imaging of the brachial or lumbosacral plexus in selected cases in an attempt to demonstrate swelling or increased signal of nerve elements which may occur in a third of MMN cases.137 In selected cases, we have felt compelled to offer a 3-month trial of IVIG when the diagnosis remains uncertain.
Serological tests for disorders of neuromuscular transmission, typically acetylcholine receptor binding, and if negative, muscle-specific kinase antibodies should be considered in any patient with painless weakness. These tests are particularly relevant in bulbar-onset cases without atrophy or fasciculations. In cases presenting with a limb-girdle pattern of weakness, associated with diminished deep tendon reflexes and/or evidence of cholinergic dysautonomia, we would obtain voltage-gated calcium channel antibodies as one means as both a sensitive and specific test for Lambert–Eaton myasthenic syndrome. EDX remains a valuable adjunct in any suspected disorder of neuromuscular transmission evaluation. Edrophonium testing remains useful in selected suspected myasthenia cases.
Kennedy disease should be considered in males with bulbar symptoms and/or proximal weakness.140–143 Like many MNDs, cramps, fasciculations, and atrophy are common. As with other spinal muscular atrophies, tremor may occur. There is an EDX evidence of sensory involvement which may or may not be evident clinically. Creatine kinase (CK) levels are frequently elevated in the two to five times the upper limits of normal range, similar to ALS. Features of impaired androgen effect such as gynecomastia occur. Needle EMG is dominated by features of chronic denervation and reinnervation with a relative paucity of ongoing denervation in most cases. The diagnosis is suspected when sensory nerve action potentials (SNAP) amplitudes are reduced on nerve conduction studies in a patient with proximal symmetric weakness and a predominantly chronic MND pattern with EDX. It is confirmed by identifying the characteristic trinucleotide repeats originating from the androgen receptor gene on the X chromosome.
Inclusion body myositis (IBM) and ALS typically affect individuals in the same age range. Both occur with a slightly greater incidence in males. Both are disorders that commonly demonstrate asymmetric limb weakness and atrophy. In addition, both frequently cause dysphagia, have similar levels of CK elevation, and may have fibrillation potentials and large MUAPs on EMG.92 However, fasciculations and fasciculation potentials are not features of IBM. In addition, the large, polyphasic MUAPs seen on EMG are frequently intermixed with myopathic units in IBM. The course in IBM is typically more indolent than ALS and the pattern of weakness usually distinctive from the segmental pattern and regional spread of ALS. Quadriceps and finger/wrist flexors are typically the most severely affected. Weakness of facial and upper esophageal muscles, neck flexors, and foot dorsiflexors are common. Unlike most myopathies, asymmetric and relatively focal patterns of weakness are common. We have observed distal interphalangeal joint (DIP) flexion weakness in the thumb with relative sparing of DIP flexion of the contiguous digits. The forearm muscles are atrophic in IBM while the hand intrinsic muscles have preserved bulk (see Chapter 33). The diagnosis of IBM is typically confirmed by distinctive muscle biopsy features although most patients with ALS do not need a biopsy to exclude IBM if attentive clinical and EDX examinations are done.
Slowly progressive LMN syndromes are not uncommon in neuromuscular clinics and may be very difficult to distinguish from PMA or LMN-D ALS at onset as it their protracted course that provides the primary means of discrimination.10,134–136 In a manner similar to PLS, 4 years has been suggested as the statute of limitations to distinguish the “benign” focal forms of MND from LMN forms of ALS.122 The benign focal forms of MND have been referred to by a wide variety of names, the most notable of which is Hirayama disease. The classic phenotype is of a sporadic disorder presenting as slowly progressive wasting and weakness of one hand in a teenage or young adult male of Pacific-rim heritage.144,145 The weakness often progresses within the C7–T1 segments for number of years and then arrests. It may or may not affect the opposite upper extremity in a similar distribution. Brachioradialis sparing is a notable clinical feature. Like MMN, deep tendon reflexes may be preserved in an affected extremity, although this may reflect the preferential C8–T1 segmental involvement with anatomic sparing of the C5–C7 segments where the most readily elicitable deep tendon reflexes reside. There is phenotypic heterogeneity in these slowly progressive LMN syndromes. There are cases with simultaneous involvement of the arms.146 Not all cases occur in those of oriental descent.147 Lower extremity syndromes exist as well which have a predilection for the posterior compartment of the leg and have been described as benign calf amyotrophy.148 The younger age of onset, the initial involvement of distal rather than proximal upper extremity muscles, and a frequent signal change within the cervical spinal cord with MR imaging serve to distinguish the symmetrical form of Hirayama disease from the bibrachial amyotrophic diplegic variant of PMA. The differential diagnosis of LMN-dominant forms of ALS also includes benign fasciculation syndrome. This has been previously addressed in the Clinical Features section.
The distal forms of spinal muscular atrophy (dSMA or hereditary motor neuropathy) would be uncommonly confused with ALS. They may be inherited in either a dominant or recessive manner. The potential resemblance to ALS originates from the occasional occurrence of UMN features, vocal cord paralysis, facial, and diaphragmatic weakness in some cases in addition to the characteristic LMN features.149,150 One such genotype, SETX, has been characterized as both a fALS and dSMA because of the potential coexistence of UMN features.150 To further confuse matters, the authors have had the personal experience of a SETX patient with a hereditary spastic paraparesis (HSP) phenotype. Other dSMA genotypes in which pyramidal signs may occur are mutations of the Berardinelli–Seip congenital lipodystrophy 2 (BSCL2), heat-shock protein B1 (HSPB1), and dynactin (DCTN1) (in which facial weakness may occur) genes, and an as of yet unidentified gene in a Jordanian cohort. dSMA associated with diaphragmatic palsy occurs in infants associated with mutations of the immunoglobulin mu–binding protein 2 (IGHMBP2 gene) or in dSMA4 whose gene has yet to be identified. Distal spinal muscular atrophy associated with vocal cord paralysis results from mutations in either the DCTN1 or transient receptor vallanoid 4 gene (TRPV4). Distal spinal muscular atrophy beginning in the upper extremities is associated with the glycyl-tRNA synthetase (GARS) as well as with BSCL2 gene mutations.150
The dSMA phenotype closely resembles and overlaps with Charcot–Marie–Tooth (CMT) disease, both being characterized by slow progression, frequent foot deformities, and a distal symmetric pattern of weakness. The difference is based largely on the presence or absence of clinical and EDX sensory loss.150 The distinction is semantic in some cases as mutations in certain genes may produce a dSMA or CMT. Mutations in HSPB1 may result in dSMA1 or 2, or CMT2F. Mutations in HSPB8 may result in dSMA1 or 2, or CMT2L. Distal spinal muscular atrophy type 5 is also allelic with CMT2D when caused by the GARS mutation. To further confound the semiology of these diseases, FIG4 mutations may produce both fALS and CMT4J phenotypes.150 In view of the cost of genetic testing and lack of commercial availability for many dSMA genotypes, the diagnosis of dSMA is typically based on clinical and EDX criteria. The reader is referred to the SMA chapter for further details.
On occasion, patients who have received radiation therapy may develop a delayed disorder that mimics MND.132,151 The disorder typically begins within a few years of radiation exposure, progresses for a period of time, and then seemingly stabilizes. In the majority of cases, the syndrome occurs in patients who have received pelvic radiation, particularly for testicular tumors. This syndrome has been presumed to have an anterior horn cell localization as the weakness usually affects both lower extremities, fasciculations are common, with sensory signs and symptoms occurs infrequently. Sphincter involvement may or may not occur. Radiation-induced plexopathy or polyradiculopathy have been suggested as alternative localizations although no single locus need be mutually exclusive. The syndrome does not appear to be solely related to the total amount of radiation delivered, or the radiation per dose, which has led to hypothesis of potential synergistic pathologies such as infectious or genetic influences.151 Diagnosis in a typical case involves the appropriate timing, deficits that remain confined to the radiation field, potentially supported by the demonstration of myokymic discharges within affected muscles.
We have rarely seen cases of a head and neck cancer presenting as a painless, progressive bulbar syndrome mimicking bulbar ALS. MR imaging of the soft tissues of the head and neck should be considered in an atypical bulbar presentation of ALS, for example, one associated with unilateral involvement of the tongue.
The differential diagnosis of PLS or UMN-D ALS phenotypes are predominantly structural and hereditary disorders, less commonly selected infectious and acquired metabolic diseases. Hereditary spastic paraparesis (HSP) and cervical spondylitic myelopathy arguably deserve the greatest consideration.79,132,152 HSP is usually readily recognized by its slow rate of progression and a phenotype which is typically a symmetric spastic paraparesis. The upper extremities may be hyperreflexic but usually display normal strength, tone, and coordination. Bulbar involvement would be distinctly uncommon.79,132,152 Pes cavus and minor proprioceptive deficits in the toes may occur and offer other discriminating features.152 As with most heritable disorders, the absence of other obviously affected family member does not exclude an HSP diagnosis. The diagnosis of HSP is also rendered more difficult in complicated forms of the disease where other neurological systems, particularly the LMNs, may be involved.153 LMN involvement has been reported in the SPG9, 10, 14, 15, 17, 20, 22, 26, 30, 38, and 41 genotypes.152 Again, in view of the cost of genetic testing and lack of commercial availability for many HSP genotypes, the diagnosis of HSP is typically made clinically with exclusion of other causes of UMN disease. The reader is referred to the chapter on hereditary spastic paraparesis for more detailed information.
Cervical spondylitic myelopathy is usually dominated by UMN features. LMN features including hand atrophy may occur but multisegmental atrophy, weakness, fasciculations, and/or evidence of active denervation should be cautiously attributed to spinal cord compression.132,154,155 Predomiantly LMN syndromes should be cautiously attributed to ALS. We have had the unfortunate experience of evaluating many cases of ALS with painless weakness associated with MR imaging evidence of spondylosis that have in retrospect undergone needless surgery.156 Nonetheless, imaging of the spinal cord should be included in any MND phenotype with prominent UMN signs and no compelling “bulbar” involvement.
There are a number of other disorders that should receive some consideration in the differential diagnosis of UMN-D or PLS presentations of MND. Primary progressive MS may present as a progressive UMN syndrome that would be addressed by MR imaging of the spinal cord and CSF examination. Dopa-responsive dystonia and stiff person syndrome may resemble progressive UMN disorders. The former diagnosis is supported by response to low-dose dopa and the latter by demonstration of antibodies against glutamic acid decarboxylase (GAD) or amphiphysin. A slowly progressive myelopathy in an young adult female may be the presenting manifestation of adrenoleukodystrophy. Usually, there are concomitant cognitive changes. Diagnosis is made by an elevated C26:C22 long-chain fatty acid ratio or by gene analysis. Retroviral infection, particularly with HTLV-1, may present as a progressive myelopathy and serological testing should be considered in symptomatic individuals at risk in residents of endemic areas or in individuals at risk by exposure through transfusion, recreational drug use, or sexual behavior.
A progressive UMN and LMN phenotype unassociated with involvement of other neurological or organ systems is almost always ALS. An ALS phenotype has been reported in association with copper deficiency.157 The individuals reported had asymmetric foot drop as one of the initial manifestations of a progressive UMN and LMN disorder. Retrospectively, there were clinical clues that could have raised suspicion of copper deficiency in these patients. Patients with suspected ALS who have large fiber sensory loss, malabsorptive risk including prior bariatric surgery, excess zinc absorption, or concomitant anemia or cytopenia should be screened for serum copper, ceruloplasmin, and zinc levels if any of the aforementioned features are present.
Most of the other discretionary tests listed in Table 6-4 are infrequently utilized by us in the evaluation of ALS suspects. Although an ALS phenotype rarely if ever occurs in association with Lyme disease, it is a frequent inquiry on the part of our patients who live in an endemic area such as ours.158 For this reason, it is our practice to obtain an ELISA screening test on many of our patients. We would not recommend this in areas where Lyme disease is uncommon. In view of the uncertain associations and rarity of occurrence, we reserve other testing for situations in which clinical context heightens index of suspicion. This would include testing for HIV, occult neoplasia or lymphoproliferative disorders (with or without) paraneoplastic antibodies, hexosaminidase A deficiency, thyrotoxicosis, heavy metal toxicity, polyglucosan disease, and dural venous malformations.132,159–170 Hexosaminidase A deficiency might be considered in a young person with MND and a concomitant spinocerebellar, dystonic or bipolar syndrome. Polyglucosan disease is a phenotypic variant of glycogen storage disease type IV with phenotypic manifestations that may include distal sensory loss, a neurogenic bladder, cerebellar dysfunction, and cognitive decline.
In the absence of genetic confirmation, ALS remains a clinical diagnosis supported by the absence of evidence of other, potentially mimicking diseases. At the time of initial evaluation, ALS may be suspected but the clinical features may not have developed sufficiently to allow a physician to feel confident in confiding their suspicions with their patient. The physician may feel conflicted as to when to have this conversation. To do so prematurely hazards the possibility of being wrong. In addition, patients may lack confidence in someone who confronts them with a diagnosis of this magnitude without the appearance of due diligence. Balanced against this is the patients’ need for answers. Perceived “foot dragging” and uncertainty may have equally deleterious effects on their trust in their physician. Disclosing the suspected diagnosis as early in the course as possible, following exclusion of reasonable differential diagnostic considerations, subsequent to demonstration of progression both within and outside of initially affected regions, would seem to be a reasonable approach. Many neurologists will not wait for a patient to fulfill EEC for definite or probable ALS before disclosing their suspicions. In view of the implications of an ALS diagnosis, considering the lack of a confirmatory test, and wishing to instill trust, patients are frequently counseled to seek a second opinion from a knowledgeable source.
LABORATORY FEATURESWith the exception of identification of known pathological mutations in known fALS genes in symptomatic patients, there are no laboratory tests that currently provide disease confirmation. Testing in ALS is done for three general reasons. The first potential goal is to identify either UMN or particularly LMN involvement when it is not obvious clinically, in a patient with either an LMN- or UMN-dominant phenotype. A second diagnostic strategy is to attempt to exclude other disorders that fall within the ALS differential diagnosis. Finally, testing may be performed in an attempt to aid in disease management and prognostication, for example, forced vital capacity measurements to identify the appropriate time to discuss positive airway pressure support, percutaneous gastrostomy placement, or end-of-life decision making.
EMG and nerve conduction studies are recommended in virtually all suspected ALS patients, even when the diagnosis is clinically unequivocal. The implications of the ALS diagnosis are such that both patient and clinician wish to be as certain as possible. EMG has the capability of confirming the existence, distribution, and relative duration of LMN degeneration in support of an ALS diagnosis.90 In addition, EDX may identify features more consistent with an alternative diagnosis as described below. The specific goals of the test are to:
1. Identify EDX abnormalities in clinically unaffected muscles to confirm a pattern consistent with ALS, that is, ongoing denervation (fibrillation potentials and sharp waves) coupled with changes implying subacute or chronic denervation and reinnervation (a reduced number of large MUAPs, MUAP instability) demonstrable in multiple muscles innervated by multiple segments (≥2 segments in limb muscles, ≥1 muscle in cranial or thoracic muscles) in more than one body region.91 Fasciculation potentials are a common and supportive but not required EEC electrodiagnostic requirement. As discussed above, we do not believe that convincing evidence has been provided to consider fasciculation potentials as a surrogate for fibrillation potentials and positive waves as a marker of ongoing denervation as proposed by the Awaji criteria.
2. Identify features that might implicate an ALS mimic or as the EEC refers to, an ALS-related disorder. Examples of this would include abnormal sensory conductions (Kennedy syndrome), a large decremental response (>20%) to 2 to 5 Hz repetitive stimulation without evidence of chronic denervation (myasthenia, Lambert–Eaton myasthenic syndrome), motor nerve conduction block or other demyelinating features (MMN), or EMG evidence of both small and large motor units with fibrillation potentials that might suggest inclusion body myositis in the appropriate clinical context.92
3. Offer insight into the rate of progression and prognosis, that is, active denervation without chronic denervation and reinnervation, motor unit variability, and a rapid decline in motor unit estimation being electrodiagnostic indicators of a more rapidly progressive course.90
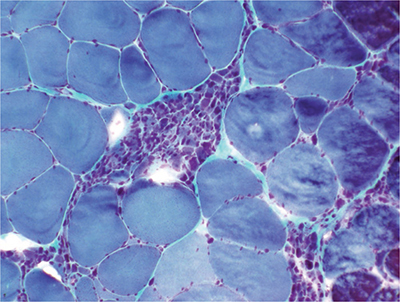
Muscle biopsy can also serve as a surrogate for EMG to confirm LMN involvement by demonstrating a pattern of denervation atrophy characterized by scattered atrophic fibers of both fiber types that are angulated in appearance, and by small groups of atrophic fibers (Fig. 6-4). Muscle biopsy is not routinely employed in ALS as it is both more invasive and less capable of demonstrating the multifocal or diffuse neurogenic features than EMG.
Figure 6-4. Muscle biopsy demonstrating small fiber-type grouped atrophy consistent with neurogenic atrophy and ALS (modified Gomori trichrome).
Unfortunately, there are no widely available and reliable surrogates for the detection of subclinical UMN involvement. A test of this nature would be an invaluable tool in patients with PMA or LMN-D presentations. Central motor conduction velocity obtained through transcranial magnetic stimulation, magnetic resonance spectroscopy, and diffusion tensor imaging are some of the methodologies utilized that have been applied with the hope of identifying subclinical cortical spinal, or cortical bulbar tract pathology.19,93–96 Single photon emission computerized tomography or positron emission tomography has also been used to demonstrate extra motor CNS involvement through reduced blood flow or overt atrophy in a frontotemporal lobar pattern (Fig. 6-5). All of these tests are utilized more for research than for clinical purposes as both their availability and accuracy are currently limited.
Figure 6-5. Preferential frontal atrophy (arrowheads) with lateral (A) and parasagittal (B) views of the cerebrum with associated enlargement of the frontal horn and caudate flattening (arrow) (C) and relative hippocampal spraing (arrow) (D) with coronal views.
The majority of laboratory tests are done in an attempt to exclude other ALS differential diagnostic considerations or to monitor disease progress. CK levels are elevated in the serum of approximately 23–75% of ALS patients.97,98 It is important to recognize this so as to not confuse an MND with a myopathy in a patient with painless weakness. Serum CK determination may be of value in the appropriate context. For example, an elevated CK value would favor ALS over myasthenia gravis in a patient presenting with bulbar symptoms. Tests of “pulmonary function” are done routinely. Their role is to aid in determination of prognosis and in timing of management decisions. For example, a declining vital capacity may suggest the need for positive airway pressure assistance, percutaneous gastrostomy, or initiation of end-of-life decision making discussions.
There have been significant additions to our knowledge regarding ALS-causing genes and an increased number of commercially available tests for fALS since the first edition of this book.99 There are now at least 13 different disease-causing gene mutations.25,43,44 The genotype for approximately half of fALS cases can now be identified. The six genes for which commercial testing is currently available include superoxide dismutase (ALS1), FUS/translated in liposarcoma (TLS) (ALS6), angiogenin (ANG) (ALS9), TAR DNA-binding protein 43 (TDP-43) (ALS10), polyphosphoinositide phosphatase (FIG4) (ALS11), and recently, the most common recognized cause of fALS, the C9ORF72 hexanucleotide expansion.
fALS mutations display varying and often incomplete penetrance.25,27 As a result, a small percentage of patients with apparent sALS will have heritable disease. Recognizing this, we refrain from offering genetic testing to recently diagnosed individuals with apparent sporadic disease. This is in keeping with European recommendations and the results of a recent survey of North American ALS clinicians and researchers.100,101 Although potentially viewed by some as being overly paternalistic, we feel that delivering the diagnosis of ALS is difficult enough without introducing the anxiety that other family members may be at risk in the absence of any intervention that can favorably alter the natural history of the disease. Should the patient inquire about the possibility of heritable disease, we provide them with a candid explanation of heritable risk. Should they request genetic testing under these circumstances, we will provide it but only after adequate genetic counseling that includes potential risks and benefits of testing results not only to the patient but other family members at risk. We do not routinely order genetic testing in a symptomatic individual with an affected first-degree relative who has already been genotyped.
If genetic testing is to be done, we order the most prevalent mutations first, SOD1 and C9ORF72. We reserve testing for less common mutations only if testing for these two genotypes is negative. If testing is performed in asymptomatic individuals, adequate genetic counseling is mandatory with a clear understanding of the implications of both a positive and a negative test result. Ideally, other family members at risk whose genetic status may be revealed by testing of the proband would be involved in the decision making as well.
One focus of current ALS research is the attempt to identify a biomarker or pattern of biomarkers that would provide a gold standard for diagnosis and potentially identify those at risk for ALS. This would provide the opportunity for early therapeutic intervention, offer insight into disease pathogenesis, and clarify nosology by defining whether MND represents one or more diseases.102–106
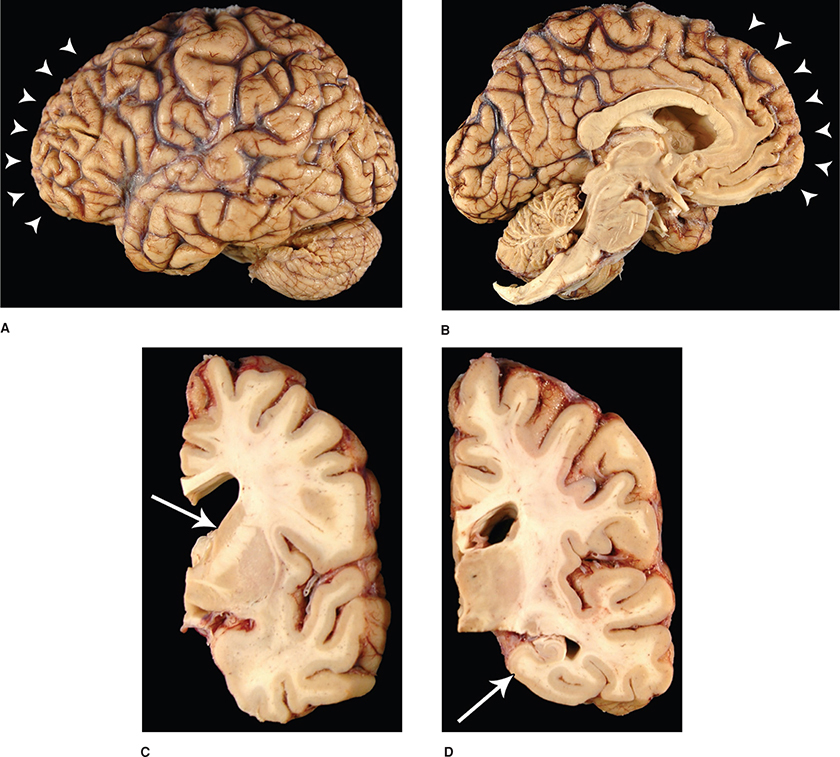
HISTOPATHOLOGYThe histopathology of ALS will be discussed from both histological and immunohistochemical perspectives. The light microscopic features of ALS in muscle are described above as they represent a potential diagnostic tool during life. The pertinent findings in the CNS however, are available on a postmortem basis only (Fig. 6-6). They consist of myelinated fiber loss in the corticospinal and corticobulbar pathways as well as loss of motor neurons within the anterior horns of the spinal cord and the motor cranial nerve nuclei at risk. As a result, ventral roots become atrophic while dorsal roots are spared. The anterior horn cell loss occurs within virtually all levels of the spinal cord with cell preservation of the intermediolateral cell columns. There is selective sparing of the third, fourth, and sixth cranial nerves as well as Onuf nucleus within the anterior horn of sacral segments 2 to 4.
Figure 6-6. Thoracic spinal cord in a patient with amyotrophic lateral sclerosis, demonstrating reduced numbers of anterior horn cells (red arrow), normal complement of neurons in intermediolateral cell column (green arrow), atrophy of ventral root (orange arrow), and normal dorsal root (blue arrow). (Luxol fast blue/hematoxylin and eosin.)
As with the majority of neurodegenerative disease, the immunohistochemical signature of ALS with or without concomitant dementia is the presence of cytoplasmic inclusions representing aggregates of misfolded proteins. In familial cases of ALS associated with SOD1 gene mutations, the inclusions consist of misfolded SOD1 protein. In the majority of sALS cases, the majority of fALS genotypes not associated with SOD1 mutations, and more than half FTLD patients without ALS, these inclusions are labeled with antibodies to ubiquitin but not SOD1. These ubiquitinated inclusions (UI) are found within the cytoplasm of relevant neurons and glial cells within the primary motor cortex, brainstem motor nuclei, spinal cord, and associated white matter tracts as well as the cingulate gyrus and dentate nuclei of the cerebellum.39,107,108 In patients with FTLD, these inclusions are found in the neocortex and hippocampus as well.107
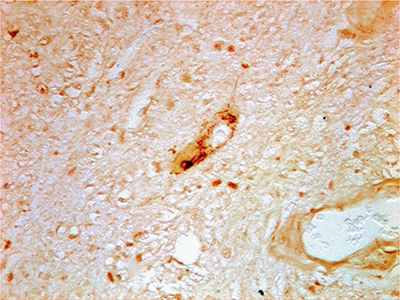
Although UI are not unique to ALS or FTLD, their location and composition may be. In 2006, it was first recognized that these UI stain with TDP-43 in virtually all sALS cases and 80% of FTLD cases associated with UI.107 UI in ALS also contain the proteins ubiquilin 2 and optineurin whose genes, when mutated, are fALS related if not causative.109,110 Ubiquitinated inclusions that occur in neurodegenerative disorders other than ALS/FTD stain for other proteins such as tau or α-synuclein rather than TDP-43. TDP-43 is a DNA/RNA binding protein translated from a gene locus on chromosome 1. TDP-43 normally shuttles between the nucleus and cytoplasm of cells although is primarily nuclear in its location.111 In ALS, either sporadic or non-SOD1 fALS, TDP-43 incorporated into UI is mislocalized to the cytoplasm of motor neurons (Fig. 6-7). Although TDP-43 is found in UI in the majority of FTLD patients, 20% remain TDP-43 negative.107 In 2009, the FUS gene/protein, also related to DNA repair and RNA microprocessing, was found to colocalize to UI in non–TDP-43 FTLD cases.25,30,107 Like TDP-43, FUS pathology in FTLD is also concentrated in the frontotemporal neocortex and hippocampus but may affect the striatum, thalamus, and brainstem as well.103 Like mutations in the TDP-43 gene, mutations in the FUS gene may result in either an ALS and/or FTD phenotype. In addition, ubiquilin 2 pathology is prominent in the hippocampus of ALS patients with (but not without) dementia.109
Figure 6-7. TDP-43 (+) skein in a motor neuron (lumbar) in a sporadic patient with amyotrophic lateral sclerosis (40× magnification—rabbit polyclonal antibody to human TDP-43, dilution 1:400). (Reproduced with permission from Michael Strong, MD and Robert Hammond, MD.)
As anticipated, the pathology and clinical phenotype do not always perfectly coincide, the latter often underestimating the former. For example, in patients with sporadic FTLD, UI/TDP-43 (+) inclusions may be found in motor neurons in the absence of any premortem evidence of MND. In addition, TDP-43 (+) inclusions are identified postmortem in the distribution characteristic of ALS as described above in patients with restricted PMA, LMN-D, and FTD/PLS phenotypes during life.112,113 These pathological observations further cement the relationship between ALS and these phenotypically overlapping disorders.
TDP-43 (+) inclusions also occur in heritable as well as sporadic forms of FTD. Specifically they have been demonstrated in mutations of the VCP, PGRN, and TARDBP genes as well as the recently discovered C9ORF72 gene on chromosome 9. Their staining patterns appear to be identical in appearance to the TDP-43 staining pattern seen with sALS and sFTD with UI.42,43,107 These associations add further support for a biological link between ALS and FTD in both their sporadic and familial forms.
One additional histological finding in ALS is the Bunina body. These are dense granular intracytoplasmic inclusions that stain for cystatin C, transferrin, and peripherin that are less commonly identified UI. They may be identified within the cytoplasm of motor neurons, are thought to be specific for ALS, and appear distinctive from but not necessary independent of TDP-43 aggregates.114,115
The vast majority of demented ALS patients will be found to have FTLD on gross inspection. Occasionally, either the patient’s phenotype, or their pathology will suggest Alzheimer disease. The association is currently considered to be coincidental and not related. The immunohistochemistry of patients with prominent memory loss is typically that of a TDP-43 proteinopathy and patients with memory loss are often found to have seemingly unrelated hippocampal sclerosis with limited plaque and tangle formation.
The histology of FTLD typically consists of linear spongiosus in the first and second layer of the cortex with prominent neuronal loss in the anterior cingulate and superior frontal gyri regardless of the makeup of the associated cytoplasmic inclusions. These spongiform changes seem to segregate demented from nondemented ALS patients. The immunohistochemical properties of cytoplasmic inclusions in FTLD are varied. In greater than half of all FTLD patients, and virtually all patients with sALS, these inclusions will immunostain with ubiquitin. In approximately 40% of FTLD, the cases, the inclusions will stain for tau.41 In patients whose inclusions stain for tau, the phenotype will not include MND but may incorporate cortical basal degeneration or progressive supranuclear palsy in addition to their FTD.
PATHOGENESISThe cause(s) of sALS remains unknown. Current hypotheses regarding disease mechanisms in sALS include oxidative damage, accumulation of toxic intracellular protein aggregates, mitochondrial dysfunction, defective axonal transport, growth factor deficiency, and/or glutamate excitotoxicity.116 These pathophysiological mechanisms may work in series or in parallel with eventual confluence. Although our knowledge of disease pathogenesis has increased, the elements of ALS biology that contribute to disease initiation, disease propagation, or represent consequence of disease remain uncertain.
It is attractive to hypothesize that ALS is a consequence of environmental exposure and genetic risk. As previously mentioned, there is considerable phenotypic and pathological overlap between sALS and fALS. Consequently, a significant proportion of ALS research in recent years has focused on disease mechanisms in familial disease with the hope that they are relevant to sporadic disease. The recognition that specific gene mutations such as TARDNP, UBQLN2, and C9ORF72 may result in both fALS and apparent sALS, and that their protein products (TDP-43 and ubiquilin 2) can be found within neuronal inclusions in sporadic as well as familial forms of the disease lends support for this construct. Unfortunately, as described above, epidemiologic studies have failed to reproducibly identify an environmental culprit. Ultimately, the biology of ALS will have to account for the identical phenotypic heterogeneity of sporadic and familial disease, the semiselective vulnerability of motor neurons, and the tendency for the disease to spread in a regional fashion.117 In that regard, one attractive hypothesis holds that misfolded protein aggregates in one cell can proselytize the normal analogous proteins of a neighboring cell to undergo the same pathological process, thus explaining the observed patterns of regional disease spread.118,119
Currently, we can only speculate as to whether ALS is a disease of singular cause capable of phenotypic diversity or the final common expression of different insults that prey upon the selected vulnerability of motor neurons. Working from the premise that sALS and fALS must share at least in part a common biology, and recognizing that similar phenotypes result from differing gene mutations, the latter paradigm that differing etiologies and disease mechanisms converge to produce a common phenotype would represent the most logical conclusion.
Since the first edition of this text, knowledge relevant to fALS knowledge has expanded at a rapid pace. As mentioned, it is the hope that insight gained from the pathophysiology of fALS will be relevant to sporadic disease as well. There are now 166 known pathological mutations of the superoxide dismutase (SOD1) gene.25,120 In addition, mutations in at least 12 additional genes are thought to be causative of fALS.25 These mutations along with a summary of their phenotypes are outlined in Table 6-2. In addition, there are 89 other ALS disease-related genes listed on the ALS online genetic database at the time of this writing, a resource for information relevant to both disease-causing and disease-related mutations.121
Currently, mutations of the SOD1 gene (ALS1) and C9ORF72 are the most common causes of fALS and constitute approximately 40–45% of cases.25,42,43 Mutations of the C9ORF72 associated with a hexanucleotide repeat mechanism may be the most significant cause of ALS and FTD found to date.42,43 Less frequent causes of fALS stem from mutations of the FUS/TLS (ALS6) and TAR DNA-binding protein (TARDBP) (TDP-43) (ALS10) genes, each representing approximately 4–6% of fALS cases. Uncommon causes of fALS include mutations of the alsin (ALS2), senataxin (SETX)(ALS4), SPG11 (ALS5), vesicle-associated membrane protein–associated protein B (VAPB) (ALS8), angiogenin (ANG) (ALS9), phosphoinositide phosphatase (FIG4) (ALS11), optineurin (OPTN) (ALS12), ataxin 2 (ATXN2) (ALS13), ubiquilin (UBQLN2) (ALS15), and VCP genes.25,28,29,42–44,62,99,107,109–111,122–128 The genes for ALS3 and ALS7 have yet to be identified.
The majority of fALS genotypes are transmitted dominantly. ALS2 and ALS5 are inherited recessively and ubiquilin 2 mutations are transmitted in an X-linked fashion. Optineurin seems unique in that both dominant and recessive inheritance patterns have been described. The penetrance of fALS is variable. SOD1 mutations were initially thought to be highly penetrant based on a bias originating from the original studies that involved a few large families in which penetrance was understandably high. In reality, less than 50% of identified ALS families have more than two identified family members.25 Currently, penetrance is estimated to be approximately 80% in SOD1 mutations by age 85. In the highly penetrant A4 V mutation, the most prevalent SOD1 mutation in North America, 90% of individuals are clinically affected by age 70. Conversely, the I113 T mutation appears to be expressed in less than 10% of patients harboring the mutation.25 This knowledge serves to blur the boundaries between sporadic disease and reinforce that a small percentage of apparent sALS patients will have heritable disease.
Further complicating our understanding of gene expression is the fact that the onset of symptoms in individuals with the same mutation may vary widely and that mutations in the same locus may behave differently in different populations. In the case of the D90 A CuZn SOD homozygotes, onset may vary from 20 to 94 years of age.25,62 This locus behaves as a recessive trait in Scandinavia where heterozygotes are asymptomatic. In North America except in those of Scandinavian descent, D90 A behaves as a dominant disorder with heterozygotes developing ALS.
The mechanisms by which the aforementioned mutations initiate and/or propagate MND and FTD remain enigmatic. A common theme in ALS is the presence of inclusions within motor neurons consisting of misfolded proteins with the implication that these inclusions confer a toxic gain of function.129 Alternatively, ALS could represent a loss of function of proteins migrating from their normal nuclear location to a pathological cytoplasmic position. As previously mentioned however, the makeup of the inclusions found in ALS is variable, with SOD1 inclusions being absent of TDP-43 staining, implicating more than one toxic mechanism.130 Although potential mechanisms remain unknown, common themes are emerging. Ubiquilin 2 is a constituent of the ubiquitin proteasome system responsible for protein degradation which may confer a toxic gain of function when mutated.44,109 Mutations of the TDP43, FUS, ANG, SETX, and C9ORF72 all appear to adversely affect RNA metabolism.25,42,43,107 For example, RNA generated by pathogenic expansions of the C9ORF72 gene is hypothesized to sequester normal RNA and proteins resulting in disruption of normal transcription.42,43 TDP-43 and FUS are both trapped into stress granules that may further hinder RNA metabolism.131 The ataxin 2 gene, large expansions of which cause the spinocerebellar ataxia type 2 phenotype, appears to be a potent modifier of TDP-43 toxicity.122–124 The mechanism of action for optineurin mutations in the genesis of ALS is uncertain.25,110,125
MANAGEMENTOnce the diagnosis has been shared with the patient and their family, we organize their management into three domains: (1) education and psychosocial support, (2) disease-specific treatment including consideration of clinical trials, and (3) supportive care for the patient and their disease, ideally provided in a multidisciplinary setting.
In the Internet age, information is readily available to patients and their families. It is the responsibility of any physician caring for an ALS patient to direct them toward reputable educational resources. To that end, the websites provided by the Muscular Dystrophy Association (www.mda.org), the ALS Association (www.alsa.org), and the Motor Neuron Disease Association (www.mndassociation.org) are recommended. Unfortunately, there are websites, for reasons altruistic or otherwise, that provide false hope or false information. www.alsuntangled.com is a web-based resource developed by ALS clinician researchers to help patients and their families sort through this potentially confusing terrain.171
In part due to the paucity of effective treatments, many patients with ALS seek advice from their physicians regarding alternative, “natural treatments” for their disorder. There is no clear correct response. A dogmatic response on the part of a physician in opposition to this approach runs the risk of losing the patient. Conversely, the physician has a responsibility to dispel what seems to be a generally held perception that “natural” is synonymous with safe. It may be helpful to emphasize to a patient that any substance that is biologically active enough to be beneficial is also biologically active enough to be harmful. “Natural” substances do not benefit from the same underlying science or undergo the same scrutiny and quality control as do pharmaceuticals and pose additional risk for those reasons alone.
In many locations, support groups are available for patients and their families as a potential resource that some but not all patients and caregivers find valuable. As the disease progresses, preferentially while the patient remains capable of communicating effectively, it is the neurologist’s duty to discuss with the patient their preferences regarding decisions that will inevitably have to be made. By doing so, the neurologist provides the patient with some semblance of control of their disease and provides respect for their autonomy regarding these difficult issues.
Patients and families frequently inquire about the potential genetic consequences of the disease. As discussed earlier, we are candid in disclosing that the possibility fALS cannot be entirely excluded in any apparent sALS patient. Fortunately, with adequate family history, it is possible to reassure the patient and biological relatives of the low statistical likelihood of heritable disease in the majority of cases.
There are no known treatments that can reverse or arrest disease progression in ALS. In 1994, Rilutek was identified as the first and only disease-specific treatment to date that favorably altered the natural history of the disease.14 Its modest benefit results in 10% slowing of disease progression by approximately 10% at its customary dose of 50 mg bid. Unfortunately, this benefit is imperceptible to the patient who neither feels nor functions better. It is an expensive drug that is usually well tolerated although some experience upper gastrointestinal side effects and increased fatigue. Reversible hepatotoxicity may occur with an associated requirement for monitoring of liver function tests. Balancing the benefits of Rilutek with its potentially significant financial burden consideration is a candid conversation that should take place with patients and their families.
There have been numerous failed ALS therapeutic trials. Current clinical trials, likely to have been completed by the time of publication, involve ceftriaxone, dexpramipexole, a combination of creatinine and tamoxifen, a combination of anakinra and riluzole, NP001, and neural-derived stem cells. Trials utilizing arimoclomol and intrathecal delivery of ISIS 333611 are available for those with fALS and identified SOD1 mutations. Patients and clinicians can be kept abreast of clinical trial availability and enrollment status through websites.172,173 Patients should be encouraged to participate in these. Not all patients will be eligible. Patients who do not meet EEC probable or definite disease criteria including those with exclusive bulbar disease, age <18 or >80, or vital capacities less than 60% of predicted are typically excluded from enrollment.
Optimal management of the patients with ALS and their families is time-consuming and requires knowledge and resources that undoubtedly surpass the capabilities of any single healthcare worker. For this reason, although potentially emotionally and physically exhausting for the patient, the multidisciplinary clinic provides a useful ALS care model. Studies have demonstrated that participation in a multidisciplinary clinic improves both quality and duration of life.55 The goals of the clinic are to anticipate and resolve problems that adversely affect the patient’s ability to function, their safety, and their comfort. During the clinic, multiple parameters including the patient’s psychosocial well-being, the presence or absence of pain, their sleep quality, “bulbar” functions, fine motor skills, ventilation, and mobility and ambulation are assessed.
The revised ALS functional rating scale (ALSFRS-R) is a validated 48-point assessment tool that can be acquired both in person and over the phone. Twelve points are signed to each of four domains assessing ventilation, bulbar function, fine motor skills, and mobility. The rate of disease progression and prognosis can be estimated, a decline of less than or greater than 0.5 points per month separating slow from fast progression.174,175
In addition to the ALSFRS-R, we routinely monitor quantitative strength using handheld dynamometers as outlined in Chapter 1 as well as measurements of ventilatory function. Although at times helpful in reinforcing the initial diagnosis, tests of ventilatory function have the greatest utility in monitoring the disease course. Declining ventilatory muscle strength not only speaks to prognosis and helps to time discussions pertaining to end-of-life decision making, it has management implications regarding the initiation of noninvasive positive airway pressure and percutaneous gastrostomy tube insertion. The most commonly used measurements are forced vital capacity, and maximal inspiratory and expiratory pressures. A drop in forced vital capacities in the supine in comparison to the sitting position of more than 10% of the predicted value suggests a disproportionate degree of diaphragmatic weakness.
Since the first edition of this book, a revised version of the ALS practice parameter endorsed by both the American Academy of Neurology and the American Association of Neuromuscular and Electrodiagnostic Medicine has been published.176,177 As an evidence-based document, it cannot adequately address all management decisions pertaining to ALS, it is nonetheless a helpful guideline for the practicing neurologist. Other management reviews attempt to fill the gaps by providing experienced, expert opinion.178 Symptomatic management of poor sleep (due to immobility, depression, urinary frequency, sleep disordered breathing), pain (immobility, joint contractures, cramps, and spasticity), sialorrhea, impaired clearance of viscous secretions, laryngospasm, impaired communication, dysphagia, impaired performance of ADLs, and disordered bowel and bladder function are typically addressed with durable medical equipment or pharmacologic treatment. These interventions are outlined in (Table 6-5). It also behooves the neurologist to become familiar with the topography and limitations of the patient’s residence and if necessary, to arrange for a home safety evaluation to identify areas of home modification that would benefit the patient.
TABLE 6-5. SYMPTOMATIC MANAGEMENT OF MND/ALS176–178

There are a number of potential interventions that are often met with resistance by patients despite their potential to enhance the quality of their lives as well as maintenance of their independence. In an attempt to overcome patient reluctance, we often utilize two strategies as well as to maintain their independence. We often introduce these concepts prior to their actual need to give the patient and their family a chance to adjust. Additionally, we emphasize the importance of compliance with the use of durable medical equipment and other intervention, the goals of fall prevention and maintenance of independence.
Percutaneous gastrostomy, noninvasive positive pressure ventilation (NIPPV), and mechanical gait aids are commonly recommended but are perceived as threatening by patients. All should be introduced as measures to improve the quality of life rather than duration of life, even though the latter is probably achieved with NIPPV and may be accomplished with gastrostomy feeding. There are no absolute criteria to determine optimal percutaneous gastrostomy timing. Loss of weight of >10% of baseline weight, doubling of eating time, demonstration of laryngeal penetration or aspiration, or recurrent coughing or choking during eating are the most common indicators utilized to prompt gastrostomy tube recommendation. We attempt to place percutaneous gastrostomy before the forced vital capacity falls below 50% of predicted to limit risk associated with the procedure.
NIPPV is typically recommended when the patient is symptomatic from shortness of breath, when disturbed sleep is attributed to hypoventilation, when symptoms attributable to hypercapnia such as morning headache or confusion occur, or when forced vital capacity decreases to less than 50% of predicted. Initial use occurs at night when hypoventilation is most likely to occur. Initial settings include an inspiratory pressure of 8 cm of water, an expiratory pressure of 4 cm of water, and a backup rate of 8 breaths per minute. A minimum of 6 hours of nocturnal use is encouraged. NIPPV appears to have the most dramatic effects on disease survival of any treatment options available. Although it is suspected that early use of NIPPV is beneficial in prolonging life expectancy, third-party payers typically require a forced vital capacity of <50% of predicted, a negative inspiratory force of less than 60 mm Hg, or evidence of carbon dioxide retention before reimbursement for NIPPV is authorized. This is problematic in patients who are symptomatic but who have yet to fulfill the aforementioned diagnostic criteria. Diaphragmatic pacing represents a newer ventilatory support modality. Although FDA approved for patients with significant but not severe ventilatory muscle weakness, the extent of its benefit(s) await ongoing study.
It is the responsibility of every physician caring for patients with ALS to participate in one or more discussions concerning end-of-life decisions, particularly regarding TAMV. Ideally, the patient should be given the autonomy to make these decisions at a time that they can still communicate effectively and before any ventilatory crisis might occur. Ideally, this discussion would occur at a time of the patient’s choosing but may need to be initiated by the physician in some cases. In the spirit of full disclosure, the physician should inform the patient of the distasteful but realistic aspects of TAMV including considerations of cost, location of care, caretaker burden, life-expectancy even with TAMV, and potential development of future dementia.
The majority of patients with ALS in the US decline TAMV consideration although some relent at the time of crisis. Cultural values regarding quality of life, the fear of being a burden, and the associated costs of long-term care, which are typically not covered by third-party payers, represent the most likely reasons why patients decline.37,38 Most patients choose to receive their terminal care at home. Hospice services either at home or in hospice facilities provide an invaluable resource. Many, if not all, patients fear that their death will involve physical suffering. Some may be intrepid enough to broach the subject, some may not. Providing patients with the reassurance that adequate palliation will be provided to avert physical discomfort is another important responsibility to fulfill. A frank discussion about the logistics and limits of how this will be accomplished is often appreciated by the patient, family, and caregivers. Typically, neurologists, through hospice nurses, provide for adequate treatment of pain, anxiety, dyspnea, nausea, and any other distressing symptom. This goal is usually achieved often with drugs such as morphine and lorazepam without suppressing ventilatory drive. Even if the latter were to occur, it is ethically acceptable under the principle of double effect as long as the primary intent is to relieve suffering. Fortunately, the vast majority of ALS patients appear to die peacefully and comfortably.
The role of exercise pertaining to its effect on both the disease progression and the preservation of function is a near universal inquiry by ALS patients. Studies done to date favorably report on the benefits of exercise done in moderation.179 We recommend low-level conditioning and nonfatiguing exercise that is safe, for their patients. For example, a rowing machine or exercise bicycle would be preferable to a treadmill for a patient with lower extremity weakness or spasticity.
In patients with presumed fALS and asymptomatic family members at risk, we encourage participation in research studies that are currently being conducted at the University of Miami. Dr. Michael Benatar and colleagues offer patients the opportunity to participate in fALS-specific therapeutic trials, and provide both groups the opportunity for counseling and genotyping at no cost. Contact information is Fals@med.miami.edu.
SUMMARYHIV and cancer are disorders that were uniformly fatal within many of our professional lifetimes. Now, in many instances, these are disorders that are controllable and at times curable. For patients with ALS, their families, and their physicians, it is important to realize that similar outcomes may be realized for ALS in the foreseeable future. Until that hope is realized, neurologists caring for patients with ALS are obligated to help maintain the quality of their patients’ life to the extent possible. When that is no longer feasible, they have an obligation to support the quality and dignity of their patients’ death as well.
1. Strong MJ. The evidence for ALS as a multisystems disorder of limited phenotypic expression. Can J Neurol Sci. 2001;28:283–298.
2. Strong MJ, Grace GM, Freedman M, et al. Consensus criteria for the diagnosis of frontotemporal cognitive and behavioral syndromes in amyotrophic lateral sclerosis. Amyotroph Lateral Scler. 2009;10:131–146.
3. Swash M, Desai J. Motor neuron disease: classification and nomenclature. Amyotroph Lateral Scler Other Motor Neuron Disord. 2000;1:105–112.
4. Gordon PH, Cheng B, Katz IB, et al. The natural history of primary lateral sclerosis. Neurology. 2006;66:647–653.
5. Gordon PH, Cheng B, Katz IB, Mitsumoto H, Rowland LP. Clinical features that distinguish PLS, upper motor neuron dominant ALS, and typical ALS. Neurology. 2009;72:1948–1952.
6. Tartaglia MC, Rowe A, Findlater K, Orange JB, Grace G, Strong MJ. Differentiation between primary lateral sclerosis and amyotrophic lateral sclerosis. Arch Neurol. 2007;64:232–236.
7. Kim WK, Liu X, Sandner J, et al. Study of 962 patients indicates progressive muscular atrophy is a form of ALS. Neurology. 2009;73:1686–1692.
8. Visser J. Van den Berg-Vos RM, Frannsen H, et al. Disease course and prognostic factors of progressive muscular atrophy. Arch Neurol. 2007;64:522–528.
9. Traynor BJ, Codd MB, Corr B, Forde C, Frost E, Hardiman OM. Clinical features of amyotrophic lateral sclerosis according to the El Escorial and Airlie House diagnostic criteria. Arch Neurol. 2000;57(8):1171–1176.
10. Rowland LR. Progressive muscular atrophy and other lower motor neuron syndromes of adults. Muscle Nerve. 2010; 41:161–165.
11. Mateen FJ, Carone M, Sorenson EJ. Patients who survive 5 years or more with ALS in Olmstead County, 1925–2004. J Neurol Neurosurg Psychiatry. 2010;81:1144–1146.
12. Mortara P, Chio A, Rosso MG, Leone M, Schiffer D. Motor neuron disease in the province of Turin, Italy, 1966–1980: survival analysis in an unselected population. J Neurol Sci. 1984;66:165–173.
13. Brooks BR. El Escorial World Federation of Neurology criteria for the diagnosis of amyotrophic lateral sclerosis. J Neurol Sci. 1994;124(suppl):96–107.
14. Bensimon G, Lacomblez L, Meininger V. A controlled trial of riluzole in amyotrophic lateral sclerosis. N Engl J Med. 1994; 330:585–591.
15. Brooks BR, Miller RG, Swash M, Munsat TL; World Federation of Neurology Research Group on Motor Neuron Diseases. El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler. 2000;1:293–300.
16. Ross MA, Miller RG, Berchert L, et al. Toward earlier diagnosis of amyotrophic lateral sclerosis. Neurology. 1998;50:768–772.
17. Chaudhuri KR, Crump SJ, Al-Sarraj S, Anderson V, Cavanagh J, Leigh PN. The validation of El Escorial criteria for the diagnosis of amyotrophic lateral sclerosis: a clinicopathological study. J Neurol Sci. 1995;129(suppl):11–12.
18. Ince PG, Evans J, Knopp M, et al. Corticospinal tract degeneration in the progressive muscular atrophy variant of ALS. Neurology. 2003;60:1252–1258.
19. Carvalho MD, Dengler R, Eisen A, et al. Electrodiagnostic criteria for diagnosis of ALS. Clin Neurophysiol. 2008;119:497–503.
20. Carvalho MD, Swash M. Awaji diagnostic algorithm increases sensitivity of El Escorial criteria for ALS diagnosis. Amyotroph Lateral Scler. 2009;10:53–57.
21. Douglass CP, Kandler RH, Shaw PJ, McDermott CJ. An evaluation of neurophysiological criteria used in the diagnosis of motor neuron disease. J Neurol Neurosurg Psychiatry. 2010;81:646–649.
22. Chen A, Weimer L, Brannagan T, Colin M, et al. Experience with the Awaji Island modifications to the ALS diagnostic criteria. Muscle Nerve. 2010;42:831–832.
23. Mills KR. Characteristics of fasciculations in amyotrophic lateral sclerosis and the benign fasciculation syndrome. Brain. 2010;133:3458–3469.
24. Benatar M, Tandan R. The Awaji criteria for the diagnosis of amyotrophic lateral sclerosis; have we put the cart before the horse? Muscle Nerve. 2011;43:461–463.
25. Andersen P, Al-Chalabi A. Clinical genetics of amyotrophic lateral sclerosis: what do we really know? Nat Rev Neurol. 2011; 7:603–615.
26. Hanby MF, Scott KM, Scotton W, et al. The risk to relative of patients with sporadic amyotrophic lateral sclerosis. Brain. 2011;134:3454–3457.
27. Donkervoort S, Siddique T. Amyotrophic lateral sclerosis overview. http://www.ncbi.nlm.nih.gov/books/NBK1450/.
28. Andersen PM, Forsgren L, Binzer M, et al. Autosomal recessive adult-onset amyotrophic lateral sclerosis associated with homozygosity for Asp90Ala CuZn-superoxide dismutase mutation: a clinical and genealogical study of 36 patients. Brain. 1996;119:1153–1172.
29. Hand C, Rouleau GA. Familial amyotrophic lateral sclerosis. Muscle Nerve. 2002;25:135–159.
30. http://www.ncbi.nlm.nih.gov/omim/?term=familial+als
31. Murphy J, Henry R, Lagmore S, Kramer JH, Miller BL, Lomen-Hoerth C. Continuum of frontal lobe impairment in amyotrophic lateral sclerosis. Arch Neurol. 2007;64:530–534.
32. Strong MJ, Lomen-Hoerth C, Caselli RJ, Bigio EH, Yang W. Cognitive impairment, frontotemporal dementia and the motor neuron diseases. Ann Neurol. 2003;54(5):S20–S23.
33. Lomen-Hoerth C, Anderson T, Miller BM. The overlap of amyotrophic lateral sclerosis and frontotemporal dementia. Neurology. 2002;59:1077–1079.
34. Murphy J, Henry R, Lomen-Hoerth C. Establishing subtypes of the continuum of frontal lobe impairment in amyotrophic lateral sclerosis. Arch Neurol. 2007;64:330–334.
35. Neary DF, Snowden JS, Gustafson L, et al. Frontotemporal lobar degeneration: a consensus on clinical diagnostic criteria. Neurology. 1998;51:1546–1554.
36. Wilson CM, Grace GN, Munoz DG, He BP, Strong MJ. Cognitive impairment in sporadic ALS: a pathologic continuum underlying a multisystem disorder. Neurology. 2001;57:651–657.
37. Yamguchi M, Hayashi H, Hiraoka K. Ventilatory support in Japan: a new life with ALS and a positive approach to living with the disease. Amyotrophic Lateral Sclerosis & Other Motor Neuron Disorders. 2001;2(4):209–211.
38. Tagami M, Kimura F, Nakajima H et al. Tracheostomy and invasive ventilation in Japanese ALS patients: decision-making and survival analysis: 1990–2010. J Neurol Sci. 2014;344 (1–2):158–164.
39. McCluskey LR, Elman LB, Martinez-Lage M, et al. Amyotrophic lateral sclerosis-plus syndrome with TAR DNA binding protein 43 pathology. Arch Neurol. 2009;66(1):121–124.
40. Wicks P, Abrahams S, Papps B, et al. SOD1 and cognitive dysfunction in familial amyotrophic lateral sclerosis. J Neurol. 2009;256(2):234–241.
41. Goldman JS, Rademakers R, Huey ED, et al. An algorithm for genetic testing of frontotemporal lobar degeneration. Neurology. 2011;76:475–483.
42. DeJesus-Hernandez M, Mackenzie IR, Boeve BF, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron. 2011;72(2):245–256.
43. Renton AE, Majounie E, Waite A, et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron. 2011;72(2):257–268.
44. Deng HX, Chen W, Hong ST, et al. Mutations in UBQLN2 cause dominant X-linked juvenile and adult-onset ALS and ALS/dementia. Nature. 2011;477:211–215.
45. Armon C. Smoking may be considered an established risk factor for sporadic ALS. Neurology. 2009;73:1693–1698.
46. Weisskopf MG, Ascherio A. Cigarettes and amyotrophic lateral sclerosis: only smoke or also fire? Ann Neurol. 2009;65(4):361–362.
47. Gallo V, Bueno-de-Mesquita HB, Vermeulen R, et al. Smoking and risk of amyotrophic lateral sclerosis: analysis of the EPIC cohort. Ann Neurol. 2009;65:378–385.
48. Nelson LM, Maguire V, Longstreth WT Jr, Matkin C. Population-based case control study of amyotrophic lateral sclerosis in western Washington State I. Cigarette smoking and alcohol consumption. Am J Epidemol. 2000;151:156–163.
49. Nelson LM, Matkin C, Longstreth WT Jr, Maguire V. Population-based case control study of amyotrophic lateral sclerosis in western Washington State II Diet. Am J Epidemol. 2000;151:164–173.
50. Worm PM. The epidemiology of motor neuron diseases: a review of recent studies. J Neurol Sci. 2001;191:3–9.
51. Cashman NR, Cudkowicz ME, Davidson MC, Pioro EP, Rosenfeld J; The ALS patient care database. Gender effects on duration and onset age of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord. 2001;2:41–42.
52. Chancellor AM, Slattery JM, Fraser H, Swingler RJ, Holloway SM, Warlow CP. Prognosis of adult-onset motor neuron disease: a prospective study based on the Scottish Motor Neuron Disease Register. J Neurol. 1993;240:339–346.
53. Bradley WG, Mash DC. Beyond Guam: the cyanobacteria/BMAA hypothesis of the cause of ALS and other neurodegenerative diseases. Amyotroph Lateral Scler. 2009;(10 suppl 2): 7–20.
54. Magnus T, Beck M, Geiss R, Puls I, Naumann M, Toyka KV. Disease progression in amyotrophic lateral sclerosis: predictors of survival. Muscle Nerve. 2002;25:709–714.
55. Albert SA, Whitaker A, Rabkin JG, et al. Medical and supportive care among people with ALS in the months before death or tracheostomy. J Pain Symptom Manage. 2009;38:546–553.
56. Traynor BJ, Alexander M, Corr B, Frost E, Hardiman O. Effect of a multidisciplinary amyotrophic lateral sclerosis (ALS) clinic on ALS survival: a population based study, 1996–2000. J Neurol Neurosurg Psychiatry. 2003;74(9):1258–1261.
57. Blexrud MD, Windebank AJ, Daube JR. Long-term follow-up of 121 patients with benign fasciculations. Ann Neurol. 1993;34:622–625.
58. Norris F, Shepherd R, Denys E, et al. Onset, natural history and outcome in idiopathic adult motor neuron disease. J Neurol Sci. 1993;118:48–55.
59. Mitsumoto H. Diagnosis and progression of ALS. Neurology. 1997;48(suppl):S2–S8.
60. Kent-Braun JA, Walker CH, Weiner MW, Miller RG. Upper and lower motor neuron function and muscle weakness in amyotrophic lateral sclerosis. Neurology. 1996;46:A470.
61. Haverkamp LJ, Appel V, Appel SH. Natural history of amyotrophic lateral sclerosis in a database population. Validation of a scoring system and a model for survival prediction. Brain. 1995;118:707–719.
62. Andersen PM. Amyotrophic lateral sclerosis associated with mutations in the CuZn superoxide dismutase gene. Curr Neurol Neurosci Rep. 2006;6:37–46.
63. Brownell B, Oppenheimer DR, Hughes JR. Central nervous system in motor neuron disease. J Neurol Neurosurg Psychiatry. 1970;33:338–357.
64. Iwanaga K, Hayashi S, Oyake M, et al. Neuropathology of sporadic amyotrophic lateral sclerosis of long duration. J Neurol Sci. 1997;146:139–143.
65. Lawyer T, Netsky MG. ALS, clinico-anatomic study of 53 cases. Arch Neurol Psychiatry. 1953;69:171–192.
66. Cudkowicz ME, McKenna-Yasek D, Chen C, Hedley-Whyte ET, Brown RH. Limited corticospinal tract involvement in amyotrophic lateral sclerosis subjects with the A4 V mutation in the copper/zinc superoxide dismutase gene. Ann Neurol. 1998;43:703–710.
67. Raaphorst J, de Visser M, van Tol MJ, et al. Cognitive dysfunction and lower motor neuron disease: executive and memory deficits and progressive muscular atrophy. J Neurol Neurosurg Psychiatry. 2011;82:170–175.
68. Katz JS, Wolfe GI, Andersson PB, et al. Brachial amyotrophic diplegia. Neurology. 1999;53:1071–1076.
69. Wijesekera LC, Mathers S, Talman P, et al. Natural history and clinical features of the flail arm and flail leg ALS variants. Neurology. 2009;72:1087–1094.
70. Pouget JY, Robert D, Sangla I, et al. Quantitative assessment of bulbar function in ALS using an objective speech evaluation. Neurology. 1996;46:A470.
71. Coon EA, Sorenson EJ, Whitwell JL, Knopman DS, Josephs KA. Predicting survival and frontotemporal dementia with motor neuron disease. Neurology. 2011;76:1886–1893.
72. Turner MR, Scaber J, Goodfellow JA, Lord ME, Marsden R, Talbot K. Diagnostic and prognosis in bulbar-onset amyotrophic lateral sclerosis. J Neurol Sci. 2010;294:81–85.
73. Portet F, Cadihac C, Touchon J, Camu W. Cognitive impairment in motor neuron disease with bulbar onset. Amyotroph Lateral Scler Other Motor Neuron Disord. 2001;2(1):23–29.
74. Lomen-Hoerth C, Murphy J, Langmore S, Kramer JH, Olney RK, Miller B. Are amyotrophic lateral sclerosis patients cognitively normal? Neurology. 2003;60:1094–1097.
75. Strong MJ, Grace GM, Orange JB, Leeper HA, Menon RS, Aere C. A prospective study of cognitive impairment in ALS. Neurology. 1999;53:1655–1670.
76. Ringholz GM, Appel SH, Bradshaw M, Cooke NA, Mosnik DM, Schulz PE. Prevalence and patterns of cognitive impairment in sporadic ALS. Neurology. 2005;65:586–590.
77. Le Forestier N, Maisonobe T, Piquard A, et al. Does primary lateral sclerosis exist? A study of 20 patients and a review of the literature. Brain. 2001;124:1989–1999.
78. Pringle CE, Hudson AJ, Munoz DG, Kiernan JA, Brown WF, Ebers GC. Primary lateral sclerosis. Clinical features, neuropathology and diagnostic criteria. Brain. 1992;115:495–520.
79. Strong MJ. Primary lateral sclerosis, hereditary spastic paraplegia and amyotrophic lateral sclerosis: discrete entities or spectrum? Amyotroph Lateral Scler Other Motor Neuron Disord. 2005;6(1):8–16.
80. Singer MA, Statland JM, Wolfe GI, Barohn RJ. Primary lateral sclerosis. Muscle Nerve. 2007;35:291–302.
81. Caselli RJ, Smith BE, Osborn D. Primary lateral sclerosis: a neuropsychological study. Neurology. 1995;45:2005–2009.
82. Josephs KA, Dickson DW. Frontotemporal lobar degeneration with upper motor neuron disease/primary lateral sclerosis. Neurology. 2007;69:1800–1801.
83. Chen R, Grand’Maison F, Strong MJ, Ramsay DA, Bolton CF. Motor neuron disease presenting as acute respiratory failure: a clinical and pathological study. J Neurol Neurosurg Psychiatry. 1996;60:455–458.
84. Shoesmith CL, Findlater K, Rowe A, Strong MJ. Prognosis of amyotrophic lateral sclerosis with respiratory onset. J Neurol Neurosurg Psychiatry. 2007;78:629–631.
85. Elamin M, Phukan J, Bede P, et al. Executive dysfunction is a negative prognostic indicator in patients with ALS without dementia. Neurology. 2011;76:1263–1269.
86. Wheaton MW, Salamone AR, Mosnik DM, et al. Cognitive impairment in familial ALS. Neurology. 2007;69:1411–1417.
87. Witgert M, Salamone AR, Strutt AM, et al. Frontal lobe mediated behavioral dysfunction in amyotrophic lateral sclerosis. Eur J Neurol. 2010;17:103–110.
88. Massman PJ, Sims J, Cooke N, Haverkamp LJ, Appel V, Appel SH. Prevalence and correlates of neuropsychological deficits in amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry. 1996;61:450–455.
89. http://www.mocatest.org/pdf_files/test/MoCA-Test-English_7_1.pdf
90. Daube JR. Electrodiagnostic studies in amyotrophic lateral sclerosis and other motor neuron disorders. Muscle Nerve. 2000;23:1488–1502.
91. Makki AA, Benatar M. The electromyographic diagnosis of amyotrophic lateral sclerosis: does the evidence support the El Escorial criteria? Muscle Nerve. 2007;35:614–619.
92. Dabby R, Lange DJ, Trojaborg W, et al. Inclusion body myositis mimicking motor neuron disease. Arch Neurol. 2001;58:1253–1256.
93. Agosta F, Chiò A, Cosottini M, et al. The present and the future of neuroimaging in amyotrophic lateral sclerosis. Am J Neuroradiol. 2010;31:1769–1777.
94. Wang S, Melhem ER, Poptani H, Woo JH. Neuroimaging in amyotrophic lateral sclerosis. Neurotherapeutics. 2011;8:63–71.
95. Mitsumoto H, Uluğ AM, Pullman SL, et al. Quantitative objective markers for upper and lower motor neuron dysfunction in ALS. Neurology. 2007;68:1402–1410.
96. van der Graff MM, Sage CA, Caan MW, et al. Upper and extra-motoneuron involvement in early motoneuron disease: a diffusion tensor imaging study. Brain. 2011;134:1211–1228.
97. Felice KJ, North WA. Creatine kinase values in amyotrophic lateral sclerosis. J Neurol Sci. 1998;160:S30–S32.
98. Chahin N, Sorenson EJ. Serum creatine kinase levels in spinal bulbar muscular atrophy and amyotrophic lateral sclerosis. Muscle Nerve. 2009;40:126–129.
99. Daoud H, Valdamanis PN, Gros-Louis F, et al. Resequencing of 29 candidate genes in patients with familial and sporadic amyotrophic lateral sclerosis. Arch Neurol. 2011;68(5):587–593.
100. Andersen PM, Borasio GD, Dengler R, et al. EFNS task force on management of amyotrophic lateral sclerosis: guidelines for diagnosis and clinical care of patients and relatives. Eur J Neurol. 2005;12:921–938.
101. Russell JA, Power B. Genetic testing in apparent sporadic ALS patients: surveyed opinions of the Northeastern ALS Consortium (NEALS) membership. Annual Meeting of the American Academy of Neurology. Honolulu, Hawaii, April 9–16, 2011.
102. Wagner KR. The need for biomarkers in amyotrophic lateral sclerosis drug development. Neurology. 2009;72:11–12.
103. Mitchell RM, Freeman WM, Randazzo WT, et al. A CSF biomarker panel for identification of patients with amyotrophic lateral sclerosis. Neurology. 2009;72:14–19.
104. Cudkowicz M, Swash M. CSF markers in amyotrophic lateral sclerosis: has the time come? Neurology. 2010;74:949–950.
105. Turner MR, Kiernan MC, Leigh PN, Talbot K. Biomarkers in amyotrophic lateral sclerosis. Lancet Neurol. 2009;8:94–109.
106. Bowser R, Lacomis D. Applying proteomics to the diagnosis and treatment of ALS and related diseases. Muscle Nerve. 2009;40:753–762.
107. Mackenzie IRA, Rademakers R, Neumann M. TDP-43 and FUS in amyotrophic lateral sclerosis and frontotemporal dementia. Lancet Neurol. 2010;9:995–1007.
108. Hodges JR, Davies RR, Xuereb JH, et al. Clinicopathological correlates and frontotemporal dementia. Ann Neurol. 2004:56(3):399–406.
109. Daoud H, Rouleau GA. A role for ubiquilin2 mutations in neurodegeneration. Nat Rev Neurol. 2011;7:599–600.
110. Del Bo R, Tiloca C, Pensato V, et al. Novel optineurin mutations in patients with familial and sporadic amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry. 2011;82:1239–1243.
111. Arai T, Hasegawa M, Akiyama H, et al. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun. 2006;351:602–611.
112. Geser F, Stein B, Partain M, et al. Motor neuron disease clinically limited to the lower motor neuron is a diffuse TDP-43 proteinopathy. Acta Neuropathol. 2011;121:509–517.
113. Kobayashi Z, Tsuchiya K, Arai T, et al. Clinicopathological characteristics of FTLD-TDP showing corticospinal tract degeneration but lacking lower motor neuron loss. J Neurol Sci. 2010;298:70–77.
114. Mizuno Y, Fujita Y, Takatama M, Okamoto K. Peripherin partially localizes in Bunina bodies in amyotrophic lateral sclerosis. J Neurol Sci. 2011;302:14–18.
115. Mori F, Tanji K, Miki Y, Kakita A, Takahashi H, Wakabayashi K. Relationship between Bunina bodies and TDP-43 inclusions in spinal anterior horn in amyotrophic lateral sclerosis. Neuropathol Appl Neurobiol. 2010;36:345–352.
116. Rothstein JR. Current hypotheses for the underlying biology of amyotrophic lateral sclerosis. Ann Neurol. 2009;65(suppl):S3–S9.
117. Ravits JM, LaSpada AR. ALS motor phenotype heterogeneity, focality, and spread: deconstructing motor neuron degeneration. Neurology. 2009;73:805–811.
118. Kuwabara S, Yokota T. Propagation: prion-like mechanisms can explain spreading of motor neuronal death in amyotrophic lateral sclerosis? J Neurol Neurosurg Psychiatry. 2011; 82(11):1181–1182.
119. Fujimura-Kiyono C, Kimura F, Ishida S, et al. Onset and spreading patterns of lower motor neuron involvements predict survival in sporadic amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry. 2011;82(11):1244–1249.
120. Siddique T, Figlewicz DA, Pericak-Vance MA, et al. Linkage of a gene causing familial amyotrophic lateral sclerosis to chromosome 21 and evidence of genetic-locus heterogeneity. N Engl J Med. 1991;324:1381–1384.
121. http://alsod.iop.kcl.ac.uk/
122. Van Damme P, Veldink JH, van Blitterswijk M, et al. Expanded ATXN2 CAG repeat size in ALS identifies genetic overlap between ALS and SCA2. Neurology. 2011;76:2066–2072.
123. Fischbeck KH, Pulst SM. Amyotrophic lateral sclerosis and spinocerebellar ataxia 2. Neurology. 2011;76:2050–2051.
124. Daoud H, Belzil V, Martins S, et al. Association of long ATXN2 CAG repeat sizes with increased risk of amyotrophic lateral sclerosis. Arch Neurol. 2011;68(6):739–742.
125. Maruyama H, Morino H, Ito H, et al. Mutations of optineurin in amyotrophic lateral sclerosis. Nature. 2010;465:223–227.
126. Valdmanis PN, Rouleau GA. Genetics of familial amyotrophic lateral sclerosis. Neurology. 2008;70:144–152.
127. Pasinelli P, Brown RH. Molecular biology of amyotrophic lateral sclerosis: insights from genetics. Nat Genet. 2006;7:710–723.
128. Shaw CE, Al-Chalabi A. Susceptibility genes in sporadic ALS: separating the wheat from the chaff by international collaboration. Neurology. 2006;67:738–739.
129. Neumann M, Kwong LK, Sampathu DM, Trojanowski JQ, Lee VM. TDP 43 proteinopathy in frontotemporal lobar degeneration and amyotrophic lateral sclerosis: protein misfolding diseases without amyloidosis. Arch Neurol. 2007;64(10):1388–1394.
130. Okamoto Y, Ihara M, Urushitani M, et al. An autopsy case of SOD-1 related ALS with TDP-43 positive inclusions. Neurology. 2011;77:1993–1995.
131. Burati E. TDP-43 and FUS in ALS/FTLD: will common pathways fit all? Neurology. 2011;77:1588–1589.
132. Rowland LP. Diagnosis of amyotrophic lateral sclerosis. J Neurol Sci. 1998;160(suppl 1):S6–S24.
133. Traynor BJ, Codd MB, Corr B, Forde C, Frost E, Hardiman O. Amyotrophic lateral sclerosis mimic syndromes: a population-based study. Arch Neurol. 2000;57:109–113.
134. Visser J, van den Berg-Vos RM, Franssen H, et al. Mimic syndromes in sporadic cases of progressive spinal muscular atrophy. Neurology. 2002;58:1593–1596.
135. Van den Berg-Vos RM, van den Berg LH, Visser J, de Visser M, Franssen H, Wokke JH. The spectrum of lower motor neuron syndromes. J Neurol. 2003;250:1279–1292.
136. Van den Berg-Vos RM, Visser J, Kalmihn S, et al. A long-term prospective study of the natural course of sporadic adult-onset lower motor neuron syndromes. Arch Neurol. 2009;66(6):751–757.
137. Nobile-Orazio E, Cappellari A, Priori A. Multifocal motor neuropathy: current concepts and controversies. Muscle Nerve. 2005;31:663–680.
138. Cats EA, Jacobs BC, Yuki N, et al. Multifocal motor neuropathy: association of anti-GM1 IgM antibodies with clinical features. Neurology. 2010;75:1961–1967.
139. Gooch CL, Amato AA. Are anti-ganglioside antibodies of clinical value in multifocal motor neuropathy? Neurology. 2010;75:1950–1951.
140. Finsterer J. Perspectives of Kennedy disease. J Neurol Sci. 2010;198:1–10.
141. Rhodes LE, Freeman BK, Auh S, et al. Clinical features of spinal and bulbar muscular atrophy. Brain. 2009;132:3242–3251.
142. Kennedy WR, Alter M, Sung JH. Proximal bulbar and spinal muscular atrophy of late onset. A sex-linked recessive trait. Neurology. 1968;18(7):671–680.
143. Parboosingh JS, Figlewicz DA, Krizus A, et al. Spinobulbar muscular atrophy can mimic ALS: the importance of genetic testing in male patients with atypical ALS. Neurology. 1997;49:568–572.
144. Sobue I, Saito N, Iida M, Ando K. Juvenile type of distal and segmental muscular atrophy of upper extremities. Ann Neurol. 1978;3:429–432.
145. Hirayama K, Tohocura Y, Tsubaki T. Juvenile muscular atrophy of unilateral extremity: a new clinical entity. Psychiatry Neurol Jpn. 1959;61:2190–2197.
146. Pradhan S. Bilaterally symmetric form of Hirayama disease. Neurology. 2009;72:2083–2089.
147. Patel DR, Knepper L, Jones HR Jr. Late-onset monomelic amyotrophy in a Caucasian woman. Muscle Nerve. 2008; 37(1):115–119.
148. Felice KJ, Whitaker CH, Grunnet ML. Benign calf amyotrophy. Arch Neurol. 2003;60:1415–1420.
149. Irobi J, De Jonghe P, Timmerman V. Molecular genetics of distal hereditary motor neuropathies. Human Mol Genet. 2004;13:R195–R202.
150. Rossor AM, Kalmar B, Greensmith L, Reilly MM. The distal hereditary motor neuropathies. J Neurol Neurosurg Psychiatry. 2012;83:6–14.
151. Ésik O, Vönöczky K, Lengyel Z, Sáfrány G, Trón L. Characteristics of radiogenic lower motor neuron disease, a possible link with a possible viral infection. Spinal Cord. 2004;42(2):99–105.
152. Fink JF. Hereditary spastic paraplegia. Curr Neurol Neurosci Rep. 2006;6:65–76.
153. Meyer T, Schwan A, Dullinger JS, et al. Early-onset ALS with long-term survival associated with spastin gene mutation. Neurology. 2005;65:141–143.
154. Mathews JA. Wasting of the small hand muscles in upper- and mid-cervical cord lesions. QJM. 1998;91:691–700.
155. Stark AJ, Kennard C, Swash MA. Hand wasting in spondylotic high cord compression: an electromyographic study. Ann Neurol. 1981;9(1):58–62.
156. Srinivasan J, Scala S, Jones HR, Saleh F, Russell JA. Inappropriate surgeries resulting from misdiagnosis of early amyotrophic lateral sclerosis. Muscle Nerve. 2006;34(3):359–360.
157. Weihl CC, Lopate G. Motor neuron disease associated with copper deficiency. Muscle Nerve. 2006;34:789–793.
158. Quereshi M, Bedlack RS, Cudkowicz ME. Lyme serology in amyotrophic lateral sclerosis. Muscle Nerve. 2009;40:626–628.
159. Henning F, Hewlett RH. Brachial amyotrophic diplegia (segmental proximal spinal muscular atrophy) associated with HIV infection. J Neurol Neurosurg Psychiatry. 2008;79:1392–1394.
160. Verma A, Berger JR. ALS syndrome in patients with HIV-1 infection. J Neurol Sci. 2006;240:59–64.
161. Drory VE, Birnbaum M, Peleg L, Goldman B, Korczyn AD. Hexosaminidase A deficiency is an uncommon cause of a syndrome mimicking amyotrophic lateral sclerosis. Muscle Nerve. 2003;28:109–112.
162. Jackson CE, Amato AA, Bryan WW, Wolfe GI, Sakhaee K, Barohn RJ. Primary hyperparathyroidism in ALS: is there are relation? Neurology. 1998;50:1795–1799.
163. Stitch O, Kleer B, Rauer S. Absence of paraneoplastic anti-neuronal antibodies in sera of 145 patients with motor neuron disease. J Neurol Neurosurg Psychiatry. 2007;78:883–885.
164. Moulignier A, Moulonguet A, Pialoux G, Rozenbaum W. Reversible ALS-like disorder in HIV infection. Neurology. 2001; 57:1995–2001.
165. Johnson WG. The clinical spectrum of hexosaminidase deficiency diseases. Neurology. 1981;31:1453–1456.
166. Younger DS. Motor neuron disease or malignancy. Muscle Nerve. 2000;23(5):658–660.
167. Gordon PH, Rowland LP, Younger DS, et al. Lymphoproliferative disorders and motor neuron disease: an update. Neurology. 1997;48:1671–1678.
168. Verma A, Berger JR. Primary lateral sclerosis with HIV-1 infection. Neurology. 2008;70(7):575–577.
169. Atkinson JL, Miller GM, Krauss WF, et al. Clinical and radiographic features of dural arteriovenous fistula, a treatable cause of myelopathy. Mayo Clin Proc. 2001;76:1120–1130.
170. Armon C, Daube JR. Electrophysiological signs of arteriovenous malformations of the spinal cord. J Neurol Neurosurg Psychiatry. 1989;52:1176–1181.
172. https://www.clinicaltrials.gov/ct2/results?term=als&Search=Search
173. http://www.alsconsortium.org/search.ph
174. Kimura F, Fujimura C, Ishida H, et al. Progression rate of ALSFRS-R at time of diagnosis predict survival time in ALS. Neurology. 2006;66:265–267.
175. Gordon PH, Cheung YK. Progression rate of ALSFRS-R at time of diagnosis predict survival time in ALS. Neurology. 2006;67:1314–1315.
176. Miller RG, Jackson CE, Karsarskis EJ, et al. Practice Parameter update: the care of the patient with amyotrophic lateral sclerosis: drug, nutritional, and respiratory therapies (an evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology. 2009;73:1218–1226.
177. Miller RG, Jackson CE, Karsarskis EJ, et al. Practice parameter update: the care of the patient with amyotrophic lateral sclerosis: multidisciplinary care, symptom management, and cognitive/behavioral impairment (an evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology. 2009;73:1227–1233.
178. Simmons Z. Management strategies for patients with amyotrophic lateral sclerosis from diagnosis through death. Neurologist. 2005;11:257–270.
179. Dal Bello-Haas V, Florence JM, Kloos D, et al. A randomized controlled trial of resistance exercise in individuals with ALS. Neurology. 2007;68:2003–2007.