 TABLE 7-1. HEREDITARY SPASTIC PARAPARESIS 2,3
TABLE 7-1. HEREDITARY SPASTIC PARAPARESIS 2,3The diagnosis of hereditary spastic paraparesis (HSP) is based on the identification of a phenotype characterized as a slowly progressive, symmetric, spastic paraparesis in which the morbidity is largely related to impaired leg control rather than weakness, with or without recognition of other family members. The prevalence of dominantly inherited HSP, at least in Ireland, is estimated at 1.27/105.1 Like many of the disorders discussed in this text, the nosology of HSP is confounded by insights generated by molecular biology. The HSP phenotype is now recognized to result from mutations involving at least 50 different genetic loci and 18 identified genes (Table 7-1).2 Despite the potential precision that a classification system based solely on gene location and gene product would provide, it remains an impractical bedside tool. Due to current limitations of genetic testing, a pragmatic classification system requires at least some consideration of clinical features. This chapter will attempt to provide a classification hybrid that addresses both clinical and genetic considerations (Table 7-1).
TABLE 7-1. HEREDITARY SPASTIC PARAPARESIS 2,3


The concept of a hereditary disorder manifesting as spasticity of the lower extremities was initially championed by Seeligmüller, Strumpell, and Lorrain in the last quarter of the 19th century. It was envisioned as a singular entity with phenotypic variation.3 The classification system still utilized today was initially promoted by Anita Harding in 1981.4 She proposed a dominantly inherited HSP dichotomy in which type I was considered to reflect an early-onset phenotype with predominant, if not exclusive, upper motor neuron (UMN) features. In contrast, type II HSP referred to those with late onset in which weakness and presumed lower motor neuron (LMN) involvement overshadowed the UMN signs. In 1983, her classification system was expanded to encompass complicated as well as uncomplicated forms of the syndrome.5 Uncomplicated HSP still refers to a syndrome of spastic paraparesis in which cavus foot deformities and mild vibratory sense loss may occur as the only other associated features. Complicated HSP is defined by involvement of additional neurological and occasionally nonneurological systems as described below (Table 7-1).
In 1996, the nosology of HSP became at the same time both enhanced and complicated with discovery of the first disease producing mutation.3 The HSPs are currently genotypically catalogued by a numerical system based on the order of individual gene discovery. Each number is prefaced by the acronym SPG which stands for spastic paraplegia gene (Table 7-1). Unlike other classification systems, subheadings distinguishing dominant from other inheritance patterns are not utilized.
 CLINICAL FEATURES
CLINICAL FEATURESIn virtually every case, the presenting symptoms relate to lower extremity spasticity which has a symmetric or near symmetric distribution. Symptom onset is typically recognized in the second or third decade but may become manifest as early as the first or as late as the seventh decade of life. Patients lose the ability to run or hop early in their course due to increased extensor tone in the lower extremities. Consequently the ability to fully flex the hip and the knee is impaired resulting in reduced stride length and difficulty running. Patients will describe dragging and stiffness of the legs and a tendency to trip on uneven ground. When observed, the legs may be noted to scissor or cross over each other due to increased adductor tone (Fig. 7-1). Circumduction (a rotational rather than linear advancement of the legs) is common in a compensatory attempt to avoid tripping. This risk results from a leg that is tonically extended at the hip and knee and from a tonic foot posture of inversion and plantar flexion (equinovarus posture). High-arched feet and hammer toe deformity are common but not invariable features of the illness. They are more likely to occur with disease onset in childhood at a time when the metatarsals remain malleable and vulnerable to the imbalance of forces produced by disproportionate involvement of specific muscle groups (Fig. 7-2).
Figure 7-1. Circumducting leg with equinovarus foot posturing in HSP.
Figure 7-2. Hammer toes and cavus deformity in HSP.
HSP morbidity results in large part from the increased lower extremity extensor tone impairing lower extremity coordination. Lower motor neuron involvement may occur but is typically overshadowed by spasticity. If weakness occurs, it typically does so in a UMN pattern, with hip flexors, knee flexors, and foot dorsiflexors being typically weaker than their respective antagonists. Hyperreflexia of the lower extremities is universal, almost always accompanied by extensor plantar responses. Hyperreflexia of the upper extremities with Hoffman’s signs and reflex spread is common as well. Significant loss of upper extremity function associated with weakness, increased tone, or impaired coordination occurs infrequently in most genotypes and should lead to consideration of an alternative diagnosis.
Mild posterior column involvement may occur with vibratory sense loss and occasionally position sense loss in the toes. Rarely is it severe enough to produce significant sensory ataxia. A strikingly positive Romberg sign should once again lead to consideration of an alternative diagnosis. Urinary frequency, urgency, and urgency incontinence are common symptoms even in uncomplicated disease. Rectal urgency and incontinence and sexual dysfunction are uncommon.
There is a wide range of associated neurological and nonneurological symptoms that can occur in complicated forms of the disease. Recognition of these additional features may aid in the identification of a specific genotype (Table 7-1). Some of the more common associated features are amyotrophy of distal limb muscles that may result from either a motor neuronopathy (SPG3A, 4, 5, 9, 10, 11, 14, 15, 17, 20, 26, 30, 38, 39, 43, 41) or peripheral neuropathy (SPG2, 5, 25, 27, 30, 31, 36, the SPOAN syndrome, and the one recognized mitochondrial mutation producing a SPG phenotype).2,6 Distal amyotrophy may initially affect either the hands or feet, hand-wasting and spastic paraparesis being referred to as Silver syndrome.7,8 Ataxia, nystagmus, dysarthria, and other features of cerebellar dysfunction occur less frequently (SPG21). Extrapyramidal manifestations are relatively uncommon as well (SPG21, 35).2,6,9,10 Cognitive changes may manifest as either mental retardation or dementia (SPG4, 11, 14, 15, 18, 21, 26, 27, 32, 35, 44, 45, 46, 47). A thin corpus callosum is a relatively common feature (SPG11, 18, 21, 35, 46, 47).2,6,10,11 Seizures, deafness, cataracts, ichthyosis, ophthalmoparesis, ocular apraxia, retinal pigmentary degeneration, and optic atrophy with visual loss are some of the other potential associated features.2,6,12
A uniform age of symptom onset and rate of disease progression are characteristic of uncomplicated HSP genotypes but exceptions to this general rule are not uncommon. The reasons for variations of disease onset and severity of affliction, both within and between families of the same genotype are not understood.13 This variability does not appear to correlate directly with the mechanism of mutation, at least within the SPG4 genotype.14 For the most part, individual families will remain segregated into either uncomplicated or complicated phenotypes but occasional families will have members with both.
DIFFERENTIAL DIAGNOSISThe differential diagnosis of HSP includes any disorder that results in UMN dysfunction of the lower extremities (Table 7-2). In our opinion, the disorder that is most likely to mimic HSP is PLS or UMN dominant ALS, either on a sporadic or inherited basis.15–18 Slow progression, symmetry, cavus foot deformity, and loss of vibration perception in the toes favors a HSP diagnosis although these clues are relative and clinical distinction may be difficult in many cases.19 Rapid progression, notable asymmetry, or impaired upper extremity or bulbar function would favor a PLS/UMN-D ALS diagnosis. The family history needs to be interpreted with caution as ALS like HSP may be hereditary, and as absence of other affected family members by no means precludes heritable disease.18 The difficulty in distinguishing between HSP and ALS was recently emphasized to us in the case of a woman in her mid-30s with 20 years of seemingly sporadic, slowly progressive, spastic paraparesis, and cavus foot deformity suggesting HSP in whom a pathological mutation in a familial ALS gene (senataxin) was identified in both her and her asymptomatic father.
TABLE 7-2. DIFFERENTIAL DIAGNOSIS OF HSP: TESTING CONSIDERATIONS
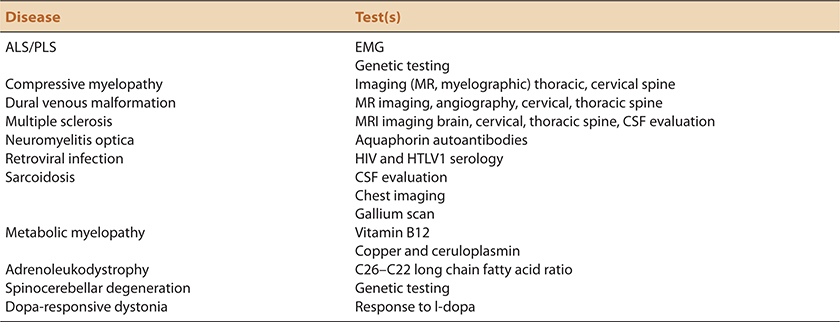
The differential diagnosis of HSP includes another category of heritable neuromuscular disease, distal spinal muscular atrophy/hereditary motor neuropathy. The latter, although dominated by LMN features, may include UMN features as well providing phenotypic and at times genotypic overlap between these two disease categories (Table 8-1).
Most metabolic or structural myelopathies affect sensory pathways more than HSP typically does. None-the-less, the differential diagnosis of HSP includes metabolic and structural disorders in which sensory signs or symptoms are limited. In consideration of its chronicity and frequency, cervical spondylotic myelopathy is a common differential diagnostic consideration. As emphasized in Chapter 6, we urge caution in attributing spastic paraparesis to cervical canal stenosis in the absence of significant spinal cord compression or abnormal cord signal at the level of compression. Other myelopathic considerations in which imaging may be abnormal include dural venous fistulas, spinal forms of multiple sclerosis, and other inflammatory disorders including such as acute disseminated encephalomyelitis, neuromyelitis optica, HIV or HTLV1 inflection, and sarcoidosis. The chronological course in these latter disorders would characteristically progress at a different and usually faster pace than HSP.
Adrenoleukodystrophy and adrenomyeloneuropathy are X-linked disorders that require consideration in young men and young adult woman. Arguably, the latter cohort provides the greatest difficultly as adrenoleukodystrophy frequently presents as a slowly progressive spastic paraparesis in young adult women. Young males typically have a more severe and multisystem phenotype producing varying combinations of adrenal insufficiency, myelopathy, neuropathy, and cognitive decline. Other leukodystrophies may be considered in the differential diagnosis of complicated forms of the disease.
Vitamin B12 and copper deficiency may affect pyramidal tracts although tend to manifest primarily as posterior column myelopathies with predominant sensory loss. Other inherited disorders with prominent myelopathic features include Friedreich ataxia and spinocerebellar ataxia type III (Machado–Joseph disease). Both are more typically clinically dominated by posterior column and/spinocerebellar ataxia respectively rather than by spasticity.
Cerebral palsy is a differential diagnostic consideration in early-onset cases of HSP. This is particularly true for SPG3A associated with mutations of the atlastin gene.20,21 Dopa-responsive dystonia may manifest itself as a progressive, spastic gait disorder of childhood. Low-dose therapy with levodopa would serve both a diagnostic and a therapeutic purpose in this disorder.
LABORATORY FEATURESGenetic testing in suspected HSP patients is emblematic of the practical problems that exist with genetic testing in general. Of more than 50 suspected recognized gene loci, 25 genes have been identified, with commercially available testing available for 14.2,6,22,23 Of these, 19 are inherited in a dominant fashion, 27 recessively, with 5 that are X-linked and one associated with a mitochondrial genome mutation. Negative genetic testing excludes neither HSP in general or in some cases, the genotype that is being tested for. Commercially utilized methodologies may not detect certain mutational mechanisms, thus providing the opportunity for a false-negative test. For example, partial deletions of the SPAST gene (SPG4 locus) may not be detected by some commercial laboratories.24
Genetic testing in HSP has the highest yield in genotypes with dominantly inherited mutation patterns. Approximately 75% of patients with dominant inheritance patterns can be currently genotyped. These are the patients however, in which a confident clinical diagnosis is the easiest to establish. Facilitating genetic analysis is the knowledge that 60% of dominantly inherited cases are attributable to four HSP types, SPG3A, 4, 6, and 31. In view of the cost considerations, we would favor initial testing for the latter three genes in suspected dominant, adult-onset cases, if genetic testing is to be performed at all. As the SPG3A (atlastin) mutation frequently manifests in childhood, we would initially avoid this test in adults as well as other infrequently occurring HSP mutations.21
The primary problem related to genetic testing occurs in apparent sporadic or recessive cases, those in which a diagnosis of HSP is the most difficult to establish clinically. Somewhere between a quarter and a half of cases with recessive inheritance patterns may be currently genotyped.2 SPG11 is thought to be responsible for approximately 50% of recessively inherited cases.2 In our opinion, this should be the first gene tested in any recessive or sporadic case with a thin corpus callosum if genotyping is performed. SPG7 is thought to be the second most common AR genotype representing ≤6% of these cases.25,26
It is important that the clinician be aware of additional nuances of HSP genetic testing. It is estimated that approximately 10–20% of apparent sporadic cases will be found to have a gene mutation more typically associated with either a dominant or recessive inheritance pattern with SPG4 and 7 being the most commonly recognized examples of this.17,25 Incomplete penetrance does occur in HSP, providing at least one reason for the aforementioned observation. Although anticipation is not a well-described phenomenon in HSP, particularly in uncomplicated forms, it has been reported to occur in SPG4.6
We strongly support the conceptual value of diagnostic confirmation of potential heritable diseases if detailed pedigree analysis is nondiagnostic. We recognize however, both the technical and financial limitations that influence routine genetic testing. This includes consideration that commercially available genetic testing is typically expensive and is commonly inadequately reimbursed by third party payers. This may create a financial burden to either the patient or the healthcare system. In view of this, and in consideration that the natural history of the disease is unlikely to be favorably altered by the knowledge of genotype, we do not routinely obtain genetic testing in HSP suspects. This practice will undoubtedly change as whole exome sequencing and the ability to distinguish benign polymorphisms from pathogenic mutations evolves.
If genotypic confirmation of the HSP diagnosis cannot be accomplished, other diagnostic testing such as imaging may be undertaken in an attempt to address other differential diagnostic considerations. Tests that may be considered are summarized in Table 7-2. Imaging in HSP is typically normal in uncomplicated cases, although atrophy of the thoracic spinal cord has been reported.3 MR imaging of the brain may identify some of the features associated with complicated forms of HSP, including atrophy of the corpus callosum, hydrocephalus, or white matter changes. This would be of more value in identifying the type of HSP rather than establishing the initial diagnosis.
HISTOPATHOLOGYHSP appears to be a “dying back myelopathy.” Affected individuals will have degeneration of both the crossed and uncrossed corticospinal tracts, most notable in the lumbosacral and thoracic segments of the spinal cord. This degeneration becomes less apparent in the cervical regions. Conversely, degeneration of the posterior columns is most evident in the fasciculus gracilis in their most centrifugal locations, that is, at the cervical–medullary junction.3 Spinocerebellar pathways are involved in some cases to a far lesser extent. Decreased numbers of anterior horn cells and/or cortical motor neurons have been reported.
In a manner similar to the clinical overlap described above, certain HSP genotypes may have TDP-43 positive inclusions, a marker more typically associated with ALS.27
PATHOGENESISThe multiple HSP genotypes suggest that there is a final common pathway by which mutations of different proteins coalesce into an identical or near-identical phenotype. Although the functions of certain HSP-related proteins are understood, the means by which they induce a fairly uniform phenotype remains unknown.2,3,28 The function of spastin (SPG4) is related to microtubule dynamics. The kinesin 5A gene (SPG10) has a role in axonal transport. Three SPG genes, paraplegin, chaperonin 60, receptor expression enhancing protein 1, and mitochondrial ATPase 6 (SPG7, 13, 31) code for mitochondrial proteins. L1 cell adhesion molecule (SPG1) plays a role in corticospinal tract development. Mutations of the proteolipid protein and the gap junction protein gamma 2 genes (SPG2, 42) result in abnormalities of myelination. The morphology of the endoplasmic reticulum is altered in mutations of the atlastin, spastin and receptor expression enhancing protein 1 genes (SPG3A, 4, 31). Disturbances of membrane trafficking, protein accumulation, and endoplasmic reticulum stress response are associated with abnormalities of strumpellin and seipin gene function (SPG8, 17).
A recurrent theme throughout this text is the increasing recognition that neuromuscular disorders historically classified as different diseases are allelic. Although this phenomenon is not as prevalent as in spinal muscular atrophy, it is relevant to HSP as well (Table 8-1). SPG3A and hereditary sensory neuropathy type I result from mutations in the atlastin gene.28 SPG17 is allelic with both hereditary motor neuropathy type V and Charcot–Marie–Tooth disease type II.6
MANAGEMENTHSP management is supportive. The goal is to maintain comfort and to the extent possible, safe and independent patient mobility. There are a number of different interventions that attempt to reduce spasticity including oral tizanidine, lioresal, dantrolene, or benzodiazepines, intrathecal baclofen, or the intramuscular injection of botulinum toxin (Table 7-3). In extreme cases, surgical release of tendons may be considered in nonambulatory patients to facilitate hygiene or improve patient comfort. The treatment of spasticity requires considerable clinical judgment. The aforementioned agents rarely produce the optimally desired beneficial effect and may create unwanted side effects. Improved ambulation, comfort, and range of motion are the desired effects. Sedation, confusion, and unmasking underlying muscle weakness that may actually add to fall risk and detract from safe mobility are to be avoided. Although spasticity hinders gait, it may also paradoxically reduce fall risk. In an individual who also has considerable underlying weakness, the increased tone of extensor muscles may represent the major source of antigravity resistance. Suppression of this tone may deprive individuals of their ability to stand.
TABLE 7-3. TREATMENT OPTIONS FOR SPASTICITY (LISTED IN APPROXIMATE ORDER OF USE)
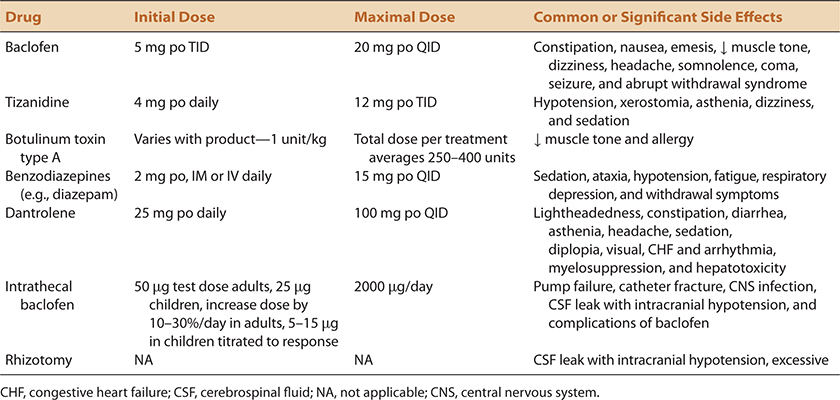
To improve tolerance, oral antispasticity drugs are typically initiated at very low doses and then titrated upward (Table 7-3). Baclofen and tizanidine are preferred as first-line agents by most. Rapid withdrawal of these agents may lead to unwanted CNS side effects, including confusion and psychosis. Dantrolene is used less frequently because of risks associated with hepatotoxicity. Benzodiazepines have less well-developed antispasticity properties and are frequently used as an adjunct rather than as a primary antispasticity treatment.
Intrathecal baclofen delivered by a programmable pump is an option if oral drugs do not provide the desired effect. The theoretical benefit is to deliver the drug directly to the afflicted end organ in small titratable doses in order to avoid the side effects commonly associated with the larger oral doses required. Intrathecal baclofen may allow certain patients who are spastic to remain ambulatory longer than their natural history would otherwise allow. A more realistic goal is to diminish refractory painful spasms or to diminish lower extremity tone to facilitate hygiene.
Injection of botulinum toxin into spastic muscles provides an alternative means to diminish muscle tone.29 Although attractive in concept because of the ability to affect only selected muscles, identification of the best dose and obtaining reimbursement for the relatively large volumes often required provide significant obstacles to its use. Many payers limit reimbursement to 400 units per session, which may be inadequate to achieve the desired goals. The effect of botulinum toxin is greatest when the toxin is delivered in proximity to the motor point. Identification of the most severely affected muscles and delivery of the lowest effective doses are the two major principles used. Repeat injections are typically required at approximately 3-month intervals.
Urinary urgency from detrusor overactivity is a common source of morbidity in HSP patients. There are a number of pharmacological agents that may ameliorate but rarely resolve this problem. The antispasticity agents, baclofen and tizanidine, may provide some relief. The mechanism of action of baclofen is thought to be as a presynaptic agonist of GABA-B receptors that is thought to have an inhibitory effect on activity of the descending bulbosacral tracts in the spinal cord. Tizanidine is an alpha adrenergic agent thought to produce presynaptic inhibition of motor neurons, potentially by reducing glutamate release, again at the spinal cord level. Typically however, the first-line treatments are muscarinic anticholinergic agents which counteract detrusor hyperreflexia mediated through parasympathetic nerve fibers traveling via the S2–4 roots and the pelvic nerve. Frequently used agents include oxybutynin, tolterodine, and trospium. Newer agents include darifenacin and solifenacin. Tricyclic antidepressants may also be utilized for their anticholinergic properties. Other, less frequently used therapies with uncertain benefit include botulinum toxin injections into the detrusor, intravesicular delivery of certain drugs including capsaicin, and S2–4 ventral root stimulation coupled with analogous dorsal rhizotomies.
Durable medical equipment and home modification can provide substantial benefit to individual patients. The reader is referred to Chapter 5 for more details. Ankle–foot orthoses are of great benefit to individual patients to prevent falls due to tripping. Ideally, they should be custom fitted to improve comfort, particularly in consideration of associated cavus foot deformities. A skilled physical therapist is an invaluable tool to decide whether a cane, Lofstran or Canadian crutches, a walker, or a wheelchair is the best solution for an individual patient. The Dashaway® walker is particularly helpful in these patients in that it diminishes the risk of falling backward more so than traditional walker designs. Power chairs and scooters may benefit some patients. Reimbursement may be problematic as payers typically require documentation that a patient is unable to propel themselves in a manual chair prior to authorization. Most HSP patients do not have this limitation. Patients who require forms of power mobility who also have trunk, upper extremity, or bulbar weakness are better served by a power chair because of the trunk support, ability to operate with a joystick control, and the ability to mount other equipment that may be beneficial to the patient. HSP patients are one group of patients where scooters, often preferred by the patient over power chairs, may be recommended.
Many patients resent the symbolism of durable medical equipment, viewing it as a “setback” and a constant reminder of their impaired condition. It may be effective to promote durable medical equipment to them as an opportunity. Specifically, it may allow them to maintain their independent mobility while minimizing the risk of falls and the potential of severe injury, the quickest and most likely threat to their independence. In patients who live in multiple-story dwellings who require access to more than one floor, stair lifts provide a safe and energy-sparing option. Patients motivated to perform daily stretching exercises claim to enjoy considerable benefit from doing so.
Like all chronic diseases, HSP patients may benefit from the resources provided by support organizations. Examples include:
• National Institute of Neurological Disorders and Stroke
• Hereditary Spastic Paraplegia Foundation, Inc., 209 Park Rd., Chelmsford MA 01824. (Phone: 703–495–9261; e-mail: community@sp-foundation.org; sp-foundation.org.)
• National Ataxia Foundation, 2600 Fernbrook Lane Suite 119, Minneapolis MN 55447. (Phone: 763–553–0020; fax: 763–553–0167, e-mail: naf@ataxia. org; www.ataxia.org.)
Genetic counseling in HSP is challenging. Although phenotypic homogeneity within families is the norm, exceptions do exist. Prenatal testing is available for certain HSP genotypes.6 HSP does not typically reduce life expectancy, the penetrance of many HSP genotypes is quite variable, and anticipation in HSP is not recognized to be a significant issue. For these reasons, we find it difficult to recommend prenatal testing in view of the risk to the fetus associated with amniocentesis or chorionic villous sampling.
SUMMARYHSP is a heritable disorder in which >50 currently recognized gene loci correlate with a fairly homogeneous phenotypic syndrome dominated by spastic paraparesis. It is a disorder that offers the opportunity to understand how semi-selective vulnerability of a single component of the nervous system can occur as a result of seemingly disparate pathophysiologies. Like other heritable disorders in which the molecular biology is providing new insights into disease mechanisms, the nosology of the HSP will undoubtedly be revised as the overlapping genetics of different heritable disorders is increasingly clarified.
1. McMonagle P, Webb S, Hutchinson M. The prevalence of “pure” autosomal dominant hereditary spastic paraplegia in the island of Ireland. J Neurol Neurosurg Psychiatry. 2002;72:43–46.
2. Fink JK. Hereditary spastic paraplegia. In: Rimoin DL, Connor JM, Pyeritz RE, Korf BR, eds. Emery and Rimoin’s Principles and Practice of Medical Genetics. 6th ed. Philadelphia, PA: Churchill Livingstone Elsevier; 2011.
3. Fink JK. Hereditary spastic paraplegia. Curr Neurol Neurosci Rep. 2006;6:65–76.
4. Harding AE. Hereditary “pure” spastic paraplegia: A clinical and genetic study of 22 families. J Neurol Neurosurg Psychiatry. 1981;44:871–883.
5. Harding AE. Classification of the hereditary ataxias and paraplegias. Lancet. 1983;1:1151–1155.
6. Fink JK. Hereditary spastic paraplegia overview. 2009. www.genetests.org.
7. Orlacchio A, Patrono C, Gaudiello F, et al. Silver syndrome variant of hereditary spastic paraplegia: a locus to 4p and allelism with SPG4. Neurology. 2008;70:1959–1966.
8. Rowland LP, Bird TD. Silver syndrome: the complexity of complicated hereditary spastic paraplegia. Neurology. 2008;70:1948–1949.
9. Simpson MA, Cross H, Proukakis C, et al. Maspardin is mutated in Mast syndrome, a complicated form of hereditary spastic paraplegia associated with dementia. Am J Hum Genet. 2003;73:1147–1156.
10. Edvardson S, Hama H, Shaag A, et al. Mutations in the fatty acid 2-hydroxylase gene are associated with leukodystrophy with spastic paraparesis and dystonia. Am J Hum Genet. 2008;83:643–648.
11. Lossos A, Stevanin G, Meiner V, et al. Hereditary spastic paraplegia with thin corpus callosum: reduction of the SPG11 interval and evidence for further genetic heterogeneity. Arch Neurol. 2006;63:756–760.
12. Dick KJ, Eckhardt M, Paisan-Ruiz C, et al. Mutation of FA2H underlies a complicated form of hereditary spastic paraplegia (SPG35). Hum Mutat. 2010;31:E1251–E1260.
13. Fink JK, Heiman-Patterson T, Bird T, et al. Hereditary spastic paraplegia advances in genetic research. Neurology. 1996;46: 1507–1514
14. Yip AG, Durr A, Marchuk DA, et al. Meta-analysis of age at onset in spastin-associated hereditary spastic paraplegia provides no evidence for a correlation with mutational class. J Med Genet. 2003;40:e106.
15. Meyer T, Schwan A, Dullinger JS, et al. Early-onset ALS with long-term survival associated with spastin gene mutation. Neurology. 2005;65:141–143.
16. Strong MJ, Gordin PH. Primary lateral sclerosis, hereditary spastic paraplegia and amyotrophic lateral sclerosis: Discrete entities or spectrum? Amyotroph Lateral Scler Other Motor Neuron Disord. 2005;6(1):8–16.
17. Brugman F, Wokke JHJ, Scheffer H, Versteeg MH, Sistermans EA, van den Berg LH. Spastin mutations in sporadic adult-onset upper motor neuron syndromes. Ann Neurol. 2005;58(6):865–869.
18. Brugman F, Wokke JHJ, Vianney de Jong JM, Franssen H, Faber CG, Van den Berg LH. Primary lateral sclerosis as a phenotypic manifestation of familial ALS. Neurology.2005;64(10):1778–1779.
19. Brugman F, Veldink JH, Franssen H, et al. Differentiation of hereditary spastic paraparesis from primary lateral sclerosis in sporadic adult-onset upper motor neuron syndromes. Arch Neurol. 2009;66(4):509–514.
20. Rainier S, Sher C, Reish O, Thomas D, Fink JK. De novo occurrence of novel SPG3A/alastin mutation presenting as cerebral palsy. Arch Neurol. 2006;63:445–447.
21. Namekawa M, Ribai P, Nelson I, et al. SPG3 A is the most frequent cause of hereditary spastic paraplegia with onset before age 10 years. Neurology. 2006;66(1):112–114.
22. Fink JK, Rainer S. Hereditary spastic paraplegia: Spastin phenotype and function. Arch Neurol. 2004;61:830–833.
23. www.athenadiagnostics.com/content/test-catalog/
24. Beetz C, Nygrem AO, Schickel J, et al. High frequency of partial SPAST deletions in autosomal dominant hereditary spastic paraplegia. Neurology. 2006;67:1926–1930.
25. Brugman F, Scheffer H, Wokke JHJ. Paraplegin mutations in apparently sporadic adult-onset upper motor neuron syndromes. Neurology. 2008;71:1500–1505.
26. Elleuch N, Depienne C, Benomar A, et al. Mutation analysis of the paraplegin gene (SPG7) in patients with hereditary spastic paraplegia. Neurology. 2006;66(5):654–659.
27. Martinez-Lage M, Molina-Porcel L, Falcone D, et al. TDP-43 pathology in a case of hereditary spastic paraplegia with a NIPA1/SPG6 mutation. Acta Neuropathol. 2012;124(2):285–291.
28. Timmerman V, Clowes VE, Reid E. Overlapping molecular pathological themes link Charcot-Marie-Tooth neuropathies and hereditary spastic paraplegia. Exp Neurol. 2013;246:14–25.
29. Comella CL, Pullman SL. Botulinum toxins in neurological disease. Muscle Nerve. 2004;29:628–644.

