 TABLE 26-1. DISORDERS OF NEUROMUSCULAR TRANSMISSION OTHER THAN AUTOIMMUNE MYASTHENIA GRAVIS
TABLE 26-1. DISORDERS OF NEUROMUSCULAR TRANSMISSION OTHER THAN AUTOIMMUNE MYASTHENIA GRAVISThis chapter describes the disorders of neuromuscular transmission (DNMT) other than myasthenia gravis (MG) (Table 26-1). The neuromuscular junction (NMJ) is a physiologically complex structure. Its ability to function optimally requires the integration of a large number of proteins including ion channels that are correctly configured and distributed. As a result of numerous potential sites of vulnerability, DNMTs may occur as a consequence of multiple, albeit infrequent disorders. Autoimmune, genetic, or toxic mechanisms may disrupt the ultrastructure or physiology of the NMJ, thus interfering with effective NMT.
TABLE 26-1. DISORDERS OF NEUROMUSCULAR TRANSMISSION OTHER THAN AUTOIMMUNE MYASTHENIA GRAVIS
Presynaptic
Lambert–Eaton myasthenic syndrome (LEMS)
Botulism and botulinum toxin
Tick paralysis (Australian)
Congenital myasthenia gravis
Choline acetyltransferase deficiency (ChAT)
Paucity of synaptic vesicles
Congenital LEMS
Toxins
Envenomation
Elapid snake species (kraits, mambas, coral snakes)
Arthropods (black and brown widow spiders, scorpions)
Marine species (cone snails, sea snakes)
Drugs
Aminoglycosides and other antibiotics
Calcium channel blocking agents (minor)
Aminopyridines
Corticosteroids
Hemicholinium-3
Synaptic/basal lamina
Congenital myasthenic syndromes
Acetylcholine esterase deficiency (COLQ)
Laminin β2 (LAMB2)
Drugs and toxins
Reversible cholinesterase inhibitors—edrophonium, pyridostigmine, and neostigmine
Irreversible—organophosphates and carbamates
Postsynaptic
Drug-induced myasthenia gravis
Penicillamine
Alpha-interferon
Congenital myasthenic syndromes
Agrin deficiency (AGRN)
AChR (subunit) deficiency with or without kinetic defect [slow (opening) or fast (opening) channel syndromes]
εAChR subunit (CHRNE)
αAChR subunit (CHRNA1)
βAChR subunit (CHRNB1)
δAChR subunit (CHRND)
γAChR subunit (Escobar syndrome)
AChR—structural or organizational defects
Dok-7 deficiency (DOK7)
MuSK deficiency (MuSK)
Rapsyn deficiency (RAPSN)
Sodium channel myasthenia (SCN4A)
Plectin deficiency (PLEC)
Glutamine-fructose-6-phosphate transaminase 1 MG (GFPT1)
Dolichyl-phosphate N-acetylglucosamine-phosphotransferase 1 MG (DPAGT1)
Drugs and toxins
d-Tubocurarine, vecuronium, and other nondepolarizing blocking agents
Succinylcholine, decamethonium, and other depolarizing blocking agents
Tetracyclines, lincomycin, and other antibiotics
 LAMBERT–EATON MYASTHENIC SYNDROME
LAMBERT–EATON MYASTHENIC SYNDROMEThe Lambert–Eaton myasthenic syndrome (LEMS) can be conceptualized as an acquired presynaptic DNMT typically presenting with symptoms of proximal lower extremity weakness and fatigue. The first single case description of LEMS was provided by Anderson in 1953. Its eponym however is credited to Edward Lambert and Lee Eaton who along with Edward Rooke described in 1956 the electrophysiological as well as clinical characteristics of the disorder in six cases.1,2 LEMS is a rare disorder with an estimated incidence of 1 and prevalence of 3.5 per million people.1–10 LEMS is largely a disease of adults although rare pediatric cases have been described [both acquired autoimmune condition and as a congenital myasthenic syndrome (CMS)].11 LEMS is paraneoplastic disorder in many cases, but can be seen as a primary autoimmune disorder without an underlying cancer. The epidemiology between those cases of LEMS associated with a malignancy differs from those cases without an underlying neoplasm. Nonneoplastic LEMS appears to have two peaks, 35 and 60 years of age, occurring far frequently in women in the younger group. Paraneoplastic LEMS peaks in incidence at age 60 with two-thirds of this group being men.2
In adult LEMS, there is a strong support for a causative relationship for autoantibodies directed against voltage-gated calcium channels (VGCC) which are detectable in the majority of afflicted individuals, regardless of the presence of an underlying malignancy.12 Paraneoplastic LEMS comprises approximately two-thirds of cases. Small-cell carcinoma of the lung (SCLC) is the underlying malignancy in approximately 90% of these cases and 50–60% of all cases.13–15 Thymic tumors, non-SCLC lung cancers, lymphoproliferative disorders, and prostate cancer are the next most common associations.2,16–19 Pancreatic, breast and ovarian carcinomas, and Wilms’ tumor may represent chance associations.12,20,21 Conversely, it is estimated that between 0.5 and 4% of all SCLC patients will develop the clinical features of LEMS and approximately 8% will develop VGCC autoantibodies.22,23 The LEMS symptoms usually precede tumor recognition. Historically, this latency has been typically estimated to be less than 1 year, but may extend beyond 5 years in rare cases. These figures may however, represent a bias generated by surveillance techniques less sensitive than those currently available. In a more recent study of 100 LEMS patients followed for a minimum of 3 and a median of 8 years, 91% of those with malignancy were identified by 3 months of symptom onset and 96% by the end of the first year.15
The phenotypic and electrophysiological characteristics of paraneoplastic and nonparaneoplastic LEMS for all intents and purposes are indistinguishable in individual cases.24 Suspicion for an underlying malignancy should increase however, if the patient is over 50 years, has a history of tobacco use, progresses rapidly, or develops weight loss, erectile dysfunction, or bulbar symptoms within 3 months of symptom onset.2 A person with all six of these characteristics has a greater than 90% chance of harboring an underlying malignancy.2 Presumably, symptoms suggestive of other paraneoplastic disorders would increase the probability of an underlying malignancy as well. In general, paraneoplastic LEMS progresses more rapidly than its nonparaneoplastic counterpart.25 Other autoimmune diseases such as rheumatoid arthritis, thyroiditis, systemic lupus erythematosus, inflammatory bowel disease, primary biliary cirrhosis, vitiligo, celiac disease, or even MG occur in approximately 25% of cases and their presence favors a nonneoplastic form of the disease.14,26,27Although the phenotype of LEMS appears homogeneous, independent of underlying cause or serology, the natural history of LEMS however, may be influenced by serotype. On average, seronegative LEMS patients appear to have a shorter life expectancy. An explanatory hypothesis for this observation is a potential therapeutic role for autoantibodies (see below).
Lack of stamina and fatigue are the most common presenting symptoms of LEMS.1,3,5,7–10,28,29 In our experience, the described morbidity often seems disproportionate to the degree of objective weakness. It has been our perspective as well that this discordance may contribute to the suspicion of a psychogenic disorder, particularly in young women. Symptomatic weakness in LEMS typically relates to functions requiring proximal, particularly lower extremity muscles and is noted in approximately 80% of patients during the course of the illness. A third of patients complain of muscle aching and stiffness during or following physical exertion. Approximately 20% of patients note that their weakness and fatigue are exacerbated by hot weather or baths. Ocular and bulbar symptoms are not as common or as severe as seen in MG but do occur.15,30,31 They typically develop later in the disease and are rarely the sole or initial manifestation.2,31,32 Symptomatic diplopia without overt ophthalmoparesis is typically transient and mild when it occurs. Ptosis is more common in our experience and is estimated to occur in a third to a half of cases. In those with cranial muscle involvement, neck flexor, extensor, and facial muscles are among the most commonly affected. Head drop has been reported as a presenting manifestation.33 Some patients develop dysarthria or dysphagia. Ventilatory muscle involvement is rare although breathing issues related to smoking, chronic lung disease, and lung cancer are not. Ventilatory failure as a rare presenting manifestation of LEMS has been described.34–36
As a presynaptic DNMT, in contrast to MG, LEMS frequently affects both nicotinic and muscarinic function with resultant cholinergic dysautonomia. This may manifest as blurred vision (impaired accommodation), xerostomia, xerophthalmia, constipation, hypohydrosis, and/or impotence.12 Xerostomia may contribute to dysphagia and dysarthria. Complaints of numbness and paresthesias in the distal extremities occur less frequently. They are not related to disordered NMT but more likely result from any of the mechanisms relevant to cancer patients. Patients with LEMS may have coexistent paraneoplastic syndromes such as sensory neuronopathy, cerebellar ataxia, and/or limbic encephalitis with frequently coexistent Hu autoantibodies.37
One potential explanation for the apparent discordance between the severity of patient symptoms and their actual strength logically extrapolates from disease pathophysiology. As brief exercise can transiently enhance neuromuscular transmission in presynaptic disorders, the patient’s strength should be ideally assessed at the initiation of contraction, not several seconds later. This transient improvement in strength usually dissipates with sustained muscle contraction. It is most readily identified in hip and shoulder girdle muscles. Repetitive squatting may be one means to demonstrate this phenomenon. Exercise may also be used to evaluate ptosis, which may be temporarily improved with sustained voluntary lid elevation in a manner opposite to MG.38
There are other potential, noteworthy observations to be made in an LEMS patient. Typical of DNMTs, muscle bulk in LEMS tends to be preserved. In advanced stages of the disease however, muscle atrophy can be observed. Sluggish pupillary reaction in response to a light stimulus or diminished sweating in response to a provocative challenge are means by which to identify cholinergic dysautonomia. Deep tendon reflexes are typically diminished or absent in LEMS. Like assessments of strength, this phenomenon may be obscured if manual muscle testing is done prior to deep tendon reflex assessment.
A small number of patients will present with what seems to be an MG/LEMS overlap syndrome.39–41 Most MG cases overlapping with LEMS are based on the presence of acetylcholine receptor (AChR) antibodies in patients who otherwise appear to have LEMS on a clinical and electrophysiological basis. As many as 13% of patients with LEMS have AChR-binding autoantibodies.42 The AChR autoantibodies may be epiphenomenal rather than pathogenic in at least some LEMS patients.42,43 Nonetheless, rare patients may exhibit clinical features of both LEMS and MG.39,42,44,46
As a disorder characterized by a subacute limb-girdle weakness and fatigue, the primary differential diagnostic considerations for LEMS are limb-girdle myopathies and MG. MG may readily be confused with LEMS, particularly if signs and symptoms of oculobulbar weakness are readily evident with LEMS or if they are inapparent in MG. As a general rule, oculobulbar signs occur early and are prominent in MG and tend to be less frequent, less severe, and occur later in the disease course in LEMS.2,31 Exceptions do exist.15,30 Botulism has similar clinical features to LEMS including the pattern of weakness and the presence of cholinergic dysautonomia. Both its typically acute onset and the clinical context in which it occurs are the usual discriminating factors. LEMS can present in childhood and CMS may first manifest itself in adulthood. Consequently, CMS deserve consideration in any LEMS suspect as well. Motor neuron diseases that produce a limb-girdle pattern of weakness such as Kennedy disease are distinguished by their chronicity, and the presence of atrophy and fasciculations. The pattern and evolution of the weakness produced by multifocal motor neuropathy are, as the name implies, usually distinctive from LEMS. Motor predominant forms of CIDP may represent a very relevant diagnostic consideration in an LEMS suspect, particularly as both abolish deep tendon reflexes and affect the autonomic nervous system. The majority of these disorders can be readily distinguished from LEMS by electrodiagnostic (EDX) testing.
Diagnostic confirmation of LEMS is obtained by electrophysiological and/or autoantibody testing. As with all DNMTs, sensory conductions are normal unless paraneoplastic sensory neuropathy, chemotherapy-induced neuropathy, or other confounding disorders coexist. H reflexes may be absent upon initial attempts at elicitation but may appear following muscle contraction, an observation more likely to be of academic interest rather than pragmatic benefit.46
In LEMS and other presynaptic DNMTs, the baseline compound muscle action potential (CMAP) amplitudes are significantly reduced in contrast to typical MG. Reduced CMAP amplitudes are in many cases widespread in the distribution. In a large study of 73 patients with LEMS (42% with lung cancer), the CMAP amplitude was reduced in the abductor digiti quinti (ADQ) in 95%, abductor pollicis in 85%, extensor digitorum brevis in 80%, and in the trapezius in only 55% of cases.47 This diffuse pattern of reduced CMAP amplitudes in the presence of normal sensory nerve action potentials (SNAPs) may provide the initial suspicion for LEMS. The CMAP response to exercise and/or repetitive stimulation along with serological testing provide confirmation.5,6,8,48–55
On occasion, the EDX pattern in LEMS may be confused with MG. LEMS patients will typically demonstrate both an incremental pattern to fast repetitive stimulation (10–50 Hz) or brief exercise as well as a decremental response to slow (2–5 Hz) repetitive stimulation (Fig. 26-1A and B).47,53–57 If a patient with LEMS is seen early enough in their illness however, their baseline CMAP amplitudes may fall within population norms. In this situation, the incremental response characteristic of a presynaptic deficit may not be either evident or sought for. The demonstration of a decrement in response to slow (2–5 Hz) repetitive stimulation, characteristic of both LEMS and MG, without demonstration of an increment may lead to misdiagnosis. If LEMS is suspected but cannot be confirmed either electrodiagnostically or serologically, the evolution into the typical presynaptic DNMT pattern may be disclosed with repeated nerve conduction studies.58,59
Figure 26-1. Incremental response to brief (10 seconds) exercise in a patient with LEMS (A) (trace 1 ulnar CMAP at baseline, trace 2 ulnar CMAP immediately after 10 seconds of isometrically resisted finger abduction, trace 3 ulnar CMAP 1 minute later). Incremental response to 20-Hz fast repetitive stimulation (B).
This decremental pattern in LEMS is similar although not necessarily identical to that described in MG. In MG, following the initial decrement between the first and fifth stimuli, there is an increase in CMAP amplitude between the fifth and tenth stimuli. The CMAP amplitudes in LEMS plateau continue to decline between the fifth and tenth responses in LEMS.60,61 In LEMS, decrement in response to 3-Hz stimulation has been demonstrated in the ADQ in 98%, APB in 98%, EDB in 84%, and trapezius in 89% of cases.47
In a cooperative patient, incremental testing can be rapidly and easily performed. A supramaximal baseline CMAP is obtained after suitable hand warming. The muscle tested is then subjected to 10–15 seconds of isometric resistance. Immediately thereafter, a second, supramaximal electrical stimulus is applied. In normal individuals, there may be a mild increase in CMAP amplitude (<40%) associated with a shorter duration and similar area under the curve (pseudofacilitation). The actual basis of this phenomenon is poorly understood. It has been postulated to represent improved motor unit synchronization due to a disproportionate increase in the conduction velocity of the slowest conducting muscle fibers.62,63 In the majority of patients with LEMS, brief exercise will produce a 100–400% increase in CMAP amplitude. It is important to recognize that patients with end-stage LEMS may fail to mount this dramatic of an incremental response.64 In individuals who cannot cooperate with isometric exercise for whatever reason, “fast” repetitive stimulation of 20 Hz or higher represents a more uncomfortable means by which to demonstrate the characteristic increment.
Abnormal insertional and spontaneous activity on needle examination such as fibrillation potentials are typically absent in LEMS.5,6,28,50,51,58,65 Abnormalities of motor unit action potential (MUAP) morphology are apparent in weak muscles if carefully assessed. Neuromuscular blockade at individual myoneural junctions effectively reduces the number of single fiber action potentials contributing to the MUAP, resulting in shorter duration and lower amplitude waveforms. Consequently, the twitch tension of motor units decline and compensatory early (increased) recruitment results. In addition, the random blockade of single myofiber action potentials desynchronizes the MUAP leading to an increased percentage of polyphasic MUAPs. Motor unit variability (instability) is readily evident in LEMS if sought for but will be less apparent with the facilitation promoted by increased MUAP firing frequencies.
Predictably, both volitional and stimulated single fiber electromyography (SFEMG) evaluations of patients with LEMS yield abnormal results.8,48,50,51,53,56,66–77 Jitter values in patients with LEMS are significantly elevated and statistically exceed that observed in MG. In essentially all NMJs examined, irrespective of muscle chosen, markedly abnormal jitter values are evident. This is disparate from MG where a spectrum of jitter values from normal to highly abnormal exists within and between individual muscles. Unlike MG, the jitter in patients with LEMS is not dependent on the degree of weakness in a particular muscle. Blocking is often more prevalent and severe in LEMS in comparison to MG. Some of the highest percentages of blocked potentials occur in LEMS.
Frequency-dependent alterations in jitter and blocking are also observed in LEMS if sought for as implied in the previous statements regarding frequency-dependent MUAP variability (instability). Specifically, at low rates of voluntary firing, jitter and blocking can be quite impressive. Further increase in the duration of muscle activation or rate of individual MUAP firing will result in reduced jitter and blocking. These observations can be quantitated by using stimulated SFEMG.72,73,76,78,79 Stimulating an intramuscular neural branch and recording a single muscle fiber potential allow jitter measurement with quantifiable stimulus rates. One study of patients with LEMS demonstrated that the jitter decreased from a mean of 150 μs at a stimulation rate of 2 Hz to about 90 μs at a firing rate of 15 Hz.70,72 Similarly, when changing the stimulus frequency from 2 to 15 Hz, the percent of blockings decreased from 70% to fewer than 10%. Distinguishing MG from LEMS by contrasting stimulated SFEMG responses is theoretically possible but impractical in most circumstances. Muscle temperature affects EDX responses in LEMS as well.79–82 Decreasing muscle temperature results in an improvement in the CMAP amplitude at rest, reduces the magnitude of decrement at low rates of stimulation, and prolongs the duration of postactivation facilitation. Like MG, the yield of EDX testing will be increased not only by ensuring that limbs are adequately warm but also by discontinuing cholinesterase inhibitors 24 hours before testing.83
As dysautonomia in LEMS is commonplace, abnormal autonomic nervous system testing is anticipated. In a series of 30 patients with LEMS, autonomic testing revealed abnormalities of sudomotor function in 83% of patients, abnormal cardiovagal reflexes in 75%, decreased salivation in 44%, and abnormal adrenergic function in 37% of tested individuals in keeping with the predominantly cholinergic dysautonomia of the disease.10,12
Antibodies directed against the P/Q-type VGCC of the motor nerve terminals are believed to be pathogenic and are highly sensitive and specific for LEMS. They are detected by immunoprecipitation of VGCC from human brain, labeled with ω-conotoxin derived from the fish-eating Conus species of snails, incubated with serum from LEMS patients.2,84 These antibodies are detectable in the serum in 98% or more of paraneoplastic and >80% of nonparaneoplastic LEMS patients.12,24,42 Conversely, as previously mentioned, it is estimated that 4% of patients with SCLC will develop VGCC autoantibodies.25 In addition, antibodies directed against the N-type VGCCs, which are located on autonomic and peripheral nerves as well as cerebellar, cortical, and spinal neurons, are present in 74% of patients with paraneoplastic LEMS and 40% of nonparaneoplastic LEMS patients.12,42
LEMS patients may harbor other autoantibodies. The SOX1 antigen was originally found as a result of antiglial nuclear antibodies cross reacting with Bergmann glia of the Purkinje cell layer of rat cerebellum. The SOX1 antigen also plays a role in the development of airway epithelia and is found in SCLC. SOX autoantibodies are of potential clinical value as they are highly specific for LEMS and SCLC. They are found in two-thirds of LEMS patients with SCLC, 12% of SCLC without LEMS, and <5% of LEMS without cancer.85,86 Despite the cerebellar location of the antigen, there is no consistent association with paraneoplastic cerebellar degeneration or other paraneoplastic disorders. These antibodies are commercially available as antiglial nuclear antibodies as part of the Mayo Clinic paraneoplastic antibody panel. As previously mentioned, some patients with paraneoplastic LEMS will also harbor Hu autoantibodies associated with a sensory ganglionopathy, cerebellar degeneration, and/or limbic encephalopathy.12,37,42 Autoantibodies against the presynaptic protein synaptotagmin have also been described in LEMS patients but have no current clinical application.2
Serological testing for VGCC and AChR autoantibodies usually accurately discriminate between LEMS and MG when there is phenotypic overlap. Nonetheless, as previously mentioned, AChR-binding antibodies are found in as many as 13% of patients with LEMS.42 Conversely, as previously mentioned, autoantibodies against the P/Q VGCCs are found in <5% of patients who have the phenotypic and electrophysiological characteristics of MG.42,43 The concurrence of both antibodies is thought to be epiphenomenal in most cases.42
Edrophonium testing in LEMS produces variable results and is not normally employed.
Surveillance for a potential underlying neoplasm should be undertaken to some extent in any patient with LEMS. There is published guidance as to how extensive the initial evaluation should be, and how frequently it should be repeated if the initial evaluation is negative.15 Although paraneoplastic LEMS is largely a disorder of older people who have smoked, we are of the opinion that every LEMS patient should have a careful physical examination, and some form of imaging. Chest x-ray is considered an insufficiently sensitive imaging procedure in this context.15 In a younger individual without a smoking history, we lobby for a total body positron emission tomographic scan (FDG-PET) while remaining aware that problems with both availability and reimbursement may be encountered. FDG-PET is likely the most sensitive means by which to detect an underlying malignancy.15 In an older patient with a smoking history, CT of the thorax will detect the majority of tumors that are initially detectable.15 If CT of the thorax is normal, FDG-PET is also suggested.15 Regarding subsequent surveillance in individuals in whom no tumor is initially detected, evaluation has been suggested at 6-month intervals for 2 years.15 Our personal preference is to apply this strategy only to those individuals with a smoking history. As contemporary data indicates that a very high percentage of individuals with initially occult neoplasms will be detected within the first year of LEMS onset, we would limit rescreening to 1 year. Once again, we prefer FDG-PET rather than CT in consideration with radiation exposure as well as the probable greater sensitivity of the former for malignancy detection outside of the central nervous system (CNS).
One reason to perform EDX in patients with limb-girdle patterns of weakness is to distinguish LEMS from myopathy and avoid muscle biopsy in these patients. If performed, muscle biopsy reveals only nonspecific type II fiber atrophy.8 On quantitative electron microscopic analysis, nerve terminals appear normal in both their size and the number of synaptic vesicles they contain.87 Similarly, the postsynaptic membrane is intact but with an increase in the postsynaptic fold area and number of secondary synaptic clefts, presumably as a compensatory mechanism in response to reduced quantal release. The total number and activation properties of individual AChRs appear normal. Freeze-fracture analysis of the presynaptic membrane demonstrates a marked decrease in the number of intramembranous proteinaceous particles, which are assumed to be P/Q VGCC. These presumptive channels are disorganized and aggregated in clumps.88–90
In summary, the preponderance of evidence suggests that LEMS is a disorder of impaired presynaptic ACh release resulting from autoantibody mediated–VGCC dysfunction. The mechanism appears to be downregulation of channels and endocytosis rather than mechanical blockade.27 As a consequence of this autoimmune assault, reduced presynaptic calcium ion concentrations occur in response to a motor nerve action potential.12,42 Quantal ACh content, that is, the number of active vesicles released in response to a nerve action potential is reduced and neuromuscular transmission compromised.12,14,27,90,92
Presynaptic function includes concentrating and storing ACh within vesicles, facilitating their movement to active release zones where they dock and fuse, thus setting the stage for ACh release into the synaptic cleft in response to a motor nerve action potential. The migration, docking, and fusion with the presynaptic plasma membrane and subsequent exocytosis into the synaptic cleft are all dependent on the complex interaction of an extensive number of proteins. Some of the more notable constituents of this complex neuromuscular transmission process include synaptobrevin and synaptotagmin (associated with the synaptic vesicles), NSF (N-ethylamide sensitive ATPase) and α-SNAP (both found in the cytosol), syntaxin and SNAP-25 (synaptic vesicle–associated 25-kDa protein), and membrane-bound VGCCs.93 Synaptobrevin, syntaxin, and SNAP-25 are collectively known as SNARE (SNAP receptor) proteins. The known specificity of tetanus and botulism toxins for SNARE proteins and the impaired ACh release that results from their exposure to the NMJ illustrate the essential function of SNARE proteins in presynaptic vesicular exocytosis.
In normal individuals, a motor nerve action potential transiently opens presynaptic VGCCs resulting in an increased motor nerve terminal intracellular calcium concentration. The effect does not achieve the magnitude that it potentially could as the duration of the nerve action potential of <1.0 ms undershoots the activation time constant of the VGCC of 1.3 ms.91 The intracellular calcium concentration peaks by 200 μs and persists for approximately 800 μs following the nerve action potential, numbers critical to the understanding of repetitive stimulation responses in DNMT.
An intracellular calcium concentration of 200–300 μM is achieved under normal circumstances. It is estimated that 60 VGCCs need to open to allow the ingress of the approximately 13,000 calcium ions to promote required for exocytosis of a solitary vesicle. In mammalian systems, the response to a motor nerve action potential is a quantal content of 50–300 whereas in LEMS, the mean is roughly 8 (3.3–15).91 Similar effects on quantal content are achieved by reducing the calcium concentration or increasing the magnesium concentration in the extracellular fluid bathing the nerve terminal.93
The facilitatory effect of intracellular calcium on ACh release initiates with its binding to synaptotagmin, subsequently with syntaxin and SNAP-25, and eventually with synaptobrevin to complete the vesicular, exocytotic cascade. Calcium is believed to be essential in cleaving the bonds holding ACh vesicles to the intraneural cytoskeletal framework which is largely composed of actin and microtubules, thus hindering the vesicle’s ability to fuse or dock with recognition proteins at the active zones.93,94
There are six types of VGCCs in mammalian systems (L, N, P, Q, R, T) distinguished by their pharmacological and biophysical properties. The predominant channel in mammalian NMJs is the P/Q channel. The P/Q channel is composed of a pore-forming α1a subunit as well as α2δ, γ, and β4a subunits. The VGCCs are normally closed by neural repolarization promoted by the opening of presynaptic voltage–gated potassium channels. Failure of these channels to close promptly results in prolonged depolarization, and certain disorders of neuromuscular hyperactivity described in Chapter 10.
In normal individuals, these P/Q-type VGCCs are present on both the granule and Purkinje cells of the cerebellum and in presynaptic motor nerve terminals. They exist as well on small-cell carcinoma cells, providing a logical substrate for an autoimmune, paraneoplastic mechanism. In support of this, autoantibodies reacting with the P/Q-type VGCCs are found in 90% of patients with LEMS.
Autoantibody binding appears to specify the α1a subunit. As a consequence, there appears to be a downregulation in the number of calcium channels, resulting in a decrease in total current flow without a reduction of current flow in individual channels.96 Complement does not appear to be involved in this process. The nerve terminal maintains a grossly normal appearance without evidence of lytic destruction.88 A causative role for these autoantibodies is supported by the induction of all of the electrophysiological, morphological, and clinical manifestation of LEMS by passive transfer of IgG from patients with paraneoplastic and nonparaneoplastic LEMS to animals, or from mother to fetus.24,96,97 In addition, similar results utilizing the serum of seronegative patients supports an autoimmune mechanism in this group as well. The trigger for this autoimmune response is unknown. In patients with cancer, it is speculated that molecular mimicry results in the presynaptic motor nerve terminal becoming the innocent bystander in an immune response initially directed at the neoplasm.90,91,98 As LEMS occurs in only a small proportion of patients harboring SCLC, a genetic predisposition is hypothesized.99 Sixty-five percent of LEMS patients have the HLA haplotype HLA-B8-DR3, lending some support for this hypothesis although its prevalence appears greatest in the younger, nonneoplastic cohort.2
Identification and treatment of an underlying neoplasm, if present, is the foundation of LEMS treatment. If the tumor can be successfully treated, the morbidity of LEMS can be substantially reduced in a number of patients with concomitant improvement in their electrophysiological studies.48,98–102 If the patient remains symptomatic, with or without successful tumor treatment, adjuvant treatment with drugs that either enhance neuromuscular transmission or address autoimmunity can be utilized.2,9,29,103–105 Like myasthenia, the treatment regimen should be individually tailored in consideration with disease severity and the degree to which it affects a patient’s lifestyle, as well as numerous other contextual features including cost, availability, and relevant comorbidities. Like MG, avoidance of drugs with known neuromuscular blocking properties is recommended.
Pharmacological treatment for LEMS is largely based on clinical experience.103 As of this writing, only 3,4 diaminopyridine (3,4 DAP) and intravenous immunoglobulin (IVIG) have been systematically studied.107
Anticholinesterase medications can be used in a manner identical to patients with MG.6,48,49,57,106 Analogous to MG, pyridostigmine provides symptomatic treatment without addressing the root cause of the disease. Electrophysiologically, its application may result in a 50–100% increase in the baseline CMAP amplitude. In our experience, the drug is effective although the response is often modest, and probably insufficient by itself in controlling the morbidity of the disease.
Guanidine, a drug that is believed to prolong the motor nerve action potential and augment quantal content, was used historically in LEMS treatment.48,52,59,107,108 A small open-label trial demonstrated that both strength and CMAP amplitude are improved in all patients tested. Unfortunately, gastrointestinal side effects and the serious risk of renal failure and bone marrow suppression have made it largely a drug of historical interest.
The aminopyridines are a class of drugs that block voltage-dependent potassium channel conductance. 3,4 DAP is the drug of choice, when available, for the symptomatic treatment of LEMS.2,48,103,106,109–112 It may be used in addition to pyridostigmine. Four prospective, randomized placebo-controlled trial of 3,4 DAP in LEMS patients (paraneoplastic and nonparaneoplastic) have identified improvements in multiple outcome measures including strength, quantitative myasthenic scales, and CMAP amplitudes.106,110,113 Our practice is to start cautiously with a dose of 10 mg t.i.d. The medication is generally well tolerated, with a few patients experiencing perioral and acral paresthesias. It is recommended that doses should not exceed 100 mg/day, as higher doses may result in seizures although its blood–brain barrier penetration is less than other aminopyridines.2,110 Cardiac conduction defects, particularly prolonged QT intervals are an additional concern and warrant EKG screening both before and during treatment. Like others, we have had no adverse cardiac experiences.2 In our experience, although beneficial, 3,4 DAP rarely restores either strength or stamina fully. It is not FDA approved and its availability is limited in the United States. It is available on a compassionate use basis through Jacobus Pharmaceuticals. Clinical trials for LEMS patients with both 3,4 DAP and an alternative formulation amifampridine are available (www.clinicaltrials.gov). The drug may also be acquired through compounding pharmacists.
An alternative pharmacological approach is attempted immunomodulation with drug treatment, plasma exchange, or intravenous immunoglobulin.2,29,48,103,109,114–119 A single crossover trial of IVIg demonstrated a benefit in strength and a decline in VGCC autoantibody titres but failed to demonstrate a statistically significant improvement in CMAP amplitudes.116 Plasma exchange has been reported to have a clinical and electrical benefit in LEMS but has never been subjected to a prospective clinical trial.117–119 Improvements in CMAP amplitudes at rest, following exercise, or in response to high rates of repetitive stimulation following plasmapheresis may be seen.117–119 The peak response is observed by about 2 weeks after the treatment, with a diminution in effectiveness by the end of 3–4 weeks. There are no controlled trials in LEMS of any of the commonly used immunomodulating agents. The most common regimen used in LEMS is probably a combination of prednisone and azathioprine which has been shown to induce clinical remission in some patients.104,116 It may improve CMAP amplitudes as well as patient strength and stamina. Case reports have suggested a benefit from rituximab.120,121
One theoretical concern with LEMS or any other paraneoplastic neuromuscular disorder is the potential risk that suppressing the patient’s immune system will have adverse effects on tumor control. One hypothesis as to why LEMS often precedes tumor detection is that the same immune response that produces LEMS simultaneously limits tumor growth. Limited data suggests that immunomodulation can be utilized in LEMS without an adverse effect on tumor control.98
CONGENITAL MYASTHENIC SYNDROMESThe CMS may be conceptualized as inherited DNMT that typically, but not universally, become evident at birth, in infancy, or in childhood. Juvenile and adult-onset cases do occur and are characteristic of specific genotypes (Tables 26-2 and 26-3).122–128 It is not uncommon for early-onset cases to go undiagnosed until later life, often misdiagnosed as seronegative myasthenia, myopathy, or spinal muscular atrophy. CMS are rare or at least uncommonly recognized conditions that are caused by specific protein deficiencies whose normal functions are requisite to successful neuromuscular transmission at presynaptic, synaptic, and/or postsynaptic locations. The majority of CMS are related to proteins unique to the NMJ resulting in a purely neuromuscular syndrome. A few of the CMS are related to proteins affecting other organ systems including cardiac and smooth muscle, skin, and kidney.128,129 Rarely, there may be CNS involvement with microcephaly, seizures ocular, and auditory abnormalities.128 Connective tissue involvement with skeletal deformities or contractures occur in some cases most notably in rapsyn deficiency, AChR γ deficiency (Escobar syndrome) and some of the more recently described, later-onset, limb-girdle syndromes associated with abnormal glycosylation.125,130–138
TABLE 26-2. CONGENITAL MYASTHENIC SYNDROMES
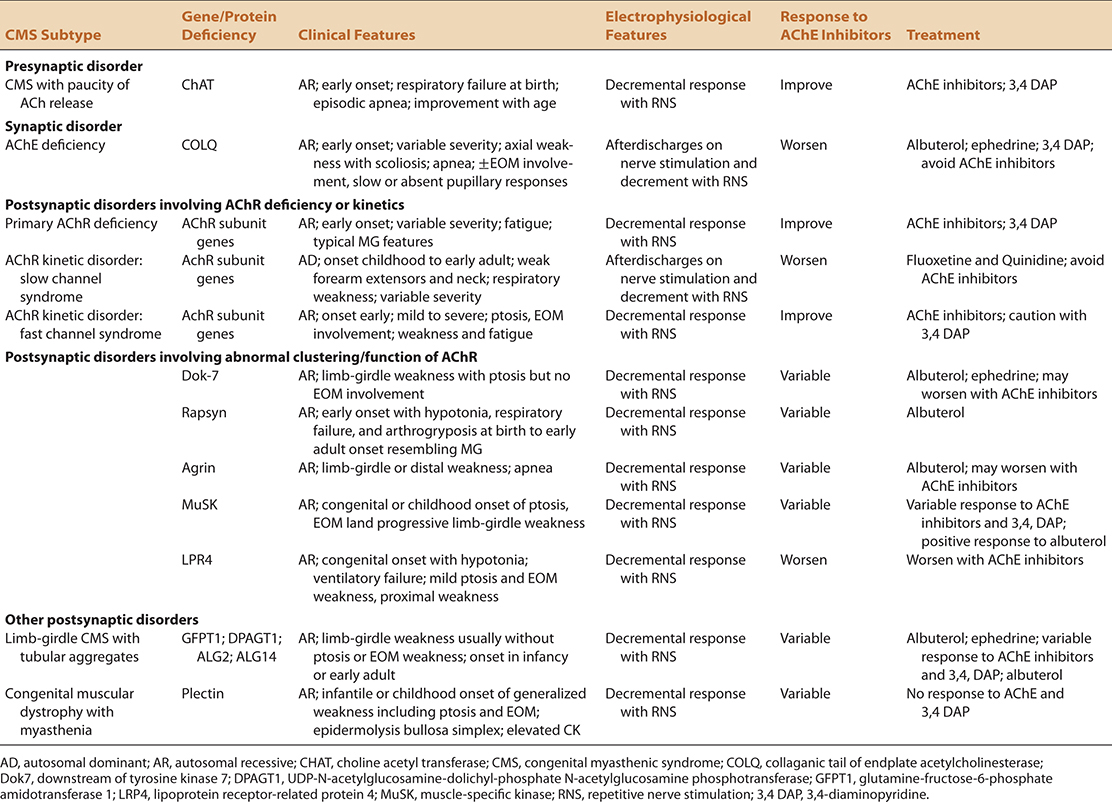
TABLE 26-3. DIAGNOSTIC CLUES USED TO IDENTIFY AND CLASSIFY CONGENITAL MYASTHENIC SYNDROMES
General clues (relative, not absolute)
• Early onset (inutero, neonatal, or infantile)
• Consanguinity, affected relatives suggesting an AR pedigree
• Seronegativity for autoimmune MG
• Refractoriness to cholinesterase inhibitors
• Failure to respond to immunomodulating agents
• Nonspecific light microscopic changes in muscle (if performed)
Epidemiology
• Ethnicity
German, Western/Central Europe
Dok-7, Rapsyn deficiencies
Brazil, Portugal, Spain, Tunisia, Algeria
AChR ε deficiency—CHRNE
Near-Eastern Jewish
Rapsyn deficiency
• Age of onset
Severe, lethal, akinetic syndrome
AChR γ deficiency
AChR α, β, δ subunit deficiencies
Rapsyn deficiency
Dok-7 deficiency
Typical onset—birth or infancy
ChAT deficiency
AChE deficiency
AChR subunit deficiencies
AChR deficiency with kinetic defect—fast channel syndrome
Rapsyn deficiency
LRP4
Variable including potential late-onset
Rapsyn deficiency (some cases)
Dok-7 deficiency
GFPT1 myasthenia (some cases)
DPAGT1 myasthenia
AChR (subunit) deficiency with kinetic defect—slow channel syndrome
MuSK deficiency
ALG2 and ALG14
• Symptoms worsened by cold
AChE deficiency
• Periodic exacerbations
Intercurrent infection
Rapsyn deficiency
ChAT deficiency
AChR (subunit) deficiency and fast channel syndrome
Pregnancy and menstruation
Dok-7
MuSK deficiency
Indeterminate reasons
DPAGT1 MG
• Dominant inheritance
AChR deficiency—kinetic disorder—slow channel
Electrophysiology
• Afterdischarges
AChE deficiency (not universal)
AChR deficiency—kinetic disorder—slow channel
Agrin deficiency
• CMAP incremental response
Congenital LEMS
• Decremental response to repetitive stimulation at 2–5 Hz
AChR (subunit) deficiency
Dok-7
ChAT deficiency (some cases)
AChE deficiency
Congenital LEMS
DPAGT1 CMS
GFPT1 CMS
ALG2/ALG14 CMS
LRP4 CMS
• CMAP decremental response to variable rates
ChAT deficiency (10 Hz RS for 5 min)
Muscle histology
• End plate myopathy
AChR deficiency—kinetic disorder—slow channel
AChE deficiency
• Tubular aggregates
GFPT1 CMS
DPAGT1 CMS
ALG2/ALG14 CMS
Response to treatment
• Refractory to or exacerbated by cholinesterase inhibitors
AChE deficiency (COLQ) (relative contraindication)
Laminin β2 myasthenia (relative contraindication)
AChR (subunit)—kinetic disorder—slow channel syndrome
Dok-7 deficiency
Agrin deficiency
Rapsyn deficiency
Plectin
LRP4 CMS
• Responsive to cholinesterase inhibitors
Laminin β2 myasthenia
GFPT1 myasthenia
DPAGT1 myasthenia
ChAT deficiency (variable)
AChR (subunit) deficiency or fast channel syndrome (partial)
CMS with a paucity of synaptic vesicles
MuSK deficiency (partial)
Congenital LEMS
• Refractory to 3,4 DAP
AChE deficiency
Congenital LEMS
Agrin defect
AChR deficiency—kinetic disorder—slow channel
AChR deficiency—kinetic disorder—fast channel syndrome (relatively contraindicated)
Dok-7 deficiency
• Responsive to 3,4 DAP
ChAT deficiency (variable)
AChR (subunit) deficiency
Rapsyn deficiency
Agrin deficiency
MuSK deficiency (limited)
Congenital LEMS
GFPT1 myasthenia
• Responsive to sympathomimetic amines (albuterol, ephedrine, salbutamol)
AChR deficiency—kinetic disorder—slow channel
AChE deficiency (some cases)
Agrin deficiency
MuSK deficiency
Dok-7 deficiency (some cases)
Laminin β2 myasthenia
DPAGT1 MG
• Responsive to open channel blockers (quinidine, quinine, fluoxetine)
Slow channel syndrome
• Responsive to guanidine
Congenital LEMS
Phenotype
• Pattern of weakness
Typical oculobulbar pattern
CMS with a paucity of synaptic vesicles
ACh receptor deficiency
MuSK deficiency
Ophthalmoparesis limited or absent
AChR deficiency with kinetic defect—slow channel syndrome
Dok-7 myasthenia
GFPT1 deficiency
Rapsyn deficiency
ChAT deficiency
Congenital LEMS
Agrin deficiency
ALG2/ALG14 CMS
Significant involvement of limb and axial muscles
ACh receptor deficiency
Dok-7 deficiency
GFPT1 MG
DPAGT1 MG
Rapsyn (some cases)
Agrin deficiency (some cases)
MuSK deficiency
ALG2/ALG14
Preferential involvement of distal muscles or distinctive muscle groups
Rapsyn (late-onset) (foot drop)
AChR deficiency—kinetic disorder—slow channel (cervical muscles, wrist/finger drop)
Agrin deficiency
GFPT1 myasthenia (scapular winging, foot drop, wrist/finger drop)
DPAGT1 MG (foot drop, wrist/finger drop)
Stridor/vocal cord paralysis
Dok-7 deficiency
Infantile hypotonia
ChAT deficiency
AChR deficiency
LRP4
Episodic apnea
ChAT deficiency
Na—channel myasthenia
Rapsyn deficiency
AChE deficiency
Dok-7 deficiency
MuSK deficiency
GFPT1
• Contractures or dysmorphic features
Rapsyn deficiency (facial deformities)
ACh receptor deficiency (particularly the gamma and fetal γ subunit)
GFPT1 myasthenia
ALG2/ALG14 CMS
• Delayed pupillary light reflex
Acetylcholinesterase deficiency
Laminin β2 myasthenia
• Systemic features
Nephrotic syndrome, ocular abnormalities Laminin β2 myasthenia
Epidermolysis bullosa simplex Plectin deficiency
The prevalence of genetically identifiable CMS is estimated at 3.8 × 106.131 Currently there are at least 18 recognized CMS and 17 recognized genotypes that are characteristically categorized by the primary anatomic region of the NMJ that is adversely affected and the identity of the mutated protein (Table 26-1).139 Of these genotypes, the most commonly occurring are postsynaptic with deficiencies of the ε subunit of the AChR, downstream of tyrosine kinase (Dok-7), and rapsyn deficiency constituting up to 75% of identifiable cases.126
CMS are phenotypically heterogeneous, even within the limited manifestations of the neonate and child. This heterogeneity exists both within and between different CMS genotypes.122,128,130,140 It is attractive to suggest that the more consistent phenotype of autoimmune MG and LEMS is a consequence of what is essentially a singular target in both conditions, the AChR and VGCC, respectively. Conversely, the CMS result from the disordered structure of nerve terminals, individual AChRs, abnormal AChR distribution, or abnormal NMT physiology at either the presynaptic, synaptic, or postsynaptic level.
There are phenotypic manifestations of CMS that serve both to suggest not only a CMS, but also in some cases implicating one or more specific CMS genotypes. These features are outlined in Tables 26-2 and 26-3. As is the case in all DNMT, the CMS phenotype is typically dominated by symptoms attributable to skeletal muscle weakness. Like autoimmune MG, oculobulbar muscles are commonly involved but sparing or limited involvement of oculobulbar musculature may occur. This pattern has been most commonly associated with choline acetyltransferase (ChAT), acetylcholinesterase (AChE, COLQ) rapsyn, agrin, Dok-7, glutamine-fructose-6-phosphate transaminase 1 (GFPT1), dolichyl-phosphate N-acetylglucosamine-phosphotransferase 1 (DPAGT1), ALG2, and ALG deficiencies.125,128–130,132,133,135–138,140,141
CMS may become evident in utero with maternal recognition of reduced fetal movement. This phenotype is most commonly associated with AChR γ subunit gene mutations and to a lesser extent with mutations of the α, β, and δ subunits, Dok-7, and rapsyn genes.125 More commonly, CMS present at birth or at infancy as a “floppy infant” with neonatal hypotonia in combination with a poor suck and weak cry, or with apneic episodes. Stridor, choking spells, and/or ventilatory difficulties are less-specific manifestations that are not uncommon particularly in this period. Contractures are another potential neonatal manifestation and have been reported in rapsyn deficiency (associated with facial deformities), ACh receptor deficiency (particularly the γ and fetal γ subunit), GFPT1, ALG2, and ALG14 deficiencies.130–133
When present, ptosis, is typically but not universally symmetric and diurnally variable. Ptosis and opthalmoparesis when present, are helpful clues in distinguishing CMS from other causes of weakness in all age groups (Fig. 26-2).123 In some patients, particularly in later onset cases, limb-girdle weakness may predominate with relative sparing of oculobulbar musculature. Historically, this was referred to as limb-girdle MG.142–145 This nomenclature has been supplanted by classification based on genotype as mutations correlating with this phenotype continue to be uncovered. Currently Dok-7, GFPT1, DPAGT1, ALG2, and ALG14 deficiencies are the most frequent causes of this syndrome.131–138,146
Figure 26-2. Ptosis, compensatory frontalis contraction, and partial ophthalmoparesis in a 15-year-old female with epsilon subunit deficiency. (Used with permission from Prof. Feza Deymeer, University of Istanbul, Istanbul, Turkey.)
Prominent scapular winging may be seen in some subtypes. CMS may affect not only proximal upper and lower extremity muscles but also can be associated with foot and finger drop. The latter may affect some digits more than others and suggest an increased likelihood of specific CMS genotypes in the appropriate context (Tables 26-2 and 26-3; Figs. 26-3 and 26-4). Despite NMJ localization, muscle atrophy is not rare in older individuals. In older individuals, limited stamina and prominent fatigue may be the predominant morbidity.
Figure 26-3. Weakness of wrist and finger extension in slow channel syndrome. (Used with permission from Prof. Feza Deymeer, University of Istanbul, Istanbul, Turkey.)
Figure 26-4. Weakness of finger extension in adult female with neonatal dysphagia and apneic episodes attributed to nonspecific myopathy. Became ventilator dependent following scoliosis surgery at age 11, and walked unassisted until age 15. Developed unilateral ptosis in adulthood. Diagnosed with CMS secondary to agrin deficiency at age 43.
Both diurnal variation and periodic exacerbation of CMS may occur, the latter commonly resulting from fever or intercurrent illness, pregnancy, menstruation, or even stress.125,126,128 Involvement of other end organs with certain CMS genotypes may produce recognizable syndromes providing an additional source of diagnostic information. Cardiac and smooth muscles are however, uncommonly involved in the majority of syndromes (Tables 26-2 and 26-3).147 We are unaware of significant dysautonomia occurring in these disorders.
The natural history of CMS is quite variable. Delayed motor milestones are the norm in those affected in infancy. Some individuals have a seemingly static course whereas insidious disease progression is anticipated in two of the syndromes. Only one of the currently recognized syndromes (slow channel syndrome) is typically inherited in a dominant fashion, the remaining disorders being autosomal recessive.148,149 Recognition of recessive inheritance pattern or parenteral consanguinity are supportive diagnostic clues for CMS although in small or fragmented families, cases may appear to be sporadic.
Because of its infrequency, CMS may not be considered in the differential diagnosis of a weak patient. Predictably this pitfall is more likely to be encountered in older individuals, particularly in those with a predominantly limb-girdle patterns of weakness. Even in an older individual with a typical MG phenotype, CMS should be given at least brief consideration in any patient with suspected seronegative myasthenia, myopathy of indeterminate cause, or spinal muscular atrophy.150
The following summarizes the notable clinical features of specific CMS. Associated genes are listed in parentheses after each disorder heading (Tables 26-2 and 26-3).
This disorder characteristically manifests as a distinctive phenotype of episodic bulbar weakness with a weak cry and poor sucking capability, and ventilatory distress with potential apnea. Ptosis is common, ophthalmoparesis is rare. The natural history is variable. Apneic episodes may begin in the neonatal period, infancy, or childhood and may be lethal. They may resolve or recur episodically even into adult life. Exacerbations may be triggered by intercurrent infection or other stress.
The single reported case of this phenotype described ptosis, ophthalmoparesis, facial weakness, and generalized fatigable weakness of the extremities that began in infancy. The molecular basis of this disorder is unknown.
Rare cases of this disorder have been described, predominantly recognized by the characteristic presynaptic EDX pattern described in the diagnostic section. The two described cases involved a severely affected neonate with hypotonia, bulbar weakness, and ventilator dependency and a young child with less-severe manifestations including delayed motor milestones without eye movement or other distinctive abnormalities.
This the most common cause of synaptic CMS, inherited in a recessive fashion. The natural history is again variable ranging from a typical neonatal disorder with severe morbidity to later-onset, more indolent cases. Early-onset cases tend to be dominated by hypotonia and delayed motor milestones, ventilatory and bulbar difficulties, associated with ptosis and ophthalmoparesis in some but not all cases. Slow pupillary responses to light are reported as a distinctive although not necessarily unique feature of this genotype. In later life, limb weakness, muscle atrophy, and skeletal deformities particularly involving the spine may become apparent. Survival into adulthood is the norm.
This is a rare, severe form of CMS. As the β2 chain of laminin is found in other tissues, mutations of the LAMB2 gene may result in Pierson syndrome as well as CMS with associated congenital nephrotic syndrome and ocular defects. The described phenotype includes delayed motor milestones with facial and limb-girdle weakness, with or without ptosis and ophthalmoparesis, pupillary and macular abnormalities, and the potential for ventilatory muscle weakness.
Despite the presynaptic site of its synthesis, the major role of agrin is to initiate AChR clustering by binding to lipoprotein receptor-related protein 4 (LRP4) resulting in phosphorylation of muscle-specific kinase (MuSK). Activated MuSK interacts in turn with Dok-7 and rapsyn to promote AChR aggregation. Agrin CMS is a rarely reported, with a mild phenotype characterized by ptosis, delayed motor milestones and mild proximal weakness. We have seen prominent wrist and finger drop when the diagnosis was delayed until adulthood (Fig. 26-5). Ophthalmoparesis is an inconsistent feature. Facial and ventilatory muscle weakness occur in some cases.
Figure 26-5. Decremental response to repetitive stimulation from extensor indicis proprius leading to diagnosis of CMS.
Subunit mutations are the most common forms of the postsynaptic CMS. The ACh channel typically consists of two α subunits, one β, one δ, and one ε, the latter which normally supplants the fetal γ subunit late in gestation. Adult subunits are encoded by the individual genes listed above. The ε subunit gene (CHRNE) is the most common subunit deficiency and the most common CMS in most series.126 This mutation tends to be more benign than other CMS as the ε subunit deficiency may be buffered to some extent by persistent fetal γ subunit function. This compensatory mechanism is not an option for other subunit mutations. Subunit mutations may adversely affect neuromuscular transmission by at least two mechanisms which may occur individually or concurrently. They may either reduce subunit expression and consequently receptor function and/or alter ACh channel kinetic properties. Kinetic alterations may produce either a slow channel (i.e., slow to close resulting in increased ionic passage) or fast channel (i.e., fast to close resulting in truncation of normal ionic passage from extracellular to intracellular compartments) syndrome. As previously implied, impaired NMT may result from either subunit deficiency and/or abnormal kinetic function and are not mutually exclusive.
The phenotype of AChR subunit mutations is again variable although more likely static than progressive. As described, mutations in the AChR ε subunit gene with reduced subunit expression typically correlates with a mild phenotype. Affected patients tend to have a nonprogressive phenotype typically manifesting as feeding problems and ptosis at birth or in infancy. Ophthalmoparesis is common although may not be present at birth (Fig. 26-2). Limb weakness occurs but ventilatory muscle involvement is rare. In contrast, non-ε AChR subunit gene mutations typically correlate with a more severe phenotype with ventilatory crises precipitated by choking and consequential shortening of life expectancy.
With a kinetic defect producing a slow channel syndrome, the clinical course is typically indolent, often sparing cranial musculature, and frequently affecting cervical muscles as well as wrist/finger extensors (Figs. 26-3 and 26-4). Spinal deformities may develop later in life. The fast channel syndrome tends to arise from the mutations in the α, δ, and ε subunit genes.166 Neonatal onset is the norm with a severe phenotype incorporating ptosis and ophthalmoparesis, bulbar and ventilatory weakness.
The Escobar syndrome results from mutations in the fetal γ subunit. As the contributions of the fetal subunit are largely dissipated by 33 weeks of gestation, neonates born with γ subunit mutations are typically born with arthrogryposis and ventilatory difficulties and do not develop a typical CMS phenotype.
This disorder most frequently presents with delayed onset in childhood but may present as late as the third decade. Although infantile onset may occur, normal motor milestones are commonly achieved before deterioration begins. Disease severity is variable, ranging from the mildly to severely symptomatic individuals. A progressive course is the most common. The typical phenotype is a limb-girdle pattern of weakness with the development of ambulatory difficulties during childhood. Ptosis may occur in later life. Ophthalmoparesis is infrequent and typically mild. Facial weakness is common. Significant bulbar symptoms including vocal cord paralysis, stridor, and poor feeding may occur in infancy. Severe disability including ventilatory failure may occur by the third decade in some cases. Worsening during pregnancy has been described.
The few reported cases of this disorder describe onset variability ranging from the neonatal period to later life. There is variability in disease severity as well. Ptosis, partial ophthalmoparesis, and mild facial weakness are commonplace. Weakness affecting proximal limbs, particularly shoulder abductors and ventilatory muscles occur in some but not all individuals.
This is one of the more common CMS, constituting approximately 15–20% of cases. The phenotype including age of onset is variable. Most cases present at birth or at infancy although cases presenting as late as the 20s occur. Neonatal arthrogryposis is common and prognathism and high-arched palate may occur. Crises may be precipitated by intercurrent infection. Ptosis, facial, jaw, and neck weakness are common whereas ophthalmoparesis is rare. Limb weakness is more common in later-onset cases. Although often proximal and symmetric, foot drop in late-onset cases is well recognized. If children survive the neonatal period, improvement with aging is not uncommon.
Predictably, a CMS patient with an LPR4 gene mutation has been recently described. The phenotype included hypotonia at birth with ventilatory and feeding difficulties. Motor milestones were delayed. As the child aged, prominent fatigue with proximal greater than distal extremity weakness became evident along with mild ptosis and ophthalmoparesis. A decremental response to repetitive stimulation was identified that responded favorably to edrophonium. Treatment with pyridostigmine however, aggravated weakness.
This disorder has been described in a single individual with recurrent apneic episodes and learning difficulties, perhaps related to hypoxic-ischemic injury.
Plectin is a protein that links different cytoskeletal elements to target organelles in different body tissues. It provides crucial support for the junctional folds of the NMJ. As a result, the phenotype includes the potential for multiorgan involvement including skin [epidermolysis bullosa simplex (EBS)], skeletal muscle (muscular dystrophy), smooth muscle (esophageal atresia), and cardiac muscle (cardiomyopathy). The phenotype typically begins in infancy as EBS and evolves into a disorder producing ptosis and ophthalmoparesis, dysphagia, facial and limb weakness associated with a decremental response to slow repetitive stimulation. Modest CK elevations may occur.
Like many of the CMS, this genotype has phenotypic heterogeneity. Onset of reported cases range from in utero recognition to 19 years of age. The course is typically slowly progressive. Neonatal cases may be arthrogrypotic, with poor bulbar function and apneic episodes. The most common phenotype is a later-onset syndrome dominated by a proximal > distal pattern of weakness. The distal muscles most commonly involved include forearm extensors, intrinsic hand muscles, and the anterior compartment of the leg. Serum CK values are elevated in 50% of cases.
Like GFPT1, DPAGT1 is an enzyme thought to be necessary for the glycosylation of the nicotinic AChR and integral to the assembly and insertion of the channel into the postsynaptic membrane. It shares other features with GFPT1 CMS including a propensity to begin beyond the neonatal period and to produce a limb-girdle pattern of weakness. It does not seem to significantly affect life expectancy and individuals in their sixth decade have been reported. Other features common to GFPT1 and DPAGT1 CMS are tubular aggregates on muscle biopsy, decremental responses to slow repetitive stimulation and responsiveness to cholinesterase inhibitors, and 3,4 DAP. Unlike GFPT1 CMS, CK values are reported to be normal. Consistent with DPAGT1’s role in glycosylation in locations other than the NMJ, mutations of this gene may also produce a severe, neonatal multisystem disorder referred to as type 2 that may include seizures, microcephaly, ventilatory distress, hypotonia, and behavioral abnormalities. The determination of phenotype appears to be based on mutation location.
Asparagine-related glycosylation plays a critical role in protein folding, transport, localization, and folding. Recessively inherited mutations in these two genes that contribute to this process have been recently reported to result in CMS that bear many similarities to DPAGT1 and GFPT1 CMS. Most affected individuals reported to date have had a limb-girdle phenotype affecting limb muscles preferentially, typically symmetrically with proximal predominance. Scapular winging may occur, contractures are commonplace and a high-arched palate has been described. Facial weakness may occur but extraocular muscle involvement is not a typical feature of the disease. Learning disability may occur. Onset age may range from infancy with hypotonia to adulthood although initial symptoms in the late first decade would seem to be the norm. Wheelchair dependency may or may not occur. A decremental response to slow repetitive stimulation and responsiveness to cholinesterase inhibitors are the norm. Tubular aggregates may be found on muscle biopsy. CK is usually normal but may be minimally elevated.
The differential diagnosis of CMS in the neonatal period is largely that of the floppy infant (Table 1-1). In the presence of ptosis and ophthalmoparesis, particular consideration should be given to transient neonatal autoimmune MG, certain congenital myopathies, mitochondrial myopathy, and myotonic muscular dystrophy (Table 1-8). Later in infancy, infantile botulism should be considered, particularly with an acute to subacute onset, and symptoms of cholinergic dysautonomia such as sluggish pupils and constipation. In childhood, autoimmune MG and rarely autoimmune LEMS require consideration. When the phenotype is dominated by a limb-girdle pattern of weakness, numerous myopathies and spinal muscular atrophy are the primary considerations. The latter may be suspected by the presence of fasciculations, tremor, or a neurogenic pattern with EDX, or if necessary, muscle biopsy.
Routine laboratory testing is of limited value in CMS. Serum CK levels are of little value in that they are characteristically normal or modestly elevated similar to other neuromuscular disorders with which CMS might be confused.124 CK testing may be helpful in distinguishing two similar phenotypes DPAGT1 and GFPT1 CMS as it is elevated in approximately half of the latter patients but is normal in all reported cases of the former to date.134 These two disorders as well as deficiencies in ALG2 and ALG14 may also be distinguished from all other CMS as they are the only CMS reported to demonstrate tubular aggregates with light microscopy and oxidative staining of a muscle biopsy specimen.132,133,138
The diagnosis of CMS, if clinically suspected, is further supported by the combination of abnormal EDX testing results and edrophonium testing when present, coupled with the absence of serological markers of autoimmune MG and LEMS. Edrophonium testing is positive in most CMS including the presynaptic disorders, AChR deficiency, and the fast-channel syndromes. End plate AChE deficiency, occasional cases of the slow-channel syndrome, and Dok-7 syndromes are notable exceptions.
At times, identification of motor unit variability, after discharges with routine motor conductions, or a decremental response to slow repetitive stimulation may lead to an unanticipated discovery of a CMS. None of these EDX findings are however, adequately sensitive or specific to allow for a definitive CMS diagnosis in many cases. Afterdischarges are typically identified in only three forms of CMS: AChE, agrin, and AChR receptor (associated with the slow-channel syndrome) deficiency. In all three, the afterdischarges represent prolonged postsynaptic depolarization. Afterdischarges may also occur with neuromyotonia, envenomation with K+ channel poisons, intoxication with organophosphate or other anticholinesterase agents, and certain muscle channelopathies.
As described below and summarized in Tables 26-2 and 26-3, not all CMS may be identified by abnormal EDX testing. Repetitive stimulation at various frequencies and stimulated SFEMG are the most frequently used techniques in the pediatric age group. Caution in interpretation is required however, as there is limited normative data in infancy.175 It would appear that in a full-term normal newborn, no decremental response to slow repetitive stimulation is demonstrable in spite of what may be an immature NMJ. A decrement in response to frequencies of 5 Hz or more has been reported in normal newborns so that techniques that utilize higher stimulation frequencies including stimulated SFEMG must be interpreted with caution.175
Decremental responses to repetitive stimulation have been described in all forms of CMS although certain forms (ChAT deficiency) require prolonged stimulation at higher rates (10 Hz) in order to provoke the decrement (Fig. 26-5).126,128,131,133,138,139 As with autoimmune MG, a decremental response in these syndromes is expected in any clinically weak muscle and in some but not necessarily all clinically unaffected muscles. Standard repetitive nerve stimulation (RNS) protocols may have to be modified in order to identify abnormal NMT in some syndromes. The decrement in AChE deficiency and the slow-channel syndrome typically becomes greater with faster rates of stimulation.126 In ChAT deficiency, 10-Hz RNS for 5 minutes may be required to identify a decremental response.125,127,152 A small increment in response to fast repetitive stimulation has been reported in ChAT deficiency as well.127 Like acquired, autoimmune LEMS, the Lambert–Eaton form of CMS is characterized by a low baseline CMAP and an incremental response of up to 2000% with 20–50-Hz repetitive stimulation.156
MUAPs that are polyphasic and smaller in both amplitude and duration may occur in CMS as a result of at least two mechanisms. Neuromuscular blockade at individual NMJs may reduce the number of single fiber action potentials that contribute to the normal MUAP size and configuration. In addition, in AChE deficiency and the slow-channel syndrome, an end plate myopathy may develop as a result of excessive end plate stimulation resulting in smaller MUAPs as well.127
A definitive diagnosis of CMS remains challenging in most cases. Historically, a CMS diagnosis was arrived at with the confluence of a characteristic phenotype in a young person with negative serological testing for autoimmune MG, EDX evidence of a DNMT, with or without a positive response to cholinesterase inhibitors and when present, a compatible family history. Confirmation was achievable only by a selected group of laboratories capable of ultrastructural analysis of NMJs and in vitro physiological testing of NMT.
Genetic testing now provides at least in principle, a more direct and efficient manner in which to not only confirm a CMS diagnosis but also to identify the genotype as well. To date, 18 CMS genotypes have been identified.139 Genetic testing in CMS however, is fraught with a number of limitations including availability and cost considerations.125 Identification of pathogenic mutations from benign polymorphisms is not always easy. Mutational analysis is also incapable of defining a kinetic defect in association with an identified subunit mutation. Kinetic defects, important in therapeutic decision making, are identified only with the use of in vitro microelectrode or single-channel patch clamp recordings.166 It is estimated that successful genotyping can occur in half to as many as 90% of CMS cases.126,127,176 If genotyping is an available option, test selection should be judicious. Considerations should be directed by the phenotypic and epidemiological clues provided in Tables 26-2 and 26-3 as well as by genotype frequency.
Light microscopic examination of muscle biopsy is of limited value in CMS but is often performed in consideration of the more likely possibility of myopathy, particularly in older individuals. Perhaps the most specific potential finding would be the presence of tubular aggregates. Although neither sensitive nor specific for CMS, they are a potential finding in limb-girdle phenotypes of CMS including GFPT1, DPAGT1, and ALG2/ALG14.124,131–133,135–138 Otherwise, light microscopic findings are of limited benefit. In the majority of cases, they neither distinguish CMS from other neuromuscular diseases nor do they aid in the definition of a specific CMS genotype.
A number of nonspecific, largely myopathic findings have been described in muscle biopsy specimens from CMS patients. Type 1 fiber predominance is perhaps the most common of these and has been described in rapsyn, Dok-7, GFPT1, and MuSK CMS.124,130,141,146,171 Other features described in other CMS including Dok-7, GFPT1, plectin, and MuSK include type 2 fiber atrophy/hypotrophy, isolated myofiber necrosis and regeneration, diminished oxidative stain uptake, ragged red fibers, increased frequency of internal nuclei, fiber splitting, and subsarcolemmal nuclear chains.124,140,141,146,171 Autophagic vacuoles that may stain with acid phosphatase and may contain glycogen have been described in DPAGT1 and GFPT1 CMS.131,132 Neurogenic features of small grouped atrophy and target formations have been described.131,132,146,156
Ultrastructural abnormalities of the NMJ occur in most CMS.125 They are not syndrome specific. Simplified junctional folds appear to be the most common abnormality in some but not all end plates in many of the disorders and are frequently associated with reduction in the number of AChRs.131,140 This finding has been reported in a number of syndromes including the slow-channel AChR kinetic disorder, and the AChE, LAMB2, rapsyn, plectin, MuSK, LRP4, GFPT1, and Dok-7 forms of CMS.125,130,131,139,140,171 The pattern of simplification of the junctional folds varies however, being focal in synaptic disorders and diffuse in many postsynaptic disorders such as AChR ε subunit or rapsyn deficiency.129 In the synaptic CMS, that is, LAMB2, agrin, and AChE deficiency, as well as in MuSK CMS, the ultrastructural findings may include reduction of the axon terminal size, partial encasement of the nerve endings by Schwann cells, widening of the primary synaptic cleft, and invasion of the synaptic cleft by the processes of Schwann cells in addition to focal simplification of postsynaptic folds.129 This observation of abnormal presynaptic morphology in a synaptic disorder reflects that the pathophysiology of individual CMS may extend beyond the primary site of involvement. In Dok-7 myasthenia, myeloid structures may populate the junctional cytoplasm. Nerve terminals ending as growth cones without AChR contact has been described in MuSK CMS.173
In AChE deficiency and the slow-channel syndrome, prolonged end plate current produces postsynaptic cationic overloading resulting in an end plate myopathy.129 The histological features of this are characterized by subsynaptic degenerative abnormalities, autophagic vacuoles, dilated sarcotubular elements, increased lipid droplets, and apoptosis of junctional nuclei occurring as a consequence of postsynaptic cationic overloading. In the presynaptic disorder, CMS with a paucity of synaptic vesicles, electron microscopy demonstrates the feature that defines the condition.127 Ultrastructural abnormalities do not occur routinely however, in all CMS. In agrin deficiency, congenital LEMS, and the fast-channel kinetic syndrome of AChR deficiency, the postsynaptic regions are reported to be ultrastructurally normal.152
The efficiency of NMT is dependent on numerous interrelated components. Optimal NMT requires adequate synthesis of ACh and its packaging and positioning of the resultant synaptic vesicles at the active zones of the presynaptic terminal. Equally important is adequate quantal content or the number of vesicles released as a result of a nerve action potential and the resultant amplitude and duration of the end plate current that the vesicular–end plate interaction produces. This is in turn dependent on synaptic metabolism of ACh that is neither inadequate nor excessive, as well as the normal positioning, clustering, positioning and kinetics of both the ACh and Na+ channels on the peaks and troughs of the end plate folds, respectively.
CMS result from single-gene mutations resulting in abnormal structure or function in one or more of these components of NMT. In each instance, there is a resulting alteration in end plate current, impaired generation of myofiber action potentials, and as a consequence, reduced strength and stamina of voluntary muscles. With the majority of genotypes, a reduction in end plate current occurs. In two of these disorders however, AChE deficiency and the slow-channel kinetic disorder of the AChR, the current is excessively prolonged.165 Although adverse pathophysiological events are typically characterized as occurring at a singular presynaptic, synaptic, or postsynaptic region, many of the CMS will have secondary consequences that affect additional NMJ loci.
In ChAT deficiency, impaired resynthesis of ACh compromises NMT by progressively depleting quantal content. This along with miniature end-plate potential (MEPP) and EPP amplitude are normal at rest but decline with repetitive stimulation at 10 Hz for 5 minutes with subsequent gradual recovery.152 CMS with a paucity of synaptic vesicles is associated with a reduced density of synaptic vesicles at the active zones. The probability of quantal release, that is, proportion of vesicles released/nerve action potential, is normal.125 Conversely, the LEMS variant of CMS is associated with reduced quantal content with a reduced probability of quantal release.125 Like its autoimmune counterpart, it is defined by the characteristic EDX pattern of a reduced CMAP amplitude at rest, a decremental response to 2–5 Hz repetitive stimulation and an incremental response to faster rates of repetitive stimulation or exercise. The exact mechanism by which this occurs is not fully understood.125 No mutations of VGCC-related proteins have been identified to date.127 A failure to respond to 3,4 DAP suggests that congenital LEMS is not a consequence of disordered calcium channels.152
With AChE deficiency, the most common of the synaptic disorders, the absence of AChE prolongs the lifetime of ACh in the synaptic space and as a consequence, the duration of the MEPP and EPP. The duration of the synaptic current outlasts the refractory period of the muscle fiber which overstimulates the postsynaptic region. Neuromuscular transmission is impaired by multiple mechanisms including loss of AChR from the degenerating junctional folds and desensitization from ACh overexposure. In addition, there are presynaptic effects with the small and often Schwann cell–encased nerve terminals associated with reduced quantal release. In addition, the excessive stimulation promotes cationic overloading resulting in an end plate myopathy.
In view of the integral role of laminin as a component of the basal lamina, and the critical role of the basal lamina in the creation and configuration of the motor end plates, LAMB2 deficiency is hypothesized to adversely affect the development of the complex end plate anatomy.127 LAMB2 deficiency also associates with abnormal nerve terminals that are both small and encased by Schwann cells. Widening of the synaptic space and junctional fold are additional consequences as is the demonstration of decreased MEPP amplitude.125,152 Agrin is bound to laminin on the synaptic basement membrane with the possibility that agrin deficiency (described below) has a synaptic as well as postsynaptic pathogenesis.151
The most common postsynaptic disorders involve mutations of AChR subunits, most commonly ε. Again, mutations may produce a deficiency and/or a kinetic disorder. With receptor deficiency, as the name implies, the AChRs at the NMJ are patchy in distribution and reduced in number with a proportionate reduction in end plate current.125,165 The pathophysiology of the AChR mutations resulting in kinetic disorders differs dependent on whether the problem is one of delayed (slow channel) or premature (fast channel) AChR closure subsequent to the generation of an initial muscle fiber action potential. The pathophysiology of the slow-channel syndrome is similar to AChE deficiency including depolarization block and the development of an end plate myopathy from excessive stimulation.125 The pathophysiology of the fast-channel syndrome includes reduced ACh affinity for the AChR, shortened duration of the EPP and as a consequence, diminished Na+ activation.165 AChR density on the postsynaptic fold is normal.177
Many of the CMS relate to mutations of genes that produce proteins integral to the proper placement and aggregation of AChRs on which optimal NMT is dependent. Agrin is secreted by the distal motor nerve terminal and binds to LRP4. The LRP4-agrin complex activates MuSK which in turn with Dok-7 stimulates rapsyn to concentrate and anchor AChR at the NMJ.93,151
CMS due to LPR4, MuSK, rapsyn, and Dok-7 can all be conceptualized as impairing NMT through impaired AChR clustering. In CMS due to rapsyn deficiency, AChR clustering is impaired, the number of AChRs per end plate reduced, and as a consequence, the amplitude of the MEPPs.125,130,178 The presumed mechanism of impaired NMT in MuSK CMS is the failure of normal MuSK to bind LPR4 and promote agrin-induced AChR clustering.125 In CMS related to Dok-7 mutations, there is loss of AChR from degenerating postsynaptic folds.141 Na+ channels are also reduced in number. Consequently, MEPP and predictably EPP amplitudes are reduced in most but not all patients.169 There are presynaptic effects as well including encasement of nerve terminals with Schwann cell processes resulting in reduced quantal content in some cases.125,169 DPAGT1, GFPT1, and ALG2/ALG14 play roles in AChR subunit glycosylation.131–133,138,179 All have been hypothesized to adversely affect NMT by impeding the normal assembly and transport of AChRs into the postsynaptic membrane resulting in simplified postsynaptic membranes with decreased end plate AChR density.127,138,180
Plectin is a versatile protein that links cytoskeletal proteins in different locations explaining its potential multisystem phenotype. One area which is concentrated is the NMJ providing at least an anatomical correlation with the CMS that may associate with plectin gene mutations. The mechanism of impaired NMT is uncertain but is associated with reduced MEPP amplitudes. Morphological changes in muscle may correlate with the limb-girdle dystrophic pattern that occurs in some patients.125 Na+ channel CMS is a rare disorder that understandably diminishes the end plate current as a result of SCN4 A mutations affecting Nav 1.4 channel function.125
Many of the CMS will respond to pharmacological treatment (Table 26-2). Unfortunately, few if any respond as dramatically as can often be achieved in patients with autoimmune MG. One benefit of accurate CMS genotyping is the ability to choose the appropriate treatment. Specific syndromes are predictably responsive or unresponsive to particular agents. In certain syndromes, for example AChE deficiency and the slow-channel syndrome associated with excessive end plate current, anticholinesterases have adverse effects and are contraindicated.
Cholinesterase inhibitors typically benefit most of the presynaptic CMS, and some of the postsynaptic disorders. Conversely, cholinesterase inhibitors are ineffective or contraindicated in others particularly those in which the end plate is overstimulated, for example, AChE deficiency and the slow-channel syndrome. The reader is referred to Tables 26-2 and 26-3 for the effect of AChE in different CMS. In children, the typical dose for pyridostigmine, which is available in syrup form, is 1 mg/kg given four to six times per day orally with the maximum dose being 7 mg/kg/day.127
Similarly, 3,4 DAP may be effective or ineffective. It has been reported to be helpful in ChAT, AChR subunit, rapsyn, agrin, and MuSK deficiencies as well as GFPT1 and DPAGT1 deficiency. Patients with AChE deficiency, both the slow- and fast-channel syndrome, congenital LEMS, and agrin deficiency do not typically respond. Patients with Dok-7 deficiency may benefit in some but not all cases. The recommended dose of 3,4 DAP is up to 1 mg/kg/day in divided doses.
Sympathomimetic amines may be effective in certain CMS, particularly those who are refractory to 3,4 DAP and cholinesterase inhibitors.180,181 Albuterol employed in divided doses of 4–18 mg/day is commonly utilized.180 These include the slow-channel syndrome, some cases of AChE, agrin, MuSK, and Dok-7 deficiency and DPAGT1 and LAMB2 CMS. The slow-channel syndrome may respond to open-channel blockers including quinidine, quinine, or fluoxetine.182
Like the majority of NM diseases, supportive treatments may be required. Parents with children with CMS associated with episodic ventilatory crises may be instructed how to deliver intramuscular neostigmine at a dose of 0.01–0.04 mg/kg.127 These households are ideally equipped with apnea and oxygen saturation monitors, and breathing aids such as Ambu bags along with the training necessary for their proper use.125 In addition, patient counseling to avoid drugs that might further impair neuromuscular transmission is advised (Table 25-3). If anesthesia is required for any purpose, neuromuscular blocking agents should be avoided. Gastrostomy tubes may be required in individual patients.
BOTULISMThe disease we know as botulism was named following an 18th-century outbreak caused by improper preparation of sausage (botolus—Latin). Botulism is a relatively unique disease in consideration with the multiple mechanisms by which it can be acquired and the clinical contexts in which these mechanisms operate. The disease may occur as either an infection or an intoxication. Either the spores (infancy) or preformed toxin (adults) may be inadvertently ingested. The latter often results as a consequence of improperly prepared foods or beverages. The organisms may incubate in wounds that facilitate growth under anaerobic conditions, particularly in the setting of parenteral drug abuse. In view of its potency, botulism represents a feared weapon in the arsenal of bioterrorists.183,184 Inadvertent toxic effects may also occur as the unintended, iatrogenic consequence of the toxin use as a therapeutic agent.
In an adult, botulism is most commonly acquired as a foodborne illness or through wounds. Foodborne botulism is a rare disease, with fewer than 35 cases reported annually in the United States. Infantile botulism is more prevalent, with 80–100 cases estimated to occur annually in the United States.185 In Canada, there were 205 foodborne cases reported in a 10-year span for an annual incidence of 0.03 × 105.186 The commercial preparation of food and the typically adverse conditions for spore formation within the adult GI tract are the primary reasons for this low incidence. Reported cases are often related to unusual regional or cultural food preparation or storage practices. Even in foodborne botulism, most reported cases are sporadic with outbreaks of more than 2–3 people being unusual.185
The morbidity of botulism is considerable, 30–67% of patients requiring intubation in different series.186,187 Mortality rates have dropped considerably from historical estimates and are estimated to be between 3 and 5% in this era of intensive care units and the availability of the heptavalent antitoxin.185,186 Although uncontrolled, one report described a statistically significant reduction in the length of hospital stay for patients receiving antitoxin.186 The mortality for infantile botulism is less. With appropriate intensive care and use of the human source antitoxin, the survival rate is estimated at nearly 100%.185 Although the effects of botulinum toxin are permanent once bound to peripheral nerve terminals, recovery occurs with adequate support, presumably on the basis of growth of new nerve terminals. Typically this recovery is measured in months and may take longer for autonomic than somatic functions. It may be dependent on serotype, type A botulinum toxin having the most protracted effect.188 Full recovery is estimated to occur in 95% of individuals if adequately supported.185 Of interest, three children born to women who developed botulism during pregnancy were unaffected by the disease.186 In addition, children born to women receiving therapeutic botulinum toxin injections have not been reported to develop adverse effects.
Botulism is commonly categorized by one of five different mechanisms of intoxication or infection. The clinical presentation of botulism is similar, regardless of the mechanism of inoculation.189–192 Foodborne botulism in adults is the classic form, first recognized in 1897, with symptoms typically occurring within 2–72 hours of ingestion of food contaminated with the preformed toxin. The initial symptoms are typically related to impaired GI motility including constipation, emesis, abdominal cramping, and/or diarrhea. Neurological impairment follows in short order. The severity of the illness is thought to be related largely to the amount of toxin ingestion. Signs and symptoms referable to motor functions of cranial nerves are virtually always the initial neurological manifestation. Signs and symptoms of dysautonomia, particularly of cholinergic function soon follow. For purposes of easy recall, the clinical manifestations have been referred to as the “dozen Ds.” They include dry mouth, diplopia, dilated pupils, droopy eyelids, droopy face, diminished gag reflex, dysphagia, dysarthria, dysphonia, difficulty lifting the head, descending paralysis, and dyspnea related to diaphragmatic paralysis.
Dysautonomic manifestations may include blurred vision from impaired accommodation, urinary retention, ileus, and postural hypotension. The latter is presumably related to impaired cholinergic release at vasomotor, preganglionic sympathetic neurons. Of potential symptoms, xerostomia, diplopia, and dysphagia are the most frequent and occur in over 90% of reported cases. Dyspnea and ventilatory muscle weakness occur in a large proportion of patients. The need for ventilator support has been reported to occur in 32–81% of patients.191,192 The duration of required mechanical ventilation is dependent on the severity of the illness and serotype of the infecting organism, with a mean of 58 days for type A and 26 days for type B botulism.191,192 Careful observation suggests that ventilatory muscle weakness precedes the recognition of limb and trunk weakness. The latter is common, typically occurring in a descending pattern affecting arms before legs, and typically although not always symmetric in distribution.190 Deep tendon reflexes may be normal or diminished initially, with progression to complete loss in severely affected individuals. The sensorium and sensory system are unaffected unless CO2 retention from ventilatory muscle weakness ensues.
Infantile botulism was first described in 1976. It is the most common form of botulism in the United States with an incidence that is approximately twice that of foodborne disease. Unlike foodborne disease, it can be conceptualized as infection, not an intoxication as the organisms colonize the vulnerable intestine of the infant. It typically affects children in the first 6 months of life and is strongly related to the use of honey which has been shown to harbor clostridial spores (particularly type B) in up to 25% of products.190 Only 20% of infantile cases are attributable to honey ingestion however.185 The clinical manifestations, in consideration of the child’s maturational age are understandably more protean than in adults. The severity of the phenotype is variable. Constipation is the usually the first and in some cases the only manifestation. Children typically develop a weak cry, a poor suck and swallowing capabilities. Excessive drooling accompanied by a weak cry is particularly worrisome. Ptosis and “smoothing” of facial expression may be noted. Hypotonia, particularly of the neck, occurs in the more severe cases. Tachycardia and urinary retention are additional dysautonomic manifestations.
Like infantile botulism, wound botulism represents an infection rather than an intoxication. It was first described in 1943 as a consequence of trauma or surgery presumably related to the anaerobic environment created by necrotic tissue. Ironically, in some cases it may have been related to the use of honey which has been applied to wounds to facilitate healing through its bactericidal and hygroscopic properties.190 Wound botulism is now however most commonly associated with recreational drug use, most commonly subcutaneous injection of black tar heroin but has been reported with subcutaneous injection of cocaine as well.185,193 This may occur as a result of abscess formation at injection sites which can be quite subtle and appear as no more than a furuncle or small area of cellulitis. As the toxin can penetrate mucous membranes as well disrupted skin, it has been associated with the inhalation of cocaine as well.190 The clinical manifestations of wound botulism are similar to foodborne botulism with the exception that gastrointestinal complaints are less common. Wound botulism is more likely to affect an individual as opposed to a group as might be anticipated in foodborne disease. The incubation period is longer in wound botulism, typically 4–14 days in comparison to hours for toxin or spore ingestion.
Hidden botulism was first described in 1977 and can be conceptualized as the adult variant of infantile botulism. The acidic milieu of the normal adult gastrointestinal tract is not normally conducive to proliferation of the Clostridium botulinum organism once introduced. Adults with abnormal gastrointestinal tracts however, due to surgery, inflammatory bowel disease, antimicrobial use, or achlorhydria may be at risk for bacterial colonization. The diagnosis in these cases is often rendered more challenging as there is no history of the more common forms of contact with either the spores or the toxin, that is, suspicious food ingestion or drug use.190
Inadvertent botulism, first described in 1997, refers largely to the unintentional consequences of therapeutic botulinum toxin use. This practice may, in some cases, result in inadvertent weakness of nearby muscles. Systemic effects of local intramuscular injections have been reported as well. These are typically occult and measurable only through SFEMG. Rarely, however, actual botulism has resulted from injection of doses well within therapeutic ranges. An additional inadvertent mechanism of botulism is through aerosolization and inhalation of the toxin, as has been reported in laboratory workers where the organism is stored and studied.190
Botulinum toxin as an instrument of terrorists would be most likely delivered as a contaminant of food preparations or in an aerosolized form. As the toxin rapidly denatures with exposure to sunlight or chlorine, contamination of public water supplies would be an unlikely strategy.183,184
Practically speaking, botulism remains a clinical diagnosis. The diagnosis should be strongly suspected with the acute onset of multiple cranial nerve abnormalities confined to the motor domain, particularly when preceded by gastrointestinal symptoms and accompanied by signs and symptoms of dysautonomia, impaired ventilation, and a descending pattern of limb weakness. The Centers for Disease Control (CDC) criteria for foodborne botulism require this phenotype occurring with identification of the toxin or the cultured organism. As identification of the organism or the toxin takes time and is imperfect, a heightened clinical index of suspicion is key. When suspected, it is important to obtain a detailed history of potential exposures early in the course as the opportunity to do so may be quickly lost if intubation is required. Even when the diagnosis is strongly supported or confirmed by ancillary testing, treatment decisions are required prior to the identification of toxin or organism.
Confirmation of foodborne botulism can be achieved by detection of the neurotoxin and identification of its subtype in serum, stool, gastric aspirate, or samples of ingested food. This opportunity dissipates rapidly however as the yield declines to <30% after 2 days. Samples need to be obtained before the administration of antitoxin which nullifies the result of the mouse bioassay.184 In all other forms of botulism except inadvertent botulism, the focus is on isolating the organism, not the toxin. In infantile or hidden botulism, the intent is to culture C. botulinum from fecal material or gastric aspirate/vomitus.186 The sensitivity of stool cultures is estimated at 60% but declines to 36% after 3 days.190 The presence of the bacillus is considered de facto evidence of botulism, as they are virtually never found in healthy individuals. In cases of suspected wound botulism, the integument should be carefully searched, not only for gross disruption and wound contamination, but also for minor bruising with or without signs of infection. Culture of these areas should be performed for anaerobic organisms. The nasal mucosa should be visualized and nasal swabs with anaerobic culture media utilized. Diagnostic criteria allow for a definite diagnosis to be made in someone with a characteristic phenotype who is epidemiologically linked to one or more other individuals who have had laboratory confirmation. This may occur in foodborne disease but is essentially nonexistent in wound botulism and other forms of the disease.
The primary differential diagnostic considerations for botulism are acute–subacute disorders that result in multiple cranial nerve deficits.184 If more than one case were to occur simultaneously, the likelihood of botulism would be significantly increased as most differential diagnostic considerations would be less apt to cluster. An outbreak of carbamate (insecticide) toxicity used on watermelons, although a somewhat different phenotype, represents a notable potential exception.194 Based on its prevalence alone, stroke is probably the primary emergency room suspicion.195 To a neurologist, Guillain–Barré syndrome (GBS) variants, tick paralysis, neuropathy related to marine intoxications, and MG are the most likely considerations. This is particularly true in consideration of the motor predominance, the cranial nerve predilection, and the associated dysautonomia that Miller Fisher syndrome and the GBS pharyngeal-cervical-brachial variants may produce.196 Any acute to subacute cause of cranial and limb weakness including disorders with an affinity for the meninges such as viral infections like polio and West Nile virus, neoplastic meningitis, sarcoidosis, marine intoxications, and Lyme disease should be considered. Many will have sensory rather than motor symptoms and are more likely to affect the limb rather than cranial nerve function. Although characterized by pharyngitis and a swollen neck, the local penetration of the diphtheria exotoxin can produce palatal paralysis, dysphonia, and dysphagia as well as a demyelinating polyneuropathy. The red, painful throat that may accompany the xerostomia of botulism may serve to confound the distinction between the two conditions. CMS would have to be considered in children although the time course should be distinctive from botulism in most cases. LEMS is also a potential consideration given that it produces muscle weakness in concert with a cholinergic dysautonomia. Cranial nerve findings are less prevalent in LEMS and the clinical context is usually different, thus allowing confident distinction from botulism in most cases.
EDX evaluation typically provides strong diagnostic support in the appropriate context.190,197–201 The pattern is typical of a presynaptic DNMT in many but not all cases. Sensory nerve conduction studies are normal. CMAP amplitudes are frequently reduced in 85% of tested nerves. A decremental response to slow repetitive stimulation (2-5 Hz) and an incremental response to fast repetitive stimulation (10-50 Hz) are frequent but not invariable findings.190,202 It is generally accepted that rates of 20 Hz or higher have greater diagnostic yields. At times however, either the decremental or incremental response may be elusive. There is a belief that EDX abnormalities in limb muscles including reduced CMAP amplitudes may be less commonly encountered in individuals whose phenotype is restricted to cranial nerve signs and symptoms. Conversely, the inability to convincingly demonstrate an incremental response also appears to correlate with very low amplitude or even absent CMAPs at baseline.201 In keeping with this, the incremental response when present tends to be of lesser magnitude than identified in LEMS. To distinguish this increment from a physiological increment, it should achieve a maximal amplitude of greater than 40% of baseline. In botulism, the increment is commonly less than 100% as opposed to LEMS in which the diagnostic threshold exceeds 100% of baseline CMAP amplitude in any given muscle.190 The duration of the increment is brief, often in the 30–60 second range when subsequent, single stimuli are delivered. It is estimated that incremental responses are demonstrable in only 60% of patients.190 Needle examination findings are often abnormal but not specific. With needle electromyography, low amplitude, short duration, polyphasic MUAPs with increased recruitment, and fibrillation potentials may be demonstrable. Increased jitter values with blocking on SFEMG are to be expected but again indicate only the existence of a neuromuscular transmission abnormality, not the cause.
The diagnosis of botulism is confirmed by the demonstration of toxin or organism in appropriately symptomatic individuals. Currently, the gold standard for the detection of botulinum toxin in foodborne botulism is the mouse bioassay, which is performed by designated BSL-3 containment facilities. It is traditionally used to confirm the presence of toxin in serum, gastric content, stool or the food that the inoculum is thought to originate from. Unfortunately, its sensitivity is insufficient to detect low toxin levels. No more than 45% of patients will test positive, depending on factors such as how rapidly the specimen is obtained and the specific serotype.185,203 The duration of toxin detection is limited and declines rapidly following exposure. The detection of botulinum toxin type B in the system may be possible for up to 12 days in contrast to type A which is estimated to persist for 4 days or less.187 In general, specimens obtained more than 7 days postexposure are unlikely to be positive.185 In addition, availability of assay result requires a minimum of 24 hours and up to 4 days. Newer methodologies utilizing functional dual coating (FDC) and polymerase chain reaction (PCR) technologies promise higher diagnostic yield.204,205 Specimens obtained for purposes of toxin identification should be refrigerated, not frozen, until they can be shipped to the CDC or limited number of state laboratories equipped to perform the assays.
Stool culture is considered the diagnostic test of choice in infantile and hidden botulism and anaerobic culture of the abscess site in wound botulism. As many patients will be constipated, acquisition of stool with the administration of a sterile water enema may be required. Cultures of nasal swabs may be helpful in rare inhalational cases related to vocational exposures or cocaine use.
Other testing is of limited diagnostic value in suspected botulism. Cerebrospinal fluid (CSF) protein if checked, is universally normal as a potentially distinguishing feature from GBS.185 Tensilon testing is generally considered to have a limited value in botulism. A positive response is rarely dramatic and occurs in only a quarter of affected individuals.184,185,206–208 Although of limited diagnostic value, assessment of forced vital capacity or negative inspiratory force are important tools for disease management purposes.
There is no role for either nerve or muscle biopsy in botulism in most if not all cases. In one autopsied case, the findings in muscle were degenerating muscle fibers and scattered angular atrophic fibers. Sural and peroneal nerves displayed no significant histopathology in this individual.209
Botulinum toxin is an exotoxin produced by the anaerobic, spore-forming bacillus C. botulinum in most cases with C. butyricum and C. baratii reported as being neurotoxigenic in India and China.185 The toxin exists in seven currently recognized serotypes, designated A to G, that have similar but not identical biological properties. In North America, disease is associated with toxin types A, B, E, and to a lesser extent type F. Type E is the most common strain causing foodborne disease in North America.185 Botulinum toxicity is substantial, rivaled by few other substances. Parenteral exposure requires a far smaller inoculum than inhalational exposure which is more potent in turn than ingestion.185
Foodborne disease has been linked to a long list (often exotic) of food types or food preparation practices that may be both geographically and culturally based.186,200 One of the more interesting and contemporary forms of foodborne botulism are outbreaks that have occurred in prison populations attributed to the production and ingestion of an illicit fermented beverage known as “pruno.”210 Despite the name, it is likely that the botulinum spores in reported outbreaks of pruno ingestion originated from potato. The C. botulinum bacillus reverts to a spore form under stress. Ideal conditions for spore germination include an anaerobic milieu, nonacidic pH, and low salt and sugar content.185 These conditions are most commonly achieved with inadequate home canning procedures without achievement of adequate duration or degree of temperatures. Outbreaks of foodborne botulism are also reported in cultures that prepare particular foods by allowing them to ferment while avoiding cooking altogether.185,186 Although spores are susceptible to heat, their elimination requires temperatures (85°C) that may be difficult to attain and maintain at higher altitudes. Outbreaks of foodborne disease appear to be more common with home canning in these regions.190 Poorly prepared or stored root vegetables such as potatoes or mushrooms appear to be frequent culprits due to their significant soil exposure where the clostridial organisms are ubiquitous.210 In view of that, it is not surprising that ingestion of botulism spores may occur commonly without causing disease. In adults, either a large inoculum (poorly prepared or stored food) or gut conditions conducive to spore germination (achlorhydria) are required for foodborne botulism to occur. Unlike the spores, the botulinum toxin itself is readily denatured by heat.
Botulinum toxin can be absorbed as an active process by the inhalational route. The toxin does not traverse unbroken skin but is bound to and can be transported across the membranes of epithelial cells including through the nasal mucosa with cocaine inhalation or aerosolization in laboratories or potentially during terrorist acts.190,211 It can also be introduced through broken skin via inadvertent wounds, or through recreational (heroin) or therapeutic (botox) injections. Once absorbed the toxin migrates to the perineuronal microcompartment in the vicinity of vulnerable cholinergic nerve endings. Only these cells have the ability to selectively accumulate the molecule and do so by receptor-mediated endocytosis and translocator mechanisms.211 The toxins then interfere with the release of ACh through slightly different mechanisms affecting the SNARE protein complex. They all gain entrance to the presynaptic terminal through endocytosis via synaptic vesicles, a “Trojan horse” effect. There botulinum toxin serotypes A, C, and E target SNAP-25 whereas botulinum toxin B, D, F, and G cleave VAMP/synaptobrevin, the net effect in each case being disruption of quantal release.188 In vitro electrophysiological studies indicate that there is a significant reduction in the EPP amplitude far below the 7–20 mV necessary to bring the myofiber from its resting membrane potential to action potential threshold. Intuitively, the frequency of MEPPs is reduced, but not MEPP amplitude.212,213
Botulism treatment involves early acquisition of diagnostic specimens and the earliest possible administration of botulinum antitoxin. It is important to have a high diagnostic suspicion of botulism in any case of acute polycranial neuropathy particularly if preceded by gastrointestinal symptoms. Botulism may represent the most important reason to obtain emergent EDX testing. Other than antitoxin administration, intensive care support, when necessary, is the other critical component of successful botulism treatment. Because of potential public health implications, it is recommended that every case of botulism be reported to the state public health department who may also aid the physician in the necessary epidemiological investigation.
Consensus opinion is that heptavalent botulinum antitoxin (HBAT), which only binds to circulating toxin, should be administered expeditiously, prior to availability of any supportive or confirmatory testing results.195 As of March 2010, HBAT is the only botulinum antitoxin available in the United States for naturally occurring noninfant botulism.214 It is available only through the CDC. Unlike prior monovalent or bivalent products, it addresses all seven serotypes. Baby botulism immune globulin (BIG) remains available for infant botulism through the California Infant Botulism Treatment and Prevention Program.214 One observational study reported that the mean length of hospitalization was 5 days shorter in adults for those who received the antitoxin.185 In infants, it has been reported to reduce the average length of hospitalization from 6 to 3 weeks.185
Botulinum toxoid was previously available as an investigational pentavalent (ABCDE) botulinum toxoid vaccine for workers at risk for occupational exposure to botulinum toxins. The CDC discontinued this product as of 2011.215
Local measures may be employed with specific botulism mechanisms. In suspected foodborne botulism, gastric lavage, or enemas may be employed if ingestion is recent in an attempt to remove as much unabsorbed toxin from the gastrointestinal tract as possible. With wound botulism, it is recommended that any potential abscess be debrided and cultured with antimicrobials being administered as required to address other potential, concomitant infections.
Pharmacologically, both pyridostigmine, guanidine, and 3,4 DAP have been used in the treatment of botulism.216 They appear to have limited benefit and have not been recommended for routine use. Avoidance of any drugs with significant NMJ blocking properties is strongly recommended (Table 25-3).
TOXINS/ENVENOMATIONSThere are many environmental intoxications or envenomations whose morbidity results largely from disordered neuromuscular transmission. Many are rare and exotic. This section will review some of the more notable examples. Table 26-1 provides a list of the more well-known NM toxins and attempts to categorize them by their presumed site of action as pre-, post-, or synaptic disorders.
Tick paralysis is a caused by exposure to the saliva of the Ixodid (hard shelled) tick family. Its history is colorful and entertaining.217 The first presumed cases were reported in southeastern Australia in 1824.217,218 It is a disease that affects multiple animal species including dogs, cats, and cattle in addition to humans.217 In Australia, its prevalence in domestic animals makes it a disease of considerable economic consequence.217 Of the two major endemic regions in North America, Dermacentor andersoni (Rocky Mountain wood tick) is the most common vector in the Pacific Northwest and Canada. Dermacentor variabilis (dog tick) is the predominant vector in the Southeastern United States where the disease is less frequently identified (Fig. 26-6). Other less common vectors include Amblyomma americanum and maculatum, and Ixodes scapularis and pacificus in the United States, and Ixodes holocyclus, the major vector in Australia.219 Like botulism, tick paralysis usually presents as isolated cases but may present with clusters as well.219 Tick paralysis is believed to be a DNMT in which the predominant effect of the toxin is to impair the presynaptic release of ACh.220,221
Figure 26-6. Dermacentor variabilis feeding. The tick has been attached for 24 hours. (Reproduced from Goldsmith LA, Katz SI, Gilchrest BA, Paller AS, Leffell DJ, Wolff K. Fitzpatrick’s Dermatology in General Medicine. 8th ed. New York, NY: McGraw-Hill; 2012.)
The phenotype of tick paralysis is strikingly similar to GBS. As the time of the tick attachment is often uncertain, the time between tick attachment and symptom onset is usually imprecise but is estimated at a mean of 5 days although experiments in sheep suggest that feeding for a week by a gravid female tick is typical.217,219 Once the patient becomes symptomatic, the disorder moves rapidly with the nadir occurring on an average of 1.5 days after initial symptom.218,219,222–234
Once the tick is identified and removed, full neurological recovery takes place at an average of 1.5 days in North America but is more protracted in Australia where tick paralysis is a more severe disease.218,219
Affected individuals, particularly children, characteristically experience a prodrome of irritability, somnolence, myalgias, asthenia, and ataxia.217 The ataxia may be so prominent in some cases as to be categorized as a cerebellar syndrome. Despite the “flu-like” prodrome, fever is not a part of the illness. Diarrhea may occur but unlike botulism, gastrointestinal symptoms are otherwise limited. Paresthesias or dysesthesia may precede the development of weakness, affecting the hands and feet in keeping with a non–length-dependent neuropathy. They may have a pruritic or burning characteristic although significant pain is uncharacteristic.217 Despite these complaints, objective sensory loss is mild if evident at all.
Like typical GBS, sensory symptoms are rapidly overshadowed by the evolution of flaccid weakness which ascends, affecting legs before arms, unlike the descending pattern of botulism. This evolution typically occurs over hours to days.217 Cranial nerve and autonomic function are typically impaired subsequent to the development of limb weakness. Bulbar involvement including hoarseness, dysphagia, and sialorrhea occur as does bifacial weakness.218 Both internal (mid-position, unreactive pupils) and external ophthalmoparesis including ptosis occur, particularly in the more severe Australian form of the disease.218 Ventilatory muscle weakness with the need for assisted mechanical ventilation is estimated to occur in 10% of patients.219 Patients are typically areflexic, but like GBS, this may not be the case initially.217,218,235 The mortality of tick paralysis has been reported at between 6 and 11% but the majority of these cases seem to have occurred in the pre-ICU era.219
As previously mentioned, tick paralysis in Australia appears to be a more severe disease.217,218,236 An atypical pattern of focal weakness, mimicking Bell palsy or a brachial plexopathy has been reported in Australian but not North American cases.217,237 Pupillary involvement occurs commonly in Australian cases but is rare in North Americans and should prompt consideration of botulism or Miller Fisher syndrome.217 Hypertension is a common manifestation of tick paralysis in Australia but not in North America. Worsening paralysis may continue following removal of the I. holocyclus tick for up to 48 hours whereas continued progression following tick removal is unusual in the United States where dramatic improvement within hours may occur.235 Prolonged need for mechanical ventilation for a week or more is not uncommon in Australia but uncharacteristic in the United States.218
Tick paralysis may be unique in that the mechanisms for the diagnosis and treatment are for all intents and purposes identical, that is, identification and removal of the tick. The definitive diagnosis involves simply clinical improvement of the characteristic syndrome temporally related to tick removal. In that spirit, the key to diagnosis is a heightened index of suspicion for tick paralysis in patients with apparent GBS, particularly in the spring and summer months, particularly with young girls with a history of outdoor exposure in the endemic areas mentioned above.219 Unfortunately, the diagnosis may be impeded in some cases as the tick may have completed feeding and detached itself prior to its identification.217 Like botulism, tick paralysis cases are usually sporadic but may occur in clusters. None of these epidemiological features are absolute however as tick paralysis has been reported in adults in 20% of cases, in nonendemic regions including urban areas, and at attachment sites other than the scalp (particularly behind the ear), the neck, and the groin.217,219,238
The major differential diagnostic consideration for tick paralysis is GBS for which tick paralysis is often misdiagnosed.219 There are relative, but no absolute differences between the two disorders. Tick paralysis tends to evolve more rapidly, has few if any demyelinating features on motor nerve conduction studies, spares sensory nerve action potentials, is less likely to be painful, and is accompanied by a normal CSF profile. Again, in consideration with this sizeable overlap, is imperative that any GBS suspect undergoes a thorough body search particularly of the scalp before the initiation of plasma exchange or IVIg. Other causes of acute motor weakness require consideration as well. These include acute myelopathies such as transverse myelitis, poliomyelitis, and other enteroviral infections, botulism, myasthenia, diphtheria, porphyria, metabolic disturbances such as severe hypokalemia or hypophosphatemia and potentially other intoxications or envenomations discussed in this chapter and other chapters affecting nerve, NMJ, or muscle.
Spinal fluid analysis is of potential value in tick paralysis as it is characteristically normal, unlike GBS or many of the other differential diagnostic considerations.217,218 As CSF protein may be normal in early GBS as well, it is not an absolute discriminator for these two diseases. Although atypical for DNMT, elevated CK has been reported in some cases.218 Edrophonium is felt to have no effect on the weakness produced by tick paralysis.217,218,235 Ironically, the greatest value of an EEG in tick paralysis is the possibility that the tick may be identified by EEG technicians during scalp electrode placement.235
The major EDX finding in tick paralysis is a reduction in CMAP amplitudes with normal SNAPs.218,223,235,239–243 This nerve conduction pattern is not pathognomonic and is consistent with any presynaptic DNMT, or a disorder of anterior horn cells, ventral roots, or motor nerves. Although minor slowing of motor conduction velocities have been reported in North American cases, they are not of any apparent diagnostic significance.217 It may become evident however with sequential testing before and after tick removal that distal latencies may be mildly affected by the disease.235,239 Theoretically, this may reflect disordered neuromuscular transmission time or potentially slowed conduction in terminal nerve twigs. Features suggesting acquired demyelination such as CMAP temporal dispersion or conduction block as occurs commonly in GBS are not generally described.235,239 We are aware of one report of proximal nerve inexcitability with normal motor conduction parameters on distal stimulation.237 This has been attributed to sodium channel dysfunction within nerve in a manner similar to certain marine toxins or the acute motor axonal neuropathy form of GBS. Neither repetitive stimulation at either low and high frequency nor exercise either brief or prolonged seem to affect CMAP amplitudes in tick paralysis in contrast to other DNMT.235,239 Needle electromyographic results are normal other than for one extreme case of a child with more than 50 attached ticks in which fibrillation potentials were identified.241
There is no apparent role for muscle or peripheral nerve biopsy.
Children, particularly girls, are three times as likely to be afflicted with tick paralysis as are adults.218,222–224,244 One hypothesis for this discrepancy is that a child’s short stature makes their head more accessible to ticks. In addition, long hair in girls is hypothesized to provide a covert location for the protracted feeding necessary to cause disease. The fact that other illnesses transmitted by the same species of tick occur more commonly in males supports this hypothesis.217 Other considerations potentially relevant to the increased incidence in children is that the toxin load is diluted in a larger adult and that the adult is more likely to find and remove the tick at an earlier stage.217 On the other hand, adult men are more likely to be affected than women presumptively because of increased exposure to wooded areas.
Tick paralysis, at least that produced by I. holocyclus, results from holocyclotoxin that is secreted into the salivary glands of the offending vector.217 The concentration of toxin has been demonstrated to increase as the tick feeds, explaining in part the latency between tick attachment and symptom onset. North American ticks are presumed to have a slightly different toxin with a similar mechanism of action. In both Northern and Southern hemisphere disease however, the toxin is believed to interfere with the presynaptic release of ACh from presynaptic terminals of NMJs and presumably autonomic neurons as well although pupillary abnormalities are the only common dysautonomic manifestation of the disease.217,220,221 In addition, there is experimental evidence implicating an additional effect on nerve conduction, hypothetically via sodium channel dysfunction providing a potential explanation for the sensory symptoms or prolonged distal latencies that may accompany the disease.217,237 Regardless of mechanism, it is unlikely to be associated with any structural injury as electrophysiological recovery can begin within hours of tick removal.237
Treatment strategies include prevention, supportive care, and tick removal. When outdoors in endemic areas, limiting exposed skin surfaces, utilizing light clothing to improve tick detection, and spraying or impregnating clothing with insect repellants such as those containing pyrethrin/pyrethroid are deterrent strategies.237 Supportive care is similar to any paralyzing neuromuscular illness including prophylaxis against deep vein thrombosis, skin breakdown, nerve compression, and surveillance for and when necessary treatment of dysautonomia and ventilatory failure.
The most important aspect of care is the identification and removal of the offending tick. The fact that there are historical cases in which the tick was found postmortem emphasizes this point.230 It is strongly recommended that ticks be removed with slow and steady pressure applied with tweezers placed as close to the skin as possible, to ensure removal of mouth parts.219 In addition, as individual ticks can harbor multiple different pathogenic organisms, it is important to avoid the expression of further material into the wound by fingertip pressure.218 Heating or covering the tick with vasoline or similar substances impervious to air is not generally recommended.217
A polyclonal antitoxin is available for the treatment of I. holocyclus. It is a canine derivative and is associated with a high incidence of adverse allergic responses including serum sickness and anaphylaxis.217,218 It is only used in severe cases. No antitoxin exists for North American disease not would it be recommended due to the rapid response to tick removal.217 It also has to be utilized rapidly to provide any benefit. Attempts have been made to develop a vaccine for tick paralysis with some reported success in the laboratory.236,245 We are unaware however of either the availability or effectiveness of a vaccine for humans.
Organophosphates and carbamates are chemicals that respectively irreversibly or reversibly inactivate AChE. They are used as insecticides or as instruments of homicide, suicide, or chemical warfare.246 Inadvertent exposure occurs, most prevalently in the developing world. The World Health Organization estimates that globally, the majority of the more than 200,000 deaths that occur each year are self-inflicted, a problem particularly prevalent in Sri Lanka.247–249 Mortality is estimated at 15–30% or more, particularly where intensive care is not readily available.248
Organophosphate toxicity produces both an acute and a delayed neurological syndrome with different mechanisms of action and different phenotypes. The neuropathy, referred to as organophosphate-induced delayed polyneuropathy or OPIDP, develops as a delayed response to toxic exposure through a different mechanism unrelated to AChE inhibition. It is discussed in Chapter 20. This section will focus exclusively on the manifestations of acute exposure. Unlike the majority of the disorders in this and the preceding chapter, the effects of acute organophosphate or carbamate exposure enhance rather than impair the effects of ACh at the NMJs and autonomic synapses through irreversible inhibition of AChE although the clinical features often evolve into manifestations of synaptic exhaustion. The phenotype of acute organophosphate toxicity is distinctive from other DNMT for two reasons. One is the initial manifestations of cholinergic excess. The other relates to the ability of organophosphates to cross the blood–brain barrier producing CNS as well as PNS cholinergic disruption.
Acute organophosphate toxicity manifests within 24 hours of exposure, typically less. Some of the more notable symptoms pertain largely to the autonomic manifestations of the disease are referred to by the acronym SLUDGE, standing for salivation, lacrimation, urination, defecation, increased gastrointestinal motility, and emesis.248,250 One report identified that 75% of affected patients have miotic pupils and CNS symptoms.251 CNS side effects more commonly occur in organophosphates than in carbamates which have less-effective CNS penetration. CNS manifestations may include agitated delirium and potentially coma, with or without seizures. Two-thirds of patients in the aforementioned series of 47 patients were noted to have hypersalivation and roughly half-experienced agitation and muscle fasciculations.251 Other muscarinic symptoms include bronchospasm, bronchorrhea, and bradycardia. Both hypotension and hypertension may occur, attributed to overstimulation of muscarinic parasympathetic and nicotinic sympathetic neurons, respectively.248 Diaphoresis may occur as well. Muscarinic symptoms may dissipate with the development of large pupils in some cases presumably due to cholinergic bombardment and postsynaptic exhaustion in a manner similar to succinylcholine effect. Muscle weakness is presumably related to the same mechanism.
In addition to paresis of limbs, ophthalmoparesis and ventilatory muscles that may require mechanical ventilation and intensive care may occur.252 In one study, approximately 20% of intoxicants required mechanical ventilation.251 Death most commonly results from respiratory or ventilatory failure that may stem from a combination of excessive pulmonary secretions, diaphragmatic or intercostal muscle weakness, or most commonly from impaired CNS ventilatory drive.248,250 The term “intermediate syndrome” has been coined to describe ventilatory failure that may suddenly occur from diaphragmatic and intercostal muscle weakness after the patient has been treated and stabilized relating to the initial symptoms of muscarinic excess.248 Assessing the patient’s ability to lift their head off the bed has been suggested as a means of indirectly monitoring the development of ventilatory muscle weakness.248
The diagnosis of organophosphate toxicity is typically based on clinical suspicion, generated by the recognition of characteristic clinical signs, smell of pesticides or solvents, and supported by characteristic EDX features and reduced butyrylcholinesterase or AChE activity in the blood.248 The initial clinical picture commonly includes miotic pupils, excessive sweating, and altered level of consciousness and hypoventilation.248
The major differential diagnosis of organophosphate toxicity is carbamate toxicity.248,253 Any acute disorder producing signs and symptoms of both muscle weakness and dysautonomia such as GBS, botulism, tick paralysis, and other phenotypically similar intoxications/envenomations should be considered.
Unlike presynaptic disorders of NMT, CMAP amplitudes at rest are typically normal in acute organophosphate poisoning.252 Repetitive stimulation at both low and high frequencies result in a decremental response.252 Like two forms of CMS, the slow-channel syndrome and AChE deficiency, nerve conduction studies in organophosphate toxicity are distinctive from most DNMT in that afterdischarges of CMAPs occur in response to a single supramaximal stimulus (Fig. 2-4) and are estimated to occur in 60% of intoxicated individuals.252 The afterdischarge, like the parent CMAP will decrement with repetitive stimulation.252
Inhibition of plasma butyrylcholinesterase, also known as pseudocholinesterase or plasma cholinesterase, aids in determining the existence but not severity of organophosphate effect. A decline in the degree of butyrylcholinesterase inhibition may signify the elimination of organophosphate from the body.250 As mentioned, AChE exists on red cells as well as at cholinergic synapses. Red cell cholinesterase levels measured in whole blood provide an additional means by which to determine not only organophosphate exposure but the severity of AChE inhibition.248 The accuracy of both assays is highly dependent on technical considerations, specifically the need to cool the patient’s blood immediately after acquisition. Methodology also exists for the detection of organophosphates in air and water samples as well as on the clothing of exposed individuals.
There is no role for nerve or muscle biopsy in acute organophosphate toxicity.
Organophosphates gain access to the human body through ingestion (usually intentional) or through inhalation or dermal exposure either of which could be accidental or intentional.246 They adversely affect AChE at synapses and on red cell membranes and additionally inhibit butyrylcholinesterase in plasma. The latter effect appears to have little or no associated morbidity.248 Organophosphates work by irreversibly preventing the ability of an AChE molecule from metabolizing ACh, by depositing a phosphoryl group at the active serine hydroxyl site of AChE at both nicotinic and muscarinic synapses.249,250 OPIDP is due to inhibition of a different enzyme, neurotoxic esterase.
Like all acutely paralyzing diseases capable of resulting in ventilatory failure, ICU care is integral to the care of many acutely intoxicated patients.248 Once stabilized, in cases where the toxin has been ingested, gastric lavage with or without activated charcoal is routinely utilized in an attempt to reduce the toxin burden.247,248 Intravenous fluids, usually normal saline, is delivered with a goal of maintaining systolic blood pressure above 80 mm Hg and urine output above 0.5 mL/kg/hour.248 Nasal oxygen is commonly administered. Positioning the patient on their left side may aid in secretion clearance, reduce risk of aspiration, and decrease pyloric emptying and toxin absorption in patients who have injected the toxin.248
The recommended pharmacological treatment of proven benefit is intravenous atropine used to lessen adverse muscarinic and CNS morbidity, atropine being capable of crossing the blood–brain barrier. Various regimens have been recommended. An initial IV bolus of 1–3 mg is suggested.248 A second bolus, double the original, is recommended if the pupil size, blood pressure, pulse, breath sounds, or sweat production do not improve in 5 minutes. Alternative regimens include either continuous IV infusion of atropine at an initial rate of 0.02–0.08 mg/kg or intermittent intravenous injections of 4 mg every 15 minutes until secretions control has been achieved have been recommended as well.251 Use of intravenous β- or calcium channel blockers have been suggested for cardioprotection if heart rates exceed 130 bpm.251 Benzodiazepines are used as a matter of routine in intubated patients and in those who have seized, and are indicated as well along with atropine for the treatment of agitation. Magnesium sulfate, α2 adrenergic agonists such as clonidine, sodium bicarbonate, butyrylcholinesterase, hemodialysis/hemofiltration and bacterially derived phospho triesterases, or hydrolases that break down organophosphates enzymatically represent suggested treatments of unproven benefit.248.
Pralidoxime is the most commonly used agent of the oxime class whose utility is relevant only in organophosphate toxicity. It has no effect on the toxicity of other carbamylated cholinesterases such as physostigmine or neostigmine. Its mechanism of action is to reactivate AChE. It does so by binding the cholinesterase molecule and by doing so, inducing a conformational change in the organophosphate molecule attached to the other end of AChE. This allows for dissociation of the otherwise irreversible bond between AChE and organophosphate.249 Unlike atropine, pralidoxime does not cross the blood–brain barrier and does not benefit CNS morbidity. The World Health Organization recommended regimen is a 30 mg/kg pralidoxime chloride bolus followed by 8 mg/kg/hour infusion.247 An alternative regimen employed in Asia is 1 g of pralidoxime every 4–6 hours for 1–3 days.254 Despite repeated evidence of a benefit in animals however, benefit in humans remains unproven with some reports suggesting a deleterious effect.247,251,254 There are numerous proposed hypotheses as to why a disparity exists in vitro and in vivo effects.251
There are numerous species of venomous arthropods, snakes, and marine species whose bite or at times ingestion may produce weakness or other symptoms referable to the neuromuscular system.255–259 Although uncommon in North America, it is estimated that there are more than 150,000 envenomation deaths that occur in the world annually as a result of venomous bites.257 The land and surrounding waters of Australia and Southeast Asia are the home for many of these species. Although many of these toxins have systemic effects as well as direct effects on peripheral nerve and muscle, the morbidity of a number of these toxins relate to adverse effects on neuromuscular transmission. As many of these disorders indirectly affect neuromuscular transmission by affecting sodium or potassium channels on presynaptic nerve terminals, separating envenomations considered as neuropathic from those whose mechanisms of action appear to be focused on NMT alone is somewhat arbitrary and artificial. As neuropathies caused by envenomations often produce sensory as well as motor consequences, this separation has some clinical validity and will be maintained throughout this text. This section, although not intended to be comprehensive, will highlight some of the more noteworthy toxins that produce neuromuscular disorders largely attributable to disordered NMT.
Serpents belonging to some but not all of the Elapid (cobras, kraits, mambas, coral snakes, sea snakes, and a number of terrestrial Australian) species produce venom that adversely affects NMT at either the presynaptic or postsynaptic level.256 Kraits (Bungarus sp.) secrete both presynaptic toxins referred to as β-neurotoxins and the postsynaptic α- or γ-neurotoxins that have curare-like effects.256,258 These actions are not mutually exclusive although it is the β-neurotoxin that is felt to be the predominant source of morbidity.258 The β-neurotoxin is a phospholipase that results in a loss of synaptophysin and a reduction in synaptic vesicles presynaptically.
The Eastern Green Mamba (Dendroaspis sp.) release two toxins, dendrotoxin and fasciculin. The former specifically binds neuronal potassium channels and prolongs depolarization in nerve terminals, thus facilitating ACh release. The latter is a cholinesterase inhibitor.260 Cobra species (Naja sp.) secrete cobrotoxin that inhibits binding of ACh at nicotinic receptors (κ-neurotoxin).258,260 Coral snakes (Micrurus sp.), the only Elapids indigenous to the United States secrete an α neurotoxin (postsynaptic).261 Vipers and rattlesnake (Crotalid sp.) envenomation may have neurological consequences, for example, generalized myokymia as a consequence of rattlesnake envenomation, but do not typically affect neuromuscular transmission.
Snake envenomation regardless of species may, and often do affect other organ systems as a result of other toxic components, particularly those that incite inflammation with a prominent local wound reaction or that have either procoagulant or anticoagulant effects. Snake venom does not cross the blood–brain barrier but can adversely affect the CNS through thrombotic or hemorrhagic complications.256 Optic neuritis, a cerebellar syndrome and a diffuse encephalomyelitis have been rarely described as a delayed complication of venomous snake bites.256 The mechanism is unknown but may represent a hypersensitivity reaction to antivenin.
The presence or absence of local reaction depends on the species and the constituency of the venom. With krait and coral snake envenomation, it tends to be negligible. Systemic symptoms typically begin within 1–4 hours but may be delayed for up to 12 hours. Initial systemic symptoms may be nonspecific including chest and abdominal discomfort and tightness, myalgias, and nausea among others. CNS symptoms are thought to result from hypoxia or hypotension as these toxins do not cross the blood–brain barrier. Although DNMT is in the exclusive domain of the Elapid species, it is not a universal consequence of envenomation.256 Symptoms referable to cholinergic excess such as fasciculations and hypersalivation may or may not occur. The following description is prototypical when weakness occurs. The pattern, like botulism, is descending. Ptosis and ophthalmoparesis typically precede facial weakness. Neck flexor weakness is a harbinger of ventilatory muscle involvement. Arms weakness may be observed to precede leg weakness if affected individuals are observed carefully.256,259 A direct myotoxic effect of neurotoxins resulting in rhabdomyolysis may occur, typically associated with β-neurotoxicity but reported with α-neurotoxicity as well.258,261 Mortality rates vary but are high without adequate medical care and death may occur within 48 hours of envenomation.258 During the Vietnam War, American soldiers referred to the multibanded krait as the two-step snake due to the exaggerated claim that death occurred within two steps of being bit. With Mamba envenomation, local swelling and nausea precede descending paralysis which includes cranial nerve palsies, ventilatory muscle and limb weakness.260
EDX evaluations have been rarely reported in Elapid envenomation.259 The pattern is one of reduced CMAP amplitudes at baseline with a mild decremental response with slow repetitive stimulation.
Treatment considerations are individualized.256 As a general rule, the involved limb should be immobilized and kept in a dependent position to limit toxin dissemination. Intubation and mechanical ventilation should be instituted early with any indication of breathing difficulties. Volume repletion should be provided and antihistamines, corticosteroids, and epinephrine considered with any indication of shock or allergic reaction to antivenin. Monitoring for and treatment of adverse procoagulant or anticoagulant effects is important. In the case of bleeding, the use of fresh frozen plasma, cryoprecipitates, and human fibrinogen concentrates is indicated. Monitoring for compartment syndrome in the vicinity of the wound is important. Fasciotomy should be undertaken cautiously however, due to considerations of hemostatic difficulties that these patients may experience. Monitoring CK levels in anticipation of possible rhabdomyolyis and myoglobinuric renal failure is recommended.258 Wound debridement may be required if local tissue necrosis ensues. If there is any doubt of the patient’s vaccination status, tetanus toxoid should be provided. Cholinesterase inhibitors may be considered if the species of snake is known and the venom recognized to be an α-neurotoxin with reversible postsynaptic blocking properties. Antivenin, delivered as soon as possible, is recommended and is felt to reduce the mortality rates of envenomation significantly. Antivenins exist in the preferable monovalent (species specific) or polyvalent forms.260 Elapid envenomation is rare in North America but does rarely occur in natural habits in the southern US and Latin America from Coral snakes or with exotic species in pet owners and zoo employees.261,262 Antivenoms for Elapids may be difficult to obtain, particularly in the United States. Valuable resources include the poison center hotline (800–222–1222) and the Association of Zoological Parks and Aquariums (301–562–0777).260 As antivenins are developed in nonhuman species, there is a significant risk of allergic response that should be monitored for and treated as necessary.
α-Latrotoxin is the active constituent of black widow and brown widow spider venom.263–266 The venom stimulates the release of a number of neurotransmitters including norepinephrine, dopamine, and acetylcholine resulting in vesicle depletion.260,267 The mechanism of depleted ACh at presynaptic terminals of the NMJ appears to be independent of the normal calcium dependent ACh-release mechanisms. Both the PNS and CNS are affected. The syndrome differs from most DNMT described in this chapter as the disordered NMT results in symptoms of neuromuscular hyperactivity. Paralysis does not occur. Local pain is a characteristic symptom of the spider bite. Initial symptoms are those of overstimulation with autonomic overactivity including vasoconstriction, and hypertension, diaphoresis and neuromuscular overactivity including painful muscle rigidity and cramping which typically begin at the bite site and spreads centrifugally. Spasms of the abdominal wall may mimic a surgical abdomen. Understandably, serum CK values may be elevated. Headache, dyspnea secondary to bronchoconstriction, emesis, priapism, lethargy, irritability, tremor, fasciculation, and/or ataxia are other common manifestations.256,260 Myocarditis is a reported, a potentially fatal manifestation.268 Treatment includes antivenom which should be used judiciously and with prophylactic antihistamine and epinephrine in view of the high rates of allergic reaction including anaphylaxis. Airway management is as always, a priority. Symptomatic use of benzodiazepines, infusions of calcium gluconate to address cramping and atropine may be considered as well as tetanus immunization.
Scorpion intoxication is also thought to manifest through an increase in presynaptic ACh release as well as a direct effect on muscle, both believed to occur as a result of impaired deactivation of sodium channels.267 The clinical picture of scorpion envenomation is dominated by muscle weakness associated with arterial hypertension, cardiac arrhythmias, myocarditis, or pulmonary edema. These latter manifestations result from the catecholamine release or direct cardiac toxicity.256 Treatment with vasodilators are commonly required to counteract the hyperadrenergic aspects of scorpion stings.
There are numerous marine species that transmit toxins to humans. The transmission may occur through bites, stings, or ingestion. Only a few of these toxins impair NMT as their primary mechanism of action.269,270
Sea snakes, formerly considered as members of the Hydrophiidae family, are now classified as Elapids and share many of the toxic properties described above. They reside almost exclusively in the warm waters of the South Pacific and Indian oceans. Symptom onset is usually 1–6 hours after the bite has occurred. The local reaction is limited. Morbidity stems from both direct myotoxic effects as well as disordered NMT. The former include myalgia aggravated with movement, trismus, and rhabdomyolysis with the risk of myoglobinuric renal failure. The latter include dysphagia, ptosis, and ophthalmoplegia, and ascending paralysis. Seizures, coma, and potentially death from ventilatory failure may occur. Identification of a specific sea snake species (52) is less likely to occur than with bites of their terrestrial cousins. Sea snake antivenom appears equally effective regardless of species. The availability is limited in the Western hemisphere but may be obtained at the Long Beach (CA) aquarium. Again, the risk of allergic reaction needs to be taken into consideration. Management is otherwise similar to that recommended for terrestrial Elapid envenomations.
The cone snail resides in habits similar to sea snakes.269 A dart-like barb coated in toxin can be fired from between the edges of its shell if handled. Envenomation with conotoxin, intended to paralyze its prey, has resulted in numerous human deaths. The conotoxins have affinity for nicotinic AChRs, neuronal calcium channels, muscle sodium channels, vasopressin receptors, and N-methyl-D-aspartate receptors. Presumably, its affinity for presynaptic calcium channels provides the basis for the paralysis it can cause.271–273 A local reaction to snail envenomation produces variable degrees of discomfort followed or accompanied by local swelling and numbness, blanching, cyanosis, and necrosis. Systemically patients may experience nausea and pruritus in addition to dysphagia, blurred vision, paralysis, and in the most severe cases ventilatory failure. Cardiovascular collapse can occur as well. Without support, death may occur as rapidly as 2 hours after envenomation. No antivenin has been developed for the cone snail.
There are multiple drugs and notable metabolic disturbances that can affect neuromuscular transmission, and multiple mechanisms by which they can do so that can occur at either a presynaptic, synaptic, or postsynaptic location.267,274–281 Those that are capable of augmenting neuromuscular transmission may be therapeutically useful in DNMT. Guanidine, rarely used now because of side effects but used historically in the treatment of LEMS, enhances NMT by inhibiting calcium egress from the presynaptic terminal and thereby increasing the probability of vesicle fusion and quantal release. Pyridostigmine, edrophonium, neostigmine, and physostigmine are used both diagnostically and therapeutically to enhance NMT by their reversible cholinesterase inhibition. They can be used to both diagnose MG, treat MG as well as other DNMT, or in case of physostigmine that crosses the blood–brain barrier, be used as a treatment for the CNS toxicity of drugs with anticholinergic properties.
When drugs that augment cholinergic function are used in normal individuals where NMT is already optimal, they may achieve either desired or undesired symptoms of cholinergic excess at muscarinic, nicotinic, and depending on blood–brain barrier penetration, CNS synapses. Conversely, with certain drugs such as succinylcholine, this effect can be of sufficient magnitude to exhaust the NMJ resulting in potentially therapeutic paralysis. This effect can be used to ensure immobility during surgery or to reduce resistance to mechanical ventilation. This same therapeutic paralytic effect can be obtained by nondepolarizing neuromuscular blockers whose mechanism of action is post- rather than presynaptic. The most notorious of these nondepolarizing neuromuscular blocking agents is curare which is a naturally occurring derivative of the plant Strychnos toxifera.276 Although first utilized as a toxin in hunting or in war, like botulinum toxin it can be used therapeutically as well as a paralyzing agent (d-tubocurarine) and was once used to augment the diagnostic yield of repetitive stimulation testing in patients with suspected MG.
There are many other drugs whose primary therapeutic target is not the NMJ but which have neuromuscular blocking properties that vary in degree. These drugs may have little or no effect on normal people with a full neuromuscular reserve but may uncover or increase morbidity in an individual with a pre-existing DNMT such as myasthenia. Finally, there are drugs, most notably penicillamine and αinterferon, that are believed to induce autoimmune myasthenia.282–300 The reader is referred to Table 25-3 for list of agents known to adversely affect neuromuscular transmission.
Although the release of ACh at presynaptic terminals is calcium dependent, an effect theoretically compromised by increased concentrations of its competing cation, magnesium, there is a paucity of information that either hypocalcemia or hypermagnesemia have significant impacts on neuromuscular transmission on most individuals. Autoantibodies (VGCC autoantibodies in LEMS) and environmental toxins (conotoxin in Cone snails) that specifically react with presynaptic VGCCs impair NMT and produce weakness. Despite that, tetany, not weakness is the typical effect of hypocalcemia and appears to result from an effect on the sarcoplasmic reticulum in muscle, not the NMJ. In addition, drugs with calcium channel blocking properties seem to have little if any significant adverse clinical or electrophysiological effects on NMT.301 Conversely, hypermagnesemia has rarely been reported as a cause of significant neuromuscular weakness by competitively inhibiting calcium entry into the nerve terminal.302–306 Serum levels of >5 mEq/L may abolish deep tendon reflexes, with generalized weakness typically present with levels >9–10 mEq/L.307 The reported EDX pattern when reported is consistent with a presynaptic DNMT, that is, reduced CMAP amplitude at rest, a decremental response to slow (2–5 Hz) repetitive stimulation and an incremental response to brief exercise or more rapid stimulation frequencies (10–50 Hz).304,306
SUMMARYNeuromuscular transmission is a complex physiological event that can be readily disrupted by numerous acquired or heritable conditions affecting one or more of its presynaptic, synaptic, or postsynaptic components. Acquired MG is the most common of these disorders. This chapter describes other, less common disorders that require an increased index of suspicion in an individual(s) presenting with painless weakness. This is particularly true when the history reveals a clinical context predisposing to one of these disorders, or when weakness is accompanied by symptoms referable to autonomic or CNS cholinergic dysfunction.
1. Lambert EH, Eaton LM, Rooke ED. Defect of neuromuscular conduction associated with malignant neoplasms. Am J Physiol. 1956;187:612–613.
2. Titulaer MJ, Lang B, Verschuuren JJ. Lambert–Eaton myasthenic syndrome: From clinical characteristics to therapeutic strategies. Lancet Neurol. 2011;10:1098–1107.
3. Eaton LM, Lambert EH. Electromyography and electric stimulation of nerves in diseases of motor unit: Observations on myasthenic syndrome associated with malignant tumors. JAMA. 1957;163(13):1117–1124.
4. Elmqvist D, Quastel DM. Presynaptic action of hemicholinium at the neuromuscular junction. J Physiol. 1965;177:463–482.
5. Elmqvist D, Lambert EH. Detailed analysis of neuromuscular transmission in a patient with the myasthenic syndrome sometimes associated with bronchogenic carcinoma. Mayo Clin Proc. 1968;43:689–713.
6. Lambert EH, Rooke ED. Myasthenic state and lung cancer. In: Brain L, Norris F, eds. The Remote Effects of Cancer on the Nervous System. New York: Grune & Stratton;. 1965:362–410.
7. Lambert EH, Lennon VA. Neuromuscular transmission in nude mice bearing oat cell tumors from Lambert–Eaton myasthenic syndrome. Muscle Nerve. 1982;5(Suppl):S39–S45.
8. O’Neill JH, Murray NM, Newsom Davis J. The Lambert–Eaton myasthenic syndrome: A review of 50 cases. Brain. 1988;111:577–596.
9. Pascuzzi RM, Kim YI. Lambert–Eaton syndrome. Semin Neurol. 1990;10:35–41.
10. Rubenstein AE, Horowitz SH, Bender AN. Cholinergic dysautonomia and Eaton–Lambert syndrome. Neurology. 1979; 29:720–723.
11. Morgan-Followell B, de Los Reyes E. Child neurology: Diagnosis of Lambert-Eaton myasthenic syndrome in children. Neurology. 2013;80(21):e220–e222.
12. Lennon VA, Kryzer TJ, Griesmann GE, et al. Calcium-channel antibodies in Lambert–Eaton myasthenic syndrome and other paraneoplastic syndromes. N Engl J Med. 1995;332:1467–1474.
13. Farrugia ME, Vincent A. Autoimmune mediated neuromuscular junction defects. Curr Opin Neurol. 2010;23:489–495.
14. Lang B, Newsom-Davis J, Wray D, Vincent A, Murray N. Autoimmune aetiology for myasthenic (Eaton-Lambert) syndrome. Lancet. 1981;2(8240):224–226.
15. Titulaer MJ, Wirtz PW, Willems LN, van Kralingen KW, Smitt PA, Verschuuren JJ. Screening for small-cell lung cancer: A follow-up study of patients with Lambert-Eaton myasthenic syndrome. J Clin Oncol. 2008;26:4276–4281.
16. Fernandez-Torron R, Arocha J, Lopez-Picazo JM, et al. Isolated dysphagia due to paraneoplastic myasthenic syndrome with anti-P/Q voltage-gated calcium-channel and anti-acetylcholine receptor antibodies. Neuromuscul Disord. 2011;21:126–128.
17. Lauritzen M, Smith T, Fischer-Hansen B, Sparup J, Olesen J. Eaton-Lambert syndrome and malignant thymoma. Neurology. 1980;30:634–638.
18. Murimoto M, Osaki T, HNagara Y, Kodate M, Motomura M, Murai H. Thymoma with Lambert-Eaton myasthenic syndrome. Ann Thorac Surg. 2010;89:2001–2003.
19. Pasquoloni E, Aubart F, Brihaye B, et al. Lambert-Eaton myasthenic syndrome and follicular thymic hyperplasia in systemic lupus erythematosus. Lupus. 2011;20:745–748.
20. Argov Z, Shapira Y, Averbuch-Heller L, Wirguin I. Lambert-Eaton myasthenic syndrome (LEMS) in association with lymphoproliferative disorders. Muscle Nerve. 1995;18:715–719.
21. Petersen CL, Hemker BG, Jacobson RD, Warwick AB, Jaradeh SS, Kelly ME. Wilms tumor presenting with Lambert-Eaton myasthenic syndrome. J Pediatr Hematol Oncol. 2013;35(4):267–270.
22. Maddison P, Newsom-Davis J, Mills KR, Souhami RL. Favourable prognosis in Lambert-Eaton myasthenic syndrome and small-cell lung carcinoma. Lancet. 1999;353:117–118
23. Wirtz PW, Lang B, Graus F, et al. P/Q-type calcium channel antibodies, Lambert-Eaton myasthenic syndrome and survival in small cell lung cancer. J Neuroimmunol. 2005;164:161–165.
24. Nakao YK, Motomura M, Fukudome T, et al. Seronegative Lambert–Eaton myasthenic syndrome: Study of 110 Japanese patients. Neurology. 2002;59:1773–1775.
25. Titulaer MJ, Klooster R, Potman M, et al. SOX antibodies in small-cell lung cancer and Lambert-Eaton myasthenic syndrome: Frequency and relation with survival. J Clin Oncol. 2009;27:4260–4267.
26. Wirtz PW, Bradshaw J, Wintzen AR, Verschuuren JJ. Associated autoimmune disease in patients with the Lambert-Eaton myasthenic syndrome. J Neuroimmunol. 2005;159:230–237.
27. Gutmann L, Crosby TW, Takamori M, Martin JD. The Eaton–Lambert syndrome and autoimmune disorders. Am J Med. 1972;53:354–356.
28. Lambert EH, Rooke ED, Eaton LM, et al. Myasthenic syndrome occasionally associated with bronchial neoplasm: Neurophysiologic studies. In: Viets HR, ed. Myasthenia Gravis. Springfield, IL: C C Thomas Publishers; 1961:362–410.
29. McEvoy KM. Diagnosis and treatment of Lambert–Eaton myasthenic syndrome. Neurol Clin. 1994;12:387–399.
30. Rudnicki SA. Lambert-Eaton myasthenic syndrome with pure ocular weakness. Neurology. 2007;68:1863–1864.
31. Wirtz PW, Sotodeh M, Nijnuis M, et al. Different distribution of muscle weakness between myasthenia gravis and the Lambert-Eaton myasthenic syndrome. J Neurol Neurosurg Psychiatry. 2002;73:766–768.
32. Burns TM, Russell JA, LaChance D, Jones HR. Oculobulbar involvement is typical with Lambert–Eaton myasthenic syndrome. Ann Neurol. 2003;53:270–273.
33. Rácz A, Giede-Jeppe A, Schramm A, Schwab S, Maihöfner C. Lambert-Eaton myasthenic syndrome presenting with a “dropped head syndrome” and associated with antibodies against N-type calcium channels. Neurol Sci. 2012;34(7):1253–1254.
34. Nicolle MW, Stewart DJ, Remtulla H, Chen R, Bolton CF. Lambert–Eaton myasthenic syndrome presenting with severe respiratory failure. Muscle Nerve. 1996;19:1328–1333.
35. Smith AG, Wald J. Acute ventilatory failure in Lambert–Eaton myasthenic syndrome and its response to 3,4-diaminopyridine. Neurology. 1996;46:1143–1145.
36. Barr CW, Claussen G, Thomas D, Fesenmeier JT, Peralman RL, Oh SJ. Primary respiratory failure as the presenting symptom in Lambert–Eaton myasthenic syndrome. Muscle Nerve. 1993;16:712–715.
37. Mason WP, Graus F, Lang B, et al. Small cell lung cancer, paraneoplastic cerebellar degeneration, and Lambert–Eaton myasthenic syndrome. Brain. 1997;120:1279–1300.
38. Breen LA, Gutmann L, Brick JF, Riggs JR. Paradoxical lid elevation with sustained upward gaze: A sign of Lambert–Eaton syndrome. Muscle Nerve. 1991;14:863–866.
39. Oh SJ, Dwyer DS, Bradley RJ. Overlap myasthenic syndrome: Combined myasthenia gravis and Eaton-Lambert syndrome. Neurology. 1987;37:1411–1414.
40. Boiardi A, Bussone G, Negri S. Alternating myasthenia and myastheniform syndromes in the same patient. J Neurol. 1979; 220:57–64.
41. Dahl DS, Sato S. Unusual myasthenic state in a teenage boy. Neurology. 1974;24:897–901.
42. Lennon VA. Serologic profile of myasthenia gravis and distinction from the Lambert–Eaton myasthenic syndrome. Neurology. 1997;48(Suppl 5):S23–S27.
43. Katz JS, Wolfe GI, Bryan WW, Tintner R, Barohn RJ. Acetylcholine receptor antibodies in the Lambert–Eaton myasthenic syndrome. Neurology. 1998;50:470–475.
44. Newsom-Davis J, Leys K, Vincent A, Ferguson I, Modi G, Mills K. Immunological evidence for the co-existence of the Lambert–Eaton myasthenic syndrome and myasthenia gravis in two patients. J Neurol Neurosurg Psychiatry. 1991;54:452–453.
45. Kim JA, Lim YM, Jang EH, Kim KK. A patient with coexisting myasthenia gravis and Lambert-Eaton myasthenic syndrome. J Clin Neurol. 2012;8(3):235–237.
46. Joy JL, Baysal AI, Oh SJ. Reflex improvement after exercise in the Eaton–Lambert syndrome. Muscle Nerve. 1987;10:671–672.
47. Tim RW, Massey JM, Sanders DB. Lambert–Eaton syndrome: Electrodiagnostic findings and response to treatment. Neurology. 2000;54:2176–2178.
48. Jablecki C. Lambert–Eaton myasthenic syndrome. Muscle Nerve. 1984;7:250–257.
49. McQuillen MP, Johns RJ. The nature of the defect in the Eaton–Lambert syndrome. Neurology. 1967;17:527–536.
50. Cruz Martinez A, Ferrer MT, Morales C, et al. Electromyography, single fiber electromyography, and necropsy findings in myasthenic syndrome associated with bronchogenic carcinoma. Electromyogr Clin Neurophysiol. 1982;22:531–548.
51. Henriksson KG, Nilsson O, Rosen I, Schiller HH. Clinical, neurophysiological and morphological findings in Eaton–Lambert syndrome. Acta Neurol Scand. 1977;56:117–140.
52. Lambert EH. Defects of neuromuscular transmission in syndromes other than myasthenia gravis. Ann N Y Acad Sci. 1966;135:367–384.
53. Oh SJ, Kim DE, Kuruoglu R, Brooks J, Claussen GW. Electrophysiological and clinical correlations in the Lambert–Eaton myasthenic syndrome. Muscle Nerve. 1996;19:903–906.
54. Oh SJ, Kurokawa K, Glaussen GC, Ryan HR Jr. Electrophysiological diagnostic criteria of Lambert–Eaton, myasthenic syndrome. Muscle Nerve. 2005;32:515–520.
55. Tim RW, Sanders DB. Repetitive nerve stimulation studies in the Lambert–Eaton myasthenic syndrome. Muscle Nerve. 1994;17:995–1001.
56. Oh SJ. The Eaton–Lambert syndrome in ocular myasthenia gravis. Arch Neurol. 1974;31:183–186.
57. Ingram DA, Davis GR, Schwartz MS, Traub M, Newland AC, Swash M. Cancer associated myasthenic (Eaton–Lambert) syndrome: Distribution of abnormality and effect of treatment. J Neurol Neurosurg Psychiatry. 1984;47:806–812.
58. Brown JC, Johns RJ. Diagnostic difficulties encountered in the myasthenic syndrome sometimes associated with carcinoma. J Neurol Neurosurg Psychiatry. 1974;37:1214–1224.
59. Scopetta C, Csali C, Vaccario ML, Provenzano C. Difficult diagnosis of Eaton–Lambert syndrome. Muscle Nerve. 1984;7:680–681.
60. Baslo MB, Deymeer F, Serdaroglu P, Parman Y, Ozdemir C, Cuttini M. Decrement pattern in Lambert-Eaton myasthenic syndrome is different from myasthenia gravis. Neuromuscul Disord. 2006;16:454–458.
61. Sanders DB, Cao L, Massey JM, Juel VC, Hobson-Webb L, Guptill JT. Is the decremental pattern in Lambert-Eaton syndrome different from that in myasthenia gravis? Clin Neurophysiol. 2014;125(6):1274–1277.
62. Desmedt JE. The neuromuscular disorder in myasthenia gravis: I. Electrical and mechanical responses to nerve stimulation in hand muscles. In: Desmedt JE, ed. New Developments in Electromyography and Clinical Neurophysiology. Basel: Karger; 1973:241–304.
63. Stalberg E, Trontelj JV. Single Fiber Electromyography. Old Woking: Mirvalle Press; 1979.
64. Oh SJ. Electromyography: Neuromuscular Transmission. Baltimore, MD: Williams & Wilkins; 1988.
65. Rooke DE, Eaton LM, Lambert EH, Hodgson CH. Myasthenia and malignant intrathoracic tumor. Med Clin North Am. 1960;44:977–988.
66. Schwartz MS, Stalberg E. Myasthenic syndrome studied with single fiber electromyography. Arch Neurol. 1975;32:815–817.
67. Schwartz MS, Stalberg E. Myasthenia gravis with features of the myasthenic syndrome. Neurology. 1975;25:80–84.
68. Cruz Martinez A, Ferrer MT, Diez Tejedor E, Perez Conde MC, Anciones B, Frank A. Diagnostic yield of single fiber electromyography and other electrophysiological technique in myasthenia gravis I. Electromyography, automatic analysis of the voluntary pattern, and repetitive nerve stimulation. Electromyogr Clin Neurophysiol. 1982;22:377–393.
69. Cruz Martinez A, Ferrer MT, Perez Conde MC, Diez Tejedor E, Barreiros P, Ribacoba R. Diagnostic yield of single fiber electromyography and other electrophysiologic techniques in myasthenia gravis II. Jitter and motor unit fiber density studies. Clinical remission and thymectomy. Electromyogr Clin Neurophysiol. 1982;22:395–417.
70. Trontelj JV, Stalberg E, Mihelin M. Jitter in the muscle fiber. J Neurol Neurosurg Psychiatry. 1990;53:49–54.
71. Trontelj JV, Stalberg E. Single motor end-plates in myasthenia gravis and LEMS at different firing rates. Muscle Nerve. 1991;14:226–232.
72. Trontelj JV, Stalberg E, Khuraibet AJ. Tetanic potentiation and depression at single motor end-plates in myasthenia and LEMS. J Neurol Sci. 1990;98(Suppl):289–290.
73. Kim DE, Claussen GC, Oh SJ. Single-fiber electromyography improvement with 3,4 diaminopyridine in Lambert–Eaton myasthenic syndrome. Muscle Nerve. 1998;21:1107–1108.
74. Oh SJ. SFEMG improvement with remission in the cancer-associated Lambert–Eaton myasthenic syndrome. Muscle Nerve. 1989;12:844–848.
75. Oh SJ, Hurwitz EL, Lee KW, Change CW, Cho HK. The single fiber EMG in the Lambert–Eaton myasthenic syndrome. Muscle Nerve. 1989;12:159–161.
76. Sadeh M, River Y, Argov Z. Stimulated single-fiber electromyography in Lambert–Eaton myasthenic syndrome before and after 3,4-diaminopyridine. Muscle Nerve. 1997;20:735–739.
77. Torbergsen T, Stalberg E, Bless JK. Subclinical neuromuscular involvement in patients with lung cancer. Acta Neurol Scand. 1984;69(Suppl 98):190–191.
78. Trontelj JV, Mihelin M, Fernandez JM, Stalberg E. Axonal stimulation for end-plate jitter studies. J Neurol Neurosurg Psychiatry. 1986;49:677–685.
79 Chaudhry V, Watson DF, Bird SJ, Cornblath DR. Stimulated single fiber electromyography in Lambert–Eaton syndrome. Muscle Nerve. 1991;14:1227–1230.
80. Ricker K, Hertel G, Stodieck S. The influence of local cooling on neuromuscular transmission in the myasthenic syndrome of Eaton and Lambert. J Neurol. 1977;217:95–102.
81. Ricker K, Hertel G, Stodieck S. Influence of temperature on neuromuscular transmission in myasthenia gravis. J Neurol. 1977;216:273–282.
82. Ward CD, Murray NM. Effect of temperature on neuromuscular transmission in the Eaton–Lambert syndrome. J Neurol Neurosurg Psychiatry. 1979;42:247–249.
83. AAEM Quality Assurance Committee. American Association of Electrodiagnostic Medicine. Practice parameter for repetitive nerve stimulation and single fiber EMG evaluation of adults with suspected myasthenia gravis of Lambert-Eaton myasthenic syndrome: Summary statement. Muscle Nerve. 2001;24:1236–1238.
84. Leys K, Lang B, Johnston I, Newsom-Davis J. Calcium channel autoantibodies in the Lambert-Eaton myasthenic syndrome. Ann Neurol. 1991;29(3):307–314.
85. Lipka AF, Verschuuren JJ, Titulaer MJ. SOX1 antibodies in Lambert-Eaton myasthenic syndrome and screening for small cell lung carcinoma. Ann N Y Acad Sci. 2012;1275:70–77.
86. Sabater L, Titulaer M, Saiz A, Verschuuren J, Güre AO, Graus F. SOX1 antibodies are markers of paraneoplastic Lambert–Eaton myasthenic syndrome. Neurology. 2008;70:924–928.
87. Engel AG, Santa T. Histometric analysis of the ultra-structure of the neuromuscular junction in myasthenia gravis and the myasthenic syndrome. Ann N Y Acad Sci. 1971;183:46–63.
88. Engel AG. Review of evidence for loss of motor nerve terminal calcium channels in Lambert–Eaton myasthenic syndrome. Ann N Y Acad Sci. 1991;635:246–258.
89. Fukunaga H, Engel AG, Osame M, et al. Paucity and disorganization of presynaptic membrane active zones in the Lambert–Eaton syndrome. Muscle Nerve. 1982;5:686–697.
90. Fukunaga H, Engel AG, Lang B, Newsom-Davis J, Vincent A. Passive transfer of Lambert–Eaton myasthenic syndrome with IgG from man to mouse depletes the presynaptic membrane active zones. Proc Natl Acad Sci USA. 1983;80:7636–7640.
91. Hughes BW, Kusner LL, Kaminski HJ. Molecular architecture of the neuromuscular junction. Muscle Nerve. 2006;33: 445–461.
92. Kim YI. Passively transferred Lambert–Eaton syndrome in mice receiving purified IgG. Muscle Nerve. 1986;9:523–530.
93. Engel AG. The neuromuscular junction. In: Aminoff M, Boller F, Swaab D, eds. Handbook of Clinical Neurology. Chapter 3, Vol. 91. Amsterdam, Netherlands: Elsevier; 2008:103–148.
94. Leveque C, Hoshino T, David P, et al. The synaptic vesicle protein synaptotagmin associates with calcium channels and is a putative Lambert–Eaton myasthenic syndrome antigen. Proc Natl Acad Sci USA. 1992;89:3625–3629.
95. Pinto A, Kazuo I, Newland C, Newson-Davis J, Lang B. The action of Lambert-Eaton myasthenic syndrome immunoglobulin B on cloned human voltage-gated calcium channels. Muscle Nerve. 2002;25:715–724.
96. Lecky BR. Transient neonatal Lambert-Eaton syndrome. J Neurol. 2006;77:1094.
97. Reuner U, Kamin G, Ramantani G, Reichmann H, Dinger J. Transient neonatal Lambert-Eaton syndrome. J Neurol. 2008;255:1827–1828.
98. Chalk CH, Murray NM, Newsom-Davis J, O’Neill JH, Spiro SG. Response of the Lambert–Eaton myasthenic syndrome of associated small-cell lung carcinoma. Neurology. 1990;40:1552–1556.
99. De Aizpurua HJ, Lambert EH, Griesmann GE, et al. Antagonism of voltage-gated calcium channels in small cell carcinomas of patients with and without Lambert–Eaton myasthenic syndrome by autoantibodies w-conotoxin and adenosine. Cancer Res. 1988;48:4719–4724.
100. Hawley RJ, Cohen MH, Saini N, Armbrustmacher VW. The carcinomatous neuromyopathy of oat cell lung cancer. Ann Neurol. 1980;7:65–72.
101. Jenkyn LR, Brooks PL, Forcier RJ, Maurer LH, Ochoa J. Remission of the Lambert–Eaton syndrome and small cell anaplastic carcinoma of the lung induced by chemotherapy and radiotherapy. Cancer. 1980;46:1123–1127.
102. Maddison P, Newsom-Davis J. Treatment for Lambert-Eaton myasthenic syndrome. Cochrane Database Syst Rev. 2005;18(2):CD003279.
103. Skeie GO, Apostolski S, Evoli A, et al. Guidelines for treatment of autoimmune neuromuscular transmission disorders. Eur J Neurol. 2010;17:893–902.
104. Maddison P. Treatment in Lambert–Eaton myasthenic syndrome. Ann N Y Acad Sci. 2012;1275:78–84.
105. Tim RW, Massey JM, Sanders DB. Lambert-Eaton myasthenic syndrome (LEMS) clinical and electrodiagnostic features and response to therapy in 59 patients. Ann N Y Acad Sci. 1988; 841:823–826.
106. Lundh H, Nilsson O, Rosen I. Treatment of Lambert–Eaton syndrome: 3,4-diaminopyridine and pyridostigmine. Neurology. 1984;34:1324–1330.
107. Oh SJ, Lee YW, Rutsky E. Eaton–Lambert syndrome: Reflex improvement with guanidine. Arch Phys Med Rehabil. 1977;58: 457–459.
108. Oh SJ, Kim KW. Guanidine hydrochloride in the Eaton–Lambert syndrome: Electrophysiological improvement. Neurology. 1973;23:1084–1090.
109. Keogh M, Sedehizadeh S, Maddison P. Treatment for Lambert-Eaton myasthenic syndrome. Cochrane Database Syst Rev. 2011;(2):CD003279.
110. Sanders DB, Massey JM, Sanders LL, Edwards LJ. A randomized trial of 3,4-diaminopyridine in Lambert–Eaton myasthenic syndrome. Neurology. 2000;54:603–607.
111. Oh SJ, Claussen GG, Hatanaka Y, Morgan MB. 3,4-Diaminopyridine is more effective than placebo in a randomized, double-blind, cross-over drug study in LEMS. Muscle Nerve. 2009;40(5):795–800.
112. McEvoy KE, Windebank AJ, Daube JR. Low PA. 3,4- diaminopyridine in Lambert-Eaton myasthenic syndrome. N Engl J Med. 1989;321(23):1567–1571.
113. Wirtz PW, Verschuuren JJ, van Dijk JG, Efficacy of 3,4- diaminopyridine and pyridostigmine in the treatment of Lambert-Eaton myasthenic syndrome: A randomized, double-blind, placebo-controlled, crossover study. Clin Pharmacol Ther. 2009;86(1):44–48.
114. Streib EW, Rothner AD. Eaton–Lambert myasthenic syndrome. Long term treatment of three patients with prednisone. Ann Neurol. 1981;10:448–453.
115. Kimura I, Ayyar DR. The Eaton–Lambert myasthenic syndrome and long-term treatment with prednisone. Tohoku J Exp Med. 1984;143:405–408.
116. Newsom-Davis J, Murray NM. Plasma exchange and immunosuppressive drug treatment in the Lambert–Eaton myasthenic syndrome. Neurology. 1984;34:480–485.
117. Bain PG, Motomura M, Newsom-Davis J, et al. Effects of intravenous immunoglobulin on muscle weakness and calcium-channel autoantibodies in the Lambert-Eaton myasthenic syndrome. Neurology. 1996;47(3):678–683.
118. Dau PC, Deny EH. Plasmapheresis and immunosuppressive drug therapy in the Eaton–Lambert syndrome. Ann Neurol. 1982;11:570–575.
119. Denys EH, Dau PC, Lindstrom JM. Neuromuscular transmission before and after plasmapheresis in myasthenia gravis and myasthenic syndrome. In: Dau PC, ed. Plasmapheresis and Immunobiology of Myasthenia Gravis. Boston, MA: Houghton Mifflin; 1979:248–257.
120. Maddison P, McConville J, Farrugia ME, et al. The use of rituximab in myasthenia gravis and Lambert-Eaton myasthenic syndrome. J Neurol Neurosurg Psychiatry. 2011;82:671–673.
121. Pellkofer HL, Voltz R, Kuempfel T. Favorable response to rituximab in a patient with anti-VGCC positive Lambert-Eaton myasthenic syndrome and cerebellar dysfunction. Muscle Nerve. 2009;40:305–308.
122. Burke G, Cossins J, Maxwell S, et al. Rapsyn mutations in hereditary myasthenia: Distinct early- and late-onset phenotypes. Neurology. 2003;61(6):826–828.
123. Beeson D, Hantai D, Lochmuller H, Engel AG. 126th International Workshop: Congenital myasthenic syndromes, 24–26 September. 2004, Naarden, the Netherlands. Neuromuscul Disord. 2005;15:498–512.
124. Müller JS, Herczegfalvi A, Vilchez JJ, et al. Phenotypical spectrum of DOK7 mutations in congenital myasthenic syndromes. Brain. 2007;130:1497–1506.
125. Engel AG. Current status of the congenital myasthenic syndromes. Neuromuscular Disorders. 2012;22:99–111.
126. Finlayson S, Beeson D, Palace J. Congenital myasthenic syndromes: An update. Pract Neurol. 2013;13:80–91.
127. Lorenzoni PJ, Scola RH, Kay CS, Werneck LC. Congenital myasthenic syndrome: A brief review. Pediatr Neurology. 2012;46:141–148.
128. Barašić N, Chaouch A, Müller JS, Lochmüller S. Genetic heterogeneity and pathophysiological mechanisms in congenital myasthenia syndromes. Eur J Pediatr Neurol. 2011;15:189–196.
129. Maselli RA, Arredondo J, Ferns MJ, Wollmann RL. Synaptic basal lamina-associated congenital myasthenic syndromes. Ann N Y Acad Sci. 2012;1275:36–48.
130. Milone M, Shen XM, Selcen D, et al. Myasthenic syndrome due to defects in rapsyn: Clinical and molecular findings in 39 patients. Neurology. 2009;73:228–235.
131. Selcen D, Shen XM, Milone M, et al. GFPT1 myasthenia. Neurology. 2013;81:370–378.
132. Finlayson S, Pallace J, Belaya K, et al. Clinical features of congenital syndrome due to mutations in DPAGT1. J Neurol Neurosurg Psychiatry. 2013;84:1119–1125.
133. Cossins J, Belaya K, Hicks D, et al. Congenital myasthenic syndromes due to mutations in ALG2 and ALG14. Brain. 2013;136:944–956.
134. Ohno K. Glycosylation defects as an emerging novel cause leading to a limb-girdle type of congenital myasthenic syndromes. J Neurol Neurosurg Psychiatry. 2013;84:1064.
135. Senderek J, Muller JS, Dusl M, et al. Hexosamine biosynthetic pathway mutations cause neuromuscular transmission defect. Am J Hum Genet. 2011;88:162–172.
136. Belaya, K, Finlayson S, Cossins J, et al. Identification of DPAGT1 as a new gene in which mutations cause a congenital myasthenic syndrome. Ann N Y Acad Sci. 2012;1275:29–35.
137. Belaya K, Finlayson S, Slater CR, et al. Mutations in DPAGT1 cause a limb-girdle myasthenic syndrome with tubular aggregates. Am J Hum Genet. 2012;91:193–201.
138. Monies DM, Al-Hindi HN, Al-Muhaizea MA, et al. Clinical and pathological heterogeneity of a congenital disorder of glycosylation manifesting as a myasthenic/myopathic syndrome. Neuromuscul Disord. 2014;24(4):353–359.
139. Ohkawara B, Cabrera-Serrano M, Nakata T, et al. LRP4 third β-propeller domain mutations cause novel congenital myasthenia by compromising agrin-mediated MuSK signaling in a position- specific manner. Hum Mol Genet. 2014;23(7):1856–1868.
140. Selcen D, Juel VC, Hobson-Webb LD, et al. Myasthenic syndrome caused by plectinopathy. Neurology. 2011;76:327–336.
141. Selcen D, Milone M, Shen XM, et al. Dok-7 myasthenia: Phenotypic and molecular genetic studies in 16 patients. Ann Neurol. 2008;64:71–87.
142. Dobkin BH, Verity MA. Familial neuromuscular disease with type 1 fiber hypoplasia, tubular aggregates, cardiomyopathy, and myasthenic features. Neurology. 1978;28:1135–1140.
143. Furui E, Fukushima K, Sakashita T, Sakato S, Matsubara S, Takamori M. Familial limb-girdle myasthenia with tubular aggregates. Muscle Nerve. 1997;20:599–603.
144. Johns TR, Campa JF, Crowley WJ, et al. Familial myasthenia with tubular aggregates. Neurology. 1971;21:449.
145. McQuillen MP. Familial limb-girdle myasthenia. Brain. 1966; 89:121–132.
146. Guergueltcheva V, Müller JS, Dusl M, et al. Congenital myasthenic syndrome with tubular aggregates caused by GFPT1 mutations. J Neurol. 2012;259:838–850.
147. Abicht A, Müller J, Lochmüller H. Congenital myasthenic syndromes. In: Pagon RA, Adam MP, Bird TD, Dolan CR, Fong CT, Stephens K, eds. GeneReviews™ [Internet]. Seattle, WA: University of Washington; http://www.ncbi.nlm.nih.gov/gtr/. 2012:1993–2013.
148. Engel AG, Ohno K, Milone M, et al. New mutations in acetylcholine receptor subunit genes reveal heterogeneity in the slow channel congenital myasthenic syndrome. Hum Mol Genet. 1996;5:1217–1227.
149. Engel AG, Lambert EH, Mulder DM, et al. A newly recognized congenital myasthenic syndrome attributed to a prolonged open time of the acetylcholine induced ion channel. Ann Neurol. 1982;11:553–569.
150. Alseth EH, Maniaol AH, Elsais A, et al. Investigation for rapsyn and Dok-7 mutations in a cohort of seronegative myasthenia gravis. Muscle Nerve. 2011;43:574–577.
151. Punga AR, Ruegg MA. Signaling and aging at the neuromuscular synapse: Lessons learnt from neuromuscular disease. Curr Opin Pharmacol. 2012;12:340–346
152. Engel AG, Shen XM, Selcen D, Sine SM. What have we learned from the congenital myasthenic syndromes. J Mol Neurosci. 2010;40:143–153.
153. Ohno K, Tsujino A, Brengman JM, et al. Choline acetyltransferase mutations cause myasthenic syndrome associated with episodic apnea in humans. Proc Natl Acad Sci USA. 2001;98:2017–2022.
154. Walls TJ, Engel AG, Nagel AS, Harper CM, Trastek VF. Congenital myasthenic syndrome associated with paucity of synaptic vesicles and reduced quantal release. Ann N Y Acad Sci. 1993;681:461–468.
155. Engel AG, Walls TJ, Nagel A, Uchitel O. Newly recognized congenital myasthenic syndromes: I. Congenital paucity of synaptic vesicles and reduced quantal release, II. High-conductance fast-channel syndrome, III. Abnormal acetylcholine receptor (AChR) interaction with acetylcholine, IV. AChR deficiency and short channel-open time. Prog Brain Res. 1990;84:125–137.
156. Bady B, Chauplannaz G, Carrier H. Congenital Lambert-Eaton myasthenic syndrome. J Neurol Neurosurg Psychiatry. 1987;50(4):476–478.
157. Engel AG, Lambert EH, Mulder DM, et al. Recently recognized congenital myasthenic syndromes: (a) end-plate acetylcholine (ACh) esterase deficiency (b) putative abnormality of the ACh induced ion channel (c) putative defect of ACh resynthesis or mobilization—clinical features, ultrastructure and cytochemistry. Ann N Y Acad Sci. 1981;377:614–616.
158. Engel AG, Lambert EH, Gomez MR. A new myasthenic syndrome with end-plate acetylcholinesterase deficiency, small nerve terminals and reduced acetylcholine release. Ann Neurol. 1977;1:315–330.
159. Hutchinson DO, Walls TJ, Nakano S, et al. Congenital acetylcholinesterase deficiency. Brain. 1993;116:633–653.
160. Jennekens FG, Hesselmans LF, Veldman H, Jansen EN, Spaans F, Molenaar PC. Deficiency of acetylcholine receptors in a case of end-plate acetylcholinesterase deficiency: A histochemical investigation. Muscle Nerve. 1992;15:63–72.
161. Maselli RA, Ng JJ, Anderson JA, et al. Mutations in LAMB2 causing a severe form of synaptic congenital myasthenic syndrome. J Med Genet. 2009;46:203–208.
162. Maselli RA. Fernandez JM, Arredondo J. et al. LG2 agrin mutation causing severe congenital myasthenic syndrome mimics functional characteristics of non-neural (z-) agrin. Hum Genet. 2012;131:1123–1135.
163. Huz´e C, Bauché S, Richard P. et al. Identification of an agrin mutation that causes congenital myasthenia and affects synapse function. Am J Hum Genet. 2009;85:155–167.
164. Burke G, Cossins J, Maxwell S, et al. Distinct phenotypes of congenital acetylcholine receptor deficiency. Neuromuscul Disord. 2004;14:356–364.
165. Ruff RL, Putecki P. Faster, slower, but never better. Mutations of the skeletal muscle acetylcholine receptor. Neurology. 2012;79:404–405.
166. Shen X-M, Ohno K, Fukudome T, et al. Congenital myasthenic syndrome caused by low-expressor fast-channel AChR δ subunit mutation. Neurology. 2002;59:1881–1889.
167. Palace J, Lashley D, Newsom-Davis J, et al. Clinical features of the DOK7 neuromuscular junction synaptopathy. Brain. 2007;130:1507–1515.
168. Selcen D, Milone M, Shen XM, et al. Dok-7 myasthenia: Clinical spectrum, endplate (EP) electrophysiology and morphology, 12 novel DNA rearrangements, and genotype–phenotype relations in a Mayo cohort of 13 patients. Neurology. 2007;68(Suppl 1):A106–A107.
169. Anderson JA, Ng JJ, Bowe C, et al. Variable phenotypes associated with mutations in Dok-7. Muscle Nerve. 2008;37:448–456.
170. Beeson D, Higuchi O, Palace J, et al. Dok-7 mutations underlie a neuromuscular junction synaptopathy. Science. 2006;313:1975–1978.
171. Maselli RA, Arredondo J, Cagney O, et al. Mutations in MUSK causing congenital myasthenic syndrome impair MuSK-Dok-7 interaction. Hum Mol Genet. 2010;19:2370–2379.
172. Mihaylova V, Salih MA, Mukhtar MM, et al. Refinement of the clinical phenotype in musk-related congenital myasthenic syndromes. Neurology. 2009;73:1926–1928.
173. Chevessier F, Faraut B, Ravel-Chapuis A, et al. MUSK, a new target for mutations causing congenital myasthenic syndrome. Hum Mol Genet. 2004;13:3229–3240.
174. Tsujino A, Maertens C, Ohno K, et al. Myasthenic syndrome caused by mutation of the SCN4 A sodium channel. Proc Natl Acad Sci USA. 2003;100:7377–7382.
175. Kosac A, Gavillet E, Whittaker RG. Neurophysiological testing in congenital myasthenic syndromes: A systematic review of published normal data. Muscle Nerve. 2013;48:711–715.
176. Scara U, Della Marina A, Abicht A. Congenital myasthenic syndromes: Current diagnostic and therapeutic approaches. Neuropediatrics. 2012;43:184–193.
177. Engel AG, Brengman J, Edvardson S, Shen X-M. Highly fatal lowaffinity fast-channel congenital myasthenic syndrome caused by a novel AChR e subunit mutation at the agonist binding site. Neurology. 2011;76(Suppl 4):A644.
178. Cheung J, Cossins J, Liu W, Belaya K, Palace J, Beeson D. Pathogenic mechanisms of RAPSN mutations in congenital myasthenic syndromes. Muscle Nerve. 2013;S11.
179. Zoltowska K, Webster R, Finlayson S, Maxwell S, Cossins J, Beeson D. GFPT1 mutations that underlie congenital myasthenic syndrome reduce AChR expression. Muscle Nerve. 2013:S10.
180. Liewluck T, Selcen D, Engel AG. Beneficial effects of albuterol in congenital endplate acetylcholinesterase deficiency and Dok-7 myasthenia. Muscle Nerve. 2011;44(5):789–794.
181. Lashley D, Palace J, Jayawant S, Robb S, Beeson D. Ephedrine treatment in congenital myasthenic syndrome due to mutations in Dok-7. Neurology. 2010;74:1517–1523.
182. Harper CM, Fukodome T, Engel AG. Treatment of slow-channel congenital myasthenic syndrome with fluoxetine. Neurology. 2003;60:1710–1713.
183. Balali-Mood M, Moshiri M, Etemad L. Medical aspects of bio-terrorism. Toxicon. 2013;69:131–142.
184. Arnon SS, Schechter R, Inglesby TV, et al. Botulinum toxin as a biological weapon. JAMA. 2001;285(8):1059–1070.
185. Sobel J. Botulism. Clin Infect Dis. 2005;41(8):1167–1173.
186. Leclair D, Fung J, Isaac-Renton JL, et al. Foodborne botulism in Canada. 1985–2005. Emerg Infect Dis. 2013;19(6):961–968.
187. Woodruff BA, Griffin PM, McCroskey LM, et al. Clinical and laboratory comparison of botulism from toxin types A, B, and E in the United States. 1975–1988. J Infect Dis. 1992;166:1281–1286.
188. Rossetto O, Megighian A, Scorzeto M, Montecucco C. Botulinum neurotoxins. Toxicon. 2013;67(1):31–36.
189. Maselli R, Bakshi N. Botulism. Muscle Nerve. 2000;23:1137–1144.
190. Cherington M. Clinical spectrum of botulism. Muscle Nerve. 1998;21:701–710.
191. Donadia JA, Gangarosa EJ, Faich GA. Diagnosis and treatment of botulism. J Infect Dis. 1971;124:108–112.
192. Schmidt-Nowara WW, Samet JM, Rosario PA. Early and late pulmonary complications of botulism. Arch Intern Med. 1983;143:451–456.
193. Rapoport S, Watkins PB. Descending paralysis resulting from occult wound botulism. Ann Neurol. 1984;16:359–361.
194. Green MA, Heumann MA, Wehr HM, An outbreak of watermelon-borne pesticide toxicity. Am J Public Health. 1987; 77: 1431–1434.
195. Forss N, Ramstead R, Bäcklund T, Lindström M, Kolho E. Difficulties in diagnosing foodborne botulism. Case Rep Neurol. 2012;4:113–115.
196. Wakerley BR, Yuki N. Pharyngeal-cervical-brachial variant of Guillain-Barré syndrome. J Neurol Neurosurg Psychiatry. 2014;85(3):339–344.
197. Cherington M, Electrophysiologic methods as an aid in diagnosis of botulism: A review. Muscle Nerve. 1982;5:S28–S29.
198. Cornblath DR, Sladky JT, Sumner AJ: Clinical electrophysiology of infantile botulism. Muscle Nerve. 1983;6:448–452.
199. Gutmann L, Bodensteiner J, Gutierrez A. Electrodiagnosis of botulism (letter). J Pediatr. 1992;121:835.
200. Kongsaengdao S, Samintarapanya K, Rusmeechan S, Sithinamsuwan P, Tanprawate S. Electrophysiological diagnosis and patterns of response to treatment of botulism with neuromuscular respiratory failure. Muscle Nerve. 2009;40(2):2 71–278.
201. Oh SJ. Botulism: Electrophysiological studies. Ann Neurol. 1977;1:481–485.
202. Cherington M. Botulism: Ten year experience. Arch Neurol. 1974;30:432–437.
203. Vasa M, Baudendistel TE, Ohikhuare CE, et al. Clinical problem-solving. The eyes have it. N Engl J Med. 2012:367(10): 938–943.
204. Jones RG, Marks JD. Use of a new functional dual coating (FDC) assay to measure low toxin levels in serum and food samples following an outbreak of human botulism. J Med Microbiol. 2013;62:828–835.
205. Mazuet C, Ezan E, Volland H, Popoff MR, Becher F. Toxin detection in patients’ sera by mass spectrometry during two outbreaks of type A botulism in France. J Clin Microbiol. 2012; 50(12):4091–4094.
206. Kuruoglu R, Cengiz B, Tokcaer A. Botulism with sensory symptoms diagnosed by neuromuscular transmission studies associated with edrophonium responsiveness. Electromyogr Clin Neurophysiol. 1996;36:477–480.
207. Athwal BS, Gale AN, Brett MM, Youl BD. Wound botulism in the UK. Lancet. 2001;357:234.
208. Hughes JM, Blumenthal JR, Merson MH, Lombard GL, Dowell VR Jr, Gangarosa EJ. Clinical features of types A and B foodborne botulism. Ann Intern Med. 1981;95:442–445.
209. Devers KG, Nine JS. Autopsy findings in botulinum toxin poisoning. J Forensic Sci. 2010;55(6):1649–1651.
210. Thurston D, Risk I, Hill MB, et al. Botulism from drinking prison-made alcohol – Utah. 2011. MMWR Morb Mortal Wkly Rep. 2012;61(39):782–784.
211. Simpson L. The life history of a botulinum toxin molecule. Toxicon. 2013;68:40–59.
212. Maselli RA, Burnett ME, Tonsgard JH. In vitro microelectrode study of neuromuscular transmission in a case of botulism. Muscle Nerve. 1992;15:273–276.
213. Maselli RA, Ellis W, Mandler RN, et al. Cluster of wound botulism in California: Clinical, electrophysiologic, and pathologic study. Muscle Nerve. 1997;20:1284–1295.
214. Centers for Disease Control and Prevention (CDC). Investigational heptavalent botulinum antitoxin (HBAT) to replace licensed botulinum antitoxin AB and investigational botulinum antitoxin E. MMWR Morb Mortal Wkly Rep. 2010;59(10):299.
215. Centers for Disease Control and Prevention (CDC). Notice of CDC’s discontinuation of investigational pentavalent (ABCDE) botulinum toxoid vaccine for workers at risk for occupational exposure to botulinum toxins. MMWR Morb Mortal Wkly Rep. 2011;60(42):1454–1455.
216. Friggeri A, Marçon F, Marciniak S, et al. 3,4-Diaminopyridine may improve neuromuscular block during botulism. Crit Care. 2013;17(5):449.
217. Edlow JA, McGillicuddy DC. Tick paralysis. Infect Dis Clin North Am. 2008;22:397–413.
218. Grattan-Smith PJ. Morris JG, Johnston HM, et al. Clinical and neurophysiological features of tick paralysis. Brain. 1997; 120:1975–1987.
219. Diaz JH. A 60 year meta-analysis of tick paralysis in the United States: A predictable, preventable, and often misdiagnosed poisoning. J Med Toxicol. 2010;6:15–21.
220. Cooper BJ, Spence I. Temperature-dependent inhibition of evoked acetylcholine release in tick paralysis. Nature. 1976; 263(5579):693–695.
221. Emmons P, McLennan H. Failure of acetylcholine release in tick paralysis. Nature. 1959;183(4659):474–475.
222. Dworkin MS, Shoemaker PC, Anderson DE. Tick paralysis: 33 human cases in Washington state, 1946–1996. Clin Infect Dis. 1999;29:1435–1439.
223. Felz MW, Smith CD, Swift TR. Brief report: A 6-year-old girl with tick paralysis. N Engl J Med. 2000;342:90–94.
224. Schaumburg HH, Herskowitz S. The weak child–a cautionary tale. N Engl J Med. 2000;342:127–129.
225. Abbott KH. Tick paralysis: A review. Proc Staff Meet Mayo Clin. 1943;18:39–64.
226. Adler K. Tick paralysis. Can Med Assoc J. 1966;94:550–551.
227. Gorman RJ, Snead OC. Tick paralysis in three children: The diversity of neurologic presentations. Clin Pediatr (Phila). 1978;17:249–251.
228. Mongan PF. Tick toxicosis in North America. J Fam Pract. 1979;5:939–944.
229. Pearn J. Neuromuscular paralysis caused by tick envenomation. J Neurol Sci. 1977;34:37–42.
230. Rose I. A review of tick paralysis. Can Med Assoc J. 1954; 70:175–176.
231. Rose I, Gregson JD. Evidence of neuromuscular block in tick paralysis. Nature. 1956;178:95–96.
232. Stanbury JB, Huyck JH. Tick paralysis: A critical review. Medicine. 1945;24:219–242.
233. Wright SW, Trott AT. North American tickborne diseases. Ann Emerg Med. 1988;17:964–972.
234. Kincaid JC. Tick bite paralysis. Semin Neurol. 1990;10:32–34.
235. Swift TR, Ignacio OJ. Tick paralysis: Electrophysiologic studies. Neurology. 1975;25(12):1130–1133.
236. Masina S, Broady KW. Tick paralysis: Development of a vaccine. Int J Parasitol. 1999;29:535–541.
237. Krishnan AV, Lin CS, Reddel SW, McGRath R, Kiernan MC. Conduction block and impaired axonal function in tick paralysis. Muscle Nerve. 2009;40:358–362.
238. Pecina CA. Tick paralysis. Semin Neurol. 2012;32:531–532.
239. Cherington M, Snyder RD. Tick paralysis: Neurophysiologic studies. N Engl J Med. 1968;278:95–97.
240. DeBusk FL, O’Connor S. Tick toxicosis. Pediatrics. 1972;50: 328–329.
241. Donat JR, Donat JF. Tick paralysis with persistent weakness and electromyographic abnormalities. Arch Neurol. 1981;38:59–61.
242. Morris HH 3rd. Tick paralysis: Electrophysiologic measurements. South Med J. 1977;70:121–123.
243. Vedanarayanan VV, Evans OB, Subramony SH. Tick paralysis in children: Electrophysiology and possibility of misdiagnosis. Neurology. 2002;59:1088–1090.
244. Schmitt N, Bowmer EJ, Gregson JD. Tick paralysis in British Columbia. Can Med Assoc J. 1969;100:417–421.
245. Stone BF, Neish AL. Tick-paralysis toxoid: An effective immunizing agent against the toxin of Ixodes holocyclus. Aust J Exp Biol Med Sci. 1984;62:189–191.
246. Nakajima T, Ohta S, Morita H, Midorikawa Y, Mimura S, Yanagisawa N. Epidemiological study of sarin poisoning in Matsumoto City, Japan. J Epidemiol. 1998;8:33–41.
247. Buckley NA, Eddleston M, Li Y, Bevan M, Robertson J. Oximes for acute organophosphate pesticide poisoning (Review). Cochrane Database Syst Rev. 2011;(2):CD005085.
248. Eddleston M, Buckley NA, Eyer P, Dawson AH. Management of acute organophosphorus pesticide poisoning. Lancet. 2008; 371:597–607.
249. Jokanović M, Prostran M. Pyridinium oximes as cholinesterase reactivators. Structure-activity relationship and efficacy in the treatment of poisoning with organophosphorus compounds. Curr Med Chem. 2009;16(17):2177–2188.
250. Janković M. Medical treatment of acute poisoning with organophosphorus and carbamate pesticides. Toxicol Lett. 2009;190(2):107–115.
251. Sungur M, Güven M. Intensive care management of organophosphate insecticide poisoning. Crit Care. 2001;5:211–215.
252. Besser R, Gutmann L, Dillman U, Weilemann LS, Hoff HC. End-plate dysfunction in acute organophosphate intoxication. Neurology. 1989;39:561–567.
253. Green MA, Heumann MA, Wehr HM, et al. An outbreak of watermelon-borne pesticide toxicity. Am J Public Health. 1987;77:1431–1434.
254. Eddleston M, Eyer P, Worek F, et al. Pralidoxime in acute organophosphate poisoning – a randomised controlled trial. PLoS Med. 2009;6(6):e10000104.
255. Kularatne SA, Senanayake N. Venomous snake bites, scorpions, and spiders. Handb Clin Neurol. 2014;120:987–1001.
256. Del Brutto OH. Neurological effects of venomous bites and stings: Snakes, spiders, and scorpions. Handb Clin Neurol. 2013;114:349–368.
257. White J. Bites and stings from venomous animals: A global overview. Ther Drug Monit. 2000;22(1):65–68.
258. Faiz A, Ghose A, Ahsan F, et al. The greater black krait (Bungarus niger), a newly recognized cause of neuro-myotoxic snake bite envenoming in Bangladesh. Brain. 2010;133:3183–3193.
259. Singh G, Pannu HS, Chawla PS, Malhotra S. Neuromuscular transmission failure due to common krait (Bungarus Caeruleus) envenomation. Muscle Nerve. 1999;22:1637–1643.
260. Tormoehlen L. Toxins and disorders of neuromuscular transmission. Annual Meeting of the American Academy of Neurology. 2013
261. Norris RL, Pfalzgraf RR, Gavin Laing G. Death following coral snake bite in the United States – First documented case (with ELISA confirmation of envenomation) in over 40 years. Toxicon. 2009;53(6):693–697.
262. Pettigrew LC, Glass JP. Neurologic complication of a coral snake bite. Neurology. 1985;35:589–592.
263. Allen C. Arachnid envenomations. Emerg Med Clin North Am. 1992;10(2):269–298.
264. Clark RF, Wethern-Kestner S, Vance MV, Gerkin R. Clinical presentation of black widow spider envenomation: A review of 163 cases. Ann Emerg Med. 1992;21:782–787.
265. Howard BD, Gundersen CB Jr. Effects and mechanisms of polypeptide neurotoxins that act presynaptically. Annu Rev Pharmacol Toxicol. 1980;20:307–336.
266. Russel FE. Venomous arthropods. Vet Hum Toxicol. 1991; 33:505–508.
267. Swift TR. Disorders of neuromuscular transmission other than myasthenia gravis. Muscle Nerve. 1981;4:334–353.
268. Golcuk Y, Velibey Y, Gonullu H, Sahin M, Kocabas E. Acute toxic fulminant myocarditis after a black widow spider envenomation: Case report and literature review. Clin Toxicol (Phila). 2013;51(3):191–192.
269. Auerbach PS. Marine envenomations. N Engl J Med. 1991;325 (7):486–493.
270. Balhara KS, Stolbach A. Marine envenomations. Emerg Med Clin North Am. 2014;32(1):223–243.
271. Kerr LM, Yoshikami D. A venom peptide with a novel presynaptic blocking action. Nature. 1984;308:282–284.
272. Olivera BM, Gray WR, Zeikus R, et al. Peptide neurotoxins from fish-hunting cone snails. Science. 1985;230:1338–1343.
273. Sano K, Enomoto K, Maeno T. Effects of synthetic ω-conotoxin, a new type of Ca2+ antagonist, on frog and mouse neuromuscular transmission. Eur J Pharmacol. 1987;141:235–241.
274. Hunter JM. New neuromuscular blocking drugs. N Engl J Med. 1995;332(95):1691–1699.
275. Gooch JL, Moore MH, Ryser DK. Prolonged paralysis after neuromuscular junction blockade: Case report and electrodiagnostic findings. Arch Phys Med Rehabil. 1993;74:1007–1011.
276. Riddings LW, Jackson CE, Rogers S, et al. Electrophysiologic evidence for neuromuscular blockade following prolonged paralysis with curare-like agents. Muscle Nerve. 1992;15:1205–1206.
277. Yee WC, Lopate G, Peeples D, et al. Postparalysis paralysis syndrome: Sequential electrodiagnostic studies. Muscle Nerve. 1992;15:1206.
278. Howard JF Jr. Adverse drug effects on neuromuscular transmission. Semin Neurol. 1990;10:89–102.
279. Atchison WD, Adgate L, Beaman CM. Effects of antibiotics on uptake of calcium into isolated nerve terminals. J Pharmacol Exp Ther. 1988;245:394–401.
280. Argov Z, Mastaglia FL. Disorders of neuromuscular transmission caused by drugs. N Engl J Med. 1979;301:409–413.
281. Klopstock T. Drug-induced myasthenic syndromes. www.medlink.com. 2009.
282. Bever CT Jr, Chang HW, Penn AS, Jaffe IA, Bock E. Penicillamine-induced myasthenia gravis: Effects of penicillamine on acetylcholine receptor. Neurology. 1982;32:1077.
283. Albers JW, Hodach RJ, Kimmel DW, Treacy WL. Penicillamine-associated myasthenia gravis. Neurology. 1980;30:1246–1249.
284. Albers JW, Beals CA, Levine SP. Neuromuscular transmission in rheumatoid arthritis, with and without penicillamine treatment. Neurology. 1981;31:1562–1564.
285. Russell AS, Lindstrom JM. Penicillamine-induced myasthenia gravis associated with antibodies to acetylcholine receptor. Neurology. 1978;28:847–849.
286. Batocchi AP, Evoli A, Servidei S, Palmisani MT, Apollo F, Tonali P. Myasthenia gravis during interferon-alpha therapy. Neurology. 1995;45:382–383.
287. Andonopoulos AP, Terzis E, Tsibri E, Papasteriades CA, Pappetropoulos T. D-Penicillamine induced myasthenia gravis in rheumatoid arthritis: An unpredictable common occurrence? Clin Rheumatol. 1994;13:568–588.
288. Bruggeman W, Herath H, Ferbert A. Follow-up and immunologic findings in drug-induced myasthenia. Med Klin. 1996;91:268–271.
289. Buchnall RC, Dixon A, St J, Glick EN, Woodland J, Zutshi DW. Myasthenia gravis associated with penicillamine treatment for rheumatoid arthritis. Br Med J. 1975;1:600–602.
290. Drosos AA, Christou L, Galanopoulou V, Tzioufas AG, Tsiakou EK. D-Penicillamine induced myasthenia gravis: Clinical, serological and genetic findings. Clin Exp Rheumatol. 1993;11:387–391.
291. Fawcett PR, McLachlan SM, Nicholson LV, Argov Z, Mastaglia FL. D-Penicillamine-associated myasthenia gravis: Immunological and electrophysiological studies. Muscle Nerve. 1982;5:328–334.
292. Liu GT, Bienfang DC. Penicillamine-induced ocular myasthenia gravis in rheumatoid arthritis. J Clin Neurol Ophthalmol. 1990;10:201–205.
293. Masters CL, Dawkikns RL, Zilko PJ, Simpson JA, Leedman RJ. Penicillamine-associated myasthenia gravis, antiacetylcholine receptor and antistriational antibodies. Am J Med. 1977;63:689–694.
294. Raynauld JP, Lee YS, Kornfeld P, Fries JF. Unilateral ptosis as an initial manifestation of D-penicillamine induced myasthenia gravis. J Rheumatol. 1993;20:1592–1593.
295. Vincent A, Newsom-Davis J, Martin V. Anti-acetylcholine receptor antibodies in D-penicillamine-associated myasthenia gravis [letter]. Lancet. 1978;1:1254.
296. Vincent A, Newsom-Davis J. Acetylcholine receptor antibody characteristics in myasthenia gravis. II. Patients with penicillamine-induced myasthenia or idiopathic myasthenia of recent onset. Clin Exp Immunol. 1982;49:266–272.
297. Lensch E, Faust J, Nix WA, Wandel E. Myasthenia gravis after interferon-alpha treatment. Muscle Nerve. 1996;19:927–928.
298. Mase G, Zorzon M, Biasutti E, et al. Development of myasthenia gravis during interferon-alpha treatment for anti-HCV positive chronic hepatitis. J Neurol Neurosurg Psychiatry. 1996;60:348–349.
299. Perez A, Perella M, Pastor E, Cano M, Secudero J. Myasthenia gravis induced by alpha-interferon therapy. Am J Hematol. 1995;49:365–366.
300. Piccolo G, Franciotta D, Versino M, Alfonsi E, Lombardi M. Poma G. Myasthenia gravis in a patient with chronic active hepatitis C during interferon-alpha treatment. J Neurol Neurosurg Psychiatry. 1996;60:348.
301. Adams RJ, Rivner MH, Salazar J, Swift TR. Effects of oral calcium antagonists on neuromuscular transmission. Neurology. 1984;34 (Suppl 1) 132–133.
302. Streib EW. Adverse effects of magnesium salt cathartics in a patient with the myasthenic syndrome (Lambert–Eaton syndrome). Ann Neurol. 1977;2:175–176.
303. Bashuk RG, Krendel DA. Myasthenia gravis presenting as weakness after magnesium administration. Muscle Nerve. 1990;13:708–712.
304. Castlebaum AR, Donofrio PD, Walker FO, Troost BT. Laxative abuse causing hypermagnesemia, quadriparesis, and neuromuscular junction defect. Neurology. 1989;39:746–747.
305. Krendel DA. Hypermagnesemia and neuromuscular transmission. Semin Neurol. 1990;10:42–45.
306. Swift TR. Weakness from magnesium containing cathartics. Muscle Nerve. 1979;2:295–298.
307. Flowers CJ Jr. Magnesium in obstetrics. Am J Obstet Gynecol. 1965;91:763–776.
308. Kaire GH. Isolation of tick paralysis toxin from Ixodes holocyclus. Toxicon. 1966;4:91–97.





