Chapter 9
Microfluidics: Basic Physics and Concepts
9.1 Chapter Overview
Microfluidics is the science of fluid flow in structures that have at least one dimension in the microscale (between 1 micrometre and 1 millimetre). Although the terms are often used interchangeably, Lab-on-a-Chip is used to describe devices that integrate several laboratory processes on a single chip, whereas Micro-Total-Analysis Systems (μTAS) are considered to integrate all laboratory processes required for an analysis on a single chip. For both cases fluid flow in one or more channel networks, fabricated into or from a solid substrate, is an essential element of the analytical or preparative function of the device.
From this chapter readers will gain an appreciation of:
9.2 Liquids and Gases
Fluids, unlike solids, do not resist shearing forces such as those acting at a solid surface. They continue to deform as long as the force is applied and assume the shape of the solid boundary, whereas solids can resist such shear and maintain an unsupported shape. As shown in Figure 9.1, fluid motion is controlled by the interaction and internal shear between fluid layers.
Figure 9.1 Fluids deform under the action of a shearing force (τ). The fluid can be considered as laminas parallel to a surface. Each fluid lamina applies a shear force τ to the next one, and is in turn sheared by those it touches.

Although they respond very differently to changes in pressure and temperature, the term ‘fluid’ includes both liquids and gases. Gases can be expanded and compressed more easily than liquids due to the lower density and larger spacing between molecules. At the molecular scale (~10−9 m) the interaction between layers involves collisions of many molecules. At the macroscale scale (>10−4 m) the physical properties of a fluid result from the statistical averages of such molecular interactions. The effects of individual molecular collisions can be ignored and we can deal with the liquid's bulk, or continuum, properties.
9.2.1 Gases
The molecules in a gas are widely spaced and interactions between them (apart from collisions) are weak, especially at low pressures. An increase of temperature increases the kinetic energy of the molecules, mass transfer between gas layers increases and viscosity increases. In gases, except for extremely high pressures, viscosity is independent of pressure. At a sufficiently low pressure, where intermolecular interactions are negligible, all gases obey the ideal gas law

where P is the pressure, V the volume, n the amount (moles) of substance of gas molecules, and T the absolute temperature. In this equation R is the gas constant given by R = kNA, where k is Boltzmann's constant = 1.38 × 10−23 J K−1, and NA is Avogadro's constant = 6.022 × 1023 mol−1. The ideal gas law follows from experiment (e.g. Boyle's Law) and Avogadro's Hypothesis (formulated in 1810) that:
Equal volumes of gases at the same temperature and pressure contain the same number of molecules.
Thus, a mole of hydrogen (2 gm) and a mole of oxygen (32 gm) at the same temperature and pressure occupy the same volume. At standard temperature and pressure (STP: 273.15 K, 100 kPa) this volume is 22.414 litres.
9.2.1.1 Mean Free Path between Molecular Collisions in a Gas
Although on average the molecules in a gas are widely spaced apart, they are in constant motion and often collide with each other. If we treat each molecule as a hard sphere of diameter d, the effective collision area for two colliding molecules can be taken as a circle of diameter 2d. The effective cross-sectional collision area Ac for a molecule is thus

The frequency of collisions between sets of two molecules will depend upon their relative velocity  of approach. For two molecules having random velocities
of approach. For two molecules having random velocities  and
and  this is given by their vector difference (see Figure 9.26 for an example)
this is given by their vector difference (see Figure 9.26 for an example)

The magnitude of the relative velocity is given by

Velocities  and
and  are random and uncorrelated, and because the same average velocity
are random and uncorrelated, and because the same average velocity  is associated with each molecule we have
is associated with each molecule we have

Over time t a collision cross-section associated with one molecule moving with an average velocity  will travel a path length
will travel a path length  . The volume of collision space swept through during this time will be
. The volume of collision space swept through during this time will be  . The mean free path length Lmfp can then be taken as the path length
. The mean free path length Lmfp can then be taken as the path length  divided by the number of molecular collisions:
divided by the number of molecular collisions:

Nv is the number of molecules per unit volume, and can be found from Avogadro's number and the ideal gas law given by Equation (9.1):

The mean free path length between collisions of molecules in a gas is therefore

In the worked Example 10.3 in Chapter 10 we find that the average distance of around 140 nm between collisions of nitrogen molecules in nitrogen gas is more than 550-times their molecular diameter (~0.25 nm) and some 40-times larger than their average molecular separation distance of 3.3 nm. This demonstrates that molecules in a gas can travel (in straight paths) over significant distances at the molecular scale before they collide with another molecule.
9.2.2 Liquids
The molecules in a liquid are closer together than for a gas, and cohesive forces such as those arising from intermolecular van der Waals interactions (see Chapter 1, Section 1.2.5) give rise to viscous effects. Glass and molten polymers are highly viscous because their large molecules get entangled. Water has a higher viscosity than liquids such as benzene or alcohols because of its network of cohesive hydrogen-bonds. An increase of temperature, and hence of molecular kinetic energy, reduces these cohesive forces and hence the viscosity. An increase of molecular kinetic energy will also facilitate an increased molecular interchange between the fluid layers, which will increase viscosity. This produces a relatively small effect and so the net result is that liquids show a reduction in viscosity for an increase in temperature. With increasing pressure the energy required for relative movement of molecules is increased, and the viscosity is increased.
9.3 Fluids Treated as a Continuum
When treated as a continuum, the properties of a fluid such as density, pressure and velocity remain constant at any defined point and changes in these properties due to molecular motions are taken to be negligible. The physical properties of fluids can be defined as continuous functions of time and space.
9.3.1 Density
This is defined as the mass contained within a unit volume, and is computed as the product of molecular mass m and the number of molecules N per unit volume V:

Molecular mass ‘m’ is the mass of the molecule given by m = Mwmu, where Mw is the molecular weight (molecular mass relative to  ) and mu is the atomic mass unit (1.6606 × 10−27 kg). We can interpret the ideal gas law of Equation (9.1) as stating that pressure is linearly proportional to the product of temperature and density.
) and mu is the atomic mass unit (1.6606 × 10−27 kg). We can interpret the ideal gas law of Equation (9.1) as stating that pressure is linearly proportional to the product of temperature and density.
9.3.2 Temperature
Temperature relates to the translational kinetic energy E of N molecules in a particular volume domain – each molecule having velocity vj and mass m:

The kinetic theory of gases uses statistical mechanics to relate the temperature to the average kinetic energy of the atoms in the system. At bulk scales the number of molecules is (almost) infinite and their average squared velocities can be assumed to follow the Maxwell distribution to be described in Section 9.3.4. In one dimension, the average kinetic energy of the molecules is related to the temperature as:

For a three-dimensional domain we have:

This relationship is an important aspect of our understanding of gas pressure.
9.3.3 Pressure
Pressure is the force imparted by collisions of molecules against a unit area of surface. Consider, as depicted in Figure 9.2, a single perfectly elastic molecule, of mass m, bouncing rapidly back and forth with velocity vx between two solid surfaces distance L apart. The motion of the particle is assumed to remain on the same path parallel to the x-axis between the two surfaces. What is the resulting force exerted on each of the two surfaces?
Figure 9.2 A perfectly elastic molecule bouncing between two solid boundaries with velocity vx along a path parallel to the x-axis.

We will use Newton's law (force = mdv/dt) in the form

The momentum lost Δp by a single molecular collision with the right-hand surface of Figure 9.2 is given by

The time Δt between successive collisions is

The force F is given by

The same force will be exerted on the left-hand surface. We now generalise the situation to a large number of molecules colliding continuously with the walls of a cubic box of side L, as shown in Figure 9.3.
Figure 9.3 A large number of molecules in motion within a cubic box and continually colliding with the internal walls.

The force acting on each of the six walls of the cubic box is:

We assume that the molecules are equally likely to be moving in any direction, so that the average value of  is the same as that of
is the same as that of  or
or  . The average squared velocity of the N molecules is given by:
. The average squared velocity of the N molecules is given by:

Defining pressure p as the force per unit area, and noting that the product of cross-sectional area A and length L gives the volume V, from Equations (9.4) and (9.5) we have:

The macroscopic pressure of a gas therefore relates directly to the average kinetic energy per molecule. From Equations (9.2) and (9.6) we have

thereby describing pressure as a function of density and kinetic energy of molecules.
From Equations (9.1) and (9.6) we have

In this equation we can replace n, the moles of gas, with n = N/NA = Nk/R, so that

to give the result quoted in Equation (9.3).
9.3.4 Maxwell Distribution of Molecular Speeds
Equation (9.6) connects the dynamics of the system at the molecular level to the macroscopic pressure, which represents a physical property averaged over a very large number of molecules. The model used to derive this equation is simplistic. It uses Newton's law and assumes that the molecules do not interact through van der Waals forces, for example, or collide with each other. The molecules are depicted as moving along straight paths between their collisions with the solid surfaces, and that they behave as perfectly elastic bodies – which they are not – during a collision process. For a system of gas particles to reach thermal equilibrium with their neighbouring molecules and enclosing walls, inelasticity is in fact required to provide the necessary energy exchanges. Maxwell understood that at equilibrium the distribution of molecular speeds remains constant with time. Also, if we ignore the effects of gravity, the molecules will on average be evenly distributed throughout containers such as that shown in Figure 9.3. Therefore, we do not require details of the position and velocity of each molecule as a function of time.
In Section 9.3.3 the term velocity was employed because both the direction and speed of molecular motion was specified. The term speed on the other hand is a scalar not a vector quantity – it describes how fast a molecule is moving but not the direction of motion. When considering an assembly of molecules moving in three-dimensional space, for any given molecular speed there will be many possible velocity vectors. We need therefore to consider

Equation (9.7) represents a sphere of radius v centred at the origin. Thus, for a particular speed v all of the possible velocity vectors lie on the surface of a sphere of radius v. As v is made larger, the sphere increases in size and the number of possible velocity vectors increases in proportion to 4πv2. However, this situation cannot go on indefinitely because at some stage there must be fewer and fewer molecules to be found proceeding to higher speeds. Our task is to found out how the speeds of all the N molecules in the cubic box are distributed in this spherical volume of velocity space (see Figure 9.4).
Figure 9.4 For a particular molecular speed v all of the possible velocity vectors lie on the surface of a sphere of radius v. The distribution function f(v) describes the probability of finding a molecule of speed between v and v + dv.

It can be shown statistically that if there are N molecules in a defined volume of gas in velocity space, the fluctuation of the number with time is of the order one part in N1/2. In Chapter 10, Example 10.3, we determine that a cubic volume 10 μm × 10 μm × 10 μm of nitrogen gas at STP contains around 1010 nitrogen molecules. The corresponding number fluctuation for even this small volume of gas is of the order one part in 105. For a macroscopic volume of gas we can therefore assume the molecular density to be smoothly distributed in both ordinary and velocity space.
We will define f(vx) to be the probability distribution function of velocities, so that f(vx) dvx is the fraction of the total number of molecules in a fixed volume to have a velocity between vx and vx + dvx. The sum of all these possible fractions must be 1:

For a volume of gas containing N molecules, the number of molecules having velocity between vx and vx + dvx is N f(vx)dvx. Extending this to three-dimensional velocity space, the number of molecules of velocity lying between vx and vx + dvx, vy and vy + dvy, and vz and vz + dvz, is:

Because the probability distribution function must depend only on the total speed of a molecule and not on the separate velocity components, Maxwell deduced that

Thus, the product of the distribution functions manifests itself as the sum of the velocity components. Solutions for Equation (9.8) take the form
(9.9) 
The number of molecules in a spherical elemental shell of volume dvxdvydvz can thus be given as:

For very small speed increments dv, the volume of velocity space between two spheres of radius v and v + dv, both centred at the origin, is equal to 4πv2dv. The fractional distribution function f(v)dv is thus the result given by Equation (9.10) multiplied by the factor 4πv2dv/N:

The sum of all the fractions of molecules in velocity space must together add up to 1:

The final result we require makes use of standard integrals to solve for Equation (9.11) together with Equation (9.3) to find the average kinetic energy per molecule:

This gives B = m/2kT and A = 4π(m/2πkT)3/2 to be used in Equation (9.11), so that Maxwell's probability distribution function for the molecular speeds is given by:

Examples of this speed distribution for nitrogen gas at three different temperatures are presented in Figure 9.5. At the lower speeds the function increases from zero at a parabolic rate to a maximum value and then decreases exponentially with increasing speed. The area under each curve must, according to Equation (9.11), equal 1.
Figure 9.5 The Maxwell speed distribution function for nitrogen gas at three different temperatures.

The maximum speed value can be obtained by differentiating Equation (9.12), equating this to zero and solving for vmax. The result we obtain is:

The mean (arithmetic average) speed  v
v is given by:
is given by:

Equation 9.13 can be used to determine the mean time between collisions of the nitrogen molecules, as well as the collision frequency (see Example 10.5 in Chapter 10). The root-mean-square speed, associated with the mean kinetic energy, is given by:

The kinetic energy E is equal to mv2/2, and so from equation (9.12) the speed distribution can also be given as

The Maxwell distribution function is only valid for atoms or molecules in the gaseous state. The laws of classical physics can be applied to particles. However, molecules or atoms in the liquid or solid state have to obey the laws of quantum statistics, in which case only certain energy values, rather than a continuous distribution, are permitted.
9.3.5 Viscosity
Viscosity is the measure of the effort required to deform a fluid and is often discussed in terms of Couette flow, corresponding to the situation shown in Figure 9.6 where a fluid is contained between a moving plate and a parallel stationary plate. The action of rubbing a cream lotion into skin corresponds to applying a shear stress to the lotion, which will then experience Couette flow. In a macro-scale Couette flow device the fluid velocity immediately next to a surface will equal the velocity of that surface. This is referred to as zero slip. If the fluid is a Newtonian fluid, such as water, the fluid velocity will change smoothly from zero at the stationary surface to the velocity of the moving surface. In other words the spatial gradient of the fluid velocity dv/dy is a constant.
Figure 9.6 Couette flow induced in a fluid contained between a moving plate and a stationary surface. For a Newtonian fluid the fluid velocity v will change smoothly from zero at the stationary surface to the velocity of the moving plate. The spatial velocity gradient dv/dx is a constant.

The dynamic viscosity η is defined as the proportionality constant between the shear stress τ applied to the fluid and the resulting rate of shear strain. The shear strain is defined as the ratio dy/dx of the lateral deformation dy to the thickness dx of the layer being displaced, and the rate of shear strain is simply (dy/dx)/dt = dv/dx (namely, the induced velocity gradient). For a Newtonian fluid we therefore have the following relationship between stress, rate of shear strain, and the dynamic viscosity:

We can interpret this relationship as indicating that for η = 1 Pa.s and τ = 1 Pa, the mobile plate moves in one second a distance equal to the thickness of the fluid layer between the plates. Values of the dynamic viscosity for some liquids and gases are given in Table 9.1.
Table 9.1 Viscosity of some liquids and gases (293 K unless specified).

The viscosity of a Newtonian fluid depends only on temperature and concentration (if diluted with another miscible fluid). For some fluids, particularly molten polymers or biological fluids such as blood, their viscosity depends also on the internal stress. These are classed as non-Newtonian fluids. Their viscosity decreases with an increase of the rate of the applied shear stress dτ/dt applied to a fluid flowing between two parallel surfaces, one moving at a constant velocity and the other one stationary, and is defined by:

where v is the velocity of the moving surface, and h is the distance between the two parallel surfaces.
Non-Newtonian fluids exhibit viscoelastic behaviour (shear-thinning) and some require an initial shear stress that must be applied before they begin to flow – as for the case of whole blood, for example. Viscoelastic fluids exhibit a relaxation time, typically ranging from milliseconds to seconds, given by the reciprocal of the critical shear rate. The critical shear rate corresponds to the shear threshold at which the viscosity begins to change or, for the case of molten polymers, where the polymer chains make the transition from a coiled to a stretched configuration.
9.4 Basic Fluidics
9.4.1 Static Fluid Pressure
The pressure exerted by a static fluid arises from the weight of that fluid and so depends only upon the fluid depth h, its density ρ, and the acceleration of gravity g:

Because the fluid pressure at a given depth h does not depend upon the total mass or total volume of the liquid, the shape of the fluid container is also not relevant. Examples of this are given in Figure 9.7.
Figure 9.7 The static fluid pressure P at a given depth h does not depend upon the total mass or total volume of the liquid, and so the shape of the vessel is irrelevant. The static fluid pressure is given as P = ρgh, where ρ is the fluid density and g the acceleration of gravity.

Pressure is measured in units of Pascals, but it is also not unusual to find pressures expressed in column height units (e.g. mm Hg), reflecting the height dependence shown in Equation (9.15).
9.4.2 Pascal's Law
Pascal's law is expressed as:
The pressure exerted anywhere in an enclosed, incompressible, static fluid is transmitted equally in all directions throughout the fluid.
This follows from the above description of static fluid pressure given by Equation (9.15), namely that the change in pressure between two elevations is due to the weight of the fluid between the elevations regardless of the geometry of the container.
Pascal's law can be interpreted to indicate that any change in pressure applied at any given point of the fluid is transmitted undiminished throughout the fluid. As shown in Figure 9.8 this makes possible a large multiplication of force and forms the basis for the operation of a hydraulic press that provides the means to lift a heavy weight with a small force.
Figure 9.8 Demonstrations of Pascal's Law: (a) The pressure in a vessel is transmitted equally throughout a fluid; (b) The operation of a hydraulic press relies on the fact that any change in pressure applied at any given point of a fluid is transmitted undiminished throughout the fluid.

In Figure 9.8a the fluid pressure P2 at the base of the vessel is P2 = P1 + ρgh. The force F2 acting on the base is given by

Because A2  A1 there is an amplification of force. This is demonstrated in the basic hydraulic press depicted in Figure 9.8b. If we ignore frictional losses
A1 there is an amplification of force. This is demonstrated in the basic hydraulic press depicted in Figure 9.8b. If we ignore frictional losses


which is equivalent to the action of a fluidic lever.
9.4.3 Laplace's Law
Laplace's law states that
The tension on the wall of a cylindrical chamber is the product of the pressure times the radius of the cylinder (or half that value for a spherical chamber).
Thus, a vessel of large radius will require a larger wall tension than one of smaller radius to withstand a given internal fluid pressure. Also, for a given vessel radius and internal pressure, a spherical vessel will have half the wall tension of a cylindrical vessel. This is demonstrated in Figure 9.9.
Figure 9.9 Laplace's Law informs us that the tension on the wall of a cylindrical vessel is twice that for a spherical vessel of the same radius and internal pressure.

An explanation of why wall tension increases with radius is given in Figure 9.10. Basically, if the fluid pressure remains constant then the inward component (Tsinθ) of the wall tension must remain the same. If the wall curvature (sinθ) is less, the total wall tension must increase in order to obtain the same inward component of tension.
Figure 9.10 Wall tension T increases with vessel radius because for a fixed internal pressure P, the counter component of the wall tension Tsinθ must equal P.

The flow of blood in arteries and veins is a good example of Laplace's law in action. The larger arteries of the body are subject to higher wall tensions than the smaller arteries having comparable blood pressures. Arteries are reinforced by fibrous bands to strengthen them against the risks of an aneurysm (capillaries with their very thin walls rely on their small radii). If an artery wall develops a weak spot and expands as a result, the expansion subjects the weakened wall to even more tension. The weakened vessel may continue to expand in what is called an aneurysm, and lead to rupture of the vessel. This is why aneurysms require prompt medical attention.
9.5 Fluid Dynamics
We will now consider the flow of fluids in channels and the forces that act on them.
9.5.1 Conservation of Mass Principle (Continuity Equation)
Fluid flowing steadily through a pipe of reducing cross-sectional area is shown in Figure 9.11. We can assume that all of the fluid mass passing through area A2 will exit the pipe and be measured as Q in units of gm s−1. If ρ2 is the density of the fluid flowing through A2 the value for Q is equal to A2ρ2v2, where v2 is the effective velocity of the fluid flow through A2.
Figure 9.11 Fluid flow through a pipe whose cross-sectional area reduces from A1 to A2. The rate of fluid flow Q through the pipe can be determined as either volumetric flow (dm3 s−1) or mass flow (gm s−1). The conservation of mass principle dictates that A1v1 = A2v2 where v1 and v2 are the average fluid velocities through A1 and A2, respectively. Because A1 > A2, then v1 < v2.

No fluid can exit or enter the pipe between areas A1 and A2, and so from the conservation of mass principle the mass of fluid crossing each section of the pipe per unit time must be the same:

This relationship, which takes the form of the equation of continuity, can be expressed as the law of conservation of mass in fluid dynamics:

We will only consider fluids that are incompressible, and so the density of the fluid will be constant (ρ1 = ρ2). The conservation of mass principle can thus be written as

or

The reduction in pipe area shown in Figure 9.11 indicates that v2 > v1.



9.5.2 Bernoulli's Equation (Conservation of Energy)
Referring to Figure 9.11, the principle of conservation of mass informs us that the fluid velocity is greatest in the part of the pipe having the smaller cross-sectional diameter. In passing from the large cross-sectional area to the smaller one the fluid velocity increases. This corresponds to an acceleration of the fluid mass, which in turn requires an unbalanced force in the form of a pressure gradient exerted on the fluid by the walls of the pipe. As depicted in Figure 9.12, the pressure P1 in the large area of the pipe must be greater than P2 in order to accelerate the fluid. Likewise, if the fluid flow is reversed, P1 must exceed P2 in order to bring about deceleration of the fluid.
Figure 9.12 Fluid flowing through a constriction. A pressure drop ΔP will occur across the constriction, with P1 > P2 because v1 < v2.

Bernoulli's principle states that, for viscous free fluid flow, an increase of the fluid velocity occurs simultaneously with a decrease in fluid pressure or a decrease in the fluid's potential energy. This principle can be applied to various types of fluid flow and quantified using various forms of what is known as Bernoulli's equation. A simple form of this equation is valid for incompressible fluids and for compressible gases moving at speeds well below the velocity of sound in a particular gas. This equation can be derived from the principle of conservation of energy, which states that in a steady fluid flow the sum of all forms of mechanical energy remains constant. along a flow line. The fluid possesses kinetic energy due to its motion, and because of its location in the earth's gravitational field it also possesses potential energy. Work is also being done on the fluid due to the static pressure acting on it. If there are no frictional losses we can apply the law of conservation of energy and write Bernoulli's equation as:

where P is the static pressure, h the height above some reference level, v the velocity, ρ the density, and g the acceleration due to gravity at any chosen elemental volume in the fluid flow line. The term (½ρv2) is known as the dynamic pressure, and the total pressure is the sum of the static pressure P and this dynamic pressure. The sum of the elevation h and static pressure head (P/ρg) is known as the hydraulic head.
A consequence of Bernoulli's principle is demonstrated in Figure 9.13, which shows an aerofoil-shaped object in flowing fluid. The fluid flows more rapidly over the top surface than over the lower surface. Faster fluid flow implies a lower pressure, so that the pressure will be greater on the bottom surface of the aerofoil and produce an upward lift force. This is the principle used in the design of aircraft wings and propeller blades, for example. An inverted version of the shape shown in Figure 9.13 will result in a downward force, which is an effect used in racing car spoilers.





9.5.3 Poiseuille's Law (Flow Resistance)
Bernoulli's principle assumes the fluid flow is not influenced by viscous forces. In fact, for the case of smooth, turbulence free, fluid flow the viscous shearing forces shown in Figure 9.1 will determine the fluid velocity profile across a channel. There will be zero fluid slip at the surfaces of the channel walls and the flow velocity will increase towards the centre line of the channel. The consequence of this is that in order to pump a viscous fluid along a channel a pressure difference ΔP must be applied between its inlet and outlet, irrespective of any changes of the channel diameter. In the 1840s Poiseuille experimentally and then theoretically derived the following relationship for fluid flow in pipes of circular-cross section:

where L is the length of the pipe, r its internal radius, and η is the dynamic viscosity of the fluid. This is also known as the Hagen-Poiseuille relationship in recognition of the contributions made by Hagen.
The flow resistance R of a channel is defined from the relationship

where Q is the volumetric flow rate. From Equations (9.17) and (9.18) the flow resistance of a channel of circular cross-section is thus given as

In practice, fluidic channels of either a rectangular or semicircular cross-section are easier to fabricate than those of circular cross-section (e.g. by placing a flat plate on top of a rectangular or rounded trench). The fluidic resistance of a rectangular channel with a high aspect ratio (i.e. width w height h) can be calculated using the formula
(9.20) 
For a channel of semicircular cross-section defined by a radius of curvature r

Thus, for any specified channel geometry, the flow resistance is directly proportional to the viscosity of the fluid. From Table 9.1 we can see that the viscosity values for gases are considerably smaller than those for liquids. Bernoulli's approximation that fluid flow is not influenced by viscous forces may thus be adequate for the flow of gases, but should not be adopted when considering the flow of liquids. For example, the flow resistance of water will be about 50-times greater than for the flow of a gas along the same channel, and this difference increases to a factor of ~105 for the flow of a highly viscous fluid such as glycerine.

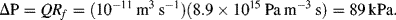

9.5.4 Laminar Flow
Figure 9.6 depicts a fluid moving in laminas with successively higher velocity. The flow velocity is zero in the vicinity of a stationary wall and increases away from the stationary wall. The fluid flow is a function of the x-coordinate and not of the y- and z-directions. This is termed as laminar flow. As depicted in Figure 9.14 we can envisage laminar flow in a pipe of circular cross-section to take the form of concentric, thin-walled, tubes of fluid whose velocities increase from zero at the pipe wall to a maximum at the centre line of the pipe. The flow is directed along the pipe's axis and there are no pressure gradients across the pipe diameter. A shear stress τ exists between each tube and increases by dτ for each tube. A pressure drop between the ends of the fluid tube is required to overcome the shear stress. It is normally assumed that the pressure declines uniformly with distance down the fluid stream, so the pressure gradient ΔP/ΔL is assumed to be constant.
Figure 9.14 Laminar flow in a cylindrical pipe can be depicted as a series of concentric ‘stream tubes’ of length ΔL whose velocities increase as a function of the distance (R-r) from the pipe wall towards the centre axis of the pipe.

Consider the elemental fluid tube shown in Figure 9.14, of length ΔL, radius r and thickness dr. If τ is the shear stress per unit area acting on the surface of this tube, the shear force Fs is given by

From Equation (9.14) τ = ηdv/dx = −ηdv/dr (x = R − r)
to give

At equilibrium this shear force will balance the force acting on the ends of the fluid tube as a result of the pressure difference ΔP:

to give

The velocity v of a fluid tube at any radius r is found by integrating between the limits v = 0 (r = R) and u = u for r = r:

from which

The fluid velocity profile across the pipe is therefore parabolic, as shown in Figure 9.15, with zero velocity at the pipe walls and a maximum velocity along the central axis (at r = 0). The maximum velocity is given as

Figure 9.15 Laminar flow exhibits a parabolic fluid velocity profile, as described by Equation (9.22). The fluid velocity is zero at the channel wall and reaches a maximum at the centre line of the channel.
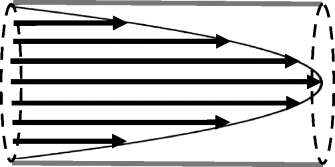
The mean velocity v is the averaged velocity in the cross-section

which corresponds to half the maximum value. The volumetric flow rate Q is given by the product of the mean velocity and the cross-sectional area:

corresponding to the Hagen-Poiseuille relationship of Equation (9.17).
We should note that the Hagen-Poiseuille relationship is derived assuming that the walls of the pipe or channel are perfectly smooth, so that the fluid flow has a unique axial component and no transverse components. If the walls are sufficiently rough to induce 3-dimensional components of fluid flow near the wall surfaces the pressure drop will tend to be greater than that predicted by Equation (9.17) and the fluid flow resistance will also be larger.
9.5.5 Application of Kirchhoff's Laws (Electrical Analogue of Fluid Flow)
Equation 9.18 can be written in the form

This takes the same form as Ohm's law that relates the current I generated along an electrical conductor of resistance Re as a result of the application of a voltage difference ΔV between the two ends of the conductor

This implies that we can employ Kirchhoff's rules, as applied to the analysis of electrical networks, to analyse fluid flow in fluidic networks.











9.6 Navier-Stokes Equations
The Navier-Stokes Equations are widely used to describe the behaviour of fluids in terms of continuous functions of space and time. They encapsulate the three conservation laws of mass, energy and momentum, and are considered in terms of flux rather than changes of their instantaneous values. In mathematical terms this is represented as partial derivatives of the dependent variables.
The calculation of fluid velocities and pressures at the macroscopic scale is based on the assumption that the fluid can be treated as a continuum. Apart from fluid velocity v and pressure P, for the most general situation that includes compressible and incompressible fluids we also require knowledge of the density ρ, viscosity η, specific heat Cp and temperature T of the fluid. Pressure and temperature characterise the energy state and number of molecules present in a given volume of fluid. If the pressure and temperature do not vary too greatly within this volume element, analytical functions can be derived that relate the density, viscosity and specific heat to the pressure and temperature. In a 3-dimensional system we are therefore left with five unknowns, namely P, T, vx, vy and vz. These five unknowns are related by a system of equations that describe:
- the conservation of mass,
- the conservation of momentum,
- the conservation of energy.
The equations describing these three conservation laws are often referred to as the Navier-Stokes equations, but it is more correct to reserve this description to the equations that describe conservation of momentum. Conservation of energy usually concerns heat flow in fluid systems in which a temperature gradient is created by an energy source or sink associated with chemical reactions or heating and cooling devices. For most microfluidic flows in lab-on-chip devices the temperature is constant, in which case the conservation of energy equation is redundant. We will thus focus on the derivations of the conservation of mass and conservation of momentum equations.
9.6.1 Conservation of Mass Equation
To simplify the situation we will consider, as depicted in Figure 9.18, a 2-dimensional element (Δx, Δy) using the Cartesian coordinate system, with fluid velocities u and v in the x- and y-directions, respectively. We will then generalise to the 3-dimensional case.
Figure 9.18 Conservation of fluid mass for a volume element ΔxΔy.

For the system of fluid flow shown in Figure 9.18 the conservation of mass is given by

Dividing by ΔxΔy we obtain

which can be written as

Defining the operator D/Dt in 3-dimensional Cartesian coordinates as

we can write Equation (9.23) in the vector form

where  is the velocity vector (u, v, w). We are concerned mainly with incompressible liquids, in which case terms such as ∂ρ/∂t, ∂ρ/∂x and Dρ/Dt are zero, and density ρ remains constant. Equations 9.23 and 9.24 thus reduce, for the 3-dimensional case, to
is the velocity vector (u, v, w). We are concerned mainly with incompressible liquids, in which case terms such as ∂ρ/∂t, ∂ρ/∂x and Dρ/Dt are zero, and density ρ remains constant. Equations 9.23 and 9.24 thus reduce, for the 3-dimensional case, to

and
(9.25b) 
9.6.2 Conservation of Momentum Equation (Navier-Stokes Equation)
The change of momentum in a fluid element is given by the balance between the inlet and outlet fluid momentum, and the tangential and normal stresses acting on that element. These are considered separately in Figures 9.19 and 9.20 for the 2-dimensional case.
Figure 9.19 Inlet and outlet fluid momentum for a fluid element in the x-direction.

Figure 9.20 The normal and tangential stresses acting on the volume element shown in Figure 9.19.

For Newtonian fluids the tangential stress τ and normal stress σ are given as

and
(9.26b) 
Summing the forces shown in Figure 9.20 in the x-direction, and using the mass conservation equations (9.25) we obtain

Combining this result with Equation (9.26) gives the Navier-Stokes equation
(9.27a) 
Extending this to 3-dimensions
(9.27b) 
(9.27c) 
and in vectoral form
(9.27d) 
where  is the velocity vector (u, v, w) and
is the velocity vector (u, v, w) and  is the force per unit volume acting on the element (Δx, Δy, Δz).
is the force per unit volume acting on the element (Δx, Δy, Δz).
9.6.3 Conservation of Energy Equation
To derive this equation we identify either a source or sink of heat SH, and specify the specific heat Cp and heat conductivity k of the liquid. The specific heat is defined as the amount of heat Q per unit mass required to raise the temperature of a substance by one degree Celsius,

The thermal conductivity of a substance is defined in terms of the quantity of heat Q conducted per unit time Δt down a unit temperature gradient ΔT in a direction normal to a surface of unit area ΔA. The heat conduction must arise only from the temperature gradient, and not from a secondary heat source or chemical reaction, for example:

The specific heat of water is 4.186 J gm−1 K−1, and its thermal conductivity is ~0.6 watts m−1 K−1.
In 3-dimensional Cartesian coordinates the conservation of energy equation is
(9.28) 
9.7 Continuum versus Molecular Model
In Chapter 10 the Knudsen Number Kn (a dimensionless parameter that compares the characteristic dimensions of a fluidic system to the molecular spacing) will be shown to be an important indicator of whether continuum or molecular physics should be used to describe fluid properties. For dimensions larger than one micron or so, we have Kn < 0.1 and the continuum model can be used (although the concept of zero fluid slip at a surface might require modification). At this scale we can assume that the fluidic domain contains an infinite number of molecules, and that it is homogeneous at all scales. As depicted in Figures 9.21 and 9.22, the fluid can be divided up or decomposed into an infinite number of identical domains and its physical properties are continuous functions of space and time. At nanometric dimensions we are obliged to consider the fluid in terms of a finite number of molecules. When divided up, even into a finite number of elements, some elements will contain molecular mass and energy and some will not. A nanometric domain has spatially discontinuous properties.
Figure 9.21 If the characteristic dimensions of a fluidic system are large compared to the molecular spacing, we can treat it as a continuum in which its properties are continuous and infinitely divisible. At nanometric dimensions the fluid is discontinuous – some elements of it will contain molecular mass and energy and some will not.

Figure 9.22 The physical properties (e.g. density, viscosity, temperature) of a continuum are continuous functions of space and time. A nanometric domain will exhibit statistical variations in its physical properties arising from a finite number of molecules in the domain.

We will now summarise the methods employed to simulate the physics of relevance to the two extreme cases of the continuum and molecular scales. This will assist an understanding of how to model the mesoscale where 0.1 < Kn < 10.
9.7.1 Solving Fluid Conservation Equations
Two widely used computational fluid dynamic methods for simulating fluid properties are outlined in Figure 9.23, and are known as the Finite Difference Method (FDM) and the Finite Element Method (FEM). With FDM, instead of derivatives being computed over infinitesimal elements, increments of finite width are used as an approximation. The partial differential (Navier-Stokes) equations can thus be replaced with simple algebraic equations, and by solving (either iteratively or by matrix inversion) yield values of fluid transport variables at discrete points in the flow field.
Figure 9.23 Two common computational methods for simulating fluid properties are the Finite Difference Method (FEM) and the Finite Element Method (FEM). With FDM simple algebraic equations yield values of fluid transport variables at discrete points in the flow field. With FEM the flow domain is divided into a finite number of cells, and the governing Navier-Stokes equations are evaluated at nodes placed at the corners or sides of these elements.

Examples of such equations, with reference to Figure 9.23, are:
Calculations at Point P:

FDM can only be used to solve fluid flows (or heat, electric current flows) having simple boundaries. More complicated situations can be solved using FEM which divides the fluid continuum into a finite number of cells or elements. Nodes placed at the corners or sides of these elements are used to fractionate and evaluate the governing equations in an integral form using weighting functions. As shown in Figure 9.24 for a heat flow simulation there is a trade off between the number of nodes chosen (hence calculations) and the resulting accuracy.
Figure 9.24 As shown in this example of a heat flow simulation using FEM, there is a trade off between the number of elements chosen (hence number of computations required) and the accuracy achieved. The heat source and sink are situated in the top right and bottom left corners, respectively.

The size of the cells or elements can vary so as to focus the most computational effort on those regions of the flow field, or any other dimensional field, where rapid changes of the physical quantity are expected. An example of this is shown in Figure 9.25 for the modelling of the electric field generated at a pin electrode. The size of the elements is made smaller around the tip of the electrode.
Figure 9.25 In this FEM model of the electric field generated by a pin electrode the size of the elements is reduced in the region where the field is expected to change the most.

An extension of FEM is the Finite Volume Method (FVM) which divides up the flow domain into elemental control volumes surrounding a node. The flow parameters are treated as fluxes between control volumes, and continuity is maintained in each element. Because of the continuum approximation these volume elements must be uniform throughout and infinitely divisible.
9.7.2 Molecular Simulations
At the nanometric scale the characteristic length of a fluid flow approaches that of the diameters of individual molecules, and so we must account for motions of individual molecules. The basic mechanism assumed in most molecular simulations is that the force acting on any molecule is determined by the movements of its neighbours, and interacts with them according to Newton's three laws of motion.
Molecular simulations rely on representative particles interacting with each other, where each particle has individual properties that determine its next position and momentum after each interaction. There are two approaches – namely deterministic (where the outcome can theoretically be worked out) or stochastic (having elements of unpredictability and chance).
9.7.2.1 Deterministic Simulations
These fall into two categories, according to whether the molecules are represented as hard or soft spheres. In the Hard Sphere Model collisions between molecules are modelled as binary interactions, occurring instantaneously, where they exchange linear momentum. No long-range interactions are assumed. The simulation advances one step at a time, to the next event or collision, and is based on the assumption that all spheres have an initial position and velocity, and that between collisions they travel at a constant speed and direction, such that their positions at any time can be calculated. This is depicted in Figure 9.26.
Figure 9.26 Molecular collisions using the Hard Sphere Model are treated as instantaneous binary interactions. Between collisions the molecules are assumed to travel in straight paths with a constant relative velocity v12, and long-range interactions are absent. See main text for further details.

In Figure 9.26 the position of a sphere at any time is given by

where ri, and vi are the position (centre) and velocity of sphere i, t0 is the start time and t1 is the new time.
The time to collision depends on the relative velocity v12 of spheres 1 and 2. If the condition v12r12 < 0 is met, then the two spheres are heading towards each other. Collision occurs when:

and the value of θ shown in Figure 9.26 determines whether or not a collision of two spheres will occur.
In the Soft Sphere Model the molecules are considered to interact by exerting van der Waals forces on each other. As described in Chapter 1 these forces are composed of long-range attractive interactions and short-range but strongly repulsive ones. These interactions occur continually, with each molecule having a ‘zone’ (of radius ~3 molecular diameters) within which other molecules are influenced, as depicted in Figure 9.27. This differs from the hard sphere model, where spheres interact only when contact is made. The resultant force arising from the repulsive and attractive force is often modelled using the Lennard-Jones 6–12 potential described in Chapter 1 and Figure 1.2.
Figure 9.27 In the Soft Sphere Model of molecular interactions each molecule interacts continually with others lying within a critical zone of radius Rc. The interactions are often modelled as short- and long-range van der Waals forces.

9.7.2.2 Stochastic Simulations
These simulations incorporate an element of randomness into the model. This is often achieved using Monte Carlo simulations involving repeated random sampling of molecular perturbations. A simple example of a stochastic procedure is given in Figure 9.28, where the value of π is calculated by ‘probing’ a square domain containing an arc. The area inside the arc is estimated from the number of points inside its constraints (squares) and the area of the square is given by the total number of points (squares + circles). An estimation of π is obtained using the following logic:

Figure 9.28 In this method for estimating the value of π, a square domain containing an arc is probed using randomly distributed test points. As the number of random sample points increases, the total number of points (squares + circles) divided by the number of points (squares) inside the arc approaches the value for π.

As an initial step in molecular dynamics modelling, the overall energy of a system of molecules is calculated. One molecule in this system is chosen at random, with a defined probability of being selected. This molecule is assigned a small perturbation, for example either of its position or speed, and the new system energy is calculated. If the new system energy is smaller than the initial system energy, this added perturbation is accepted. If the new system energy increases, the perturbation is accepted according to a probability law (e.g. one based on Boltzmann statistics). Rejected perturbations are ignored. This procedure is repeated until the system reaches ‘equilibrium’.
9.7.3 Mesoscale Physics
The mesoscale region, defined as a scale between the micro- and molecular-scale, covers the change in physics between the continuum approximation and discontinuous molecular models. The lower limit of the mesoscale can be taken to be around 100 atomic or molecular diameters. The upper limit, corresponding to where the continuum approximating laws are violated, is not so well defined. For example, a rarefied gas might invalidate the use of continuum physics up to scales of ~10 μm, whereas for a dense liquid the continuum laws could be valid down to scales below 1 μm. Travis et al. [1] have modelled the velocity profile and heat flux profile of an atomic liquid in a narrow channel, using molecular dynamics and Navier-Stokes equations. For a channel width of 5.1 molecular diameters the two simulations of the velocity profiles differed significantly. The heat flux profile did not agree with that predicted by Navier-Stokes hydrodynamics, but exhibited significant oscillations located about one molecular diameter from the walls. However, classical Navier-Stokes behaviour was approached for a channel width greater than 10 molecular diameters.
As an example of experimental investigations at the boundary between the micro- and mesoscale, Pfahler et al. [2] constructed three channels of rectangular cross-section ranging in area from 7200 to 80 square microns. In the relatively large flow channels the experimental observations were in rough agreement with predictions from the Navier-Stokes equations but significant deviations were found for the smallest of the channels. Mala and Li [3] studied water flow through microtubes with diameters ranging from 50 to 254 μm. Results in rough agreement with conventional continuum theory were obtained for the large diameters, but not for the smaller diameters.
Either a top-down or a bottom-up approach can be adopted to model the mesoscale.
9.7.3.1 Top-Down Approach
Gases are well understood in terms of kinetic theories and are thus better suited than fluids to describe the approach from continuum physics to molecular dynamics. We will progress down in scale from where the fluid can be considered to be continuous, infinitely divisible and in thermodynamic equilibrium. In the next chapter we will see that the first stage of the breakdown of the continuum approximation for gases occurs at a Knudson number greater than 0.001, where areas of high gradient such as boundaries, cannot maintain the continuous distribution of macroscopic properties. This is a result of the deviation from thermodynamic equilibrium, where there are insufficient collisions in the system for the energy to propagate smoothly in areas near boundaries. The low number of molecular collisions with the boundary means that the velocity and temperature of the solid and fluid are no longer the same at the interface. This causes a violation of the no-slip condition at a moving surface, as well as the no-jump-in temperature condition, assumed in continuum physics. To account for this initial violation we can describe the slip and no-slip conditions by relating the difference in velocity between the wall and fluid (ufluid − uwall) to the strain rate at the wall (∂u/∂y)wall:

with Ls being the slip length. This slip condition can be included in the continuum approximation, using either a simulated or experimentally observed value for Ls. For normal continuum conditions Ls is so small that the fluid and wall move at the same speed (no-slip condition), but as the Knudson number of the system increases above 0.001 the slip effects become more pronounced. The amount of slip that is allowed depends on the roughness of the surface over which the fluid is flowing and the interaction rate between the fluid and solid molecules. We can expect this model to fail as the surface roughness of the wall approaches the mean free path of the fluid.
The transition between the continuum and molecular regions for liquids goes through the same stages as for gases, but there is no parameter to act as a guide throughout the transition. The Knudson number cannot be defined, as there is no concept of mean free path for liquid flows – the molecules are in a constant state of collision and move over much shorter distances (comparable to the molecular diameter). As shown in Figure 9.29, computational modelling can be used in which finite element meshes are systematically reduced in scale to ‘handshake’ a molecular dynamic region, either using Monte Carlo simulations [4], or avoiding such simulations [5]. Nie et al. [6] have developed a hybrid multiscale method for simulating micro- and nanoscale fluid flows. Continuum Navier-Stokes equations are used in one flow region and molecular dynamics in another. The spatial coupling between continuum equations and molecular dynamics is achieved through constrained dynamics in an overlap region.
Figure 9.29 In the top-down approach to model the interface between the micro- and mesoscale, a finite element mesh is scaled down to meet molecular sites.

9.7.3.2 Bottom-Up Approach
A serial approach can be adopted, where a very small molecular simulation builds up to describe the physical relationships behind a large-scale fluid system. Such approaches can require supercomputers operating in parallel. An extreme example of this is the global simulation described by Clementi [7], which begins with the building of molecular liquids from nuclei and electrons using quantum mechanics, proceeding next to multibody interaction potentials, again by quantum mechanics, followed by the application of Monte Carlo and molecular dynamics to study the motions and collective properties of water molecules. This is then extended to the realm of fluid dynamics by considering a flow along a channel with or without obstacles, to finally consider the tidal movements in Buzzards Bay, New England!
However, in general the main issue faced with bottom-up methods is the scaling up of information from the molecular level and the removal of degrees of freedom from the system to minimise computational effort. One approach is to use the scheme depicted in Figure 9.30. The simulation involves two layers. One layer contains molecular information and covers the whole system. This is overlaid with an equivalent finite element mesh that gradually scales up, from nodes corresponding to atomic sites close to the region of interest, to elements containing many molecules. Because the large-scale elements contain many molecules, their energies local to each other have approximately similar values. In a method known as the quasi-continuun technique the approximation is made that the local neighbourhood of molecules can be represented by the value of just one molecule [8]. This can dramatically reduce the number of computations required in the simulation – but can only be applied to the coarse FE meshes, and breaks down when the effective mesh element contains only a few molecules.
Figure 9.30 In this bottom-up approach to mesoscale simulation, a mesh of atomic sites at constant density is covered by a finite element mesh that gradually scales up from the atomistic to a quasi-continuum scale.

9.8 Diffusion
From Equation (9.3) we know that a molecule in thermal equilibrium with a surrounding fluid of absolute temperature T has an average kinetic energy of 3 kT/2, and thus an average velocity (3 kT/m)1/2 associated with motion along each of the three axes in a 3-dimensional volume. Diffusion is the random migration of molecules or small particles from multiple collisions arising from the kinetic motion of neighbouring molecules. A schematic of this process is shown in Figure 9.31, where a cluster of gas molecules is shown occupying the corner of an otherwise empty container. As an oversimplification, only to demonstrate the process involved, we will assume that the time τ and average mean free path length between collisions remains constant. The rate of randomising collisions is thus 1/τ. After a sufficiently large number n of such collisions the molecules will become evenly distributed in the container (after time nτ).
Figure 9.31 Randomising collisions, at a rate of 1/τ s−1, result in the even distribution of molecules through a process that is called diffusion.

From their independent analyses of Brownian motion (the buffeting of macroscopic particles through collisions with fluid molecules) Einstein and Smoluchowski derived the following expression for the diffusion coefficient D

where  is the mean free path length given in Equation (9.2). An excellent discussion of the origins and validity of this so-called Einstein-Smoluchowski equation has been given by Isla [9]. From the worked Examples 10.3 and 10.5 of Chapter 10 we find that for nitrogen gas at room temperature and atmospheric pressure
is the mean free path length given in Equation (9.2). An excellent discussion of the origins and validity of this so-called Einstein-Smoluchowski equation has been given by Isla [9]. From the worked Examples 10.3 and 10.5 of Chapter 10 we find that for nitrogen gas at room temperature and atmospheric pressure  = 14.4 × 10−10 m, and τ = 3.1 × 10−10 s. From Equation (9.29) we obtain an estimate for D of 3.3 × 10−9 m2 s−1. Einstein also demonstrated that for macroscopic particles exhibiting Brownian motion in a fluid, the particle's diffusion coefficient can be expressed as
= 14.4 × 10−10 m, and τ = 3.1 × 10−10 s. From Equation (9.29) we obtain an estimate for D of 3.3 × 10−9 m2 s−1. Einstein also demonstrated that for macroscopic particles exhibiting Brownian motion in a fluid, the particle's diffusion coefficient can be expressed as

where ‘a’ is the particle's effective hydrodynamic radius and η is the fluid viscosity. This is called the Stokes-Einstein equation, and its origin and validity has also been discussed by Isla [9]. For particles suspended in water, the effective hydrodynamic radius is defined as the radius of a rigid uncharged sphere which exhibits the same hydrodynamic behaviour as the solvated molecule in solution. This should therefore include water of hydration which is too firmly bound to the particle's surface to participate in the viscous shearing process as it moves through the aqueous medium. Equation (9.30) was derived on the assumption that the solute molecule is large compared to the solvent. Nevertheless the equation has been experimentally confirmed for suspended particles with radii as small as 5 nm, and for large colloidal particles with suspension volume fractions up to 3%. It can also provide good approximations for the diffusion of molecular species in water. Thus sucrose (a ≈ 0.5 nm) can be estimated to have a diffusion coefficient in water (η = 1 × 10−3 Pa) at 298 K of ~3.9 × 10−10 m2 s−1, which can be favourably compared to the value of 5.2 × 10−10 m2 s−1 cited in Table 10.4 of Chapter 10. Approximate values of diffusion coefficients for some biologically relevant particles are given in Table 9.2.
Table 9.2 Approximate diffusion coefficients for some biologically relevant particles in water at 293 K. Values for the mean diffusion distance (diffusion layer thickness), defined by Equation (9.31), are given for time intervals of 1ms and 10 s.

A description of particle diffusion can be made in terms of a one-dimensional random walk, often described in terms of the ‘drunken sailor’ problem outlined in Figures 9.32 and 9.33. At each new step forward the drunken sailor is equally likely to stagger one step to the left as he is to the right. We can use this analogy to describe the resulting random direction that a molecule follows after colliding with another molecule.
Figure 9.32 The probability distribution for a 1-dimensional random (drunken sailor) walk is given by the factorials of the binomial coefficients as displayed by the rows in Pascal's triangle (Pascal's pyramid for 3D).

Figure 9.33 The probability distribution for a 1-dimensional random walk after time t = 4τ. After a period of 4τ the probability of the sailor standing straight ahead of his original location is 6/16 = 0.375.

After a number of random steps the spatial distribution of particles along a one-dimensional axis takes the form of a probability distribution described by the factorials of the binomial coefficients. Applying Stirling's approximation for these factorials, then for a sufficiently large number of thermal collisions we can represent the probability distribution as a Gaussian or normal distribution. In one-dimension the probability P(x)dx of finding a particle between x and x + dx at time t is given by Isla [9] as:

The mean displacement  of the particle is thus given by
of the particle is thus given by

An alternative way to derive this equation is to employ Equation (9.29) as follows:

to give

We can consider  as the mean diffusion length for a molecule interacting through collisions with neighbouring molecules. Values for this diffusion length are given in Table 9.2 for times of 1 ms and 10 s. We can see that small sugar molecules like glucose will diffuse a distance of around 1 μm after 1 ms, and 0.1 mm after 10 s. These can be significant distances in microfluidic systems. For particles of the size of bacteria, however, the corresponding diffusion lengths are much less at 20 nm and 2 μm, respectively.
as the mean diffusion length for a molecule interacting through collisions with neighbouring molecules. Values for this diffusion length are given in Table 9.2 for times of 1 ms and 10 s. We can see that small sugar molecules like glucose will diffuse a distance of around 1 μm after 1 ms, and 0.1 mm after 10 s. These can be significant distances in microfluidic systems. For particles of the size of bacteria, however, the corresponding diffusion lengths are much less at 20 nm and 2 μm, respectively.
Diffusion of molecules and particles tends to occur down their concentration gradient – also referred to as diffusion gradients (see Figure 9.34).
Figure 9.34 Molecules tend to diffuse down a concentration gradient (also termed as a diffusion gradient).

This diffusion process can be described by Fick's 1st equation of diffusion:

which states that the net flux Jx (moles m−2 s−1) of diffusing molecules or particles is proportional to the concentration gradient and diffusion constant of the molecule/particle (the negative sign indicates that the molecules diffuse down the concentration gradient). Unless the concentration gradient is artificially maintained (e.g. with a continuous source and sink of the molecules or particles) the factor ∂C/∂x will change as a function of time. This leads to Fick's 2nd equation:

This equation can be used (with the appropriate boundary conditions) to determine how a nonuniform distribution of molecules or particles will redistribute itself as a function of time. Diffusion along a microfluidic channel is effectively a one-dimensional problem. In this case the solutions of Fick's 2nd equation are:

An example of the application of Equations (9.32) is given in Figure 9.35, to show how a 0.1 nL aliquot of 1 M methanol (D = 1.6 × 10−5 cm2 s−1) diffuses after its injection into a channel, of height and width 100 μm, filled with stationary water.
Figure 9.35 The temporal concentrations of methanol (1 M, 0.1 nL, aliquot injected at time t = 0 into location x = 0) as it diffuses along a channel of static water of width and height 100 μm.

As shown in Figure 9.35, within a period of 5 seconds the leading edges of the diffusing methanol reach a distance of ~250 μm either side of the injection port, and this extends 500 μm after 10 seconds. For a fixed location along the channel, the concentration of diffusing methanol rises and then falls, rather like a tidal wave passing a buoy (see Figure 9.36).
Figure 9.36 At any fixed location along the channel the concentration of diffusing methanol shown in Figure 9.35 appears as a passing wave.

In Chapter 10 we will learn that it is very difficult, if not impossible, to induce turbulent flow in channels of micron-scale dimensions. The flow is inevitably laminar, of the form shown in Figure 9.15. In Figure 9.37 a fluidic Y-junction (equivalent to the T-junction of Figure 9.16) is used to flow together two liquids into a third channel of diameter 100 μm. A practical question to ask is how long should the third channel be to achieve complete mixing of the two liquids, and to what extent is mixing influenced by the rate of fluid flow? In the absence of mechanical stirring, the only way for the merging liquid streams to mix is through the diffusion of their constituent molecules across the interface between the travelling liquids. The profile of this interface will broaden and dissipate with time along the channel rather like the concentration profiles shown in Figure 9.35. If a flow rate of 5 μL per minute is chosen, then as shown in Figure 9.37 no discernable mixing of the two fluid streams occurs after a distance of 2.5 mm along the third channel. A 10-fold reduction of the flow rate to 0.5 μL per minute does result in some mixing of the two fluid streams, but it is clear that the channel length will need to extend much further before complete mixing occurs. The long channel lengths required for the thorough mixing of laminar flow streams can be accommodated in lab-on-chip designs using the serpentine geometry shown in Figure 9.37.
Figure 9.37 Modelling (top) of liquid streams flowing together via a Y-junction into a channel of radius 100 μm. Mixing of the fluids is evident for a fluid flow rate of 0.5 μL/min, but not at 5 μL/min. A serpentine geometry (bottom) is often used in lab-on-chip devices to accommodate the long channel lengths required for the mixing of laminar fluid streams.

9.9 Surface Tension
Surface tension is a significant and useful force in microfluidic devices. The origin of the surface force known as surface tension is shown in Figure 9.38, which depicts a free surface between liquid and air. A molecule in the fluid bulk experiences mutually attractive forces with neighbouring molecules. Van der Waals forces are usually the most dominant, and for aqueous solutions hydrogen-bond forces are also significant. A molecule at the surface is attracted by a reduced number of neighbours and so is in an energetically unfavourable state. The creation of a new surface is thus energetically costly, and fluids will act to minimise their surface area. This is the driving force for small volumes of fluid to assume a spherical shape, as for example trickles of water breaking up into spherical drops to minimise the total surface area.
Figure 9.38 The molecular origin of surface tension is the fact molecules in bulk liquid experience mutual attractive forces. The net attractive force on a surface molecule is less, and so a surface molecule is in an unfavourable energy state.

If U is the total cohesive energy per molecule in the fluid bulk, then this is halved to a value of U/2 for a molecule located at a flat surface. The surface tension created per unit area of surface is directly related to this cohesive energy reduction. For a characteristic molecular dimension R, the effective molecular area is R2 and the surface tension is U/(2R2). Surface tension is thus directly proportional to the intermolecular attraction and inversely proportional to the molecular size. Surface tension values for some common fluid–air interfaces are given in Table 9.3. Of note in this table is the fact that water has a significantly larger surface tension than alcohol, soap solutions and oils, reflecting not only the relatively small size of the water molecule but also the cohesive energy supplied by hydrogen-bonds in water. Liquid metals (e.g. mercury) exhibit the highest surface tension values.
Table 9.3 Values of surface tension T (liquid surface with air) at 20 °C.

Surface tension T is defined as the ratio of the surface force F to the length d along which the force acts (T = F/d) and thus has units of force per unit length (equivalent to energy per unit area) and acts tangentially to the free surface. Because its action is confined to the interface it does not appear in the Navier-Stokes equations, which deal with pressure gradients within a fluid bulk. A total force γdl will act on a surface line element dl. If the surface line element is a closed loop, and the surface tension uniform, the net surface tension force acting on the loop is zero. If surface tension gradients are present, a net force on the surface element may result and distort it through an induced flow of surface liquid. Surface tension gradients can arise from the presence of a surfactant.
9.9.1 Surfactants
Chemical compounds that lower the surface tension of a liquid are known as surfactants. Detergents, soap, fatty acids and fatty alcohols are common examples. They can be used to stabilise mixtures of oil and water, for example, by reducing the surface tension at the interface between the oil and water molecules. Their molecular structures often consist of a hydrophilic head and a hydrophobic tail group, so that their location at a free liquid surface can be energetically favourable. Gradients in surfactant concentration result in surface tension gradients. A simple, but instructive demonstration of this can be observed by coating one end of a matchstick with soap or liquid detergent. When placed carefully on water the matchstick will be propelled across the surface away from the dissolving surfactant.
9.9.2 Soap Bubble
The pressure difference between the inside and outside of a soap bubble can be derived by representing the bubble as two hemispheres. This is depicted in Figure 9.39. The net pressure acting to push the top hemispheres away is balanced by the surface tension T acting around the circumference of the lower hemisphere (noting that there is an outer and inner surface to the bubble). If we ignore the film thickness compared to the bubble radius r, and the weight of fluid comprising the bubble film, then the net upward and downward forces are given by:

Figure 9.39 The pressure difference (Pi − Po) between the inside and outside of a soap bubble can be derived by representing the bubble as two hemispheres. The net pressure acting on the upper hemisphere is balanced by the surface tension T acting around the circumference of the lower hemisphere.

Equating these two forces we obtain the result:

The 1/r relationship in this equation indicates that it is more difficult to inflate a small balloon than a larger one. This is of anatomical relevance. Oxygen exchange in the lungs takes place across the membranes of small balloon-like structures called alveoli. Their surface tension is lowered nearly 15-fold by a surfactant coating so that a very small pressure differential is sufficient to inflate them with air. Elastic recoil of the alveoli assists with their deflation (exhalation).
If instead of a soap bubble we consider an air bubble in a liquid (or a fluid droplet in air) there is just the one (outer) surface to take into account. Accordingly, the pressure difference is halved: Pi − Po = 2T/r.
9.9.3 Contact Wetting Angle
A drop of liquid on a solid surface is shown in Figure 9.40. As shown in this figure, there are three interfaces, namely the solid–liquid, the liquid–air and the solid–air interface. A line on the solid surface (the xy-plane) defines the boundary separating these three interfacial areas. The contact angle θ is defined as the angle formed at this three phase boundary between the tangent to the liquid surface and the xy-plane. A tension exists in each interface, and different values for these result in different liquids adopting different contact angles relative to different solid surfaces. An equilibrium situation exists when the horizontal components of the surface free energies balance. From Figure 9.41 the equilibrium condition is readily seen to be described by the following relationship, known as Young's equation:

where TS-A, TS-L and TL-A are the surface tensions at the solid–liquid, the liquid–air and the solid–air interfaces, respectively, and θ is the contact angle defined above.
Figure 9.40 The contact angle θ of a drop of liquid on a solid surface is defined as the angle formed between the tangent to the liquid surface and the xy-plane at the boundary between the three interfaces (liquid-solid, liquid-air, solid-air). A summary of the factors influencing the contact angle in terms of the surface energy and wettability of the surface is given in Table 9.4.

Figure 9.41 At equilibrium the horizontal components of the surface free energies balance, and this is expressed in the form of Young's equation.

A liquid with low surface tension (low surface energy) resting on a solid surface of higher surface energy will spread out on the surface forming a contact angle θ less than 90°. The liquid is said to wet the surface – if the liquid is water we say the surface is hydrophilic. If the surface energy of the liquid exceeds that of the solid, the liquid will form a bead and θ will have a value between 90° and 180°. In this case we have a nonwetting liquid relative to the surface, corresponding to a hydrophobic surface when considering aqueous liquids. These aspects are summarised in Table 9.4.
Table 9.4 A summary of the parameters associated with the contact angle of a liquid with a solid surface.
| >90° | Contact angle | <90° |
| Low | Solid's surface free energy | High |
| Poor (hydrophobic) | Wettability | Good (hydrophilic) |
| Poor | Adhesiveness | Good |
Surface wetting is an important aspect of printing. The surface energy of the ink when wet should be lower than that of the surface to be printed. In this case the contact angle will be low and the ink will spread evenly and adhere strongly to the surface. However, if the surface energy of the ink is greater than that of the surface the contact angle will be large – the ink will form globules and not spread evenly.
9.9.4 Capillary Action
In a sufficiently narrow capillary of circular cross-section (radius a), the interface between a fluid and the capillary surface forms a meniscus that is a portion of the surface of a sphere, and has radius R given by:

This geometrical relationship is shown in Figure 9.42. The contact angle θ depends on the free surface energies of the fluid and the capillary surface with which it is in contact. The pressure jump ΔP across this surface is:

Figure 9.42 The contact angle θ between a sphere and a tangent plane is the angle between the normal to the sphere at the point of tangency and to the plane perpendicular to the z-axis.

The pressure difference is thus given by:

To maintain hydrostatic equilibrium, the induced capillary pressure is balanced by a change in height (h) of the fluid. As shown in Figure 9.43, this height change can be positive or negative, depending on whether the contact angle is less or greater than 90°. At equilibrium we have the condition:

where ρ is the density of the liquid in the capillary and g is the gravitational acceleration constant. From Equation (9.34) we obtain the capillary height h as:
(9.35) 
Figure 9.43 To maintain hydrostatic equilibrium the capillary pressure is balanced by the height h of a fluid in a capillary. The height change depends on the magnitude of the contact angle.

Capillarity manifests itself in many ways. Paper towels absorb water through capillarity. When burning a candle, the melted wax rises up the wick due to capillarity. In biology, though blood is pumped throughout the body, capillary action distributes blood in the smallest blood vessels – called capillaries. Many species of water-walking insects utilise the high water tension of water. They deflect the free surface of water, thus generating curvature forces that bear their weight, which is typically of the order 1~10 mg.
9.9.5 Practical Aspects of Surface Tension for Lab-on-Chip Devices
- The energy associated with surface tension can be used to drive liquids through microfluidic devices. By treating the surfaces of microchannels to be hydrophilic, water will be driven through any sized channel (typical of lab-on-chip devices) without any applied pressure. This flow is driven by the attractive energy between the water and the channel wall surface.
- Air bubbles pose a big problem in microfluidic devices because of their small radius of curvature r. The relationship Pi − Po = 2T/r, derived in Section 9.9.2 for a bubble in a fluid, informs us that the smaller the radius, the larger will be the pressures required to remove them from a fluidic channel. In the initial wetting of a hydrophilic device, air can become trapped where a wide channel narrows down to a smaller one, for example. Air bubbles can also form after the device is wet, if air spontaneously comes out of solution. Water has a very high surface tension, about 3-times higher than other liquids (see Table 9.3). A strategy to reduce air bubbles is to initially wet the device with a liquid, such as alcohol, that has a lower surface tension. Then water can be fed in behind the other liquid without exposing any air/water interface. This reduces by a factor of around three the force due to surface tension that must be overcome to push air bubbles through a constriction.
9.1. A syringe pump, exerting an excess pressure of 0.1 bar (10 kPa) between the inlet and outlet of an open-ended fluidic micromixer chip, produces a volumetric water flow of 100 μL/min. Calculate the resistance to water flow of this device.
(Give your answer in units of Pa m−3 s.)
9.2.
9.3.
Figure 9.44 The microfluidic Y-connector system for self-study Problem 9.3.

9.4. The figure below depicts the top view of a toy boat that uses soap as the propelling agent. The soap is contained in the shaded box at the rear of the boat, and the arrow indicates the direction of motion of the boat.

Explain the origins of the net propulsive force acting on the boat when it is placed on water.
9.5. Calculate the height to which water will rise in a glass tube of radius 50 μm. The following values are to be used:

1. Travis, K.P., Todd, B.D. and Evans, D.J. (1997) Departure from Navier-Stokes hydrodynamics in confined liquids. Physical Review E, 55 (4), 4288–4295.
2. Pfahler, J., Harley, J. and Bau, B. (1989) Liquid transport in micron and submicron channels. Sensors & Actuators, 22, 431–434.
3. Mala, Gh.M. and Li, D. (1999) Flow characteristics of water in microtubes. International Journal of Heat and Fluid Flow, 20, 142–148.
4. Garcia, A.L., Bell, J.B., Crutchfield, W.Y. and Alder, B.J. (1999) Adaptive mesh and algorithm refinement using direct simulation Monte Carlo. Journal of Computational Physics, 154, 134–155.
5. Abraham, F.F. (2000) Dynamically spanning the length scales from the quantum to the continuum. International Journal of Modern Physics, 11 (6), 1135–1148.
6. Nie, X.B., Chen, S. and Robbins, W.N.E. (2004) A continuum and molecular dynamics hybrid method for micro- and nano-fluid flow. Journal of Fluid Mechanics, 500, 55–64.
7. Clementi, E. (1988) Global scientific and engineering simulations on scalar, vector and parallel LCAP-type supercomputers. Philosophical Transactions of the Royal Society of London. Series A: Mathematical and Physical Sciences, 326, 445–470.
8. Ransing, R., Dyson, P., Williams, P.M. and Williams, P.R. (2008) Chapter 2, in Fluid Properties at Nano/Meso Scale, John Wiley & Sons, Ltd, Chichester.
9. Isla, M.A. (2004) Einstein-smoluchowski diffusion equation: a discussion. Physica Scripta, 70, 120–125.
Bruus, H. (2008) Theoretical Microfluidics, Oxford University Press.
Nguyen, N.-T. and Wereley, S.T. (2008) Fundamentals and Applications of Microfluidics, 2nd edn, Artech House Inc., Boston.
Ransing, R., Dyson, P., Williams, P.M. and Rhodri Williams, P. (2008) Fluid Properties at Nano/Meso Scale, John Wiley & Sons, Ltd, Chichester.