HISTORIC PERSPECTIVE
Human Longevity
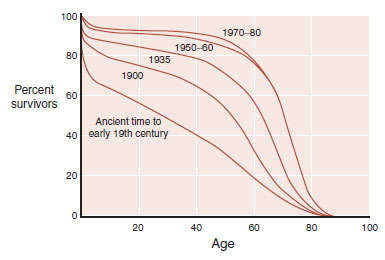
Although historic records indicate that older people have always existed, old age was once rare. Before the 20th century, few people lived beyond 50 years. Now, 95% of the children born in developed countries live past that age (Figure 24.1). Changes in health care, sanitation, and nutrition (to name a few) have had a profound impact on life expectancy. The ultimate result is that more and more people are living to ripe old ages. With more people living into their geriatric years, the aging-related central nervous system (CNS) disorders are becoming common.
All nerve cells are affected by aging. Sensory and motor skills decline with age. Neurodegenerative disorders such as Parkinson’s disease, amyotrophic lateral sclerosis, and Huntington’s disease become more prevalent as people get older. Cellular and molecular changes that accumulate over time render neurons vulnerable to damage. Most likely, the damage results from a combination of genetic vulnerability and environmental hits.
FIGURE 24.1 The percentage of people living beyond the age of 60 years has increased dramatically in the last two centuries. (Adapted from Kandel ER, Schwartz JH, Jessell TM, eds. Principles of Neural Science. 4th ed. New York, NY: McGraw-Hill; 2000.)
The percentage of people living beyond the age of 60 years has increased dramatically in the last two centuries. (Adapted from Kandel ER, Schwartz JH, Jessell TM, eds. Principles of Neural Science. 4th ed. New York, NY: McGraw-Hill; 2000.)
POINT OF INTEREST
The maximum human life span is approximately 125 years and has not changed in over 100,000 years. Cell culture studies suggest that each species has a biologic clock that influences its life span. For example, fibroblasts will only divide a limited number of times before dying. The number of divisions is related to life span (see table).

The important point is that life span and age-related illnesses such as dementia are likely to be controlled by different mechanisms.
Dementia, the progressive deterioration of cognitive skills, is perhaps the most worrisome development for all of us. There are numerous causes of dementia, including cerebral vascular accidents, alcoholism, and infections. Alzheimer’s disease (AD) is the most common cause of dementia. The surge in dementia cases as the baby boomers age is expected to overwhelm the health care system unless some intervention is discovered.
The disease we now call Alzheimer’s was first discussed when Alois Alzheimer presented a case in 1906 of a woman with the early onset of dementia. Her symptoms started in middle age with a change in personality and mild memory impairment. She was institutionalized when she became paranoid and unmanageable. Alzheimer repeatedly examined the woman as he followed up her deteriorating clinical course. Four and a half years after her initial symptoms, she was bedridden in a fetal position until she died.
The autopsy revealed gross atrophy of the cortex without localized foci. With the application of the new staining methods (see Figure 1.4), Alzheimer found sclerotic plaques scattered throughout the cortex, especially in the upper layers. Additionally, he noted that many of the cortical neurons were reduced to dense bundles of neurofibrils. Alzheimer thought that his description of plaques and neurofibrillary tangles in a patient with “presenile dementia” was a new and unique condition.
In fact, Alzheimer’s finding was cognitive loss associated with the following:
1. Cortical atrophy
2. Plaques outside the neurons
3. Tangles inside the neurons
These findings have become the description of the dementia that bears his name. Figure 24.2 shows a schematic representation of what Alzheimer might have seen when he looked through his microscope.
ALZHEIMER’S DISEASE
Surprisingly, what Alzheimer saw roughly a 100 years ago remains the focus of current research. However, the application of modern technology has greatly advanced the understanding of the pathophysiology of atrophy, plaques, and tangles.
Cortical Atrophy
The most striking feature of the Alzheimer’s brain is the dramatic shrinkage of the cortical tissue secondary to neuronal cell death. AD is a bit like losing hair. It starts years before it is actually noticed and progresses slowly. In some people, it starts sooner and proceeds faster. Furthermore, almost everyone experiences some hair loss with aging.
Brain volume loss is also a “normal” feature of aging. Brain volume peaks in adolescence and then declines as much as 0.2% to 0.5% per year. Patients with AD experience accelerated neural loss. Likewise, some people are genetically predisposed to early-onset AD.
Examination of the AD brain at autopsy shows extensive atrophy. Figure 24.3 compares two views of normal brains with AD brains. The enlargement of the ventricles and sulci in combination with the decreased tissue is easily recognized—and a bit unsettling for those of us in middle age.
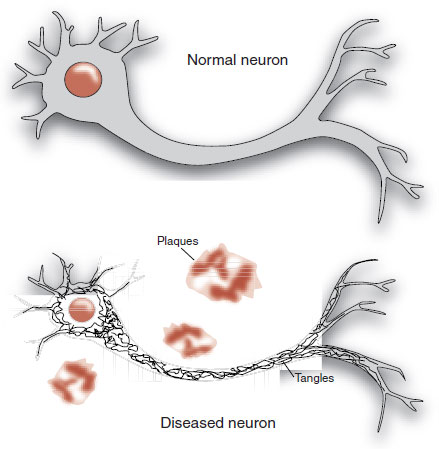
FIGURE 24.2 Alzheimer’s disease includes the constellation of neuronal shrinkage, plaques, and neurofibrillary tangles.
FIGURE 24.3 Gross examination (A) and coronal slices (B) show the extensive shrinkage of the brain from Alzheimer’s disease. (A courtesy of George Grossberg and the St. Louis University Alzheimer’s Brain Bank.)
Brain imaging, although not yet diagnostic for early AD, can document the volume loss and contrast the changes for those with and without AD. Figure 24.4 shows the results of sequential magnetic resonance imaging on one patient destined to develop familial AD. Note how his brain atrophy proceeds faster than in a healthy elderly control. It is also of interest that the symptoms of AD did not appear until significant brain tissue was lost.
The decrease in energy metabolism secondary to the extensive neuronal damage can be seen in functional imaging studies such as positron emission tomography (PET). Figure 24.5 shows the marked reduction in glucose metabolism in a patient with AD compared with a healthy control. The difference is so prominent that some have suggested that PET could be used to differentiate patients with AD from those normally aging. A large European study including more than 500 subjects found a 93% sensitivity and specificity for separating mild to moderate AD from normal controls. Unfortunately, they were not as effective at separating AD from other forms of dementia, and PET is even less helpful in diagnosing patients with mild cognitive impairment and determining whether they will go on to develop AD.
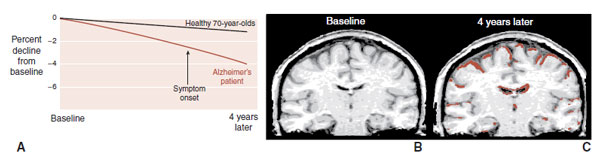
FIGURE 24.4 A. Sequential magnetic resonance imaging (MRI) scans show the aggressive brain atrophy in a patient with Alzheimer’s disease (AD) compared with healthy geriatric controls. B. MRI in a patient with AD at baseline and 4 years later (C). Brown overlay represents tissue loss compared with baseline. (Adapted from Fox NC, Schott JM. Imaging cerebral atrophy: normal ageing to Alzheimer’s disease. Lancet. 2004;363(9406):392-394.)
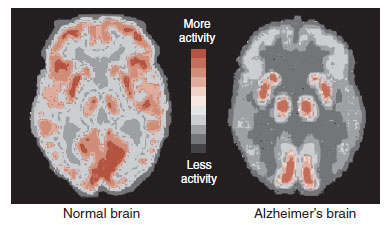
FIGURE 24.5 Positron emission tomography (PET) images showing glucose metabolism in a normal brain compared with an Alzheimer’s disease brain. Note the reduced activity in the frontal and temporal regions of the AD brain. (Adapted from Mattson MP, Magnus T. Ageing and neuronal vulnerability. Nat Rev Neurosci. 2006;7[4]:278–294.)
Amyloid Plaques
The extracellular deposits that Alzheimer saw are called amyloid plaques, which is a bit of a misnomer. They are actually aggregates of fibrous protein and not amyloid at all. It was not until 1984 that the primary component of the plaques was found to be a small protein called amyloid-β or A-β. (To add to the confusion, the most common term used in the literature is beta-amyloid or β-amyloid.) Specifically, it is a long 42-amino-acid chain called A-β-42 that seems to be the real culprit, although there may be others that are more noxious.
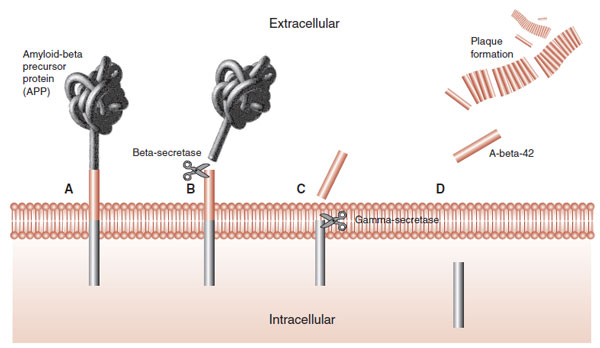
FIGURE 24.6 Amyloid plaques are formed from the cleavage of amyloid-β precursor protein into smaller proteins that clump together. (Adapted from Wolfe MS. Shutting down Alzheimer’s. Sci Am. 2006;294(5):72-79.)
A-β is cleaved from a larger molecule called amyloid-β precursor protein (APP). APP is a large protein protruding through the cell wall (Figure 24.6). It is found in cells throughout the body, but is prominent in neurons. The functions of APP are not fully understood, but may include regulating neuronal survival, neural neurite outgrowth, and synaptic plasticity.
APP is cleaved into smaller portions by at least two enzymes called β- and γ-secretase. The final cleavage results in the generation of A-β-42 (and others) that coalesces into long filaments. It is the clumping of the filaments that forms the “amyloid” plaques. Pharmacologic inhibition of the activity of the secretase enzymes has been of interest to those seeking ways to slow down the development of plaques.
Amyloid Hypothesis
Many believe that amyloid plaques are the source of the problem with AD. Two lines of reasoning suggest this is true. First, some people carry a genetic predisposition for early-onset AD usually developed before the age of 65 years. In all cases where they have identified the gene, the genetic abnormality causes an increased production of A-β. The toxicity of A-β is the other evidence that supports the amyloid hypothesis. A-β is toxic to neurons grown in Petri dishes. Furthermore, A-β can impair the development of long-term potentiation as well as the memory for a maze in rodents.
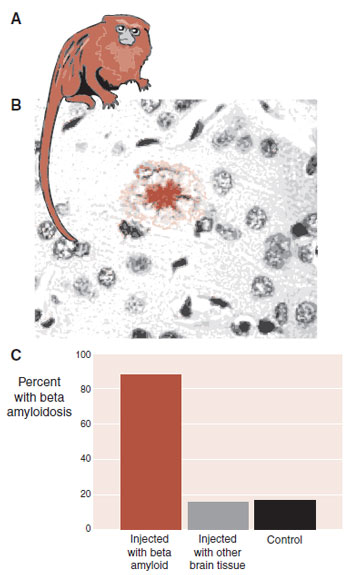
Recently, researchers from England have reported the results of a long-term study with marmoset monkeys. The monkeys received cerebral injections of A-β or other brain tissue that did not contain β-amyloid. Because the monkeys died, their brains were analyzed for amyloid plaques (Figure 24.7). Monkeys that were injected with A-β were much more likely to have cerebral amyloidosis at autopsy. These results not only show the toxic effects of A-β but also imply that the presence of A-β seeds the progression.
The exact mechanisms of the toxicity of A-β remain murky. Some research suggests that it is the soluble form of the protein that causes the damage. Other research suggests that it is not the A-β-42 protein, but different A-β proteins as yet unidentified. Furthermore, it is not clear why the plaques coalesce in the first place. Some evidence hints that the genetic forms of AD result from an overproduction of APP, whereas the more common sporadic cases result from the failure to clear the excess A-β. Clearly, efforts to treat the disorder would benefit from better understanding of these issues.
GENE EXPRESSION
In previous chapters we have discussed how the addition of methyl groups to the DNA structure can silence gene expression, which, in turn, can produce psychiatric symptoms. The opposite may be happening with AD; that is, the awakening of improper gene expression through demethylation. There is some indirect evidence that demethylation of the DNA sequence coding for β-secretase results in increased production of that enzyme. This could result in greater production of A-β and a faster progression of the disease.
FIGURE 24.7 Marmoset monkeys (A) can develop cerebral amyloid plaques (B). Monkeys injected with A- were much more likely to develop amyloidosis in the next 3 to 4 years (C). (Adapted from Ridley RM, Baker HF, Windle CP, et al. Very long-term studies of the seeding of beta-amyloidosis in primates. J Neural Transm. 2006;113:1243–1251.)
Imaging Amyloid
The first step in any treatment approach is an accurate diagnosis of the condition. Historically, the gold standard for the diagnosis of AD has been at autopsy—a bit late to start treatment. As noted earlier, imaging studies are poor at differentiating AD from other forms of dementia. More recently, researchers have been looking at ways to detect the presence of amyloid deposits in subjects even before symptoms appear. In 2012, the FDA approved florbetapir F18 (Amyvid) for PET imaging of the brain in cognitively impaired adults. Florbetapir binds to amyloid in the brain and gives an estimate of the density of the plaques. Figure 24.8 shows the results of a scan comparing a healthy control with a patient with AD. With this scan, unlike the PET scan in Figure 24.5, the subject with the disease lights up when the florbetapir attaches to the amyloid deposits.
The usefullness of a florbetapir PET scan is to identify reversible causes of cognitive decline, other than AD, such as depression. The ultimate utility of such a scan—identifying early stages of the disease so that treatment can be started prior to substantial neural cell loss—awaits the development of effective treatments.
Neurofibrillary Tangles
The final major pathology of AD is the intracellular neurofibrillary tangles. The neurofibrillary tangles come from the proteins on the microtubules of the neuron. The microtubules are the internal cytoskeleton that provides structure for the cell and, more importantly, transports essential molecules and organelles from the cell body to the synapses. Damage to the microtubules causes the peripheral aspects of the neuron to effectively starve.
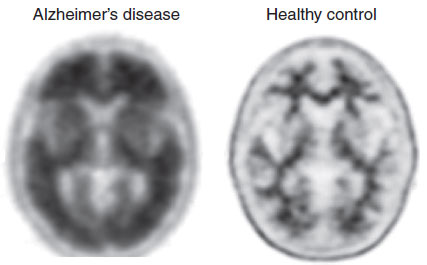
FIGURE 24.8 A positive scan (left) shows high uptake of florbetapir F18 in the cortical gray matter, indicating the presence of amyloid plaques. A negative scan (right) is paradoxically a healthier brain. (Images courtesy of Avid Radiopharmaceuticals, a wholly owned subsidiary of Eli Lilly and Company. © Eli Lilly and Company.)
The tau proteins bind to the microtubules and provide stability. The problem seems to start with the hyperphosphorylation of the tau proteins (Figure 24.9). Too many phosphates attached to the tau proteins cause them to detach from the microtubules. It is these detached proteins that clump together and form the neurofibrillary tangles, which in turn clog the neuron’s axons and dendrites and cause the cell to die. What causes the hyperphosphorylation remains unclear but seems to be initiated by β-amyloid—possibly soluble A-β that diffuses across the cell wall.
Alzheimer’s Disease Progression
AD is a relentlessly progressive disorder. There are no remissions. The first signs are marked by subtle decline in memory. As the disease advances, changes in personality and language skills develop. Eventually even motor functions are impaired. Understanding the spread of the pathology of AD gives a greater appreciation of the changing clinical picture.
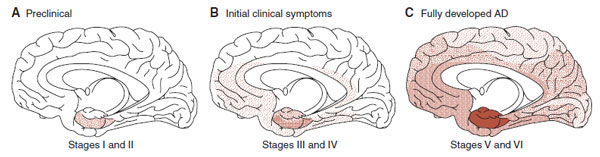
The progression of the disease can be staged by the development and progression of neurofibrillary tangles. In a landmark analysis of more than 2,500 brains in Germany over 10 years, Braak documented the insidious evolution of AD shown in Figure 24.10. The initial stages start in the entorhinal cortex of the hippocampus. From there the disease spreads into the temporal and frontal cortex. The final stages involve the entire brain, with the greatest deposits remaining in the regions where it all started.
PREVENTION AND TREATMENT
The number of cases of AD is expected to quadruple in the next 40 years if nothing is done to prevent or treat the disease. Current treatments can temporarily improve cognition in those affected, but do nothing to alter the underlying pathophysiology of the condition. Recent studies have been disappointing. Encouraging preliminary studies have failed in larger trials. The optimistic predictions of just a few years ago have not borne fruit.
CEREBRAL SPINAL FLUID
Tau protein is increased in the cerebral spinal fluid (CSF) of patients with AD. The diagnostic accuracy of the disease may be enhanced from measurements of specific proteins in the CSF, as well as better imaging studies—in patients with signs of cognitive decline.
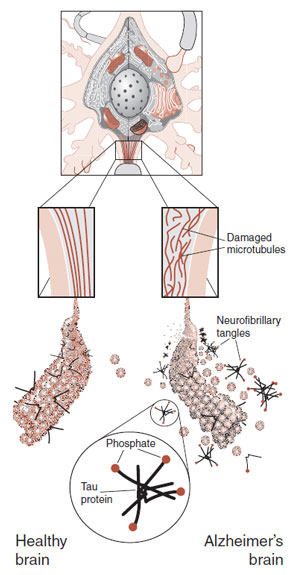
FIGURE 24.9 Hyperphosphorylation of the tau proteins produces neurofibrillary tangles that damage the microtubules. The result is impaired axonal transport and ultimately cell death.
Perhaps even more discouraging is that we do not even know which pathology of AD is most detrimental: amyloid plaques, neurofibrillary tangles, or some upstream event that precedes the development of plaques and tangles. Those of us on the other side of our life curve hope that the scientific community will develop effective interventions in the near future. We will review several of the favorable prospects.
FIGURE 24.10 The spread of neurofibrillary tangles in Alzheimer’s disease (AD). (Adapted from Braak H, Braak E. Frequency of stages of Alzheimer-related lesions in different age categories. Neurobiol Aging. 1997;18(4):351-357.)
Vaccine
Immunizations against childhood diseases have transformed the risks of growing up. The possibility of using vaccines to treat and/or prevent AD is an exciting application of this old intervention, which could likewise revolutionize growing old. The trick is to get the immune system to attack the amyloid plaques without irritating other parts of the brain or inciting an inflammatory response in the CNS.
The basic plan is laid out in Figure 24.11. Peripheral injections of A-β-42 are ingested by antigen-presenting cells. A-β antigens are presented to T cells, which in turn activate B cells. Anti-Aβ antibodies produced by the B cells attack the amyloid plaques as well as soluble Aβ. Microglial cells then clean the plaques through phagocytosis.
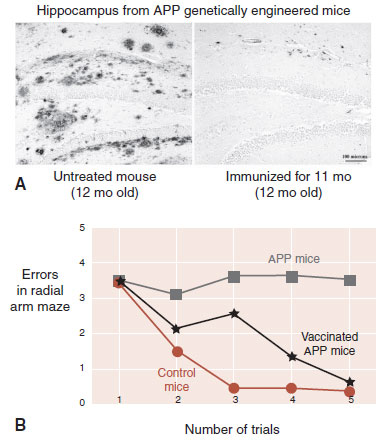
The vaccine was initially tested in mice genetically engineered to overexpress APP (APP mice). Such mice will develop amyloid plaques and memory loss by the age of 12 months. They have become the accepted animal model of AD. Immunotherapy with APP mice has produced extraordinary results (Figure 24.12). Clearing of amyloid plaques and preservation of cognitive functions have been repeatedly documented in animal studies.

FIGURE 24.11 A. Aβ is injected peripherally. B. The Aβ molecules are ingested and presented to T cells by the antigen-presenting cells. C. T cells activate B cells, which produce anti-Aβ antibodies. D. The anti-Aβ antibodies attack Aβ in the amyloid plaques and are cleared by microglia. (Adapted from Schenk D, Hagen M, Seubert P. Current progress in beta-amyloid immunotherapy. Curr Opin Immunol. 2004;16(5):599-606.)
FIGURE 24.12 A. Slices of amyloid-β precursor protein (APP) mice hippocampi show the accumulation of amyloid plaques without and with the Aβ vaccination. B. Vaccinated mice display memory similar to control mice and superior to the APP mice. (A from Lemere CA, Maier M, Jiang L, et al. Amyloid-beta immunotherapy for the prevention and treatment of Alzheimer disease: lessons from mice, monkeys, and humans. Rejuvenation Res. 2006;9(1):77-84, courtesy of Cynthia A. Lemere. B adapted from Morgan D, Diamond DM, Gottschall PE, et al. A beta peptide vaccination prevents memory loss in an animal model of Alzheimer’s disease. Nature. 2000;408(6815):982-985.)
In 2001, clinical trials of a synthetic version of the β-amyloid protein for use as a vaccine were started in humans. Preliminary safety studies were completed without a hitch. Unfortunately, the larger phase II study had to be stopped after several months when 6% of the participants developed an excessive inflammatory response (meningoencephalitis). Further analysis has suggested that T-cell activation may have been the problem.
Follow-up studies on the 372 subjects who were immunized have found some encouraging results. Those individuals who did mount an antibody response to A-β showed subtle signs of improved memory and cognitive skills. Furthermore, postmortem studies of a few patients have documented clearing of the amyloid plaques in some regions of their brains.
In 2012, a group in Sweden published what appears to be the next step in the vaccine story. The vaccine, called CAD106, is designed to mount an antibody response against a small portion of the Aβ molecule without activating the T cells (humeral response but not cellular). A small double-blind phase 1 study of 58 patients with mild to moderate AD was conducted to establish safety and tolerability. While almost every patient developed minor side effects (sore throat, headache, fatigue, etc.), none developed meningoencephalitis. (Whew!) The ultimate effects on cognition or amyloid deposits will need to be measured in larger studies. Furthermore, there are other vaccines also in development, so this exciting treatment option continues to move forward.
The neuronal loss from AD might be prevented or at least limited with appropriate stimulation from growth factor proteins. Animal studies suggest that growth factor proteins may be useful for treating neurodegenerative diseases such as AD. Specifically, nerve growth factor (NGF, see Figure 1.9) has been shown to prevent cholinergic degeneration and improve memory in animals. The problem arises in choosing a method to deliver the NGF to the brain. The molecule is too large to cross the blood–brain barrier. Likewise, direct infusion into the ventricles results in excessive stimulation and intolerable side effects, for example, pain and glial cell infiltration.
ANTIPSYCHOTICS
Alzheimer’s original patient was ultimately admitted to his ward because of her uncontrollable psychotic symptoms. Currently, most clinicians would quickly place such a patient on a new-generation antipsychotic medication. Yet, the use of antipsychotic medications for AD is not without problems. A meta-analysis looked at the effect of second-generation antipsychotic medications on cognitive decline in patients with AD. They found that the medications actually worsened the cognitive decline when compared with placebo (see table).

Adapted from Schneider LS, et al. Am J Ger Psych. 2006:14:19–210.
An alternative method of delivery entails hijacking the DNA in autologous fibroblasts using retroviral vectors. Such fibroblasts can be induced to express NGF. In turn, they can be placed directly into the brain of the subject and deliver NGF within a few millimeters of the nucleus basalis of Meynert, where cholinergic neuronal degeneration occurs. Because the fibroblasts are autologous, they do not activate an immune response.
Researchers at the University of California in San Diego completed a phase I trial with eight patients with probable AD. Fibroblasts producing NGF were injected into the subjects’ cholinergic basal forebrain. Two patients had significant complications from the surgery, but with five of the remaining six, cognition stabilized or improved during the 6 to 18 months after injection. PET scans showed increased activity.
Although this treatment will not halt the development of amyloid plaques or neurofibrillary tangles, it does show that NGFs may play a role in reducing the symptoms of AD. Additionally, the study highlights that unique mechanisms can be utilized to deliver NGFs to specific regions of the brain.
A larger randomized, double-blind, placebo-controlled phase 2 trial commenced in 2009 seeking to recruit 50 patients. Patients in the placebo arm of this ambitious study are required to undergo sham neurosurgery. As much as we would not wish to inflict sham neurosurgery on anyone, this is no other way to accurately assess the efficacy of the treatment. The study should be completed in 2014.
Brain Reserve
Postmortem studies have found that a substantial proportion of people have the histopathology of AD, but not the cognitive failings of dementia. Prospective studies suggest the number may be as high as 40%. Some believe that this is due to brain reserve; that is, greater neural substrate buffers against the clinical expression of the disease (similar to starting with more adipose tissue during times of famine). Indeed, prospective studies have found that individuals who have the plaques and tangles at death, but were not demented, had greater number of neurons in the frontal, parietal, and temporal cortices.
One of the most remarkable studies on this topic was the Nun Study, in which the correlation between early verbal skills and later cognitive impairment was determined. The researchers completed extensive cognitive assessments of the nuns older than 75 years in their retirement. In their early 20s, the nuns had completed an autobiographic essay when they entered the order. These essays were blindly graded for linguistic ability. The nuns with low idea density and low grammatical complexity in their autobiographies written 50 years earlier were 15 times more likely to have low cognitive scores in late life. In other words, cognitive skills in early life prevent cognitive impairment later on.
A follow-up study has taken this analysis one step further by including postmortem brain examination. This study included 156 individuals in whom the researchers could correlate educational background, level of cognition before death, and amyloid load in the brain at autopsy. The results are shown in Figure 24.13. The individuals with greater years of education had less cognitive impairment even with increasing amyloidosis. The authors concluded that education is associated with factors that somehow reduce the effect of amyloid on cognition. They estimated that the difference between 15 and 22 years of education is equivalent to approximately 2.6 years of amyloid progression. We hope this information provides some relief for the sort of person reading this book.
“Use it or lose it” is one of the mantras resulting from this line of research, the implication being that exercising the brain is neuroprotective against the pathology of AD. Clearly, the research described here shows that smarter, more educated people have greater brain resilience. What we do not know is whether these people were born with a larger reserve of neural substrate or whether a lifetime of cognitive enrichment–stimulated neuronal growth protected their brains from the effects of AD.
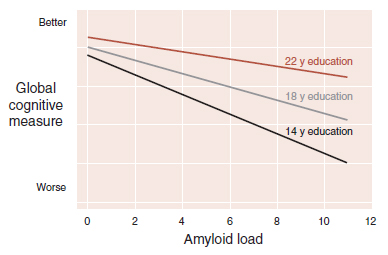
FIGURE 24.13 The cognitive impairment associated with amyloidosis is reduced in people with more education. (Adapted from Bennett DA, Schneider JA, Wilson RS, et al. Education modifies the association of amyloid but not tangles with cognitive function. Neurology. 2005;65[6]:953–955.)
Caloric Restriction
Caloric restriction is known to enhance longevity (see LONGEVITY, page 158). Dietary restriction may also retard the effects of AD. Studies with animals suggest that low-calorie diets may protect the brain in the following ways:
1. Limiting oxidative stress
2. Reducing DNA damage
3. Increasing brain-derived neurotrophic factor production
A recent study shows the profound effects that calorie restriction can have on amyloid deposits in nonhuman primates. A colony of squirrel monkeys was raised on a diet reduced by 30% and compared with a freely eating control group. As the monkeys died of natural causes, their temporal cortical tissues were measured for β-amyloid, APP, and the secretase enzymes.
The monkeys on dietary restriction showed no change in the amount of APP molecule, but a remarkable reduction in A-β. The activity of β- and γ-secretase enzymes (Figure 21.6) was no different between the two groups. However, α-secretase was almost 100% more active in the diet-restricted group. α-Secretase is an enzyme that cuts the APP molecule in a manner that limits the production of β-amyloid. In other words, caloric restriction enhances the activity of an enzyme that prevents the buildup of amyloid plaques.
Although caloric restriction may be effective in altering the buildup of β-amyloid, it seems unlikely to be utilized by the large number of people at risk for AD. Indeed, the industrial societies that are at most risk for AD are also the ones struggling with the obesity epidemic. However, if treatments that reduce hunger or enhance satiety can be developed, these could be reasonable pharmacologic approaches to forestalling AD.
Exercise
Four clinical trials presented at the Alzheimer’s Association International Conference in 2012 suggest that regular exercise can improve memory and preserve brain functions in the elderly. One such study from the University of Pittsburgh randomized 120 sedentary elderly adults to aerobic exercise or a stretching control group. Both groups demonstrated improvements in spatial memory at 6 months and 1 year. However, the exercise group had a 2% increase in their hippocampal volume at 1 year while the stretching group showed a slight decline.
Exercise is a low-cost, easily accessible treatment intervention that may preserve brain structures and maintain memory. Apparently, what is good for the heart is also good for the brain.
CABERNET SAUVIGNON
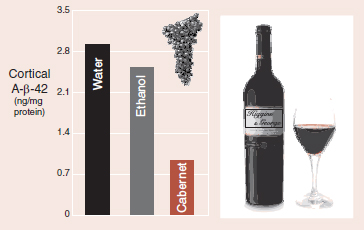
Alzheimer’s modeled mice showed significant reductions in cortical β-amyloid with daily Cabernet Sauvignon. (Adapted from Wang J, Ho L, Zhao Z, et al. Moderate consumption of Carernet Sauvignon attenuates AB neuropathology in a mouse model of Alzheimer’s disease. FASEB J. 2006;20:2313-2320.)
It is with great pleasure that we report the results of a study showing the cerebral benefits of moderate doses of red wine. Mice altered to overexpress APP were given daily supplements of California Cabernet Sauvignon, ethanol, or water. The wine group had not only superior memory function but also less β-amyloid in their cortex (figure).
We think that anyone who has finished reading every word of this book should celebrate with a little California Cabernet Sauvignon. Heck, it will give a little tweak to the nucleus accumbens, as well as promote β-amyloid clearance—all in moderation of course.
1. One hundred and twenty-five years
a. Life expectancy.
b. Fibroblast duration.
c. Maximum cell divisions.
d. Life span.
2. Alzheimer’s identified all of the following, except
a. Cortical atrophy.
b. Amyloid plaques.
c. β-Amyloid.
d. Neurofibrillary tangles.
3. All of the following are increased in AD, except
a. Amyloid-β precursor protein (APP).
b. A-β-42.
c. Ventricular size.
d. CSF tau protein.
4. Neurofibrillary tangles are composed of
a. Damaged microtubules.
b. Extracellular protein tangles.
c. Excessively phosphorylated tau.
d. Untransportable cellular products.
5. The goal of Alzheimer’s vaccine is to increase which of the following?
a. A-β-42.
b. T-cell activation.
c. Memory performance.
d. Anti-Aβ antibodies.
6. Increases with calorie restriction
a. Amyloid-β precursor protein (APP).
b. α-Secretase.
c. β-Secretase.
d. γ-Secretase.
See Answers section at the end of the book.