Simple and complex renal cysts
•Simple renal cysts: do not communicate with any part of the nephron or the renal pelvis. They are mainly confined to the renal cortex, are filled with clear fluid, and contain a membrane composed of a single layer of flattened or cuboidal epithelium. They can be single or multiple, ranging from a few millimetres to several centimetres in diameter. They can be unilateral or bilateral and often affect the lower pole of the kidney.
•Parapelvic cysts: describe simple parenchymal cysts located adjacent to the renal pelvis or hilum.
Prevalence
Increases with age, the precise prevalence depending on the method of diagnosis. On CT, 20% of adults have renal cysts by age 40y, and 33% by the age of 60.1 At post-mortem, 50% of subjects aged >50y have simple cysts. Cysts do not usually increase in size with age but may increase in number. ♂ and ♀ are affected equally.
Aetiology
Both congenital and acquired causes have been suggested. Chronic dialysis is associated with the formation of new simple cysts.
Presentation
Simple cysts are most commonly diagnosed as an incidental finding following a renal USS) or CT performed for other purposes. The majority are asymptomatic; however, very large cysts may present as an abdominal mass or cause dull flank or back pain. Acute severe loin pain may follow bleeding into a cyst (causing sudden distension of the wall). Rupture (spontaneous or following renal trauma) is rare. Rupture into the pelvicalyceal system can produce haematuria. Infected cysts (rare) present with flank pain and fever. Very occasionally, large cysts can cause obstruction and hydronephrosis.
Differential diagnosis
•RCC (4–7% of RCCs are cystic).
•Early autosomal dominant polycystic kidney disease (ADPKD—diffuse, multiple, or bilateral cysts, associated with hepatic cysts.
•Complex renal cysts (i.e. those which contain blood, pus, or calcification).
Investigation
Renal USS
Simple cysts are round or spherical, have a smooth and distinct outline, and are ‘anechoic’ (no echoes within the cyst, i.e. sound waves are transmitted through the cyst). USS using microbubble contrast agents can improve diagnostic accuracy. Evidence of calcification, septation, irregular margins, or clusters of cysts requires further investigation (renal triphasic CT). In the absence of these features, no further investigation is required.
CT
See Table 8.1 for Bosniak’s classification of the appearance of simple and complex cysts.
Simple cysts are seen as round, smooth-walled lesions with homogenous fluid in the cavity (with a typical density of –10 to +20 Hounsfield units) and with no enhancement after contrast (enhancement implies that it contains vascular tissue or communicates with the collecting system, i.e. that it is not a simple cyst). Hyperdense cysts have a density of +20–90 Hounsfield units, do not enhance with contrast media, and are <3cm in diameter.
Biopsy
Image-guided cyst aspiration or biopsy can be used to help diagnose indeterminate cysts and prevent unnecessary surgery.
Treatment
A simple cyst (type I: round or spherical, smooth wall, distinct outline, and no internal echoes) requires no further investigation, no treatment, and no follow-up. In the rare situation where the cyst is thought to be the cause of symptoms (e.g. back or flank pain), treatment options include percutaneous aspiration ± injection of sclerosing agent or open or laparoscopic surgical excision of the cyst wall. In the rare event of cyst infection, percutaneous drainage and antibiotics are indicated.
Cysts with features on USS suggesting possible malignancy (calcification, septation, irregular margins) should be investigated by CT with contrast.
Table 8.1 Bosniak’s classification of CT appearance of simple and complex cysts
| Type |
Description |
Approximate % of such cysts which are malignant2 |
Treatment |
| I |
Simple benign cyst with smooth margins, no contrast enhancement, no septation, no calcification |
<2% |
None; no follow-up required |
| II |
Benign cyst with smooth margins; few thin septae; minimal calcification; no contrast enhancement; <3cm |
|
Observation—repeat USS looking for increase in size or development of malignant features* |
| IIF |
 number of thin septae; thickening and/or minimal enhancement of septae; may contain calcium, but no enhancement. Includes non-enhancing, high-attenuation >3cm cysts number of thin septae; thickening and/or minimal enhancement of septae; may contain calcium, but no enhancement. Includes non-enhancing, high-attenuation >3cm cysts |
19%
(II and IIF combined) |
Follow-up with USS (or CT). Type IIF cysts have greater malignant potential than type II cysts |
| III |
Irregular margins; moderate calcification; thick septation (septae >1mm thick); enhancement |
33% |
Surgical exploration ± partial nephrectomy |
| IV |
Cystic malignant lesion; irregular margins and/or solid enhancing elements |
93% |
Radical nephrectomy |
* Bosniak suggested follow-up scans at 6 months and 1y. If the lesion remained stable after this time, it is considered benign.
References
1Laucks SP Jr, McLachlan MS (1981). Aging and simple cysts of the kidney. Br J Radiol 54:12–14.
2Warren KS, McFarlane J (2005). The Bosniak classification of renal cystic masses. BJU Int 95:939–42.
Calyceal diverticulum
A calyceal diverticulum is a spherical outpouching of the renal collecting system (specifically from a calyx) which protrudes into the corticomedullary region of the kidney. It communicates with the renal calyx via a narrow neck or channel. It is lined by a transitional cell epithelium and is covered by a thin layer of renal cortex. They range from only a few millimetres to many centimetres in size.
Aetiology
The exact aetiology of calyceal diverticula is unknown. Some may be congenital. Acquired calyceal diverticula can develop after obstruction of a calyceal infundibulum or following blunt renal trauma.
Presentation
They are usually asymptomatic and are discovered incidentally on an IVU, most commonly seen in upper pole calyces. Symptoms may result from the development of a stone or infection within the diverticulum, presumably caused by urinary stasis.
Investigation
On IVU, a calyceal diverticulum appears as a rounded collection of contrast medium next to a papilla, although often, the connecting channel is too narrow to be clearly seen. They can be identified on CT, MRI, and USS; however, the distinction between a renal cyst and an obstructed calyx may be difficult on unenhanced images.
Treatment
Stones that form within the calyceal diverticulum may be treated by flexible ureteroscopy and laser lithotripsy or, if large, by percutaneous nephrolithotomy (PCNL) if percutaneous access is possible. Endoscopic dilatation or incision of the neck of the diverticulum may be attempted at the time of stone surgery to prevent recurrence, and this technique can also be employed if the diverticulum is thought to be the cause of recurrent urinary infection. Open surgery has also been used to remove stones and to deroof calyceal diverticula. Extracorporeal shock wave lithotripty (ESWL) therapy is not helpful. ESWL may result in stone fragmentation, but it may be difficult for the stone fragments to get out of the diverticulum and they may simply reform into a larger stone.
Medullary sponge kidney (MSK)
Definition
A congenital cystic disorder of the kidneys characterized by dilatation of the distal CDs associated with the formation of multiple cysts and diverticula within the medulla of the kidney.
Prevalence
Difficult to know, as it may be asymptomatic (diagnosed on an IVU performed for other reason or at post-mortem). Estimated to affect between 1 in 5000 to 1 in 20 000 people in the general population; 1 in 200 in those under going IVU (a select population). In 75% of cases, both kidneys are affected.
Pathology
The renal medulla resembles a sponge in cross-section due to dilated CDs in the renal papillae and the development of numerous small cysts. This is associated with urinary stasis and the formation of small calculi within the cysts. Some report a familial inheritance. It can be associated with other congenital or inherited disorders, including hemihypertrophy and Beckwith–Wiedemann syndrome.*
Presentation
The majority of patients are asymptomatic. When symptoms do occur, they include ureteric colic, renal stone disease (calcium oxalate ± calcium phosphate), UTI, and haematuria (microscopic or macroscopic). Up to 50% have hypercalciuria due to renal calcium leak or GI calcium absorption. Renal function is normal, unless obstruction occurs (secondary to renal pelvis or ureteric stones).
Differential diagnosis
Other causes of nephrocalcinosis (deposition of calcium in the renal medulla, e.g. TB, hyperparathyroidism, healed papillary necrosis, multiple myeloma).
Investigation
•MSU: dipstick ± culture. Check for UTI and treat according to sensitivities.
•Biochemistry: 24h urinary calcium may be elevated (hypercalciuria). Detection of hypercalciuria requires further investigation to exclude other causes (i.e. raised PTH levels indicate hyperparathyroidism).
•Imaging: IVU is the principle method for diagnosing MSK, although CT and USS may also be used. The characteristic radiological features of MSK, as seen on IVU, are enlarged kidneys associated with dilatation of the distal portion of the CDs, along with numerous associated cysts and diverticula (the dilated ducts are said to give the appearance of ‘bristles on a brush’). The CDs may become filled with calcifications, giving an appearance described as a ‘bouquet of flowers’ or ‘bunches of grapes’ (Fig. 8.1).
Treatment
Asymptomatic MSK disease requires no treatment. General measures to reduce urine calcium levels help reduce the chance of calcium stone formation (high fluid intake, vegetarian diet, low salt intake, consumption of fruit and citrus fruit juices). Thiazide diuretics may be required for hypercalciuria resistant to dietary measures and are designed to lower urine calcium concentration. Intrarenal calculi are often small and, as such, may not require treatment, but if indicated, this can take the form of ESWL or flexible ureteroscopy and laser treatment. Ureteric stones are again usually small and will therefore pass spontaneously in many cases with a period of observation. Recurrent UTI may need prophylactic antibiotics. Renal function tends to remain stable in the long term. Rarely, recurrent infection and nephrocalcinosis may lead to the complication of renal impairment.
Acquired renal cystic disease
A cystic degenerative disease of the kidney, with ≥5 cysts visualized on CT scan. By definition, this is an acquired condition, as opposed to ADPKD which is inherited (in an autosomal dominant fashion). It is predominantly associated with chronic and end-stage renal failure and, as such, is commonly found in patients undergoing haemodialysis or peritoneal dialysis. Over one-third of patients develop acquired renal cystic disease (ARCD) after 3y of dialysis. Clinically important because it may cause pain and haematuria and is associated with the development of benign and malignant renal tumours. The ♂:♀ ratio is 3:1.
Pathology
Usually multiple bilateral cysts found mainly within the cortex of small, contracted kidneys. Cysts vary in size (average 0.5–1cm) and are filled with a clear fluid which may contain oxalate crystals. They usually have cuboidal or columnar epithelial linings and are in continuity with renal tubules (and therefore cannot be defined as simple cysts). Atypical cysts have a hyperplastic lining of epithelial cells, which may represent a precursor for tumour formation. Renal transplantation can cause regression of cysts in the native kidneys.
Aetiology
The exact pathogenesis is unknown, but several theories have been proposed. Obstruction or ischaemia of renal tubules may induce cyst formation. Renal failure may predispose to the accumulation of toxic endogenous substances or metabolites, alter the release of growth factors, and result in changes in sex steroid production or cause cell proliferation (secondary to immunosuppressive effects) which result in cyst formation.
Associated disorders
There is an risk of benign and malignant renal tumours. The chance of developing RCC is ~20%, 3–6 times greater than the general population (♂ > ♀). When on dialysis, RCC usually develops within the first 10y of treatment.
Presentation
Flank pain; UTI; visible haematuria; renal colic (stone disease); hypertension.
Investigation
This depends on the presenting symptoms.
•For suspected UTI: culture urine.
•For haematuria: urine cytology, flexible cystoscopy, and renal USS. On USS, the kidneys are small and hyperechoic, with multiple cysts of varying size, many of which show calcification. If the nature of the cysts cannot be determined with certainty on USS, arrange a renal CT.
Treatment
Persistent macroscopic haematuria can become problematic, exacerbated by heparinization (required for haemodialysis). Options include transferring to peritoneal dialysis, renal embolization, or nephrectomy (acceptable as these patients already on dialysis by definition have non-functioning kidneys). Infected cysts, which develop into abscesses, require percutaneous or surgical drainage. Radical nephrectomy is indicated for renal masses with features suspicious of malignancy. Smaller asymptomatic masses require surveillance. Patients with ARCD on long-term dialysis should also be considered for renal surveillance with ultrasonography or CT.
Autosomal dominant polycystic kidney disease
Definition
An autosomal dominant inherited disorder involving multiple expanding renal parenchymal cysts (Fig. 8.2).
Epidemiology
Incidence is 0.1–0.5%; 95% are bilateral. ADPKD can affect children and adults, although symptoms usually occur between ages 30 and 50y. ADPKD accounts for 10% of all renal failures (which usually manifest at >40y old).
Pathology
The kidneys reach an enormous size due to multiple fluid-filled cysts and can easily be palpated on abdominal examination. Expansion of the cysts results in ischaemic atrophy of the surrounding renal parenchyma and obstruction of normal renal tubules. End-stage renal failure occurs at around age 50y.
Associated disorders
Ten to 30% incidence of circle of Willis berry aneurysms (associated with subarachnoid haemorrhage), cysts of the liver (33%), pancreas (10%), spleen (<5%), and seminal vesicles, mitral valve prolapse, aortic root dilatation, aortic aneurysms, and diverticular disease. Of note, the incidence of renal adenoma is ~20%; however, the risk of RCC is the same as the general population.
Aetiology
Two genes have been identified in ADPKD. The PKD1 gene is localized on the short arm of chromosome 16 (16p13.3) and accounts for 85% of cases. The PKD2 gene is on the long arm of chromosome 4 (4q21) and causes 15% of cases. A third gene PKD3 is also implicated. Pathogenesis theories include intrinsic basement membrane abnormalities, tubular epithelial hyperplasia (causing tubular obstruction and basement membrane weakness), and alterations in the supportive extracellular matrix due to defective proteins, all of which may cause cyst formation.
Presentation
•Positive family history.
•Hypertension (75%).
•Palpable abdominal masses.
•Flank pain (due to mass effect, infection, stones, or following acute cystic distension due to haemorrhage or obstruction).
•Haematuria (visible or non-visible).
•UTI.
•Renal failure which may present with lethargy, nausea, vomiting, anaemia, confusion, and seizures.
Differential diagnosis
Other forms of renal cystic disease: multiple simple cysts, autosomal recessive polycystic kidney disease (ARPKD), familial juvenile nephronophthisis, medullary cystic disease (see  pp. 706–707).
pp. 706–707).
Multiple renal cysts are also found in other autosomal dominant conditions:
•TS: has TSC1 and 2 gene mutations on chromosomes 9 and 16. It presents with adenoma sebaceum, epilepsy, learning difficulties, polycystic kidneys, and renal tumours (angiomyolipomas and, more rarely, RCC).
•VHL syndrome: has a VHL tumour suppressor gene mutation on the short arm of chromosome 3 (3p25) which causes HIF to increase the levels of growth factors [platelet-derived growth factor (PDGF), TGF-α, VEGF), which can stimulate the formation of haemangioblastomas (cerebellar and retinal) and RCC. VHL syndrome also includes renal, pancreatic, and epididymal cysts, and phaeochromocytoma.
Investigation
This depends on the presenting symptoms:
•Adult patients with a family history of ADPKD: first counsel the patient on the implications of a positive diagnosis. USS*, CT, and MRI of the renal tract are useful for initial diagnosis and investigation of complications. On USS, the kidneys are small and hyperechoic, with multiple cysts of varying size, many of which show calcification. If the nature of the cysts cannot be determined with certainty on USS, arrange a renal CT. Genetic testing can be done if imaging is equivocal or when a definite diagnosis is required in a young patient.
•For suspected UTI: culture urine.
•For haematuria: urine cytology, flexible cystoscopy, and renal USS.
•Renal failure: refer for management by a nephrologist. Renal failure may be associated with anaemia, although conversely, ADPKD can cause erythropoietin production and polycythaemia.
Treatment
The aim is to preserve renal function for as long as possible (monitor and control hypertension and UTI). Infected cysts should be drained. Persistent, heavy haematuria can be controlled by embolization or nephrectomy. Progressive renal failure requires dialysis and ultimately renal transplantation.
Due to the high risk of inheritance of ADPKD, offsprings should be fully counselled and offered genetic testing or USS screening at an appropriate time.
Vesicoureteric reflux in adults
VUR is the retrograde flow of urine from the bladder into the upper urinary tract with or without dilatation of the ureter, renal pelvis, and calyces (see pp. 692–695).
Pathophysiology
Reflux is normally prevented by low bladder pressures, efficient ureteric peristalsis, and the ability of the VUJ to occlude the distal ureter during bladder contraction. This is assisted by the ureters passing obliquely through the bladder wall (the ‘intramural’ ureter) which is 1–2cm long. Normal intramural ureteric length to ureteric diameter ratio is 5:1. VUR of childhood tends to resolve spontaneously with increasing age, because as the bladder grows, the intramural ureter lengthens.
Classification
•Primary: a primary anatomical (and therefore functional) defect where the intramural length of the ureter is too short (ratio <5:1).
•Secondary: to some other anatomical or functional problem.
•BOO (BPO, DSD, urethral stricture, missed PUVs), leading to elevated bladder pressures.
•Poor bladder compliance or intermittently elevated pressures of neuropathic detrusor overactivity (due to neuropathic disorders,* e.g. SCI, spina bifida).
•Iatrogenic reflux. A relatively common cause would be direct ureteric reimplantation into the bladder without using an antirefluxing technique. Other causes include: ureteric meatotomy, i.e. incision of the ureteric orifice for removal of ureteric stones stuck at the VUJ; following incision of a ureterocele; following TURP or TURBT; and post-pelvic RT.
•Inflammatory conditions affecting function of the VUJ—TB, schistosomiasis, UTI.
Associated disorders
•VUR is commonly seen in duplex ureters (the Weigert–Meyer law)** and associated with PUJ obstruction. Cystitis can cause VUR through bladder inflammation, reduced bladder compliance, pressures, and distortion of the VUJ. Coexistence of UTI with VUR can cause pyelonephritis. Reflux of infected urine under high pressure may lead to reflux nephropathy, resulting in renal scarring, hypertension, and renal impairment—although this is much less common in adults, compared to children. More commonly, this would manifest as loin pain in adults.
Presentation
•VUR may be asymptomatic. It may only be detected incidentally during investigations performed for other reasons such as videourodynamics, IVU, or renal USS.
•Loin pain (sometimes associated with a full bladder or immediately after micturition).
•UTI symptoms.
Investigation
The definitive test for the diagnosis of VUR is cystography, which may be apparent during bladder filling or during voiding [micturating cystourethrography (MCUG)]. Where clinically indicated, urodynamics establishes the presence of voiding dysfunction. If there is radiographic evidence of reflux nephropathy, check BP and the urine for proteinuria, measure serum creatinine, and arrange a 99mTc-DMSA renogram study to assess for renal cortical scarring and determine the split renal function.
Management
VUR is harmful to the kidney in the presence of infected urine and/or where bladder pressures are markedly elevated (due to severe BOO, poor compliance, or high-pressure OAB contractions). In the absence of these factors, VUR is not harmful, at least in the short term (months). Subsequent management depends on:
•The presence and severity of symptoms.
•The presence of recurrent, proven urinary infection.
•The presence of already established renal damage, as indicated by radiological evidence of reflux nephropathy, hypertension, impaired renal function, or proteinuria.
Primary VUR
•For the patient with primary VUR and recurrent UTIs with no symptoms between infections, no hypertension, and good renal function: treat the UTIs when they occur; consider low-dose antibiotic prophylaxis if UTIs occur frequently (>3 per year). If UTIs are regularly associated with systemic symptoms (acute pyelonephritis, rather than uncomplicated cystitis), then ureteric reimplantation is indicated.
•For the patient with primary VUR and objective evidence of deterioration in the affected kidney: ureteric reimplantation.
•Reflux into a non-functioning kidney (<10% function on DMSA scan) with recurrent UTIs and/or hypertension: nephroureterectomy.
•Primary reflux with severe recurrent loin pain: ureteric reimplantation.
Secondary VUR
•VUR into a transplanted kidney: no treatment is necessary.
•VUR in association with the neuropathic bladder: treat the underlying cause—relieve BOO, improve bladder compliance (options: intravesical BTX injections, augmentation cystoplasty, sacral deafferentation).
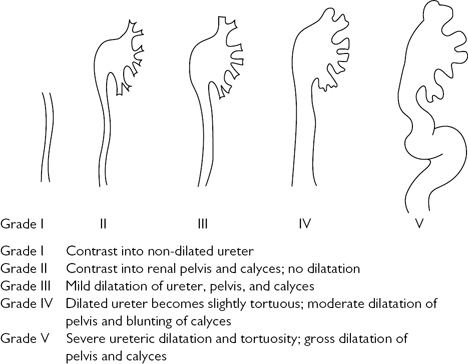
•VUR with no symptoms, no UTI, no high bladder pressures, and no BOO: for grade I–II reflux, monitor for infection, hypertension, and evidence of deterioration in the appearance and function of the kidneys. For grades III–V, some urologists would recommend ureteric reimplantation or an endoscopic injection of bulking agent at the ureteric orifice (for VUR grading) (Fig. 8.3).
Pelviureteric junction obstruction in adults
Definition
PUJO is an obstruction of the proximal ureter at the junction with the renal pelvis, resulting in a restriction of urine flow (see pp. 702–703)—known as ‘uretero-pelvic junction obstruction’ (UPJO) in North America.
Aetiology
Congenital
•Intrinsic: smooth muscle defect results in an aperistaltic segment of the ureter at the PUJ. The ureter can insert high on the renal pelvis (which may be a primary abnormality or secondary to the pelvic dilatation).
•Extrinsic: compression from the lower renal pole (‘aberrant’) vessel over which the PUJ runs. It is unlikely that these vessels are the primary cause of the obstruction. It is more probable that PUJO leads to a dilated PUJ and ballooning of the renal pelvis over the lower pole vessels, which may thus contribute to, but is not the primary cause of, the obstruction.
Acquired
PUJ stricture secondary to ureteric manipulation (e.g. ureteroscopy); trauma from passage of calculi; fibroepithelial polyps; TCC of the urothelium at the PUJ; external compression of the ureter by retroperitoneal fibrosis or malignancy.
Presentation
Flank pain precipitated by diuresis (high fluid intake, especially after consumption of alcohol); flank mass; UTI; haematuria (after minor trauma). It may also be associated with VUR.
Investigation
•Blood test: for renal function (U&E, eGFR).
•MSU: to exclude infection.
•Renal USS: shows renal pelvis dilatation in the absence of a dilated ureter.
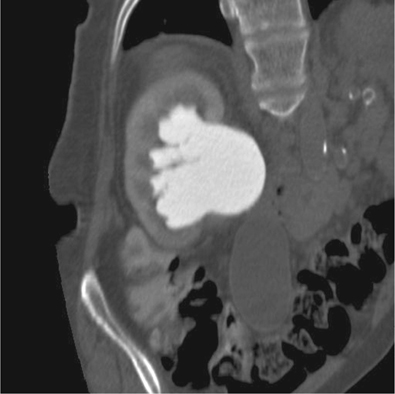
•IVU: demonstrates a delay of excretion of contrast and a dilated pelvicalyceal system (Fig. 8.4).
•CT: shows a dilated renal pelvis and non-dilated ureter. Also helpful in excluding a small, radiolucent stone, urothelial TCC, or retroperitoneal pathology, which may be the cause of the obstruction at the PUJ (Fig. 8.5).
•MAG3 renogram (with administration of furosemide to establish maximum diuresis): is the definitive diagnostic test for PUJO. Radioisotope accumulates in the renal pelvis, and following IV furosemide, it continues to accumulate (a ‘rising’ curve). Also useful as it provides split renal function.
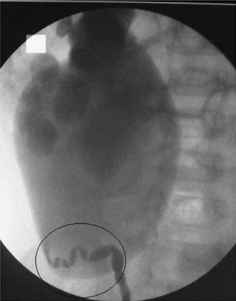
•Retrograde pyelography: to establish the exact site of the obstruction—often performed at the time of PUJ repair to avoid introducing infection into an obstructed renal pelvis (Fig. 8.6).
Management
Surgery is indicated for recurrent episodes of bothersome pain, renal impairment, where a stone has developed in the obstructed kidney, and where infection has supervened (an acutely infected obstructed kidney in a septic patient will require nephrostomy insertion). In the absence of symptoms, consider watchful waiting with serial MAG3 renograms. If renal function remains stable and the patient remains free of symptoms, there is no need to operate. A non-functioning, ‘burnt-out’ kidney with PUJO may require nephrectomy to avoid the complication of pyonephrosis.
Pyeloplasty
(See pp. 816–817.)
•Laparoscopic pyeloplasty: dismembered pyeloplasty is the most commonly performed technique using transperitoneal, retroperitoneal, or robotic-assisted approaches. Success rates are ~95%.
•Open pyeloplasty: success rates of 95%. Common techniques include dismembered or Anderson–Hynes pyeloplasty. The narrowed area of the PUJ is excised, and the proximal ureter is spatulated and anastomosed to the renal pelvis. Alternative techniques include flap pyeloplasty (Culp) and Y–V-plasty (Foley).
A double J ureteric stent is left for 6wk post-operatively, which can be removed with flexible cystoscopy as an outpatient procedure.
Endopyelotomy (or pyelolysis)
A minimally invasive technique to treat PUJO, but tends not to be offered as first-line therapy other than in older or frail patients. It can be utilized after pyeloplasty has failed. A full-thickness incision is made through the obstructing proximal ureter from within the lumen of the ureter down into the peripelvic and periureteral fat, using a sharp knife or Holmium:YAG laser. The incision is stented for 4wk to allow re-epithelialization of the PUJ. Generally not used for PUJO of >2cm in length. The incision may be made percutaneously or by a retrograde approach via a rigid or flexible ureteroscope or by using a specially designed endopyelotomy balloon—the Acucise® technique. Here, an angioplasty-type balloon (over which runs a cautery wire) is inflated across the PUJ. An electrical current heats the wire, and this cuts through the obstructing ring of tissue at the PUJ.
The presence of a combination of PUJO and a renal stone that is suitable for PCNL is an indication for combined PCNL and percutaneous endopyelotomy.
Success rates in terms of relieving obstruction: percutaneous endopyelotomy, 60–100% (mean 70%); cautery wire balloon endopyelotomy, 70%; ureteroscopic endopyelotomy, 80%.
Follow-up
Repeat MAG3 renogram is usually performed 3 months post-operatively. If there has been no improvement, this can be repeated 6–12 months post-operatively. Failure may need redo surgery or endopyelotomy.
Anomalies of renal fusion and ascent: horseshoe kidney and ectopic kidney
Abnormalities of renal fusion and ascent occur in weeks 6–9 of gestation, when the embryonic kidney is ‘ascending’ to its definitive lumbar position in the renal fossa (‘ascending’ as a result of rapid caudal growth of the embryo).
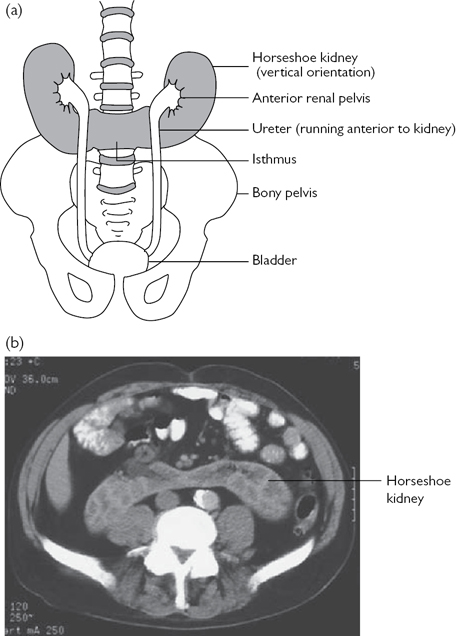
Horseshoe kidney
Commonest example of renal fusion. Prevalence 1 in 400. ♂:♀ ratio 2:1. The kidneys lie vertically (instead of obliquely) and are joined at their lower poles (in 95%) by midline parenchymal tissue (the isthmus). The inferior mesenteric artery obstructs the ascent of the isthmus. Consequently, the horseshoe kidney lies lower in the abdomen (L3 or L4 vertebral level). Normal rotation of the kidney is also prevented, and therefore, the renal pelvis lies anteriorly, with the ureters also passing anteriorly over the kidneys and isthmus (but entering the bladder normally). Blood supply is variable, usually from one or more renal arteries or their branches or from branches off the aorta or inferior mesenteric artery (Fig. 8.7).
A proportion of individuals with horseshoe kidneys have associated congenital abnormalities (Turner’s syndrome, trisomy 18, genitourinary anomalies, ureteric duplication), VUR, PUJ obstruction, and renal tumours (including Wilms’ tumours).
Most patients with horseshoe kidneys remain asymptomatic; however, infection and calculi may develop and cause symptoms. The diagnosis is usually suggested on renal USS and confirmed by IVU (calyces of the lower renal pole are seen to point medially and lie medially in relation to the ureters) or CT. Renal function is usually normal.
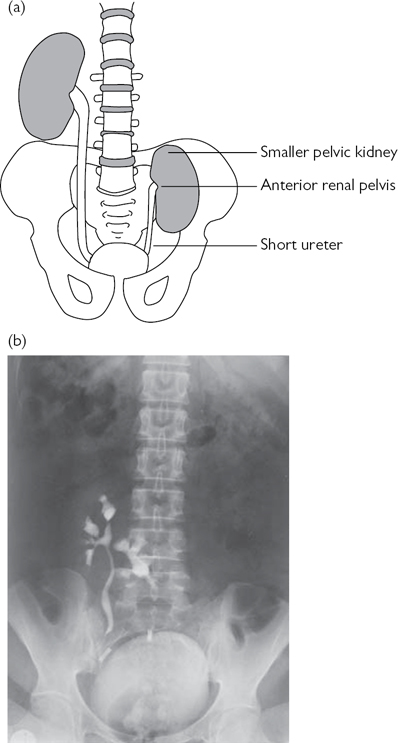
Ectopic kidney
The kidney fails to achieve its normal position and may be located in the thorax, abdomen, lumbar region (in iliac fossa), or pelvis (on the contralateral side or crossed). The prevalence of renal ectopia is 1 in 900, with both sexes affected equally. The left kidney is affected more often than the right, and bilateral cases are seen in <10%. The affected kidney is smaller, with the renal pelvis positioned anteriorly (instead of medially), and the ureter is short but enters the bladder normally. Pelvic kidneys occur in 1 in 2000–3000 and lie opposite the sacrum and below the aortic bifurcation and are supplied by adjacent (aberrant) vessels (Fig. 8.8). Renal ectopia has an risk of congenital anomalies, including contralateral renal agenesis and genital malformations.
Most are asymptomatic. Diagnosis is made on renal USS, IVU, or renography. Complications include hydronephrosis [secondary to VUR, VUJ obstruction (VUJO), and PUJO], stones, and infection.
Anomalies of renal number and rotation: renal agenesis and malrotation
Renal agenesis
Unilateral renal agenesis is the absence of one kidney due to embryological abnormality or absence of the ureteric bud. This results in failure of the ureteric bud to contact the metanephric blastema, with failed induction of nephrogenesis. The incidence is 1 in 1000; left side > right, ♂ > ♀. Absence of a kidney may also be caused by involution of a multicystic dysplastic kidney in utero or postnatally. Many patients are asymptomatic; however, it is associated with Turner’s syndrome and cardiac, respiratory, GI, and musculoskeletal abnormalities. Associated genitourinary anomalies include absence of the ipsilateral ureter, abnormal trigone, VUR, PUJO, VUJO, uterine abnormalities (unicornuate—one side has failed to develop; bicornuate—partially divided uterus; didelphys—double uterus), vaginal agenesis, anomalies of the seminal vesicles, and absence of the vas deferens. Often discovered as an incidental finding on USS performed for other reasons or during investigation of associated abnormalities. Long-term follow-up of renal function, urinalysis, and BP should be considered.
Bilateral renal agenesis is rare and incompatible with life. It is associated with complete ureteric atresia, bladder hypoplasia or absence, intrauterine growth retardation, pulmonary hypoplasia, and oligohydramnios (reduced amniotic fluid), causing characteristic ‘Potter’ facial features (blunted nose, low-set ears, depression on the chin) and limb abnormalities.
Malrotation
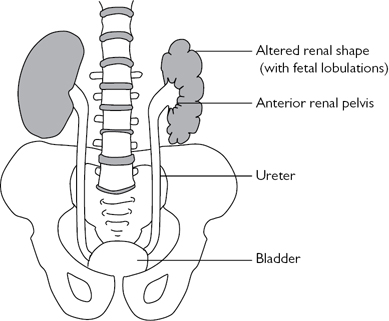
The kidney is located in a normal position, but the renal pelvis fails to rotate to the normal medial orientation. Often seen with horseshoe kidneys and renal ectopia and associated with Turner’s syndrome. The incidence is ~1 in 1000, with a ♂:♀ ratio of 2:1. The renal shape may be altered (flattened, oval, triangular, or elongated), and the kidney retains its fetal lobulated outline (Fig. 8.9). It is associated with deposition of fibrous tissue around the renal hilum, which can produce symptoms due to ureteric or PUJ obstruction (causing hydronephrosis, infection, or stone formation). Most patients, however, remain asymptomatic. The diagnosis is made on USS, IVU, or retrograde pyelography.
Upper urinary tract duplication
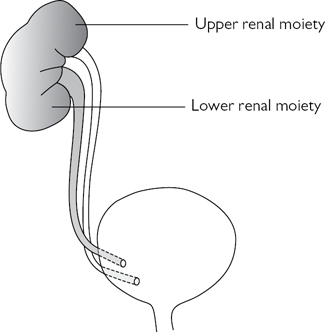
Definitions
A duplex kidney has an upper renal moiety and a lower renal moiety, each with its own separate pelvicalyceal system and ureter. The two ureters may join to form a single ureter at the PUJ (bifid system) (Fig. 8.10) or more distally (bifid ureter) before entering the bladder through one ureteric orifice. Alternatively, the two ureters may pass down individually to the bladder (complete duplication) (Fig. 8.11). In this case, the Weigert–Meyer rule states that the upper moiety ureter always opens onto the bladder medially and inferiorly to the ureter of the lower moiety, thereby predisposing to ectopic placement of the ureteric orifice and obstruction (due to the longer intramural course of the ureter through the bladder wall). The lower moiety ureter opens onto the bladder laterally and superiorly, reducing the intramural ureteric length which predisposes to VUR (in up to 85%) (Fig. 8.12).
Epidemiology
Ureteric duplication occurs in 1 in 125 individuals. The ♀:♂ ratio is 2:1. Unilateral cases are commoner than bilateral cases, with right and left sides affected equally. Risk of other congenital malformations is .
Embryology
In duplication, two ureteric buds arise from the mesonephric duct (week 4 of gestation). The ureteric bud situated more distally (lower moiety ureter) enters the bladder first and so migrates a longer distance, resulting in the superior and lateral position of the ureteric orifice. The proximal bud (upper moiety ureter) has less time to migrate, and consequently, the ureteric orifice is inferior and medial (ectopic) (see pp. 698–699). Interaction of each ureteric bud with the same metanephric tissue creates separate collecting systems within the same renal unit. With bifid ureters, a single ureteric bud splits after it has emerged from the mesonephric duct.
Complications
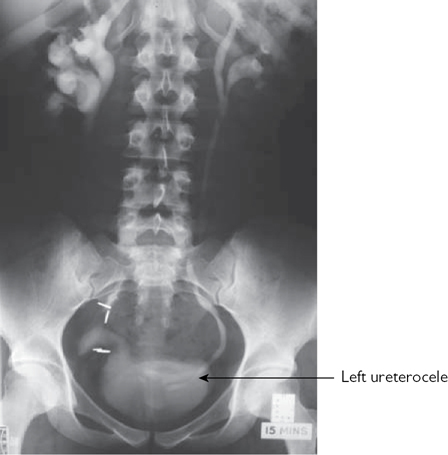
Ectopic ureters are associated with upper renal moiety hydronephrosis (secondary to obstruction), renal hypoplasia or dysplasia (maldevelopment of the kidney correlating with the degree of ectopic displacement of the ureteric orifice),1 and ureteroceles (Fig. 8.10). Lower moiety ureters are prone to reflux, resulting in hydroureter and hydronephrosis. Bifid ureters can get urine continuously, passing from one collecting system to the other (yo-yo reflux), causing urinary stasis (and predisposing to infection).
Presentation
Symptoms of UTI, flank pain, or an incidental finding.
Investigation
•Renal USS: demonstrates ureteric duplication ± dilatation and hydronephrosis.
•IVU:  contrast excretion from the renal upper pole ± hydronephrosis (which may displace the lower pole downwards and outwards, producing a ‘drooping lily’ appearance). Contrast in a ureterocele gives the appearance of a ‘cobra head’ (Fig. 8.10).
contrast excretion from the renal upper pole ± hydronephrosis (which may displace the lower pole downwards and outwards, producing a ‘drooping lily’ appearance). Contrast in a ureterocele gives the appearance of a ‘cobra head’ (Fig. 8.10).
•MCUG: will determine whether reflux is present.
•Enhanced CT and MRI: reveals detailed anatomical information.
• 99mTc-DMSA renogram: assesses individual renal moiety function.
Management
Uncomplicated complete or incomplete ureteric duplication does not require any intervention. In symptomatic patients, the aim is to reduce obstruction and reflux and to improve function. Where renal function is reasonable, common sheath ureteric reimplantation (where a cuff of bladder tissue is taken that encompasses both duplicated ureters) can treat both conditions. A poorly functioning renal moiety (i.e. upper moiety associated with an ectopic ureter and/or reflux or lower moiety associated with a ureterocele) may require heminephrectomy and ureterectomy. Where both renal moieties have poor function or dysplasia, nephroureterectomy is indicated.
Reference
1Mackie GG, Stephens FD (1975). Duplex kidneys: a correlation of renal dysplasia with position of the ureteric orifice. J Urol 114:274–80.
* Beckwith–Wiedemann syndrome: a growth disorder characterized by macroglossia, macrosomia, visceromegaly, Wilms’ tumour, neuroblastoma, omphalocele, and renal anomalies.
* USS diagnostic criteria. Patients at 50% risk for developing ADPKD are: ≥2 unilateral or bilateral cysts if aged <30y; two cysts in each kidney in patients aged 30–59y; four cysts in each kidney in patients aged >60y.
* Neuropathic disorders cause VUR because they lead to intermittently or chronically raised bladder pressure (due to BOO, poor compliance, and/or detrusor overactivity).
** The lower renal moiety ureter inserts into the bladder in a higher and more lateral location, as compared to the upper moiety ureter, which inserts distally and medially, i.e. nearer the bladder neck. The lower moiety ureter has a shorter intramural length and therefore can be prone to reflux. The upper moiety ureter has a longer intramural length and tends to be at risk of obstruction.