Chapter 8
Step-Growth (Condensation) Polymerization
8.1 Introduction
Polymerization reactions may be divided into two categories according to the mechanism by which the chains grow. In step-growth polymerization, also known as condensation polymerization, chains of any lengths x and y can combine to form longer chains:

In chain-growth (also known as addition or free-radical) polymerization, a chain of any length x can only add a monomer molecule to continue its growth:

These two mechanisms generally require different organic functional groups, carboxylic acids, amines, alcohols, etc., for step-growth and alkenes for chain-growth. Another distinction that you may have picked up on in Chapter 2 is that the molecular weight of the monomer and the repeat unit are generally the same for chain-growth polymers, but the repeat unit is smaller (due to the condensation of water) for most step-growth polymers. This is not always true, since some condensation polymerizations do not split out a water molecule (see Examples 2.4F and 2.4Q). This chapter covers step-growth polymerization, while chain-growth polymerization is considered in Chapters 9 and 10.
Regardless of the type of polymerization reaction, quantitative treatments are usually based on the assumption that the reactivity of the functional group at a chain end is independent of the length of the chain, x and y in Equations (8.1a) and (8.1b). Experimentally, this is an excellent assumption for x's greater than five or six. Since most polymers must develop x's on the order of a hundred (or much greater) to be of practical value, this assumption introduces little error while enormously simplifying the mathematical treatment. With these basic concepts in mind, we proceed to a more detailed and quantitative treatment of polymerization reactions.
8.2 Statistics of Linear Step-Growth Polymerization
Consider the two equivalent linear step-growth reactions, assuming difunctional monomers and stoichiometric equivalence:

(8.2b) 
where A and B represent the complementary functional groups. It is important to note that x is used here to denote the number of monomer residues or structural units in the chains, rather than repeat units (which would be either [BR′B-ARA] or [ARB]1).
Each of the polymer molecules above contains a total of x A groups:

so x still represents the degree of polymerization with either repeat unit listed above.
Considering a volume of the reaction mixture at a certain time during polymerization, we can use statistical analysis to find the polymer chain length and average molecular weight. For this analysis, we will let


The total probability of finding a molecule with x A groups (reacted and unreacted) is equal to the mole or number fraction of those molecules present in the reaction mass nx/N. This, in turn, is equal to the probability of finding a molecule with x − 1 reacted A groups and one unreacted A group. Since the overall probability is the product of the individual probabilities,

This is the so-called most probable distribution. It results from the random nature of the reaction between chains of different lengths, which will produce polymers of various lengths at any time during the reaction. As the reaction proceeds with time, the conversion increases to yield longer chains. The distribution is plotted in Figure 8.1 for several conversions, p. Note that at any conversion, the shorter chains are always more numerous, that is, the longer the chain, the fewer the chain. Also, the fraction of short chains decreases with conversion, while the fraction of long chains increases with conversion, because increasing conversion hooks together shorter chains to form longer chains.
Figure 8.1 Number- (mole-) fraction distributions for linear step-growth polymers at different conversions (Eq. (8.3)).

Because each growing polymer chain has two functional groups (one at each end), step-growth polymerizations can involve reactions between monomers, oligomers, or polymers of any length, x:

This is an unique characteristic of step-growth polymerizations and contrasts with chain-growth polymerizations (Chapter 9) that add only monomers (1-mers) to growing chains (Eq. (8.1b)).
8.3 Number-Average Chain Lengths
Since a distribution of chain lengths always arises in polymerization reactions, the average chain length is often of interest. Each molecule in the reaction mass has, on an average, one unreacted A group. If No is the original number of molecules present in the reaction mass, there are No unreacted A groups present at the start of the reaction. At some time later during the reaction there are fewer molecules (N), and therefore N unreacted A groups are present, so No − N of the A groups have reacted. Thus,

Because the No original monomer molecules are distributed among the N molecules present in the reaction mass, the average number of monomer residues per molecules,  , is given by
, is given by

Combining Equations (8.4) and (8.5) gives

This rather simple conclusion was reached by W.H. Carothers, the discoverer of nylon and one of the founders of polymer science, in the 1930s. (The same result may be obtained in a more laborious fashion by inserting Equation (8.3) into Equation 5.24.) Its importance becomes obvious when it is realized that typical linear polymers must have 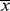 n values on the order of at least 100 to achieve useful mechanical properties. This requires a conversion of at least 99%, assuming difunctional monomers in perfect stoichiometric equivalence. Such high conversions are almost unheard in most organic reactions, but are necessary to achieve high molecular weight condensation polymers.
n values on the order of at least 100 to achieve useful mechanical properties. This requires a conversion of at least 99%, assuming difunctional monomers in perfect stoichiometric equivalence. Such high conversions are almost unheard in most organic reactions, but are necessary to achieve high molecular weight condensation polymers.
Since condensation reactions are reversible, many step-growth polymerizations would reach equilibrium at low conversions if the low molecular weight product of the reaction (usually condensed water) were not efficiently removed (e.g., by heat and vacuum or by a second reaction) to drive the reaction to high conversions.
 n of the polymer to No, the moles of monomer charged to the reactor, and M, the total moles of water evolved since the start of the reaction.
n of the polymer to No, the moles of monomer charged to the reactor, and M, the total moles of water evolved since the start of the reaction.

So far, we have only discussed polymerizations where there is a stoichiometric balance of A and B groups (which is always true for monomers such as hydroxy acids, as shown in Example 8.1). However, if the number of A and B groups is not equal, the length of the chains formed will be reduced (even at 100% conversion!). As an analogy to explain the result of an imbalanced stoichiometric feed, assume that each person in a classroom is a monomer—girls are AA's and boys are BB's. They have to link hands girl–boy–girl, etc. If there are an equal number of boys and girls, a complete ring can be made with all hands connected. However, if there are fewer girls in the class, then there will be several B hands that are left unlinked, even when all of the girls' A hands are connected. This causes a serious drop in the molecular weight for condensation polymers and is of utmost importance in designing reactors for step-growth polymerizations. We can use similar (but more involved) reasoning for Example 8.1 to derive

where r = NA0/NB0 is the stoichiometric ratio of the functional groups present (A is taken to be the limiting reactant, so r is always less than or equal to 1, and p represents the fraction of A groups reacted).
To examine the effect of stoichiometric imbalance, consider the limiting case of complete conversion, p = 1, for which Equation (8.7) reduces to
(8.8)
When A and B groups are supplied in stoichiometric equivalence (r = 1), all the reactant molecules are combined in a single molecule of essentially infinite molecular weight. Reducing r to 0.99 reduces  to 199, and for r = 0.95, all the way down to 39. This is an extremely important concept! To reach the large chain lengths needed for useful materials, step-growth polymerizations must have functional groups supplied at or very close to stoichiometric equivalence. Considering the usual industry purity levels and precision of weight techniques, this is not always easy to achieve. One way to avoid this problem is to use a difunctional monomer of the type ARB, with the stoichiometric equivalence built into the monomer. This is not always feasible or possible, so many step-growth polymerization reactions also require careful reactor design. For the production of nylon 6/6, for example, where adipic acid (ARA) and hexamethylene diamine (BR′B) are used as the monomers (see Example 2.4E), the monomers form an ionic salt of a perfect 1:1 ratio (“nylon salt”), which is carefully purified by crystallization before polymerization. Another option for forming nylons is to use interfacial polymerization, with each monomer in a separate liquid phase (see Chapter 12). Also, chain length may sometimes be pushed up despite an initial stoichiometric imbalance by eliminating excess amount of a reactant. For example, if a polyester is made from a diacid plus and an excess amount of glycol, the reaction must stop at a point when all the chains are capped by OH groups. If the glycol is volatile enough to be driven off by applying heat and vacuum, however, the chains may combine further with the elimination of excess amount of glycol:
to 199, and for r = 0.95, all the way down to 39. This is an extremely important concept! To reach the large chain lengths needed for useful materials, step-growth polymerizations must have functional groups supplied at or very close to stoichiometric equivalence. Considering the usual industry purity levels and precision of weight techniques, this is not always easy to achieve. One way to avoid this problem is to use a difunctional monomer of the type ARB, with the stoichiometric equivalence built into the monomer. This is not always feasible or possible, so many step-growth polymerization reactions also require careful reactor design. For the production of nylon 6/6, for example, where adipic acid (ARA) and hexamethylene diamine (BR′B) are used as the monomers (see Example 2.4E), the monomers form an ionic salt of a perfect 1:1 ratio (“nylon salt”), which is carefully purified by crystallization before polymerization. Another option for forming nylons is to use interfacial polymerization, with each monomer in a separate liquid phase (see Chapter 12). Also, chain length may sometimes be pushed up despite an initial stoichiometric imbalance by eliminating excess amount of a reactant. For example, if a polyester is made from a diacid plus and an excess amount of glycol, the reaction must stop at a point when all the chains are capped by OH groups. If the glycol is volatile enough to be driven off by applying heat and vacuum, however, the chains may combine further with the elimination of excess amount of glycol:

On the other hand, reducing r by the deliberate introduction of an excess amount of one of the monomers or of some monofunctional material provides a convenient means of limiting chain length when desired.
8.4 Chain Lengths on a Weight Basis
The previous development of the distribution of chain lengths (Chapter 5) in a linear step-growth polymer, although perfectly legitimate, is in some ways misleading, because it describes the number of molecules of a given chain length present and equally counts both monomer units and chains containing many hundreds of monomer units, that is, each is one molecule. For example, in a mixture consisting of one monomer molecule and one 100-mer, the number or mole fraction of each is 1/2. Another way of looking at it is to inquire about the relative weights of the various chain lengths present. On this basis, the weight fraction of monomer in the mixture is only 1/101. (This assumes that the monomer and the repeat unit have the same molecular weight.) By neglecting the weight of the small molecule that splits out in condensation reactions, we can most simply obtain the weight-fraction distribution of chain lengths by combining Equations 5.33 (8.3) and (8.6) to get

This “most-probable” weight fraction distribution is shown in Figure 8.2. Although the smaller molecules (monomers, dimers, trimers, etc.) are the most numerous, their combined weight is an insignificant portion of the total weight. It should be noted that step-growth polymerization typically results in very few monomers, as most of them react to form at least dimers or trimers. This is an important point of contrast with chain-growth polymerization (Chapter 9), where only large molecular weight polymers and monomers exist as the conversion increases. In the figure, the peak is at x = −1/(ln p) ≈ 1/(1 − p), that is, the x-mer present in greatest weight is approximately  . The neglect of a molecule of condensation (reacted and unreacted A groups do not have the same weight, but each of them is part of one monomer residue) in this derivation can lead to significant errors at low x's. The exact solution is quite complex [1].
. The neglect of a molecule of condensation (reacted and unreacted A groups do not have the same weight, but each of them is part of one monomer residue) in this derivation can lead to significant errors at low x's. The exact solution is quite complex [1].
Figure 8.2 Weight-fraction distributions for linear step-growth polymers (Eq. (8.9)).

The weight-average chain length,  , can be obtained by inserting the number distribution, Equation (8.3) into Equation 6.23 and integrating at constant p:
, can be obtained by inserting the number distribution, Equation (8.3) into Equation 6.23 and integrating at constant p:

(Evaluation of the above integrals is a good exercise for mathematical masochists!) Combining Equations (8.10) and (8.6) gives an expression for the polydispersity index in terms of conversion:
(8.11)
8.5 Gel Formation
If a step-growth polymerization batch contains some monomer with a functionality f > 2 and if the reaction is carried to a high enough conversion, a cross-linked network or gel may be formed (Figure 8.3). In this context, a gel is defined as a molecule of essentially infinite molecular weight, extending throughout the reaction mass. In the production of thermosetting polymers, the reaction must be terminated short of the conversion at which a gel is formed or the product could not be molded or processed further (crosslinking is later completed in the mold). Hence, the prediction of this gel point conversion is of great practical importance.
Figure 8.3 Time course of reaction of multifunctional A to difunctional B monomer. First small polymers are formed that are not crosslinked, but as enough A and B groups react, a tight network forms.

The multifunctional monomer is represented by Af for example,

while the reaction mixture usually also consists of difunctional monomers of the form ARA and BR′B. In a gel network, the multifunctional monomer acts as a branch unit. These branch units are connected to chains, which are linear segments of difunctional units, that lead either to another branch unit or to an unreacted end (Figure 8.4).
Figure 8.4 A growing polymer chain with two branch points indicating the position of the trifunctional monomers that have been added.

The branching coefficient α is defined as the probability that a given functional group on a branch unit is connected to another branch unit by a chain (rather than to an unreacted end).
Further analysis is based on two assumptions. (1) All functional groups are equally reactive. This might not always be true. For example, the secondary (middle) hydroxyl group in glycerin (HOCH2–CH(OH)–CH2OH) is probably not as reactive as the primary (end) hydroxyls, due to steric hindrances. (2) Furthermore, intramolecular condensations (or polymer back-biting, i.e., reactions between A and B groups on the same molecule) do not occur. In other words, each reaction between an A and a B reduces the number of molecules in the reaction mass by one.
Consider the reaction:

where
ρ = fraction of the total A groups in the branch units Af
(1 − ρ) = fraction of the total A groups in the difunctional AA molecules
pA = probability of finding a reacted A group
pB = probability of finding a reacted B group
The probabilities of finding each of the numbered types of bonds in the polymer shown on the right of the above reaction are tabulated below.
| Bond | Probability of Formation | |
| 1 | pA | probability of A reacting with B |
| 2 | pB(1 − ρ) | probability of B reacting with A on AA |
| 3 | pA | probability of A reacting with B |
| 4 | pB ρ | probability of B reacting with A on Af |
Therefore, the probability of finding the chain shown, which, counting from left to right, contains 1-type one bond, i-type two bonds, i-type three bonds, and 1-type four bond is:

From the standpoint of connecting branch units, i may have any value between zero and infinity. Thus, to obtain the probability of finding a chain of any i that connects the branch units, we must sum over all i to find the branching coefficient:
(8.12)
Now,
(8.13)
(8.14)
(8.15)
From the stoichiometry of the reaction, the number of reacted A groups must be equal to the number of reacted B groups:
(8.16)
Therefore,
(8.17)
and

Each of the terminal branch units in the polymer molecule on the right above has (f − 1) unreacted functional groups. If at least one of these unreacted A groups is then connected to another branch unit, a gel is formed. Since all the unreacted A groups on branch units are statistically identical, all the branch units in the reaction mass will be connected together by chains, which is our definition of a gel. (In general, there will still be some monomer and linear chains floating throughout the gel network, i.e., the reaction mass is not a single molecule at the gel point.) This occurs when  , that is, when the product of the probability of a chain connecting a given functional group to another branch unit and the number of remaining unreacted functional groups becomes a certainty. Thus, the critical value of the branching coefficient for gelation is:
, that is, when the product of the probability of a chain connecting a given functional group to another branch unit and the number of remaining unreacted functional groups becomes a certainty. Thus, the critical value of the branching coefficient for gelation is:

Combining Equations (8.18) and (8.19) gives the gel-point conversion, ρc.

Experimentally, the gel-point conversion for the system in Example 8.2 was found to be pA = 0.765. This was done by noting the conversion at which bubbles ceased to rise in the reaction mass. The difference between the experimental and the theoretical results can be accounted for by some intramolecular condensation (back-biting) and the lower reactivity of the secondary hydroxyls at the center of the glycerin molecules. Also, Bobalek et al. [3] showed that as large molecules grew within the reaction mass, their molecular weights became high enough to cause them to precipitate from the solution before a true, infinite gel network was formed (a particle just 1 μm in diameter has a molecular weight in the order of 1011 g/mol). Higher conversions were needed experimentally to link these precipitated particles together to prevent the rise of bubbles.
Regardless of the reasons, however, the theoretical calculation usually gives the lower limit, or most conservative value, for the gel-point conversion, giving a margin of safety in practice. An apparent exception can sometimes occur. In concentrated solutions, physical entanglements of chains can cause a polymer to behave as a crosslinked gel at conversions lower than that predicted by this theory. Also, in the preparation of unsaturated polyester resins (see Example 2.3), the double bond that would be assumed inert in the condensation reaction participates in crosslinking reactions during condensation polymerization. This, in effect, makes the value of f much greater than that for condensation alone, which results in premature gelling. Additional crosslinking is normally counteracted by incorporating an inhibitor (see Chapter 9) for addition reactions during the polycondensation (step-growth) reaction.








8.6 Kinetics of Polycondensation
We now know something about the chemistry of forming step-growth polymers and about the relation of the length of the polymer chains with the reaction conversion; but how long do these reactions occur? The kinetics of polycondensation reactions is similar to that of ordinary condensation reactions. Since the average chain length is related to conversion in linear polycondensation by Equation (8.7), and the conversion is given as a function of time by the kinetic expression,  is directly related to the reaction time and can thus be controlled by limiting the reaction time. Similarly, the time to reach a gel point is related by the rate expression and Equations (8.18) and (8.19). The rate of reaction depends on several parameters, including the concentrations of monomer species, as demonstrated in Example 8.4, and the temperature (through usual Arrhenius behavior, in that an increase in temperature raises kinetic rate constants and speeds up the reaction rate, see Chapter 9).
is directly related to the reaction time and can thus be controlled by limiting the reaction time. Similarly, the time to reach a gel point is related by the rate expression and Equations (8.18) and (8.19). The rate of reaction depends on several parameters, including the concentrations of monomer species, as demonstrated in Example 8.4, and the temperature (through usual Arrhenius behavior, in that an increase in temperature raises kinetic rate constants and speeds up the reaction rate, see Chapter 9).





Two cautions are in order about the preceding example. First, by writing an irreversible rate expression, we have assumed that any molecule of condensation is being continuously and efficiently removed from the reaction mass so that there is no depolymerization. Second, not all step-growth reactions are of second order. Some polyesterifications, for example, are catalyzed by their own acid groups and are, therefore, first order in hydroxyl concentration, second order in acid, and third order overall. The rate may also be proportional to the concentration of an added catalyst (usually acids or bases for polycondensations), if used.
References [4] and [5] provide a more comprehensive and detailed look at step-growth polymerizations, extending and applying many of the concepts introduced here.
Notes
1. Some of the molecules formed in this type of reaction will be capped with two A groups. For each of these, however, stoichiometric equivalence requires that there should be one capped with two B groups, so Equation (8.2a) represents the average reaction.
Problems
 , when NB moles of B are added per mole of AA or BB.
, when NB moles of B are added per mole of AA or BB. ,
,  , and PDI for the reaction mixture?
, and PDI for the reaction mixture?

| A3 | 1 mol |
| AA | 1 mol |
| B | 1 mol |
| BB | 2 mol |
 to time for a linear polyesterification that is catalyzed by its own acid groups, that is, second order in COOH and first order in OH. Assume an irreversible reaction at constant volume and [A]o = [B]o.
to time for a linear polyesterification that is catalyzed by its own acid groups, that is, second order in COOH and first order in OH. Assume an irreversible reaction at constant volume and [A]o = [B]o.1. Grethlein, H.E., Ind. Eng. Chem. Fund. 8, 206 (1969).
2. Flory, P.J., Principles of Polymer Chemistry, Cornell University Press, Ithaca, NY, 1953.
3. Bobalek, E.G., et al., J. Appl. Polym. Sci. 8, 625 (1964).
4. Gupta, S.K. and A. Kumar, Reaction Engineering of Step-Growth Polymerizations, Plenum, New York, 1987.
5. Odian, G., Principles of Polymerization, Wiley, New York, 1991.