Chapter 10
Advanced Polymerization Methods
10.1 Introduction
The previous two chapters covered two of the primary methods for forming polymers: step-growth (or condensation) polymerization and free-radical addition (or chain) polymerization. Of course, there are other ways to make really big polymers, and some of these techniques have gained commercial importance due to their capabilities to form more stereoregular (e.g., isotactic or syndiotactic) polymers, to decrease a polymer's polydispersity, or to provide other methods to carefully control the polymer structure. This chapter will introduce the basic concepts for some of the more common advanced polymerization techniques: cationic, anionic, heterogeneous catalyzed (specifically the Ziegler–Natta catalyst system), metallocene-catalyzed, ring-opening metathesis, and atom transfer radical polymerizations.
10.2 Cationic Polymerization [1–4]
Strong Lewis acids, that is, electron acceptors, are often capable of initiating addition polymerization of monomers with electron-rich substituents adjacent to the double bond. Cationic catalysts are most commonly metal trihalides such as AlCl3 or BF3. These compounds, although electrically neutral, are short of two electrons for having a complete valence shell of eight electrons. Traces of a cocatalyst, usually water, are usually required to initiate polymerization, first by grabbing a pair of electrons from the cocatalyst:

The leftover proton (the cation) is thought to be the actual initiating species, abstracting a pair of electrons from the monomer and leaving a cationic chain end that reacts with additional monomer molecules (similar to the propagation step in free-radical polymerization).

An important point is that the gegen or counter ion (here, [BF3OH]−) is electrostatically held near the growing chain end and therefore can exert a steric influence on the addition of monomer units. Termination is thought to occur by a disproportionation-like reaction that regenerates the catalyst complex. This complex, therefore, is a true catalyst, unlike free-radical initiators:

These reactions proceed very rapidly at low temperatures. For example, the polymerization of isobutylene illustrated above is carried out commercially at −150 °F. The average chain length actually formed increases as the temperature is lowered.
Cationic initiation is successful only with monomers having electron-rich substituents adjacent to the double bond (like isobutylene), including

None of these monomers can be polymerized to high molecular weight with free-radical initiators, therefore cationic polymerization plays an important role in commercial production of these polymers.
10.3 Anionic Polymerization [5–7]
Addition polymerization may also be initiated by anions. Anionic polymerization has achieved tremendous commercial importance in the past few decades because of its ability to control molecular structure during polymerization, allowing the synthesis of materials that were previously difficult or impossible to obtain. A variety of anionic initiators has been investigated but the organic alkali-metal salts are perhaps the most common, as illustrated below for the polymerization of styrene with n-butyllithium:

The anionic (−) chain end then propagates the chain by adding another monomer molecule. Again, the gegen ion can sterically influence the reaction.
Sodium and lithium metals were used to polymerize butadiene in Germany during World War II. After the war, in the United States, it was discovered that under appropriate conditions, dispersions of lithium could lead to largely cis-1,4 addition of butadiene and isoprene (the latter being the synthetic counterpart of natural rubber). In these processes, a metal atom first reacts with the monomer to form an anion radical:

These anion radicals then react in either of two ways. One may react with another atom of lithium


and/or two may rapidly undergo radical recombination.
Either way, the result is a dianion that propagates a chain from each end. Other dianionic initiators have also been developed [5–8].
There are a couple of very interesting aspects to these reactions. First, the rates of initiation and propagation vary with the monomer, gegen ion, and solvent. In general, the reactions proceed more rapidly in polar solvents as the species are more highly ionized. (The polarity of the solvent also strongly influences the stereospecific nature of the polymer.) In many important cases, the rate of initiation is comparable to the rate of propagation, unlike free-radical addition, in which ri  rp. This means that the initiator (in this case, it is an initiator rather than a catalyst) promptly starts growing chains. Second, in the absence of impurities, there is no termination step. The chains continue to grow until the monomer supply is exhausted. The ionic chain end is perfectly stable and the growth of the chains will resume if more monomer is added. For this reason, these materials are aptly termed “living” polymers and if additional monomer (or a different monomer) is later added, the reaction continues. However, proton-donating impurities such as water or acids quickly kill (terminate) them:
rp. This means that the initiator (in this case, it is an initiator rather than a catalyst) promptly starts growing chains. Second, in the absence of impurities, there is no termination step. The chains continue to grow until the monomer supply is exhausted. The ionic chain end is perfectly stable and the growth of the chains will resume if more monomer is added. For this reason, these materials are aptly termed “living” polymers and if additional monomer (or a different monomer) is later added, the reaction continues. However, proton-donating impurities such as water or acids quickly kill (terminate) them:

This unique mechanism has a number of practically important consequences.
10.3.1 Block Copolymerization
If a second monomer is introduced after the initial monomer, charge is exhausted and the living chains resume propagation with the second monomer, neatly giving a block copolymer. Monomers can be alternated as desired to give AB (diblock) or ABA (triblock) or even more complicated block structures, conceivably even including three or more different monomers.
10.3.2 Synthetic Flexibility
Anionic polymerization allows the synthesis of all sorts of interesting and useful molecules. For example, bubbling carbon dioxide through a batch of living chains followed by exposure to water produces a carboxyl-terminated polymer:


Similarly, ethylene oxide gives hydroxyl-terminated chains:


Note that if these reagents are added to a lithium-initiated dianion, both ends of the chain will be capped with the functional group. Such chains are macro diacids or diols. Carboxyl- or hydroxyl-terminated chains may then participate in the usual step-growth polymerizations (see Chapter 8).
Once the monomer is exhausted in a reaction, if a tetrafunctional monomer such as divinyl benzene (DVB), H2C=CHϕHC=CH2, is added to a batch of living chains, it couples to itself and to the living chains. If it is assumed that a DVB molecule can react with another DVB only at one end (there are probably good steric reasons why this should be so), the following type of star polymer (or dendrimer) structures result with linear branches radiating from a DVB core (Figure 10.1).
Figure 10.1 Schematic showing the attachment of several linear polymers to a core that contains several DVB units linked together. The cartoon on the right depicts how a multifunctional core can attach many polymer arms to make a star polymer.

It is conceivable that at this point, a second difunctional monomer could be added giving a star polymer with two different kinds of branches, but normally, the reactor would be opened and the reaction terminated. From the structure above, there are (b − 1) moles of DVB per mole of star branches, where b is the average number of branches per star. Since the number of moles of star molecules is equal to the moles of living chains over the average number of branches, and with an initiator such as n-BuLi (n-butyl lithium), each molecule starts one living chain; to make a b-branch star polymer, one must add
(10.1) 
moles of DVB, where Io is the moles of initiator charged to the reactor.
Living chains can also be linked by, for example, dichloro compounds, instantly doubling their chain lengths:

With multifunctional linking agents, this technique can be used to form star polymers with various numbers of branches. It would be nice if all the functional groups on such compounds took part in the linking reaction, but sometimes steric factors lower the linking efficiency. Multifunctional initiators can also be used to grow star polymers. Various initiators, linking agents, and other reagents for anionic polymerization have been reviewed [5–8].
10.3.3 Monodisperse Polymers
Anionic polymerization is also quite useful at making polymers with narrow size distributions. Consider a batch reactor containing, say, a 20% solution of styrene in the solvent tetrahydrofuran (THF). A charge of n-BuLi is suddenly added. In this fairly polar solvent, the n-BuLi ionizes immediately and completely and with ri ≈ rp promptly starts chains growing––one chain for each molecule of n-BuLi. In the absence of terminating impurities, the number of growing chains remains constant and they compete on an even basis for the available monomer. They will therefore have essentially the same length, giving a nearly monodisperse polymer. Not only can anionic polymerization be used to synthesize essentially monodisperse homopolymers but the blocks in the block copolymers formed this way can also tailor to be monodisperse.
As with other modes of polymerization, the number-average chain length is given by the moles of monomer polymerized over the moles of chains present. With an initiator like n-BuLi, each molecule starts a single chain, therefore,

(If the volume is constant, the moles may be bracketed to give concentrations.) Statistically, the distribution of chain lengths is obtained by answering the following question: given Mo − M marbles and Io buckets, what will be the distribution of marbles among the buckets if the marbles are thrown completely randomly into the buckets? The result is the Poisson distribution from statistics:

(As written above, the distribution does not include the initiator residue.) To a good approximation, the polydispersity index of this distribution is1
(10.4) 
from which it is seen that even at moderate  values, the polymer is essentially monodisperse. Figure 10.2 compares the Poisson distribution with the most probable distribution, Equations 8.3 and 9.42, for
values, the polymer is essentially monodisperse. Figure 10.2 compares the Poisson distribution with the most probable distribution, Equations 8.3 and 9.42, for  values of 100. Because of their sharpness, the number and weight distributions are almost identical.
values of 100. Because of their sharpness, the number and weight distributions are almost identical.
Figure 10.2 Comparison of Poisson and most-probable number-fraction distributions of chain lengths for  .
.

As in the case of step-growth polymerization, the average chain length increases continuously with conversion (Eq. (10.2)). Unlike step-growth, the reaction mass consists only of monomer molecules and polymer chains of essentially a single length. Equations (10.2) and (10.3) characterize the polymer present.
 at 100% conversion.
at 100% conversion.



10.4 Kinetics of Anionic Polymerization [5, 6, 9]
A general description of anionic polymerization kinetics is complicated by the associations that may occur, particularly in nonpolar (hydrocarbon) solvents. The rate of propagation is proportional to the product of the monomer concentration and the concentration of active living chains [ ]:
]:
(10.5) 
With negligible association (e.g., in THF solvent or hydrocarbons at n-BuLi concentrations less than 10−4 mol), each initiator molecule starts a growing chain, and in the absence of terminating impurities, the number of active living chains equals the number of initiator molecules added,
(10.6) 
(10.7) 
or making use of the definition of conversion X (Eq. 9.14) and assuming that the volume of the reaction mass is linear in the conversion (Eq. 9.17), we get
(10.8) 
Unlike the case for free-radical addition, this can be readily integrated for a batch reactor ([I]o is constant) to give

but it is still not easier to get X as an explicit function of t.
Because anionic polymerizations are generally carried out in rather dilute solutions in inert solvents, volume changes with conversion tend to be much smaller than when undiluted monomer is polymerized. This often justifies the neglect of volume change (ε = 0) for which Equation (10.9) becomes

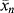 to time. Neglect volume change.
to time. Neglect volume change.
In n-BuLi polymerizations at high concentrations in nonpolar solvents, the chain ends are present largely as inactive dimers, which dissociate slightly according to the equilibrium

where
(10.11) 
The concentration of active chains is then
(10.12) 
Now it takes two initiator molecules to make one inactive dimer, therefore
(10.13) 
The rate of polymerization then becomes
(10.14) 
The low value of K, reflecting the presence of most chain, ends in the inactive associated state (dimers) giving rise to low rates of polymerization in nonpolar solvents. At very high concentrations, association may be even greater and the rate is essentially independent of [I]o.
10.5 Group-Transfer Polymerization
In the 1980s, the DuPont Company developed and patented [10] a new type of polymerization that mechanistically is similar to anionic polymerization. Group-transfer polymerization (GTP) has been defined as “polymerization of α,β-unsaturated esters, ketones, nitriles, or amides, initiated by silyl ketene acetals [11].” It has most commonly been used to polymerize acrylate and methacrylate monomers with the aid of anionic catalysts (they are true catalysts here), such as the bifluoride ion, [FHF]−, or bioxyanions. GTP is illustrated below for the polymerization of methyl methacrylate (MMA) with silyl ketene acetal (SKA):
Initiation

Propagation

The initiating functionality is transferred to the growing end of the chain as each new monomer unit is added. They are living chains as in anionic addition and can likewise be used to produce monodisperse polymers, block copolymers, and, with the addition of appropriate reagents, chains with desired terminal groups. They can also control stereoregularity in the chain.
Unlike with anionic addition, chain transfer can occur:


As in anionic addition polymerization, chain length can be reduced by using more initiators, but because these initiators are rather expensive, it is often preferable to use a chain-transfer agent instead. Also, at low monomer concentrations, termination can occur through cyclization of the chain end.
10.6 Atom Transfer Radical Polymerization
A more recent development in living polymerizations that has shown great promise for tailoring polymer structure and molecular weight is atom transfer radical polymerization (ATRP). The discovery of ATRP in the 1990s [12] has led to the broadening of living polymerizations, which can include a wide range of monomer types (including vinyl, (meth)acrylates, styrenes, and epoxies). This is largely due to ATRP's robust method of synthesis that is less sensitive to the presence of impurities than anionic polymerization. The reaction proceeds through the use of a relatively inexpensive copper complex as the catalyst (Figure 10.3), which is normally bound to a ligand chemical, such as pyridine. The reaction produces high molecular weight, narrowly disperse polymers. In addition to the catalyst, ATRP requires an initiator, most often an alkyl halide (e.g., bromides and chlorides). As with free-radical polymerization, the molecular weight of the resulting polymer is inversely proportional to the amount of initiator used. One unique characteristic of the ATRP initiator is that multiple functionalities (i.e., multiple halide groups) can influence the structure of the polymer; for instance, an initiator with six Br atoms can grow up to six polymer chains from the same initiator molecule, creating a star polymer.
Figure 10.3 Metal (often copper) ligand (M/L) structure to support ATRP reactions. Here, Pm and Pm+c are polymers with length m and m+c, respectively, X is typically a halide atom, such as Cl or Br, and the k's are kinetic rate constants for activation (ka), deactivation (kda), and termination (kt). Reprinted from [16] with permission from Elsevier.

In ATRP, there are reactive and dormant polymer species in equilibrium during the polymerizations, which alternate between halide-capped polymers (dormant) and growing (reactive) polymers with a free radical on the end. The choice of catalyst controls this equilibrium which in turn influences the polymerization rate and the distribution of chain lengths. The mechanism offers flexibility to conduct reactions in bulk, solution, or emulsions/suspensions, just as free-radical polymerizations. Due to the capability to polymerize a large range of monomers with an inexpensive catalyst in a reactor, where purity is nearly as important as in anionic polymerizations, ATRP continues to grow in popularity. For further information, review articles written by the inventors are available [12, 16].
10.7 Heterogeneous Stereospecific Polymerization [13--15]
Many monomers are gases at or near atmospheric temperature and pressure and can be polymerized using solid catalysts (hence, multiple phases in a reactor or heterogeneous reactions). In the early 1950s, Karl Ziegler in Germany observed that certain heterogeneous catalysts based on transition metals would polymerize ethylene to a linear, high-density material (HDPE, Chapter 5) at low pressure and temperature (compared to the existing free-radical process for low-density polyethylene). Giulio Natta in Italy showed that these catalysts would produce highly stereospecific poly α-olefins ([H2C–CHR]x, where R = CnHm) (notably polypropylene) and polydienes. Ziegler and Natta shared the 1963 Nobel Prize in chemistry for their work. Almost simultaneously, scientists at Amoco and Phillips in the United States developed heterogeneous catalysts based on Mo and Cr that also produced linear polyethylene under mild conditions.
One example of a Ziegler–Natta catalyst system is titanium tetrachloride, TiCl4, and aluminum triethyl, Al(C2H5)3. Vanadium and cobalt chlorides are also used as is Al(C2H5)2Cl. When the substances are mixed in an inert solvent, a crystalline solid is obtained along with a highly colored supernatant (deep violet or brown). It is known that the reaction involves the reduction of the titanium to a lower valence state, probably +2, since TiCl3 also forms an effective catalyst. The supernatant liquid alone will polymerize α olefins, but the resulting polymers show little stereospecificity. Commercial catalyst systems are based on the solid, either alone or on a support such as SiO2 or MgCl2. A “Phillips” catalyst typically consists of CrO3 supported on silica or alumina.
Polymerization with Ziegler–Natta catalysts is thought to occur at active sites formed by interaction of the metal alkyl with a metal chloride on the surface of the metal chloride crystals. Monomer is chemisorbed at the site (thus accounting for its specific orientation when added to the chain), and propagation occurs by insertion of the chemisorbed monomer into the metal-chain bond at the active site.

The chain thus grows out from the site like a hair from the scalp.
Hydrogen is used as a chain-transfer agent in these reactions:

Chain transfer with the metal alkyl component in Ziegler–Natta systems has been identified as

and transfer to monomer may also occur.
Most commercial processes based on these catalysts are carried out with the monomers either in the gas phase or in the liquid phase at temperatures such that the polymer precipitates as it is formed. Solid, porous catalyst particles in the range of 10–100 µm are introduced to the reactor. As polymer begins to form, they rapidly break up into many smaller (0.01–1 µm) fragments (primary crystallites) on which the polymer continues to grow. The reaction mass therefore consists of a suspension (not an emulsion—there is no surfactant) of macroparticles, which are in turn made up of an agglomeration of microparticles, each of which surrounds a primary crystallite (Figure 10.4). According to Ray and coworkers [17], the macroparticle usually remains intact so that one is generated from each catalyst particle fed to the reactor. The primary catalyst crystallites are distributed more or less uniformly throughout the ultimate macroparticle.
Figure 10.4 Mechanism of particle formation in Ziegler–Natta polymerization [17].

If you are not yet convinced that these are complex systems, consider the following. The catalysts consist of two or more components, the nature and relative amounts of which influence the rate of polymerization. Each catalyst particle contains a multiplicity of surface sites on which polymer can grow and they may differ in activity (e.g., due to different crystal faces). Sites may be deactivated so that their number decreases with time. The monomer must be chemisorbed on the catalyst surface before it can be added to a chain. And before it even gets to the catalyst surface it must diffuse from the bulk fluid to the surface of the macroparticle through the interstices (pores) between the microparticles and finally through the layer of solid polymer that coats the catalyst fragment within each microparticle. Furthermore, the heat of polymerization is liberated at the surface of the catalyst fragments that must flow outward. This could give rise to significant temperature gradients, complicating the interpretation of experimental data and limiting the validity of isothermal rate expressions. It should not be surprising, therefore, that kinetic equations are significantly more complex to describe this reaction than those for free-radical and anionic addition polymerizations. However, the relevance of this catalyst system is of great industrial importance as tens of billions of pounds of polymer are produced with these catalyst systems each year. The following discussion illustrates some of the approaches that have been taken to describe the kinetics in systems with two-component Ziegler–Natta catalysts such as the TiCl4–Al(C2H5)3 system mentioned above.
The mechanism developed to describe the reaction is based on four assumptions [18]:
Let C represent the number of catalytic sites on the catalyst surface, presumably associated with the Ti. Before a chain can grow on a site, however, the site must be activated by chemisorption of the Al(C2H5)3 on an adjacent site. Therefore,
(10.15) 
where Cp* is the number of growing chains stuck to the catalyst surface and θA is the fraction of the surface covered by chemisorbed alkyl.
The rate of polymerization is given by

where ks is the insertion rate constant, which characterizes the rate of insertion of chemisorbed monomer into the Ti-chain bond, and θM is the fraction of the surface covered by chemisorbed monomer.
Monomer and metal alkyl compete for chemisorption on the uncovered fraction of the active surface, θM = (1 − θM − θA)


where S represents a surface site:
(10.17) 
(10.18) 
(10.19) 
(10.20) 
[Ms] and [As] are the concentrations of monomer and alkyl at the surface. If we assume steady-state surface concentrations of both monomer and alkyl and equate the rates of adsorption and desorption of each we get

and

where KM = kM+1/kM−1 and KA = kA+1/kA−1 are the equilibrium constants for monomer and alkyl adsorption onto the catalyst. Equations (10.21) and (10.22) are Langmuir–Hinshelwood absorption isotherms, which when inserted into Equation (10.16) gives [19]2:

Equation (10.23) appears to give a reasonable description of experimental results. For example, the activity of a catalyst can be greatly enhanced kby ball milling and/or dispersing it on the surface of a support such as MgCl2, both of which increase the exposed surface area, and therefore C. A maximum in rate is generally observed as the alkyl concentration is increased. Actually, almost all experiments show the rate to be the first order in monomer concentration, and Equation (10.23) is not linear in monomer. This simply means that KM[Ms] 1, which is generally agreed to be the case.
With a little creative algebra, Equation (10.23) can be put into a simpler and more convenient form:

where kp = ksKM is a propagation rate constant and  is interpreted as the number of active growing chains, that is, those that possess an open adjacent site for the chemisorption of monomer or alkyl. Again, because
is interpreted as the number of active growing chains, that is, those that possess an open adjacent site for the chemisorption of monomer or alkyl. Again, because
(10.25) 
is a function of monomer concentration, Equation (10.24) is not the first order in monomer unless KM[Ms] 1, which generally seems to be the case. Equation (10.24) is the form usually used by chemical engineers to describe Ziegler–Natta polymerizations.
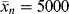 . (c) Also, estimate how long the reaction must be carried out under these conditions to achieve a catalyst yield of 10,000 g polymer/g catalyst (which is desirable to avoid a costly catalyst removal step). [Ms] is kept constant by bubbling gas-phase monomer into the reactor as polymerization proceeds.
. (c) Also, estimate how long the reaction must be carried out under these conditions to achieve a catalyst yield of 10,000 g polymer/g catalyst (which is desirable to avoid a costly catalyst removal step). [Ms] is kept constant by bubbling gas-phase monomer into the reactor as polymerization proceeds. is given by the average number of monomer units in a chain (
is given by the average number of monomer units in a chain (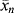 ) times the number of growing chains (C*) divided by the rate at which monomer is added to all chains (rp):
) times the number of growing chains (C*) divided by the rate at which monomer is added to all chains (rp):
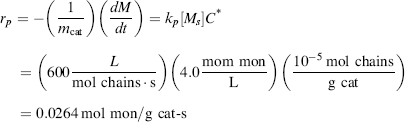

Unfortunately, things are not always as simple as the last example in real life. For one thing, catalyst activity usually drops with time (due to fouling) even in the absence of polymerization. Catalyst half-lives typically are in the order of minutes to hours. This is explained by a progressive deactivation of the sites responsible for polymerization, C*, and is described quantitatively with an nth-order deactivation law:

Currently, second-order deactivation n = 2 seems to be the most popular, but n = 1 and n = 3 have also been used to fit data.
 at
at 
 at
at 



Another complication is the possibility of mass-transfer limitations on the reaction rate. And, because the polymer layer grows, mass transfer resistance will increase during a heterogeneous polymerization; it is quite possible that the polymerization will start out reaction-limited and change to mass transfer-limited as the conversion increases. To polymerize, monomer must first diffuse from the bulk fluid surrounding the macroparticles, where its concentration is [Mb], to the catalytic surface within the microparticles, where its concentration is [Ms]. Where the inherent rate of reaction on the catalytic surface and/or the resistance to diffusion are high, the overall reaction rate can be determined by the physics of diffusion rather than the chemistry of the reaction.
Another way of putting this is that [Ms] < [Mb]. To use Equation (10.23) or 10.24, [Ms] must be known. Relating [Ms] to [Mb] is a complex problem in mass transfer and is beyond the scope of this chapter. It is treated and explained in the literature [20, 21]. Suffice it to say that to make calculations, you need to know the geometry of the particles, the mass-transfer coefficient at the surface of the macroparticles, the effective diffusivity for the monomer in the interstices (pores) between the microparticles, and the diffusivity of the monomer through the layer of polymer coating the primary catalyst particles (in addition to knowing the kinetic parameters of the reaction).
Calculations show that rates can be mass-transfer limited, depending on the activity of the catalyst and the physical properties of the system. One rather unusual result is that the resistance to mass transfer can decrease with time, causing an increase in rate (at constant [Mb]). This increase, when combined with a deactivating catalyst, gives rise to a maximum in the polymerization rate with time. The parameters in Example 10.5 represent a case that is not mass-transfer limited, therefore we can assume that [Ms] ≈ [Mb].
If all the polymer-producing sites on a catalyst surface had the same activity and were exposed to the same monomer concentration, and if chain transfer occurred randomly, we would expect the polymer to have a most-probable distribution of chain lengths (Eq. 9.47) with a polydispersity index of 2.0 [22]. Polymers produced with these heterogeneous catalysts typically have large polydispersity indices (from 3 to 20), however. Two reasons have been advanced for these broad distributions: (1) mass-transfer resistance, which causes with monomer concentration to vary with location and perhaps time in the particles, and (2) a range of site activities (kp's) on the catalyst surface. Different types of sites have been identified on the surfaces [22].
Recent calculations show that while mass-transfer limitations can contribute somewhat to a broadening of the distribution, a range of site activities is needed to account for the observed polydispersities. Furthermore, the different sites must have a pretty wide range of activities [20]. To examine the effects of site heterogeneity, Equation (10.24) must be generalized to
(10.27) 
where N is the number of different types of sites and
(10.28) 
You can get large polydispersity indices with only two types of catalyst sites, provided that they have a large enough difference (at least an order of magnitude) in activities. However, this results in a bimodal (two-peaked) molecular weight distribution, which is not generally seen in practice. In some applications, narrow molecular weight distributions are desirable. Relatively small polydispersity indices in the range of three to four have been reported for systems where the catalyst is supported on MgCl2 (which could be due to selective promotion of certain types of sites) or when additives are used (which may selectively poison certain types of sites) [21]. Single-site (e.g., EXXPOL metallocene) catalysts have also been developed to avoid the problem of high polydispersity [22, 23].
The question of temperature gradients in the particles has also been addressed. The conclusion is that they are generally not significant, except in the very early stages of gas-phase polymerizations, where temperatures may get high enough to melt the polymer [24].
In practice, Ziegler–Natta catalyst systems are difficult to work with. Great care must be exercised in their preparation and use, since they are easily poisoned by water, among other things. They are pyrophoric (spontaneously burst into flame on contact with oxygen) and are used in close proximity to large amounts of flammable monomers and solvents. Therefore, they can present a significant safety hazard both in the laboratory and in the plant.
10.8 Grafted Polymer Surfaces
Grafting a polymer onto a surface is a great way to change the surface properties of a material. This can be done to improve the compatibility between layers in a composite material, to reduce surface fouling, or to improve biocompatibility (among many potential applications). Because living polymerizations can be used for good control of polymer properties, they are often used in graft polymerizations.
The grafting method normally follows one of two patterns (Figure 10.5): grafting to or grafting from. In the first method, oligomers or polymers are pre made (and can easily be characterized for molecular weight and polydispersity) and attached to a surface through any number of chemistries (several condensation reactions can be used here). In grafting from method, the surface must be activated (i.e., form a free-radical or growing living polymer complex) through which monomer can add. In this second technique, the grafts are more difficult to analyze, since the molecular weight and polydispersity cannot be determined without destroying the surface (or at least removing the grafts). Although this drawback may seem to favor grafting to techniques, a balancing disadvantage of the grafting to technique is that the large oligomers or polymers may have significant steric hindrances or diffusive resistance in reaching the surface for binding. This results in a surface that has a lower graft coverage (graft density). Grafting techniques are discussed further by Bhattarcharya and Misra [25].
Figure 10.5 Depiction of methods to form a surface grafted with polymers. (a) Grafting from a surface by growing a polymer chain from surface functional groups (F). (b) Grafting to a surface by attaching a premade polymer to functional groups (F) on the surface.

10.9 Summary
The various advanced polymerization methods described in this chapter highlight some of the more important mechanisms that can be used for polymerizations. Living polymerizations offer the ability for more precise control of polymer structures, including block, graft, and star polymers, whereas heterogeneous catalysis (such as the Ziegler–Natta polymerizations) offers the benefit of improved stereoregularity. It is beyond the scope of this textbook to go beyond an introduction of these techniques, but these polymerizations (and others such as reversible addition fragmentation chain transfer (RAFT) polymerizations and nitroxide-mediated polymerization (NMP)) are treated in greater depth elsewhere [25–27].
Problems
 for the product.
for the product.
1. Obtain an expression for the rate of polymerization.




 , (n/N), (w/W) (the number- and weight-fraction distributions of chain lengths), and the polydispersity index
, (n/N), (w/W) (the number- and weight-fraction distributions of chain lengths), and the polydispersity index 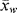 /
/ . Does any of these look familiar?
. Does any of these look familiar? in terms of concentrations and rate constants. Assume sites of uniform activity.
in terms of concentrations and rate constants. Assume sites of uniform activity.Notes
1. There are important practical limitations of not being able to add or mix reagents instantaneously and uniformly, and the almost inevitable presence of some terminating impurities. In practice, these always cause some additional broadening of the distribution.
2. It should be noted that when dealing with heterogeneous reactions such as these, rates are usually given per unit moles or mass of catalyst rather than per unit of reactor volume, as is the case for homogeneous reactions. In Equation (10.23), for example, the rate would typically be in mol monomer/mol Ti (or TiCl4) s.
1. Plesch, P.H. (ed.), The Chemistry of Cationic Polymerization, MacMillan, New York, 1963.
2. Russell, K.E. and G.J. Wilson. Cationic polymerizations, in Polymerization Processes, C.E. Schildknecht and I. Skeist (eds), Wiley, New York, 1977, Chapter 10.
3. Percec, V., Recent developments in cationic polymerization, in Applied Polymer Science, 2nd ed., R.W Tess and G.W. Poehlein (eds), American Chemical Society, Washington, DC, 1985, Chapter 5.
4. Odian, G., Principles of Polymerization, 4th ed., John Wiley and Sons, New York, 2004, Chapter 5-2.
5. Morton, M., Anionic Polymerizations: Principles and Practice, Academic, New York, 1983.
6. Swarc, M., Living Polymerizations and Mechanisms of Anionic Polymerization, Springer, Berlin, Germany, 1983.
7. McGrath, J.E. (ed.), Anionic Polymerization: Kinetics, Mechanisms and Synthesis, American Chemical Society, Washington, DC, 1981.
8. Franta, E. and P. Rempp, Polym. Prepr. 20(1), 5 (1979).
9. Odian, G., op. cit. Chapter 5-3.
10. United States patents 4,414,372 and 4,417,034.
11. Stinson, S.C., Chem. Eng. News, April 27, 1983, p. 43.
12. Keii, T., Kinetics of Ziegler-Natta Polymerization, Chapman Hall, London, U.K., 1972.
13. Chien, J.C.W. (ed.), Coordination Polymerization, Academic, New York, 1975.
14. Karol, F.J., Coordinated anionic polymerization and polymerization mechanisms, in Applied Polymer Science, 2nd ed., R.W. Tess and G.W. Poehlein (eds), American Chemical Society, Washington, DC, 1985, Chapter 4.
15. Matyjaszewski, K. and J. Xia, Chem. Rev. 101(9), 2921 (2001).
16. Coessens, V., T. Pintauer, and K. Matyjaszewski, Prog. Polym. Sci. 26(3), 337 (2001).
17. Floyd, S., T. Heiskanen, and W.H. Ray, Chem. Eng. Progr. 85(11), 56 (1988).
18. Tait, P.J.T., Chem. Technol. 5, 688 (1975).
19. Keii, T., et al., Makromol. Chem. 183, 2285 (1982).
20. Floyd, S., et al., J. Appl. Polym. Sci. 33, 1021 (1987).
21. Floyd, S., et al., J. Appl. Polym. Sci. 32, 2935 (1986).
22. Anon., Mod. Plast.68(7),61 (1991); Leaversuch, R.D., Mod. Plast.68(10), 46 (1991); Anon., Chem. Eng. News, Dec. 23, 1991, p. 16.
23. Anon., Chem. Eng. Prog. 87(10), 21 (1991).
24. Zucchini, U. and G. Cecchin, Adv. Polym. Sci. 51, 101 (1983).
25. Bhattarcharya, A. and B.N. Misra, Prog. Polym. Sci. 29(8), 767 (2004).
26. Matyjaszewski, K. Controlled/Living Radical Polymerizations: From Synthesis to Materials, ACS Symposium Series 944, American Chemical Society, Washington, DC, 2006.
27. Buchmeiser, M. Metathesis Polymerization, Advances in Polymer Science Series, Springer-Verlag, New York, 2009.