13
Blood Disorders
Lloyd E. Damon, MD
Charalambos Babis Andreadis, MD, MSCE
ANEMIAS
 General Approach to Anemias
General Approach to Anemias
Anemia is present in adults if the hematocrit is below 41% (hemoglobin less than 13.6 g/dL [135 g/L]) in males or below 36% (hemoglobin less than 12 g/dL [120 g/L]) in females. Congenital anemia is suggested by the patient’s personal and family history. The most common cause of anemia is iron deficiency. Poor diet may result in folic acid deficiency and contribute to iron deficiency, but bleeding is the most common cause of iron deficiency in adults. Physical examination demonstrates pallor. Attention to physical signs of primary hematologic diseases (lymphadenopathy; hepatosplenomegaly; or bone tenderness, especially in the sternum or anterior tibia) is important. Mucosal changes such as a smooth tongue suggest megaloblastic anemia.
Anemias are classified according to their pathophysiologic basis, ie, whether related to diminished production (relative or absolute reticulocytopenia) or to increased production due to accelerated loss of red blood cells (reticulocytosis) (Table 13–1), and according to red blood cell size (Table 13–2). A reticulocytosis occurs in one of three pathophysiologic states: acute blood loss, recent replacement of a missing erythropoietic nutrient, or reduced red blood cell survival (ie, hemolysis). A severely microcytic anemia (mean corpuscular volume [MCV] less than 70 fL) is due either to iron deficiency or thalassemia, while a severely macrocytic anemia (MCV greater than 120 fL) is almost always due to either megaloblastic anemia or to cold agglutinins in blood analyzed at room temperature. A bone marrow biopsy is generally needed to complete the evaluation of anemia when the blood laboratory evaluation fails to reveal an etiology, when there are additional cytopenias present, or when an underlying primary or secondary bone marrow process is suspected.
Table 13–1. Classification of anemia by red blood cell (RBC) pathophysiology.
Decreased RBC production (relative or absolute reticulocytopenia)
Hemoglobin synthesis lesion: iron deficiency, thalassemia, anemia of chronic disease, hypoerythropoietinemia
DNA synthesis lesion: megaloblastic anemia, folic acid deficiency, DNA synthesis inhibitor medications
Hematopoietic stem cell lesion: aplastic anemia, leukemia
Bone marrow infiltration: carcinoma, lymphoma, fibrosis, sarcoidosis, Gaucher disease, others
Immune-mediated inhibition: aplastic anemia, pure red cell aplasia
Increased RBC destruction or accelerated RBC loss (reticulocytosis)
Acute blood loss
Hemolysis (intrinsic)
Membrane lesion: hereditary spherocytosis, elliptocytosis
Hemoglobin lesion: sickle cell, unstable hemoglobin
Glycolysis lesion: pyruvate kinase deficiency
Oxidation lesion: glucose-6-phosphate dehydrogenase deficiency
Hemolysis (extrinsic)
Immune: warm antibody, cold antibody
Microangiopathic: disseminated intravascular coagulation, thrombotic thrombocytopenic purpura, hemolytic-uremic syndrome, mechanical cardiac valve, paravalvular leak
Infection: Clostridium perfringens, malaria
Hypersplenism
Table 13–2. Classification of anemia by mean red blood cell volume (MCV).
Microcytic
Iron deficiency
Thalassemia
Anemia of chronic disease
Lead toxicity
Zinc deficiency
Macrocytic (Megaloblastic)
Vitamin B12 deficiency
Folate deficiency
DNA synthesis inhibitors
Macrocytic (Nonmegaloblastic)
Aplastic anemia
Myelodysplasia
Liver disease
Reticulocytosis
Hypothyroidism
Bone marrow failure state (eg, aplastic anemia, marrow infiltrative disorder, etc)
Copper deficiency
Normocytic
Kidney disease
Non-thyroid endocrine gland failure
Copper deficiency
Mild form of most acquired microcytic or macrocytic etiologies of anemia
IRON DEFICIENCY ANEMIA
ESSENTIALS OF DIAGNOSIS

 Iron deficiency: serum ferritin is less than 12 ng/mL (27 pmol/L) or less than 30 ng/mL (67 pmol/L) if also anemic.
Iron deficiency: serum ferritin is less than 12 ng/mL (27 pmol/L) or less than 30 ng/mL (67 pmol/L) if also anemic.
Caused by bleeding unless proved otherwise.
Responds to iron therapy.
General Considerations
Iron deficiency is the most common cause of anemia worldwide. The causes are listed in Table 13–3. Aside from circulating red blood cells, the major location of iron in the body is the storage pool as ferritin or as hemosiderin in macrophages.
Table 13–3. Causes of iron deficiency.
Deficient diet
Decreased absorption
Autoimmune gastritis
Celiac disease
Helicobacter pylori gastritis
Hereditary iron-refractory iron deficiency anemia
Zinc deficiency
Increased requirements
Pregnancy
Lactation
Blood loss (chronic)
Gastrointestinal
Menstrual
Blood donation
Hemoglobinuria
Iron sequestration
Pulmonary hemosiderosis
Idiopathic
The average American diet contains 10–15 mg of iron per day. About 10% of this amount is absorbed in the stomach, duodenum, and upper jejunum under acidic conditions. Dietary iron present as heme is efficiently absorbed (10–20%) but nonheme iron less so (1–5%), largely because of interference by phosphates, tannins, and other food constituents. The major iron transporter from the diet across the intestinal lumen is ferroportin, which also facilitates the transport of iron to apotransferrin in macrophages for delivery to erythroid progenitor cells in the bone marrow prepared to synthesize hemoglobin. Hepcidin, which is increasingly produced during inflammation, negatively regulates iron transport by promoting the degradation of ferroportin. Small amounts of iron—approximately 1 mg/day—are normally lost through exfoliation of skin and gastrointestinal mucosal cells.
Menstrual blood loss plays a major role in iron metabolism. The average monthly menstrual blood loss is approximately 50 mL but may be five times greater in some individuals. Women with heavy menstrual losses must absorb 3–4 mg of iron from the diet each day to maintain adequate iron stores, which is not commonly achieved. Women with menorrhagia of this degree will almost always become iron deficient without iron supplementation.
In general, iron metabolism is balanced between absorption of 1 mg/day and loss of 1 mg/day. Pregnancy and lactation upset the iron balance, since requirements increase to 2–5 mg of iron per day. Normal dietary iron cannot supply these requirements, and medicinal iron is needed during pregnancy and lactation. Decreased iron absorption can also cause iron deficiency, such as in people affected by celiac disease (gluten enteropathy), and it also commonly occurs after gastric resection or jejunal bypass surgery.
The most important cause of iron deficiency anemia in adults is chronic blood loss, especially menstrual and gastrointestinal blood loss. Iron deficiency demands a search for a source of gastrointestinal bleeding if other sites of blood loss (menorrhagia, other uterine bleeding, and repeated blood donations) are excluded. Prolonged aspirin or nonsteroidal anti-inflammatory drug use may cause it even without a documented structural lesion. Celiac disease, even when asymptomatic, can cause iron deficiency through poor absorption in the gastrointestinal tract. Zinc deficiency is another cause of poor iron absorption. Chronic hemoglobinuria may lead to iron deficiency, but this is uncommon. Traumatic hemolysis due to a prosthetic cardiac valve and other causes of intravascular hemolysis (eg, paroxysmal nocturnal hemoglobinuria) should also be considered. The cause of iron deficiency is not found in up to 5% of cases.
Pure iron deficiency might prove refractory to oral iron replacement. Refractoriness is defined as a hemoglobin increment of less than 1 g/dL (10 g/L) after 4–6 weeks of 100 mg/day of elemental oral iron. The differential diagnosis in these cases (Table 13–3) includes malabsorption from autoimmune gastritis, Helicobacter pylori gastric infection, celiac disease, and hereditary iron-refractory iron deficiency anemia. Iron-refractory iron deficiency anemia is a rare autosomal recessive disorder due to mutations in the transmembrane serine protease 6 (TMPRSS6) gene, which normally down-regulates hepcidin. In iron-refractory iron deficiency anemia, hepcidin levels are normal to high and ferritin levels are high despite the iron deficiency.
Clinical Findings
A. Symptoms and Signs
The primary symptoms of iron deficiency anemia are those of the anemia itself (easy fatigability, tachycardia, palpitations, and dyspnea on exertion). Severe deficiency causes skin and mucosal changes, including a smooth tongue, brittle nails, spooning of nails (koilonychia), and cheilosis. Dysphagia due to the formation of esophageal webs (Plummer-Vinson syndrome) may occur in severe iron deficiency. Many iron-deficient patients develop pica, craving for specific foods (ice chips, etc) often not rich in iron.
B. Laboratory Findings
Iron deficiency develops in stages. The first is depletion of iron stores without anemia followed by anemia with a normal red blood cell size (normal MCV) followed by anemia with reduced red blood cell size (low MCV). The reticulocyte count is low or inappropriately normal. Ferritin is a measure of total body iron stores. A ferritin value less than 12 ng/mL (27 pmol/L) (in the absence of scurvy) is a highly reliable indicator of reduced iron stores. Note that the lower limit of normal for ferritin generally is below 12 ng/mL (27 pmol/L) in women due to the fact that the normal ferritin range is generated by including healthy menstruating women who are iron deficient but not anemic. However, because serum ferritin levels may rise in response to inflammation or other stimuli, a normal or elevated ferritin level does not exclude a diagnosis of iron deficiency. A ferritin level less than 30 ng/mL (67 pmol/L) almost always indicates iron deficiency in anyone who is anemic. As iron deficiency progresses, serum iron values decline to less than 30 mcg/dL (67 pmol/L) and transferrin (the iron transport protein) levels rise to compensate, leading to transferrin saturations of less than 15%. Low transferrin saturation is also seen in anemia of inflammation, so caution in the interpretation of this test is warranted. Isolated iron deficiency anemia has a low hepcidin level, not yet a clinically available test. As the MCV falls (ie, microcytosis), the blood smear shows hypochromic microcytic cells. With further progression, anisocytosis (variations in red blood cell size) and poikilocytosis (variation in shape of red cells) develop. Severe iron deficiency will produce a bizarre peripheral blood smear, with severely hypochromic cells, target cells, and pencil-shaped or cigar-shaped cells. Bone marrow biopsy for evaluation of iron stores is rarely performed. If the biopsy is done, it shows the absence of iron in erythroid progenitor cells by Prussian blue staining. The platelet count is commonly increased, but it usually remains under 800,000/mcL (800 × 109/L).
Differential Diagnosis
Other causes of microcytic anemia include anemia of chronic disease (specifically, anemia of inflammation), thalassemia, lead poisoning, and congenital X-linked sideroblastic anemia. Anemia of chronic disease is characterized by normal or increased iron stores in bone marrow macrophages and a normal or elevated ferritin level; the serum iron and transferrin saturation are low, often drastically so, and the total iron-binding capacity (TIBC) (the blood’s capacity for iron to bind to transferrin) and transferrin are either normal or low. Thalassemia produces a greater degree of microcytosis for any given level of anemia than does iron deficiency and, unlike virtually every other cause of anemia, has a normal or elevated (rather than a low) red blood cell count as well as a reticulocytosis. In thalassemia, red blood cell morphology on the peripheral smear resembles severe iron deficiency.
Treatment
The diagnosis of iron deficiency anemia can be made either by the laboratory demonstration of an iron-deficient state or by evaluating the response to a therapeutic trial of iron replacement. Since the anemia itself is rarely life-threatening, the most important part of management is identification of the cause—especially a source of occult blood loss.
A. Oral Iron
Ferrous sulfate, 325 mg once daily or every other day on an empty stomach, is a standard approach for replenishing iron stores. As oral iron stimulates hepcidin production, once daily or every other day dosing maximizes iron absorption compared to multiple doses per day, and with fewer side effects. Nausea and constipation limit compliance with ferrous sulfate. Extended-release ferrous sulfate with mucoprotease is a well-tolerated oral preparation. Taking ferrous sulfate with food reduces side effects but also its absorption. An appropriate response to oral iron is a return of the hematocrit level halfway toward normal within 3 weeks with full return to baseline after 2 months. Iron therapy should continue for 3–6 months after restoration of normal hematologic values to replenish iron stores. Failure of response to iron therapy is usually due to noncompliance, although occasional patients may absorb iron poorly, particularly if the stomach is achlorhydric. Such patients may benefit from concomitant administration of oral ascorbic acid. Other reasons for failure to respond include incorrect diagnosis (anemia of chronic disease, thalassemia), celiac disease, and ongoing blood loss that exceeds the rate of new erythropoiesis. Treatment of H pylori infection, in appropriate cases, can improve oral iron absorption.
B. Parenteral Iron
The indications are intolerance of or refractoriness to oral iron (including those with iron-refractory iron deficiency anemia), gastrointestinal disease (usually inflammatory bowel disease) precluding the use of oral iron, and continued blood loss that cannot be corrected, such as chronic hemodialysis. Historical parenteral iron preparations, such as high-molecular-weight iron dextran, were problematic due to long infusion times (hours), polyarthralgia, and hypersensitivity reactions, including anaphylaxis. Current parenteral iron preparations coat the iron in protective carbohydrate shells or contain low-molecular-weight iron dextran, are safe, and can be administered over 15 minutes to 1 hour. Most iron deficient patients need 1–1.5 g of parenteral iron; this dose corrects for the iron deficit and replenishes iron stores for the future.
Ferric pyrophosphate citrate (Triferic) is an FDA-approved additive to the dialysate designed to replace the 5–7 mg of iron that patients with chronic kidney disease tend to lose during each hemodialysis treatment. Ferric pyrophosphate citrate delivers sufficient iron to the marrow to maintain hemoglobin and not increase iron stores; it may obviate the need for intravenous iron in hemodialysis patients.
When to Refer
Patients should be referred to a hematologist if the suspected diagnosis is not confirmed or if they are not responsive to oral iron therapy.
Auerbach M et al. Treatment of iron deficiency in the elderly: a new paradigm. Clin Geriatr Med. 2019 Aug;35(3):307–17. [PMID: 31230732]
Camaschella C. Iron deficiency. Blood. 2019 Jan 3;133(1):30–9. [PMID: 30401704]
Powers JM et al. Disorders of iron metabolism: new diagnostic and treatment approaches to iron deficiency. Hematol Oncol Clin North Am. 2019 Jun;33(3):393–408. [PMID: 31030809]
ANEMIA OF CHRONIC DISEASE
ESSENTIALS OF DIAGNOSIS
Mild or moderate normocytic or microcytic anemia.
Normal or increased ferritin and normal or reduced transferrin.
Underlying chronic disease.
General Considerations
Many chronic systemic diseases are associated with mild or moderate anemia. The anemias of chronic disease are characterized according to etiology and pathophysiology. First, the anemia of inflammation is associated with chronic inflammatory states (such as inflammatory bowel disease, rheumatologic disorders, chronic infections, and malignancy) and is mediated through hepcidin (a negative regulator of ferroportin) primarily via elevated IL-6, resulting in reduced iron uptake in the gut and reduced iron transfer from macrophages to erythroid progenitor cells in the bone marrow. This is referred to as iron-restricted erythropoiesis since the patient is iron replete. There is also reduced responsiveness to erythropoietin, the elaboration of hemolysins that shorten red blood cell survival, and the production of other inflammatory cytokines that dampen red cell production. The serum iron is low in the anemia of inflammation. Second, the anemia of organ failure can occur with kidney disease, liver failure, and endocrine gland failure. Erythropoietin is reduced and the red blood cell mass decreases in response to the diminished signal for red blood cell production; the serum iron is normal (except in chronic kidney disease where it is low due to the reduced hepcidin clearance and subsequent enhanced degradation of ferroportin). Third, the anemia of older adults is present in up to 20% of individuals over age 85 years in whom a thorough evaluation for an explanation of anemia is negative. The anemia is a consequence of (1) a relative resistance to red blood cell production in response to erythropoietin, (2) a decrease in erythropoietin production relative to the nephron mass, (3) a negative erythropoietic influence of higher levels of chronic inflammatory cytokines in older adults, and (4) the presence of various somatic mutations in myeloid genes typically associated with myeloid neoplasms. The latter condition is now referred to as clonal cytopenias of undetermined significance, which has a 1–1.5% per year rate of transformation to a myeloid neoplasm, such as a myelodysplastic syndrome. The serum iron is normal.
Clinical Findings
A. Symptoms and Signs
The clinical features are those of the causative condition. The diagnosis should be suspected in patients with known chronic diseases. In cases of significant anemia, coexistent iron deficiency or folic acid deficiency should be suspected. Decreased dietary intake of iron or folic acid is common in chronically ill patients, many of whom will also have ongoing gastrointestinal blood losses. Patients undergoing hemodialysis regularly lose both iron and folic acid during dialysis.
B. Laboratory Findings
The hematocrit rarely falls below 60% of baseline (except in kidney failure). The MCV is usually normal or slightly reduced. Red blood cell morphology is usually normal, and the reticulocyte count is mildly decreased or normal.
1. Anemia of inflammation—In the anemia of inflammation, serum iron and transferrin values are low, and the transferrin saturation may be extremely low, leading to an erroneous diagnosis of iron deficiency. In contrast to iron deficiency, serum ferritin values should be normal or increased. A serum ferritin value less than 30 ng/mL (67 pmol/L) indicates coexistent iron deficiency. Anemia of inflammation has elevated hepcidin levels; however, no clinical test is yet available. A particular challenge is the diagnosis of iron deficiency in the setting of the anemia of inflammation, in which the serum ferritin can be as high as 200 ng/mL (450 pmol/L). The diagnosis is established by a bone marrow biopsy with iron stain. Absent iron staining indicates iron deficiency, whereas iron localized in marrow macrophages indicates pure anemia of inflammation. However, bone marrow biopsies are rarely done for this purpose. Two other tests all support iron deficiency in the setting of inflammation: a reticulocyte hemoglobin concentration of less than 28 pg or a soluble serum transferrin receptor (units: mg/L) to log ferritin (units: mcg/L) ratio of 1–8 (a ratio of less than 1 is virtually diagnostic of pure anemia of chronic disease). A functional test is hemoglobin response to oral or parenteral iron in the setting of inflammation when iron deficiency is suspected. A note of caution: certain circumstances of iron-restricted erythropoiesis (such as malignancy) will partially respond to parenteral iron infusion even when the iron stores are replete due to the immediate distribution of iron to erythropoietic progenitor cells after the infusion.
2. Other anemias of chronic disease—In the anemias of organ failure and of older adults, the iron studies are generally normal. The anemia of older persons is a diagnosis of exclusion. Clonal cytopenias of undetermined significance are diagnosed by sending a blood sample for myeloid gene sequencing.
Treatment
In most cases, no treatment of the anemia is necessary and the primary management is to address the condition causing the anemia of chronic disease. When the anemia is severe or is adversely affecting the quality of life or functional status, then treatment involves either red blood cell transfusions or parenteral recombinant erythropoietin (epoetin alfa or darbepoetin). The FDA-approved indications for recombinant erythropoietin are hemoglobin less than 10 g/dL and anemia due to rheumatoid arthritis, inflammatory bowel disease, hepatitis C, zidovudine therapy in HIV-infected patients, myelosuppressive chemotherapy of solid malignancy (treated with palliative intent only), or chronic kidney disease (estimated glomerular filtration rate of less than 60 mL/min). The dosing and schedule of recombinant erythropoietin are individualized to maintain the hemoglobin between 10 g/dL (100 g/L) and 12 g/dL (120 g/L). The use of recombinant erythropoietin is associated with an increased risk of venothromboembolism and arterial thrombotic episodes, especially if the hemoglobin rises to greater than 12 g/dL (120 g/L). There is concern that recombinant erythropoietin is associated with reduced survival in patients with malignancy. For patients with end-stage renal disease receiving recombinant erythropoietin who are on hemodialysis, the anemia of chronic kidney disease can be more effectively corrected by adding soluble ferric pyrophosphate to their dialysate than by administering intravenous iron supplementation.
When to Refer
Referral to a hematologist is not usually necessary.
Cappellini MD et al. Iron deficiency across chronic inflammatory conditions: international expert opinion on definition, diagnosis, and management. Am J Hematol. 2017 Oct;92(10):1068–78. [PMID: 28612425]
Lanier JB et al. Anemia in older adults. Am Fam Physician. 2018 Oct 1;98(7):437–42. [PMID: 30252420]
Weiss G et al. Anemia of inflammation. Blood. 2019 Jan 3;133(1):40–50. [PMID: 30401705]
THE THALASSEMIAS
ESSENTIALS OF DIAGNOSIS
Microcytosis disproportionate to the degree of anemia.
Positive family history.
Lifelong personal history of microcytic anemia.
Normal or elevated red blood cell count.
Abnormal red blood cell morphology with microcytes, hypochromia, acanthocytes, and target cells.
In beta-thalassemia, elevated levels of hemoglobin A2 and F.
General Considerations
The thalassemias are hereditary disorders characterized by reduction in the synthesis of globin chains (alpha or beta). Reduced globin chain synthesis causes reduced hemoglobin synthesis and a hypochromic microcytic anemia because of defective hemoglobinization of red blood cells. Thalassemias can be considered among the hyperproliferative hemolytic anemias, the anemias related to abnormal hemoglobin, and the hypoproliferative anemias, since all of these factors play a role in pathogenesis. The hallmark laboratory features are small (low MCV) and pale (low mean corpuscular hemoglobin [MCH]) red blood cells, anemia, and a normal to elevated red blood cell count (ie, a large number of the small and pale red blood cells are being produced). Although patients often exhibit an elevated reticulocyte count, generally the degree of reticulocyte output is inadequate to meet the degree of red blood cell destruction (hemolysis) occurring in the bone marrow and the patients remain anemic.
Normal adult hemoglobin is primarily hemoglobin A, which represents approximately 98% of circulating hemoglobin. Hemoglobin A is formed from a tetramer of two alpha- globin chains and two beta-globin chains—and is designated alpha2beta2. Two copies of the alpha-globin gene are located on each chromosome 16, and there is no substitute for alpha-globin in the formation of adult hemoglobin. One copy of the beta-globin gene resides on each chromosome 11 adjacent to genes encoding the beta-like globins delta and gamma (the so-called beta-globin gene cluster region). The tetramer of alpha2delta2 forms hemoglobin A2, which normally composes 1–3% of adult hemoglobin. The tetramer alpha2gamma2 forms hemoglobin F, which is the major hemoglobin of fetal life but which composes less than 1% of normal adult hemoglobin.
The thalassemias are described as thalassemia trait when there are laboratory features without significant clinical impact, thalassemia intermedia when there is an occasional red blood cell transfusion requirement or other moderate clinical impact, and thalassemia major when the disorder is life-threatening and the patient is transfusion-dependent. Most patients with thalassemia major die of the consequences of iron overload from red blood cell transfusions.
Alpha-thalassemia is due primarily to gene deletions causing reduced alpha-globin chain synthesis (Table 13–4). Each alpha-globin gene produces one-quarter of the total alpha-globin quantity, so there is a predictable proportionate decrease in alpha-globin output with each lost alpha-globin gene. Since all adult hemoglobins are alpha containing, alpha-thalassemia produces no change in the proportions of hemoglobins A, A2, and F on hemoglobin electrophoresis. In severe forms of alpha-thalassemia, excess beta chains may form a beta-4 tetramer called hemoglobin H. In the presence of reduced alpha chains, the excess beta chains are unstable and precipitate, leading to damage of red blood cell membranes. This leads to both intramedullary (bone marrow) and peripheral blood hemolysis.
Table 13–4. Alpha-thalassemia syndromes.
Beta-thalassemias are usually caused by point mutations rather than deletions (Table 13–5). These mutations result in premature chain termination or in problems with transcription of RNA and ultimately result in reduced or absent beta-globin chain synthesis. The molecular defects leading to beta-thalassemia are numerous and heterogeneous. Defects that result in absent beta-globin chain expression are termed beta0, whereas those causing reduced but not absent synthesis are termed beta+. In beta+ thalassemia, the degree of reduction of beta-globin synthesis is consistent within families but is quite variable between families. The reduced beta-globin chain synthesis in beta-thalassemia results in a relative increase in the proportions of hemoglobins A2 and F compared to hemoglobin A on hemoglobin electrophoresis, as the beta-like globins (delta and gamma) substitute for the missing beta chains. In the presence of reduced beta chains, the excess alpha chains are unstable and precipitate, leading to damage of red blood cell membranes. This leads to both intramedullary (bone marrow) and peripheral blood hemolysis. The bone marrow demonstrates erythroid hyperplasia under the stimuli of anemia and ineffective erythropoiesis (intramedullary destruction of the developing erythroid cells). In cases of severe thalassemia, the marked expansion of the erythroid compartment in the bone marrow may cause severe bony deformities, osteopenia, and pathologic bone fractures.
Table 13–5. Beta-thalassemia syndromes.

Clinical Findings
A. Symptoms and Signs
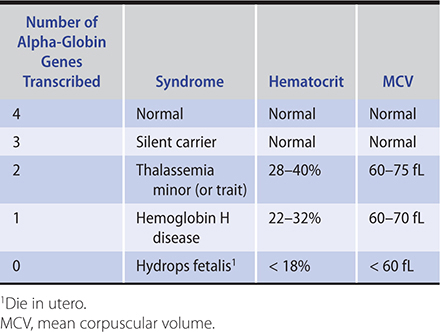
The alpha-thalassemia syndromes are seen primarily in persons from southeast Asia and China and, less commonly, in blacks and persons of Mediterranean origin (Table 13–4). Normally, adults have four copies of the alpha-globin chain. When three alpha-globin genes are present, the patient is hematologically normal (silent carrier). When two alpha-globin genes are present, the patient is said to have alpha-thalassemia trait, a form of thalassemia minor. In alpha-thalassemia-1 trait, the alpha gene deletion is heterozygous (alpha –/alpha –) and affects mainly those of Asian descent. In alpha-thalassemia-2 trait, the alpha gene deletion is homozygous (alpha alpha/– –) and affects mainly blacks. These patients are clinically normal and have a normal life expectancy and performance status, with a mild microcytic anemia. When only one alpha globin chain is present (alpha –/– –), the patient has hemoglobin H disease (alpha-thalassemia-3). This is a chronic hemolytic anemia of variable severity (thalassemia minor or intermedia). Physical examination might reveal pallor and splenomegaly. Affected individuals usually do not need transfusions; however, they may be required during transient periods of hemolytic exacerbation caused by infection or other stressors or during periods of erythropoietic shutdown caused by certain viruses (“aplastic crisis”). When all four alpha-globin genes are deleted, no normal hemoglobin is produced and the affected fetus is stillborn (hydrops fetalis). In hydrops fetalis, the only hemoglobin species made is gamma and is called hemoglobin Bart’s (gamma4).
Beta-thalassemia primarily affects persons of Mediterranean origin (Italian, Greek) and to a lesser extent Asians and blacks (Table 13–5). Patients homozygous for beta-thalassemia (beta0/beta0 or some with beta+/beta+) have beta-thalassemia major (Cooley anemia). Affected children are normal at birth, but after 6 months, when hemoglobin synthesis switches from hemoglobin F to hemoglobin A, severe anemia develops that requires transfusion. Numerous clinical problems ensue, including stunted growth, bony deformities (abnormal facial structure, pathologic bone fractures), hepatosplenomegaly, jaundice (due to gallstones, hepatitis-related cirrhosis, or both), and thrombophilia. The clinical course is modified significantly by transfusion therapy, but transfusional iron overload (hemosiderosis) results in a clinical picture similar to hemochromatosis, with heart failure, cardiac arrhythmias, cirrhosis, endocrinopathies, and pseudoxanthoma elasticum (calcification and fragmentation of the elastic fibers of the skin, retina, and cardiovascular system), usually after more than 100 units of red blood cells have been transfused. Iron overloading occurs because the human body has no active iron excretory mechanism. Before the application of allogeneic stem cell transplantation and the development of more effective forms of iron chelation, death from iron overload usually occurred between the ages of 20 and 30 years.
Patients homozygous for a milder form of beta-thalassemia (beta+/beta+, but allowing a higher rate of beta-globin synthesis) have beta-thalassemia intermedia. These patients have chronic hemolytic anemia but do not require transfusions except under periods of stress or during aplastic crises. They also may develop iron overload because of periodic transfusion. They survive into adult life but with hepatosplenomegaly and bony deformities. Patients heterozygous for beta-thalassemia (beta/beta0 or beta/beta+) have beta-thalassemia minor and a clinically insignificant microcytic anemia.
Prenatal diagnosis is available, and genetic counseling should be offered and the opportunity for prenatal diagnosis discussed.
B. Laboratory Findings
1. Alpha-thalassemia trait—These patients have mild or no anemia, with hematocrits between 28% and 40%. The MCV is strikingly low (60–75 fL) despite the modest anemia, and the red blood count is normal or increased. The peripheral blood smear shows microcytes, hypochromia, occasional target cells, and acanthocytes (cells with irregularly spaced spiked projections). The reticulocyte count and iron parameters are normal. Hemoglobin electrophoresis is normal. Alpha-thalassemia trait is thus usually diagnosed by exclusion. Genetic testing to demonstrate alpha-globin gene deletion is available.
2. Hemoglobin H disease—These patients have a more marked anemia, with hematocrits between 22% and 32%. The MCV is remarkably low (60–70 fL) and the peripheral blood smear is markedly abnormal, with hypochromia, microcytosis, target cells, and poikilocytosis. The reticulocyte count is elevated and the red blood cell count is normal or elevated. Hemoglobin electrophoresis will show a fast-migrating hemoglobin (hemoglobin H), which comprises 10–40% of the hemoglobin. A peripheral blood smear can be stained with supravital dyes to demonstrate the presence of hemoglobin H.
3. Beta-thalassemia minor—These patients have a modest anemia with hematocrit between 28% and 40%. The MCV ranges from 55 fL to 75 fL, and the red blood cell count is normal or increased. The reticulocyte count is normal or slightly elevated. The peripheral blood smear is mildly abnormal, with hypochromia, microcytosis, and target cells. In contrast to alpha-thalassemia, basophilic stippling is present. Hemoglobin electrophoresis shows an elevation of hemoglobin A2 to 4–8% and occasional elevations of hemoglobin F to 1–5%.
4. Beta-thalassemia intermedia—These patients have a moderate anemia with hematocrit between 17% and 33%. The MCV ranges from 55 fL to 75 fL, and the red blood cell count is normal or increased. The reticulocyte count is elevated. The peripheral blood smear is abnormal with hypochromia, microcytosis, basophilic stippling, and target cells. Hemoglobin electrophoresis shows up to 30% hemoglobin A, an elevation of hemoglobin A2 up to 10%, and elevation of hemoglobin F from 6% to 10%.
5. Beta-thalassemia major—These patients have severe anemia, and without transfusion the hematocrit may fall to less than 10%. The peripheral blood smear is bizarre, showing severe poikilocytosis, hypochromia, microcytosis, target cells, basophilic stippling, and nucleated red blood cells. Little or no hemoglobin A is present. Variable amounts of hemoglobin A2 are seen, and the predominant hemoglobin present is hemoglobin F.
Differential Diagnosis
Mild forms of thalassemia must be differentiated from iron deficiency. Compared to iron deficiency anemia, patients with thalassemia have a lower MCV, a normal or elevated red blood cell count (rather than low), a more abnormal peripheral blood smear at modest levels of anemia, and usually a reticulocytosis. Iron studies are normal or the transferrin saturation or ferritin (or both) are elevated. Severe forms of thalassemia may be confused with other hemoglobinopathies. The diagnosis of beta-thalassemia is made by the above findings and hemoglobin electrophoresis showing elevated levels of hemoglobins A2 and F (provided the patient is replete in iron), or beta-gene sequencing. The diagnosis of alpha-thalassemia is made by exclusion since there is no change in the proportion of the normal adult hemoglobin species or confirmed by alpha gene deletion studies. The only other microcytic anemia with a normal or elevated red blood cell count is iron deficiency in a patient with polycythemia vera.
Treatment
Patients with mild thalassemia (alpha-thalassemia trait or beta-thalassemia minor) require no treatment and should be identified so that they will not be subjected to repeated evaluations and treatment for iron deficiency. Patients with hemoglobin H disease should take folic acid supplementation (1 mg/day orally) and avoid medicinal iron and oxidative drugs such as sulfonamides. Patients with severe thalassemia are maintained on a regular transfusion schedule (in part to suppress endogenous erythropoiesis and therefore bone marrow expansion) and receive folic acid supplementation. Splenectomy is performed if hypersplenism causes a marked increase in the transfusion requirement or refractory symptoms. Patients with regular transfusion requirements should be treated with iron chelation (oral or parenteral) in order to prevent or delay life-limiting organ damage from iron overload.
Allogeneic stem cell transplantation is the treatment of choice for beta-thalassemia major and the only available cure. Children who have not yet experienced organ damage from iron overload do well, with long-term survival in more than 80% of cases. Autologous gene therapy is showing promise for thalassemia major.
When to Refer
All patients with thalassemia intermedia or major should be referred to a hematologist. Any patient with an unexplained microcytic anemia should be referred to help establish a diagnosis. Patients with thalassemia minor or intermedia should be offered genetic counseling because offspring of thalassemic couples are at risk for inheriting thalassemia major.
Cappellini MD et al. New therapeutic targets in transfusion-dependent and -independent thalassemia. Hematology Am Soc Hematol Educ Program. 2017 Dec 8;2017(1):278–83. [PMID: 29222267]
Porter J. Beyond transfusion therapy: new therapies in thalassemia including drugs, alternate donor transplant, and gene therapy. Hematology Am Soc Hematol Educ Program. 2018 Nov 30;2018(1):361–70. [PMID: 30504333]
Srivastava A et al. Cure for thalassemia major—from allogeneic hematopoietic stem cell transplantation to gene therapy. Haematologica. 2017 Feb;102(2):214–23. [PMID: 27909215]
Taher AT et al. Thalassaemia. Lancet. 2018 Jan13;391(10116):155–67. [PMID: 28774421]
Thompson AA et al. Gene therapy in patients with transfusion-dependent β-thalassemia. N Engl J Med. 2018 Apr 19;378(16):1479–93. [PMID: 29669226]
VITAMIN B12 DEFICIENCY
ESSENTIALS OF DIAGNOSIS
Macrocytic anemia.
Megaloblastic blood smear (macro-ovalocytes and hypersegmented neutrophils).
Low serum vitamin B12 level.
General Considerations
Vitamin B12 belongs to the family of cobalamins and serves as a cofactor for two important reactions in humans. As methylcobalamin, it is a cofactor for methionine synthetase in the conversion of homocysteine to methionine, and as adenosylcobalamin for the conversion of methylmalonyl-coenzyme A (CoA) to succinyl-CoA. Vitamin B12 comes from the diet and is present in all foods of animal origin. The daily absorption of vitamin B12 is 5 mcg.
The liver contains 2–5 mg of stored vitamin B12. Since daily utilization is 3–5 mcg, the body usually has sufficient stores of vitamin B12 so that it takes more than 3 years for vitamin B12 deficiency to occur if all intake or absorption immediately ceases.
Since vitamin B12 is present in foods of animal origin, dietary vitamin B12 deficiency is extremely rare but is seen in vegans—strict vegetarians who avoid all dairy products, meat, and fish (Table 13–6). Pernicious anemia is an autoimmune illness whereby autoantibodies destroy gastric parietal cells (that produce intrinsic factor) and cause atrophic gastritis or bind to and neutralize intrinsic factor, or both. Abdominal surgery may lead to vitamin B12 deficiency in several ways. Gastrectomy will eliminate the site of intrinsic factor production; blind loop syndrome will cause competition for vitamin B12 by bacterial overgrowth in the lumen of the intestine; and surgical resection of the ileum will eliminate the site of vitamin B12 absorption. Rare causes of vitamin B12 deficiency include fish tapeworm (Diphyllobothrium latum) infection, in which the parasite uses luminal vitamin B12; pancreatic insufficiency (with failure to inactivate competing cobalamin-binding proteins [R-factors]); severe Crohn disease, causing sufficient destruction of the ileum to impair vitamin B12 absorption; and perhaps prolonged use of proton pump inhibitors.
Table 13–6. Causes of vitamin B12 deficiency.
Dietary deficiency
Decreased production or absorption of intrinsic factor
Pernicious anemia (autoimmune)
Gastrectomy
Helicobacter pylori infection
Competition for vitamin B12 in the gut
Blind loop syndrome
Fish tapeworm (rare)
Pancreatic insufficiency
Proton pump inhibitors
Decreased ileal absorption of vitamin B12
Surgical resection
Crohn disease
Transcobalamin II deficiency (rare)
Clinical Findings
A. Symptoms and Signs
Vitamin B12 deficiency causes a moderate to severe anemia of slow onset; patients may have few symptoms relative to the degree of anemia. In advanced cases, the anemia may be severe, with hematocrits as low as 10–15%, and may be accompanied by leukopenia and thrombocytopenia. The deficiency also produces changes in mucosal cells, leading to glossitis, as well as other vague gastrointestinal disturbances such as anorexia and diarrhea. Vitamin B12 deficiency also leads to a complex neurologic syndrome. Peripheral nerves are usually affected first, and patients complain initially of paresthesias. As the posterior columns of the spinal cord become impaired, patients complain of difficulty with balance or proprioception, or both. In more advanced cases, cerebral function may be altered as well, and on occasion dementia and other neuropsychiatric abnormalities may be present. It is critical to recognize that the nonhematologic manifestations of vitamin B12 deficiency can be manifest despite a completely normal complete blood count.
Patients are usually pale and may be mildly icteric or sallow. Typically, later in the disease course, neurologic examination may reveal decreased vibration and position sense or memory disturbance (or both).
B. Laboratory Findings
The diagnosis of vitamin B12 deficiency is made by finding a low serum vitamin B12 (cobalamin) level. Whereas the normal vitamin B12 level is greater than 210 pg/mL (155 pmol/L), most patients with overt vitamin B12 deficiency have serum levels less than 170 pg/mL (126 pmol/L), with symptomatic patients usually having levels less than 100 pg/mL (74 pmol/L). The diagnosis of vitamin B12 deficiency in low or low-normal values (level of 170–210 pg/mL [126–155 pmol/L]) is best confirmed by finding an elevated level of serum methylmalonic acid (greater than 1000 nmol/L) or homocysteine. Of note, elevated levels of serum methylmalonic acid can be due to kidney disease.
The anemia of vitamin B12 deficiency is typically moderate to severe with the MCV quite elevated (110–140 fL). However, it is possible to have vitamin B12 deficiency with a normal MCV from coexistent thalassemia or iron deficiency; in other cases, the reason is obscure. Patients with neurologic symptoms and signs that suggest possible vitamin B12 deficiency should be evaluated for that deficiency despite a normal MCV or the absence of anemia. In typical cases, the peripheral blood smear is megaloblastic, defined as red blood cells that appear as macro-ovalocytes, (although other shape changes are usually present) and neutrophils that are hypersegmented (six [or greater]-lobed neutrophils or mean neutrophil lobe counts greater than four). The reticulocyte count is reduced. Because vitamin B12 deficiency can affect all hematopoietic cell lines, the white blood cell count and the platelet count are reduced in severe cases.
Other laboratory abnormalities include elevated serum lactate dehydrogenase (LD) and a modest increase in indirect bilirubin. These two findings reflect the intramedullary destruction of developing abnormal erythroid cells.
Bone marrow morphology is characteristically abnormal. Marked erythroid hyperplasia is present as a response to defective red blood cell production (ineffective erythropoiesis). Megaloblastic changes in the erythroid series include abnormally large cell size and asynchronous maturation of the nucleus and cytoplasm—ie, cytoplasmic maturation continues while impaired DNA synthesis causes retarded nuclear development. In the myeloid series, giant bands and meta-myelocytes are characteristically seen.
Differential Diagnosis
Vitamin B12 deficiency should be differentiated from folic acid deficiency, the other common cause of megaloblastic anemia, in which red blood cell folic acid is low while vitamin B12 levels are normal. The bone marrow findings of vitamin B12 deficiency are sometimes mistaken for a myelodysplastic syndrome (MDS) or even acute erythrocytic leukemia. The distinction between vitamin B12 deficiency and myelodysplasia is based on the characteristic morphology and the low vitamin B12 and elevated methylmalonic acid levels.
Treatment
Initially, patients with vitamin B12 deficiency are usually treated with parenteral therapy. Intramuscular or subcutaneous injections of 100–1000 mcg of vitamin B12 are adequate for each dose (with the higher dose recommended initially). Replacement is usually given daily for the first week, weekly for the next month, and then monthly for life. The vitamin deficiency will recur if patients discontinue their therapy. Oral or sublingual methylcobalamin (1 mg/day) may be used instead of parenteral therapy once initial correction of the deficiency has occurred. Oral or sublingual replacement is effective, even in pernicious anemia, since approximately 1% of the dose is absorbed in the intestine via passive diffusion in the absence of active transport. It must be continued indefinitely and serum vitamin B12 levels must be monitored to ensure adequate replacement. For patients with neurologic symptoms caused by vitamin B12 deficiency, long-term parenteral vitamin B12 therapy is recommended, though its superiority over oral vitamin B12 therapy has not proven conclusively. Because some patients are concurrently folic acid deficient from intestinal mucosal atrophy, simultaneous folic acid replacement (1 mg daily) is advised for the first several months of vitamin B12 replacement.
Patients respond to therapy with an immediate improvement in their sense of well-being. Hypokalemia may complicate the first several days of therapy, particularly if the anemia is severe. A brisk reticulocytosis occurs in 5–7 days, and the hematologic picture normalizes in 2 months. Central nervous system symptoms and signs are potentially reversible if they have been present for less than 6 months. Red blood cell transfusions are rarely needed despite the severity of anemia, but when given, diuretics are also recommended to avoid heart failure because this anemia develops slowly and the plasma volume is increased at the time of diagnosis.
When to Refer
Referral to a hematologist is not usually necessary.
Green R. Vitamin B(12) deficiency from the perspective of a practicing hematologist. Blood. 2017 May 11;129(19):2603–11. [PMID: 28360040]
Wolffenbuttel BHR et al. The many faces of cobalamin (vitamin B12) deficiency. Mayo Clin Proc Innov Qual Outcomes. 2019 May 27;3(2):200–14. [PMID: 31193945]
FOLIC ACID DEFICIENCY
ESSENTIALS OF DIAGNOSIS
Macrocytic anemia.
Megaloblastic blood smear (macro-ovalocytes and hypersegmented neutrophils).
Reduced folic acid levels in red blood cells or serum.
Normal serum vitamin B12 level.
General Considerations
“Folic acid” is the term commonly used for pteroylmonoglutamic acid. Folic acid is present in most fruits and vegetables (especially citrus fruits and green leafy vegetables). Daily dietary requirements are 50–100 mcg. Total body stores of folic acid are approximately 5 mg, enough to supply requirements for 2–3 months.
The most common cause of folic acid deficiency is inadequate dietary intake (Table 13–7). Alcoholic or anorectic patients, persons who do not eat fresh fruits and vegetables, and those who overcook their food are candidates for folic acid deficiency. Reduced folic acid absorption is rarely seen, since absorption occurs from the entire gastrointestinal tract. However, medications such as phenytoin, trimethoprim-sulfamethoxazole, or sulfasalazine may interfere with its absorption. Folic acid absorption is poor in some patients with vitamin B12 deficiency due to gastrointestinal mucosal atrophy. Folic acid requirements are increased in pregnancy, hemolytic anemia, and exfoliative skin disease, and in these cases the increased requirements (5–10 times normal) may not be met by a normal diet.
Table 13–7. Causes of folic acid deficiency.
Dietary deficiency
Decreased absorption
Celiac disease
Medications: phenytoin, sulfasalazine, trimethoprim-sulfamethoxazole
Concurrent vitamin B12 deficiency
Increased requirement
Chronic hemolytic anemia
Pregnancy
Exfoliative skin disease
Excess loss: hemodialysis
Inhibition of reduction to active form
Methotrexate
Clinical Findings
A. Symptoms and Signs
The clinical features are similar to those of vitamin B12 deficiency. However, isolated folic acid deficiency does not result in neurologic abnormalities.
B. Laboratory Findings
Megaloblastic anemia is identical to anemia resulting from vitamin B12 deficiency. A red blood cell folic acid level below 150 ng/mL (340 nmol/L) is diagnostic of folic acid deficiency. Whether to order a serum or a red blood cell folate level remains unsettled since there are few, if any, data to support one test over the other. Usually the serum vitamin B12 level is normal, and it should always be measured when folic acid deficiency is suspected. In some instances, folic acid deficiency is a consequence of the gastrointestinal mucosal megaloblastosis from vitamin B12 deficiency.
Differential Diagnosis
The megaloblastic anemia of folic acid deficiency should be differentiated from vitamin B12 deficiency by the finding of a normal vitamin B12 level and a reduced red blood cell (or serum) folic acid level. Alcoholic patients, who often have nutritional deficiency, may also have anemia of liver disease. Pure anemia of liver disease causes a macrocytic anemia but does not produce megaloblastic morphologic changes in the peripheral blood; rather, target cells are present. Hypothyroidism is associated with mild macrocytosis and also with pernicious anemia.
Treatment
Folic acid deficiency is treated with daily oral folic acid (1 mg). The response is similar to that seen in the treatment of vitamin B12 deficiency, with rapid improvement and a sense of well-being, reticulocytosis in 5–7 days, and total correction of hematologic abnormalities within 2 months. Large doses of folic acid may produce hematologic responses in cases of vitamin B12 deficiency, but permit neurologic damage to progress; hence, obtaining a serum vitamin B12 level in suspected folic acid deficiency is paramount.
When to Refer
Referral to a hematologist is not usually necessary.
Achebe MM et al. How I treat anemia in pregnancy: iron, cobalamin, and folate. Blood. 2017 Feb 23;129(8):940–9. [PMID: 28034892]
Green R et al. Megaloblastic anemias: nutritional and other causes. Med Clin North Am. 2017 Mar;101(2):297–317. [PMID: 28189172]
Sobczyńska-Malefora A et al. Laboratory assessment of folate (vitamin B9) status. J Clin Pathol. 2018 Nov;71(11):949–56. [PMID: 30228213]
HEMOLYTIC ANEMIAS
The hemolytic anemias are a group of disorders in which red blood cell survival is reduced, either episodically or continuously. The bone marrow has the ability to increase erythroid production up to eightfold in response to reduced red cell survival, so anemia will be present only when the ability of the bone marrow to compensate is outstripped. This will occur when red cell survival is extremely short or when the ability of the bone marrow to compensate is impaired.
Hemolytic disorders are generally classified according to whether the defect is intrinsic to the red cell or due to some external factor (Table 13–8). Intrinsic defects have been described in all components of the red blood cell, including the membrane, enzyme systems, and hemoglobin; most of these disorders are hereditary. Hemolytic anemias due to external factors are immune and microangiopathic hemolytic anemias and infections of red blood cells.
Table 13–8. Classification of hemolytic anemias.
Intrinsic
Membrane defects: hereditary spherocytosis, hereditary elliptocytosis, paroxysmal nocturnal hemoglobinuria
Glycolytic defects: pyruvate kinase deficiency, severe hypophosphatemia
Oxidation vulnerability: glucose-6-phosphate dehydrogenase deficiency, methemoglobinemia
Hemoglobinopathies: sickle cell syndromes, thalassemia, unstable hemoglobins, methemoglobinemia
Extrinsic
Immune: autoimmune, lymphoproliferative disease, drug-induced, idiopathic
Microangiopathic: thrombotic thrombocytopenic purpura, hemolytic-uremic syndrome, disseminated intravascular coagulation, valve hemolysis, metastatic adenocarcinoma, vasculitis, copper overload
Infection: Plasmodium, Clostridium, Borrelia
Hypersplenism
Burns
Certain laboratory features are common to all hemolytic anemias. Haptoglobin, a normal plasma protein that binds and clears free hemoglobin released into plasma, may be depressed in hemolytic disorders. However, the haptoglobin level is influenced by many factors and is not always a reliable indicator of hemolysis, particularly in end-stage liver disease (its site of synthesis). When intravascular hemolysis occurs, transient hemoglobinemia ensues. Hemoglobin is filtered through the renal glomerulus and is usually reabsorbed by tubular cells. Hemoglobinuria will be present only when the capacity for reabsorption of hemoglobin by renal tubular cells is exceeded. In the absence of hemoglobinuria, evidence for prior intravascular hemolysis is the presence of hemosiderin in shed renal tubular cells (positive urine hemosiderin). With severe intravascular hemolysis, hemoglobinemia and methemalbuminemia may be present. Hemolysis increases the indirect bilirubin, and the total bilirubin may rise to 4 mg/dL (68 mcmol/L) or more. Bilirubin levels higher than this may indicate some degree of hepatic dysfunction. Serum LD levels are strikingly elevated in cases of microangiopathic hemolysis (thrombotic thrombocytopenic purpura, hemolytic-uremic syndrome) and may be elevated in other hemolytic anemias.
PAROXYSMAL NOCTURNAL HEMOGLOBINURIA
ESSENTIALS OF DIAGNOSIS
Episodic hemoglobinuria.
Thrombosis is common.
Suspect in confusing cases of hemolytic anemia with or without pancytopenia.
Flow cytometry demonstrates deficiencies of CD55 and CD59.
General Considerations
Paroxysmal nocturnal hemoglobinuria (PNH) is a rare acquired clonal hematopoietic stem cell disorder that results in abnormal sensitivity of the red blood cell membrane to lysis by complement and therefore hemolysis. Free hemoglobin is released into the blood that scavenges nitric oxide and promotes esophageal spasms, male erectile dysfunction, kidney damage, and thrombosis. Patients with significant PNH live about 10–15 years following diagnosis; thrombosis is the primary cause of death.
Clinical Findings
A. Symptoms and Signs
Classically, patients report episodic hemoglobinuria resulting in reddish-brown urine. Hemoglobinuria is most often noticed in the first morning urine due to the drop in blood pH while sleeping (hypoventilation) that facilitates this hemolysis. Besides anemia, these patients are prone to thrombosis, especially within mesenteric and hepatic veins, central nervous system veins (sagittal vein), and skin vessels (with formation of painful nodules). As this is a hematopoietic stem cell disorder, PNH may appear de novo or arise in the setting of aplastic anemia or myelodysplasia with possible progression to acute myeloid leukemia (AML). It is common that patients with idiopathic aplastic anemia have a small PNH clone (less than 2%) on blood or bone marrow analysis; this should not be considered true PNH per se, especially in the absence of a reticulocytosis or thrombosis.
B. Laboratory Findings
Anemia is of variable severity and frequency, so reticulocytosis may or may not be present at any given time. Abnormalities on the blood smear are nondiagnostic but may include macro-ovalocytes and polychromasia. Since the episodic hemolysis is mainly intravascular, urine hemosiderin is a useful test. Serum LD is characteristically elevated. Iron deficiency is commonly present, related to chronic iron loss from hemoglobinuria.
The white blood cell count and platelet count may be decreased and are always decreased in the setting of aplastic anemia. The best screening test is flow cytometry of blood erythrocytes, granulocytes, or monocytes to demonstrate deficiency of CD55 and CD59. The proportion of erythrocytes deficient in these proteins might be low due to the ongoing destruction of affected erythrocytes. The FLAER assay (fluorescein-labeled proaerolysin) by flow cytometry is more sensitive. Bone marrow morphology is variable and may show either generalized hypoplasia or erythroid hyperplasia or both. The bone marrow karyotype may be either normal or demonstrate a clonal abnormality.
Treatment
Many patients with PNH have mild disease not requiring intervention. In severe cases and in those occurring in the setting of myelodysplasia or previous aplastic anemia, allogeneic hematopoietic stem cell transplantation may prove curative. In patients with severe hemolysis (usually requiring red cell transfusions) or thrombosis (or both), treatment with eculizumab is warranted. Eculizumab is a humanized monoclonal antibody against complement protein C5 given every 2 weeks. Binding of eculizumab to C5 prevents its cleavage so the membrane attack complex cannot assemble. Eculizumab improves quality of life and reduces hemolysis, transfusion requirements, fatigue, and thrombosis risk. Eculizumab increases the risk of Neisseria meningitidis infections; patients receiving the antibody should undergo meningococcal vaccination (including vaccines for serogroup B) and take oral penicillin (or equivalent) meningococcal prophylaxis. Ravulizumab is a longer-acting version of eculizumab and is given every 8 weeks. Iron replacement is indicated for treatment of iron deficiency when present, which may improve the anemia while also causing a transient increase in hemolysis. For unclear reasons, corticosteroids are effective in decreasing hemolysis.
When to Refer
Most patients with PNH should be under the care of a hematologist.
Devos T et al. Diagnosis and management of PNH: review and recommendations from a Belgian expert panel. Eur J Haematol. 2018 Dec;101(6):737–49. [PMID: 30171728]
Patriquin CJ et al. How we treat paroxysmal nocturnal hemoglobinuria: a consensus statement of the Canadian PNH Network and review of the national registry. Eur J Haematol. 2019 Jan;102(1):36–52. [PMID: 30242915]
GLUCOSE-6-PHOSPHATE DEHYDROGENASE DEFICIENCY
ESSENTIALS OF DIAGNOSIS
X-linked recessive disorder seen commonly in American black men.
Episodic hemolysis in response to oxidant drugs or infection.
Bite cells and blister cells on the peripheral blood smear.
Reduced levels of glucose-6-phosphate dehydrogenase between hemolytic episodes.
General Considerations
Glucose-6-phosphate dehydrogenase (G6PD) deficiency is a hereditary enzyme defect that causes episodic hemolytic anemia because of the decreased ability of red blood cells to deal with oxidative stresses. G6PD deficiency leads to excess oxidized glutathione (hence, inadequate levels of reduced glutathione) that forces hemoglobin to denature and form precipitants called Heinz bodies. Heinz bodies cause red blood cell membrane damage, which leads to premature removal of these red blood cells by reticuloendothelial cells within the spleen (ie, extravascular hemolysis).
Numerous G6PD isoenzymes have been described. The usual isoenzyme found in American blacks is designated G6PD-A and that found in whites is designated G6PD-B, both of which have normal function and stability and therefore no hemolytic anemia. Ten to 15 percent of American blacks have the variant G6PD isoenzyme designated A–, in which there is both a reduction in normal enzyme activity and a reduction in its stability. The A– isoenzyme activity declines rapidly as the red blood cell ages past 40 days, a fact that explains the clinical findings in this disorder. More than 150 G6PD isoenzyme variants have been described, including some Mediterranean, Ashkenazi Jewish, and Asian variants with very low enzyme activity, episodic hemolysis, and exacerbations due to oxidizing substances including fava beans. Patients with G6PD deficiency seem to be protected from malaria parasitic infection, have less coronary artery disease, and possibly have fewer cancers and greater longevity.
Clinical Findings
G6PD deficiency is an X-linked disorder affecting 10–15% of American hemizygous black males and rare female homozygotes. Female carriers are rarely affected—only when an unusually high percentage of cells producing the normal enzyme are X-inactivated.
A. Symptoms and Signs
Patients are usually healthy, without chronic hemolytic anemia or splenomegaly. Hemolysis occurs episodically as a result of oxidative stress on the red blood cells, generated either by infection or exposure to certain medications. Medications initiating hemolysis that should be avoided include dapsone, methylene blue, phenazopyridine, primaquine, rasburicase, toluidine blue, nitrofurantoin, trimethoprim/sulfamethoxazole, sulfadiazine, pegloticase, and quinolones. Other medications, such as chloroquine, quinine, high-dose aspirin, and isoniazid, have been implicated but are less certain as offenders since they are often given during infections. Even with continuous use of the offending medication, the hemolytic episode is self-limited because older red blood cells (with low enzyme activity) are removed and replaced with a population of young red blood cells (reticulocytes) with adequate functional levels of G6PD. Severe G6PD deficiency (as in Mediterranean variants) may produce a chronic hemolytic anemia.
B. Laboratory Findings
Between hemolytic episodes, the blood is normal. During episodes of hemolysis, the hemoglobin rarely falls below 8 g/dL (80 g/L), and there is reticulocytosis and increased serum indirect bilirubin. The peripheral blood cell smear often reveals a small number of “bite” cells—cells that appear to have had a bite taken out of their periphery, or “blister” cells. This indicates pitting of precipitated membrane hemoglobin aggregates (ie, Heinz bodies) by the splenic macrophages. Heinz bodies may be demonstrated by staining a peripheral blood smear with cresyl violet; they are not visible on the usual Wright-Giemsa–stained blood smear. Specific enzyme assays for G6PD reveal a low level but may be falsely normal if they are performed during or shortly after a hemolytic episode during the period of reticulocytosis. In these cases, the enzyme assays should be repeated weeks after hemolysis has resolved. In severe cases of G6PD deficiency, enzyme levels are always low.
Treatment
No treatment is necessary except to avoid known oxidant medications.
Belfield KD et al. Review and drug therapy implications of glucose-6-phosphate dehydrogenase deficiency. Am J Health Syst Pharm. 2018 Feb 1;75(3):97–104. [PMID: 29305344]
Georgakouli K et al. Exercise in glucose-6-phosphate dehydrogenase deficiency: harmful or harmless? A narrative review. Oxid Med Cell Longev. 2019 Apr 4;2019:8060193. [PMID: 31089417]
SICKLE CELL ANEMIA & RELATED SYNDROMES
ESSENTIALS OF DIAGNOSIS
Recurrent pain episodes.
Positive family history and lifelong history of hemolytic anemia.
Irreversibly sickled cells on peripheral blood smear.
Hemoglobin S is the major hemoglobin seen on electrophoresis.
General Considerations
Sickle cell anemia is an autosomal recessive disorder in which an abnormal hemoglobin leads to chronic hemolytic anemia with numerous clinical consequences. A single DNA base change leads to an amino acid substitution of valine for glutamate in the sixth position on the beta-globin chain. The abnormal beta chain is designated betas and the tetramer of alpha-2betas-2 is designated hemoglobin S. Hemoglobin S is unstable and polymerizes in the setting of various stressors, including hypoxemia and acidosis, leading to the formation of sickled red blood cells. Sickled cells result in hemolysis and the release of ATP, which is converted to adenosine. Adenosine binds to its receptor (A2B), resulting in the production of 2,3-biphosphoglycerate and the induction of more sickling, and to its receptor (A2A) on natural killer cells, resulting in pulmonary inflammation. The free hemoglobin from hemolysis scavenges nitric oxide causing endothelial dysfunction, vascular injury, and pulmonary hypertension.
The rate of sickling is influenced by the intracellular concentration of hemoglobin S and by the presence of other hemoglobins within the cell. Hemoglobin F cannot participate in polymer formation, and its presence markedly retards sickling. Factors that increase sickling are red blood cell dehydration and factors that lead to formation of deoxyhemoglobin S (eg, acidosis and hypoxemia) either systemic or local in tissues. Hemolytic crises may be related to splenic sequestration of sickled cells (primarily in childhood before the spleen has been infarcted as a result of repeated sickling) or with coexistent disorders such as G6PD deficiency.
The betaS gene is carried in 8% of American blacks, and 1 of 400 American black children will be born with sickle cell anemia; prenatal diagnosis is available when sickle cell anemia is suspected. Genetic counseling should be made available to patients.
Clinical Findings
A. Symptoms and Signs
The disorder has its onset during the first year of life, when hemoglobin F levels fall as a signal is sent to switch from production of gamma-globin to beta-globin. Chronic hemolytic anemia produces jaundice, pigment (calcium bilirubinate) gallstones, splenomegaly (early in life), and poorly healing skin ulcers over the lower tibia. Life-threatening severe anemia can occur during hemolytic or aplastic crises, the latter generally associated with viral or other infection caused by immunoincompetence from hyposplenism or by folic acid deficiency causing reduced erythropoiesis.
Acute painful episodes due to acute vaso-occlusion from clusters of sickled red cells may occur spontaneously or be provoked by infection, dehydration, or hypoxia. Common sites of acute painful episodes include the spine and long appendicular bones. These episodes last hours to days and may produce low-grade fever. Acute vaso-occlusion may cause strokes due to sagittal sinus venous thrombosis or to bland or hemorrhagic central nervous system arterial ischemia and may also cause priapism. Vaso-occlusive episodes are not associated with increased hemolysis.
Repeated episodes of vascular occlusion especially affect the heart, lungs, and liver. The acute chest syndrome is characterized by acute chest pain, hypoxemia, and pulmonary infiltrates on a chest radiograph and must be distinguished from an infectious pneumonia. Ischemic necrosis of bones may occur, rendering the bone susceptible to osteomyelitis due to salmonellae and (somewhat less commonly) staphylococci. Infarction of the papillae of the renal medulla causes renal tubular concentrating defects and gross hematuria, more often encountered in sickle cell trait than in sickle cell anemia. Retinopathy similar to that noted in diabetes mellitus is often present and may lead to visual impairment. Pulmonary hypertension may develop and is associated with a poor prognosis. These patients are prone to delayed puberty. An increased incidence of infection is related to hyposplenism as well as to defects in the alternate complement pathway.
On examination, patients are often chronically ill and jaundiced. There is often hepatomegaly, but the spleen is not palpable in adult life. The heart may be enlarged with a hyperdynamic precordium and systolic murmurs and, in some cases, a pronounced increase in P2. Nonhealing cutaneous ulcers of the lower leg and retinopathy may be present.
B. Laboratory Findings
Chronic hemolytic anemia is present. The hematocrit is usually 20–30%. The peripheral blood smear is characteristically abnormal, with sickled cells comprising 5–50% of red cells. Other findings include reticulocytosis (10–25%), nucleated red blood cells, and hallmarks of hyposplenism such as Howell-Jolly bodies and target cells. The white blood cell count is characteristically elevated to 12,000–15,000/mcL, and reactive thrombocytosis may occur. Indirect bilirubin levels are high.
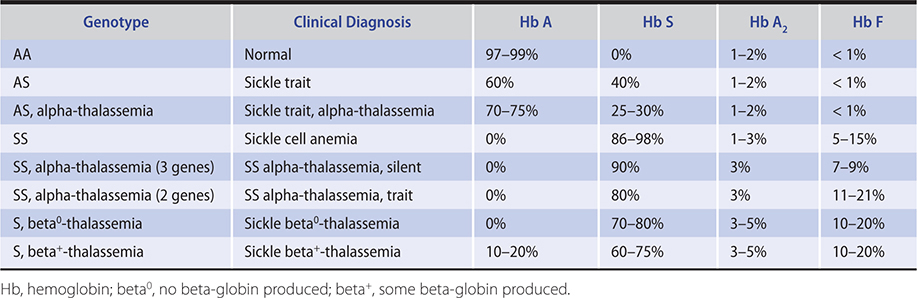
The diagnosis of sickle cell anemia is confirmed by hemoglobin electrophoresis (Table 13–9). Hemoglobin S will usually comprise 85–98% of hemoglobin. In homozygous S disease, no hemoglobin A will be present. Hemoglobin F levels are sometimes increased, and high hemoglobin F levels (15–20%) are associated with a more benign clinical course. Patients with S-beta+-thalassemia and SS alpha-thalassemia also have a more benign clinical course than straight sickle cell anemia (SS) patients.
Table 13–9. Hemoglobin distribution in sickle cell syndromes.
Treatment
When allogeneic hematopoietic stem cell transplantation is performed before the onset of significant end-organ damage, it can cure more than 80% of children with sickle cell anemia who have suitable HLA-matched donors, with a reasonably good quality of life. Transplantation remains investigational in adults. Other therapies modulate disease severity: hydroxyurea increases hemoglobin F levels epigenetically. Hydroxyurea (500–750 mg orally daily) reduces the frequency of painful crises in patients whose quality of life is disrupted by frequent vaso-occlusive pain episodes (three or more per year). Long-term follow-up of patients taking hydroxyurea demonstrates it improves overall survival and quality of life with little evidence for secondary malignancy. The use of omega-3 (n-3) fatty acid supplementation may also reduce vaso-occlusive episodes and reduce transfusion needs in patients with sickle cell anemia. L-glutamine has been shown to favorably modulate sickle pain crises and acute chest syndrome. Finally, a monoclonal antibody (crizanlizumab-tmca) reduces vaso-occlusive episodes by 50%. It blocks P-selectin on activated endothelial cells and thus disrupts the adverse interactions of platelets, red blood cells, and leukocytes with the endothelial wall.
Supportive care is the mainstay of treatment for sickle cell anemia. Patients are maintained on folic acid supplementation (1 mg orally daily) and given transfusions for aplastic or hemolytic crises. When acute painful episodes occur, precipitating factors should be identified and infections treated if present. The patient should be kept well hydrated, given generous analgesics, and supplied oxygen if hypoxic. Pneumococcal vaccination reduces the incidence of infections with this pathogen while hydroxyurea and L-glutamine reduce hospitalizations for acute pain. Angiotensin-converting enzyme inhibitors are recommended in patients with microalbuminuria.
Exchange transfusions are indicated for the treatment of severe or intractable acute vaso-occlusive crises, acute chest syndrome, priapism, and stroke. Long-term transfusion therapy has been shown to be effective in reducing the risk of recurrent stroke in children. Phenotypically matched transfused red blood cells are recommended to reduce the risk of red blood cell alloimmunization. It has been recommended that children with SS who are aged 2–16 years have annual transcranial ultrasounds and, if the Doppler velocity is abnormal (200 cm/s or greater), the clinician should strongly consider beginning transfusions to prevent stroke. Iron chelation is needed for those on chronic transfusion therapy.
Prognosis
Sickle cell anemia becomes a chronic multisystem disease, leading to organ failure that may result in death. With improved supportive care, average life expectancy is now between 40 and 50 years of age.
When to Refer
Patients with sickle cell anemia should have their care coordinated with a hematologist and should be referred to a Comprehensive Sickle Cell Center, if one is available.
When to Admit
Patients should be admitted for management of acute chest syndrome, for aplastic crisis, or for painful episodes that do not respond to outpatient interventions.
Bauer DE et al. Curative approaches for sickle cell disease: a review of allogeneic and autologous strategies. Blood Cells Mol Dis. 2017 Sep;67:155–68. [PMID: 28893518]
Blair HA. Crizanlizumab: first approval. Drugs. 2020 Jan;80(1):79–84. [PMID: 31933169]
Kutlar A et al. Effect of crizanlizumab on pain crises in subgroups of patients with sickle cell disease: a SUSTAIN study analysis. Am J Hematol. 2019 Jan;94(1):55–61. [PMID: 30295335]
Niihara Y et al. A phase 3 trial of l-glutamine in sickle cell disease. N Engl J Med. 2018 Jul 19;379(3):226–35. [PMID: 30021096]
Rees DC et al. How I manage red cell transfusions in patients with sickle cell disease. Br J Haematol. 2018 Feb;180(4):607–17. [PMID: 29377071]
Thein SL et al. How I treat the older adult with sickle cell disease. Blood. 2018 Oct 25;132(17):1750–60. [PMID: 30206116]
SICKLE CELL TRAIT
People with the heterozygous hemoglobin genotype AS have sickle cell trait. These persons are hematologically normal, with no anemia and normal red blood cells on peripheral blood smear. Hemoglobin electrophoresis will reveal that approximately 40% of hemoglobin is hemoglobin S (Table 13–9). People with sickle cell trait experience more rhabdomyolysis during vigorous exercise but do not have increased mortality compared to the general population. They may be at increased risk for venous thromboembolism. Chronic sickling of red blood cells in the acidotic renal medulla results in microscopic and gross hematuria, hyposthenuria (poor urine concentrating ability), and possibly chronic kidney disease. No treatment is necessary but genetic counseling is recommended.
Liem RI. Balancing exercise risk and benefits: lessons learned from sickle cell trait and sickle cell anemia. Hematology Am Soc Hematol Educ Program. 2018 Nov 30;2018(1):418–25. [PMID: 30504341]
Pecker LH et al. The current state of sickle cell trait: implications for reproductive and genetic counseling. Hematology Am Soc Hematol Educ Program. 2018 Nov 30;2018(1):474–81. [PMID: 30504348]
SICKLE THALASSEMIA
Patients with homozygous sickle cell anemia and alpha-thalassemia have less vigorous hemolysis and run higher hemoglobins than SS patients due to reduced red blood cell sickling related to a lower hemoglobin concentration within the red blood cell and higher hemoglobin F levels (Table 13–9). The MCV is low, and the red cells are hypochromic.
Patients who are compound heterozygotes for betas and beta-thalassemia are clinically affected with sickle cell syndromes. Sickle beta0-thalassemia is clinically very similar to homozygous SS disease. Vaso-occlusive crises may be somewhat less severe, and the spleen is not always infarcted. The MCV is low, in contrast to the normal MCV of sickle cell anemia. Hemoglobin electrophoresis reveals no hemoglobin A but will show an increase in hemoglobins A2 and F (Table 13–9).
Sickle beta+-thalassemia is a milder disorder than homozygous SS disease, with fewer pain episodes but more acute chest syndrome than sickle beta0-thalassemia. The spleen is usually palpable. The hemolytic anemia is less severe, and the hematocrit is usually 30–38%, with reticulocytes of 5–10%. Hemoglobin electrophoresis shows the presence of some hemoglobin A and elevated hemoglobins A2 and F (Table 13–9). The MCV is low.
AUTOIMMUNE HEMOLYTIC ANEMIA
ESSENTIALS OF DIAGNOSIS
Acquired hemolytic anemia caused by IgG autoantibody.
Spherocytes and reticulocytosis on peripheral blood smear.
Positive antiglobulin (Coombs) test.
General Considerations
Autoimmune hemolytic anemia is an acquired disorder in which an IgG autoantibody is formed that binds to a red blood cell membrane protein and does so most avidly at body temperature (ie, a “warm” autoantibody). The antibody is most commonly directed against a basic component of the Rh system present on most human red blood cells. When IgG antibodies coat the red blood cell, the Fc portion of the antibody is recognized by macrophages present in the spleen and other portions of the reticuloendothelial system. The interaction between splenic macrophages and the antibody-coated red blood cell results in removal of red blood cell membrane and the formation of a spherocyte due to the decrease in surface-to-volume ratio of the surviving red blood cell. These spherocytic cells have decreased deformability and are unable to squeeze through the 2-mcm fenestrations of splenic sinusoids and become trapped in the red pulp of the spleen. When large amounts of IgG are present on red blood cells, complement may be fixed. Direct complement lysis of cells is rare, but the presence of C3b on the surface of red blood cells allows Kupffer cells in the liver to participate in the hemolytic process via C3b receptors. The destruction of red blood cells in the spleen and liver designates this as extravascular hemolysis.
Approximately one-half of all cases of autoimmune hemolytic anemia are idiopathic. The disorder may also be seen in association with systemic lupus erythematosus, other rheumatic disorders, chronic lymphocytic leukemia (CLL), or lymphomas. It must be distinguished from drug-induced hemolytic anemia. When penicillin (or other medications, especially cefotetan, ceftriaxone, and piperacillin) coats the red blood cell membrane, the autoantibody is directed against the membrane-drug complex. Fludarabine, an antineoplastic, causes autoimmune hemolytic anemia through its immunosuppression; there is defective self- versus non–self-immune surveillance permitting the escape of a B-cell clone, which produces the offending autoantibody.
Clinical Findings
A. Symptoms and Signs
Autoimmune hemolytic anemia typically produces an anemia of rapid onset that may be life-threatening. Patients complain of fatigue and dyspnea and may present with angina pectoris or heart failure. On examination, jaundice and splenomegaly are usually present.
B. Laboratory Findings
The anemia is of variable degree but may be very severe, with hematocrit of less than 10%. Reticulocytosis is present, and spherocytes are seen on the peripheral blood smear. In cases of severe hemolysis, the stressed bone marrow may also release nucleated red blood cells. As with other hemolytic disorders, the serum indirect bilirubin is increased and the haptoglobin is low. Approximately 10% of patients with autoimmune hemolytic anemia have coincident immune thrombocytopenia (Evans syndrome).
The antiglobulin (Coombs) test forms the basis for diagnosis. The Coombs reagent is a rabbit IgM antibody raised against human IgG or human complement. The direct antiglobulin (Coombs) test (DAT) is performed by mixing the patient’s red blood cells with the Coombs reagent and looking for agglutination, which indicates the presence of antibody or complement or both on the red blood cell surface. The indirect antiglobulin (Coombs) test is performed by mixing the patient’s serum with a panel of type O red blood cells. After incubation of the test serum and panel red blood cells, the Coombs reagent is added. Agglutination in this system indicates the presence of free antibody (autoantibody or alloantibody) in the patient’s serum.
The direct antiglobulin test is positive (for IgG, complement, or both) in about 90% of patients with autoimmune hemolytic anemia. The indirect antiglobulin test may or may not be positive. A positive indirect antiglobulin test indicates the presence of a large amount of autoantibody that has saturated binding sites on the red blood cell and consequently appears in the serum. Because the patient’s serum usually contains the autoantibody, it may be difficult to obtain a “compatible” cross-match with homologous red blood cells for transfusions since the cross-match indicates the possible presence (true or false) of a red blood cell “alloantibody.”
Treatment
Initial treatment consists of prednisone, 1–2 mg/kg/day orally in divided doses. Patients with DAT-negative and DAT-positive autoimmune hemolysis respond equally well to corticosteroids. Transfused red blood cells will survive similarly to the patient’s own red blood cells. Because of difficulty in performing the cross-match, possible “incompatible” blood may need to be given. Decisions regarding transfusions should be made in consultation with a hematologist and a blood bank specialist. Death from cardiovascular collapse can occur in the setting of rapid hemolysis. In patients with rapid hemolysis, therapeutic plasmapheresis should be performed early in management to remove autoantibodies. If prednisone is ineffective or if the disease recurs on tapering the dose, splenectomy should be considered, which may cure the disorder. Patients with autoimmune hemolytic anemia refractory to prednisone and splenectomy may also be treated with a variety of agents. Treatment with rituximab, a monoclonal antibody against the B cell antigen CD20, is effective in some cases. The suggested dose is 375 mg/m2 intravenously weekly for 4 weeks. Rituximab is used in conjunction with corticosteroids as initial therapy in some patients with severe disease. Danazol, 400–800 mg/day orally, is less often effective than in immune thrombocytopenia but is well suited for long-term use because of its low toxicity profile. Immunosuppressive agents, including cyclophosphamide, vincristine, azathioprine, mycophenolate mofetil, alemtuzumab (an anti-CD52 antibody), or cyclosporine, may also be used. High-dose intravenous immune globulin (1 g/kg daily for 2 days) may be effective in controlling hemolysis. The benefit is short-lived (1–3 weeks), and the medication is very expensive. The long-term prognosis for patients with this disorder is good, especially if there is no other underlying autoimmune disorder or lymphoproliferative disorder. Treatment of an associated lymphoproliferative disorder will also treat the hemolytic anemia.
When to Refer
Patients with autoimmune hemolytic anemia should be referred to a hematologist for confirmation of the diagnosis and subsequent care.
When to Admit
Patients should be hospitalized for symptomatic anemia or rapidly falling hemoglobin levels.
Brodsky RA. Warm autoimmune hemolytic anemia. N Engl J Med. 2019 Aug 15;381(7):647–54. [PMID: 31412178]
Hill A et al. Autoimmune hemolytic anemia. Hematology Am Soc Hematol Educ Program. 2018 Nov 30;2018(1):382–9. [PMID: 30504336]
Hill QA et al. Defining autoimmune hemolytic anemia: a systematic review of the terminology used for diagnosis and treatment. Blood Adv. 2019 Jun 25;3(12):1897–906. [PMID: 31235526]
COLD AGGLUTININ DISEASE
ESSENTIALS OF DIAGNOSIS
Increased reticulocytes on peripheral blood smear.
Antiglobulin (Coombs) test positive only for complement.
Positive cold agglutinin titer.
General Considerations
Cold agglutinin disease is an acquired hemolytic anemia due to an IgM autoantibody (called a “cold agglutinin”) usually directed against the I/i antigen on red blood cells. These IgM autoantibodies characteristically will react poorly with cells at 37°C but avidly at lower temperatures, usually at 0–4°C (ie, “cold” autoantibody). Since the blood temperature (even in the most peripheral parts of the body) rarely goes lower than 20°C, only cold autoantibodies reactive at relatively higher temperatures will produce clinical effects. Hemolysis results indirectly from attachment of IgM, which in the cooler parts of the circulation (fingers, nose, ears) binds and fixes complement. When the red blood cell returns to a warmer temperature, the IgM antibody dissociates, leaving complement on the cell. Complement lysis of red blood cells rarely occurs. Rather, C3b, present on the red blood cells, is recognized by Kupffer cells (which have receptors for C3b), and red blood cell sequestration and destruction in the liver ensues (extravascular hemolysis). In some cases, the complement membrane attack complex forms, lysing the red blood cells (intravascular hemolysis).
Most cases of chronic cold agglutinin disease are idiopathic. Others occur in association with Waldenström macroglobulinemia, lymphoma, or CLL, in which a monoclonal IgM paraprotein is produced. Acute postinfectious cold agglutinin disease occurs following mycoplasmal pneumonia or viral infection (infectious mononucleosis, measles, mumps, or cytomegalovirus [CMV] with autoantibody directed against antigen i rather than I).
Clinical Findings
A. Symptoms and Signs
In chronic cold agglutinin disease, symptoms related to red blood cell agglutination occur on exposure to cold, and patients may complain of mottled or numb fingers or toes, acrocyanosis, episodic low back pain, and dark-colored urine. Hemolytic anemia is occasionally severe, but episodic hemoglobinuria may occur on exposure to cold. The hemolytic anemia in acute postinfectious syndromes is rarely severe.
B. Laboratory Findings
Mild anemia is present with reticulocytosis and rarely spherocytes. The blood smear made at room temperature shows agglutinated red blood cells (there is no agglutination on a blood smear made at body temperature). The direct antiglobulin (Coombs) test will be positive for complement only. Serum cold agglutinin titer will semi-quantitate the autoantibody. A monoclonal IgM is often found on serum protein electrophoresis and confirmed by serum immunoelectrophoresis. There is indirect hyperbilirubinemia and the haptoglobin is low during periods of hemolysis.
Treatment
Treatment is largely symptomatic, based on avoiding exposure to cold. Splenectomy and prednisone are usually ineffective (except when associated with a lymphoproliferative disorder) since hemolysis takes place in the liver and blood stream. Rituximab is the treatment of choice. The dose is 375 mg/m2 intravenously weekly for 4 weeks. Relapses may be effectively re-treated. High-dose intravenous immunoglobulin (2 g/kg) may be effective temporarily, but it is rarely used because of the high cost and short duration of benefit. Patients with severe disease may be treated with cytotoxic agents, such as bendamustine (plus rituximab), cyclophosphamide, fludarabine, or bortezomib, or with immunosuppressive agents, such as cyclosporine. As in warm IgG-mediated autoimmune hemolysis, it may be difficult to find compatible blood for transfusion. Red blood cells should be transfused through an in-line blood warmer.
Berentsen S et al. Bendamustine plus rituximab for chronic cold agglutinin disease: results of a Nordic prospective multicenter trial. Blood. 2017 Jul 27;130(4):537–41. [PMID: 28533306]
Berentsen S et al. Novel insights into the treatment of complement-mediated hemolytic anemias. Ther Adv Hematol. 2019 Sep 9;10:2040620719873321. [PMID: 31523413]
Go RS et al. How I treat autoimmune hemolytic anemia. Blood. 2017 Jun 1;129(22):2971–9. [PMID: 28360039]
APLASTIC ANEMIA
ESSENTIALS OF DIAGNOSIS
Pancytopenia.
No abnormal hematopoietic cells seen in blood or bone marrow.
Hypocellular bone marrow.
General Considerations
Aplastic anemia is a condition of bone marrow failure that arises from suppression of, or injury to, the hematopoietic stem cell. The bone marrow becomes hypoplastic, fails to produce mature blood cells, and pancytopenia develops.
There are a number of causes of aplastic anemia (Table 13–10). Direct hematopoietic stem cell injury may be caused by radiation, chemotherapy, toxins, or pharmacologic agents. Systemic lupus erythematosus may rarely cause suppression of the hematopoietic stem cell by an IgG autoantibody directed against it. However, the most common pathogenesis of aplastic anemia appears to be autoimmune suppression of hematopoiesis by a T-cell-mediated cellular mechanism, so called idiopathic aplastic anemia. In some cases of idiopathic aplastic anemia, defects in maintenance of the hematopoietic stem cell telomere length (eg, dyskeratosis congenita) or in DNA repair pathways (eg, Fanconi anemia) have been identified and are likely linked to both the initiation of bone marrow failure and the propensity to later progress to myelodysplasia, PNH, or AML. Complex detrimental immune responses to viruses can also cause aplastic anemia.
Table 13–10. Causes of aplastic anemia.
Autoimmune: idiopathic, systemic lupus erythematosus
Congenital: defects in telomere length maintenance or DNA repair (dyskeratosis congenita, Fanconi anemia, etc)
Chemotherapy, radiotherapy
Toxins: benzene, toluene, insecticides
Medications: chloramphenicol, gold salts, sulfonamides, phenytoin, carbamazepine, quinacrine, tolbutamide
Post-viral hepatitis (A, B, C, E, G, non-A through -G)
Non-hepatitis viruses (EBV, parvovirus, CMV, echovirus 3, others)
Pregnancy
Paroxysmal nocturnal hemoglobinuria
Malignancy: large granular lymphocytic leukemia (T-LGL)
EBV, Epstein-Barr virus; CMV, cytomegalovirus.
Clinical Findings
A. Symptoms and Signs
Patients come to medical attention because of the consequences of bone marrow failure. Anemia leads to symptoms of weakness and fatigue, neutropenia causes vulnerability to bacterial or fungal infections, and thrombocytopenia results in mucosal and skin bleeding. Physical examination may reveal signs of pallor, purpura, and petechiae. Other abnormalities such as hepatosplenomegaly, lymphadenopathy, or bone tenderness should not be present, and their presence should lead to questioning the diagnosis.
B. Laboratory Findings
The hallmark of aplastic anemia is pancytopenia. However, early in the evolution of aplastic anemia, only one or two cell lines may be reduced.
Anemia may be severe and is always associated with reticulocytopenia. Red blood cell morphology is unremarkable, but there may be mild macrocytosis (increased MCV). Neutrophils and platelets are reduced in number, and no immature or abnormal forms are seen on the blood smear. The bone marrow aspirate and the bone marrow biopsy appear hypocellular, with only scant amounts of morphologically normal hematopoietic progenitors. The prior dictum that the bone marrow karyotype should be normal (or germline if normal variant) has evolved and some clonal abnormalities or other genetic aberrations may be present even in the setting of idiopathic aplastic anemia.
Differential Diagnosis
Aplastic anemia must be differentiated from other causes of pancytopenia (Table 13–11). Hypocellular forms of myelodysplasia or acute leukemia may occasionally be confused with aplastic anemia. These are differentiated by the presence of cellular morphologic abnormalities, increased percentage of blasts, or abnormal karyotype in bone marrow cells typical of MDS or acute leukemia. Hairy cell leukemia has been misdiagnosed as aplastic anemia and should be recognized by the presence of splenomegaly and by abnormal “hairy” lymphoid cells in a hypocellular bone marrow biopsy. Pancytopenia with a normocellular bone marrow may be due to systemic lupus erythematosus, disseminated infection, hypersplenism, nutritional (eg, vitamin B12 or folate) deficiency, or myelodysplasia. Isolated thrombocytopenia may occur early as aplastic anemia develops and may be confused with immune thrombocytopenia.
Table 13–11. Causes of pancytopenia.
Primary bone marrow disorders
Aplastic anemia
Myelodysplasia
Acute leukemia
Chronic idiopathic myelofibrosis
Infiltrative disease: lymphoma, myeloma, carcinoma, hairy cell leukemia, etc
Non–primary bone marrow disorders
Hypersplenism (with or without portal hypertension)
Systemic lupus erythematosus
Infection: tuberculosis, HIV, leishmaniasis, brucellosis, CMV, parvovirus B19
Nutritional deficiency (megaloblastic anemia)
Medications
Cytotoxic chemotherapy
Ionizing radiation
CMV, cytomegalovirus.
Treatment
Mild cases of aplastic anemia may be treated with supportive care, including erythropoietic (epoetin or darbepoetin) or myeloid (filgrastim or sargramostim) growth factors, or both. Red blood cell transfusions and platelet transfusions are given as necessary, and antibiotics are used to treat infections.
Severe aplastic anemia is defined by a neutrophil count of less than 500/mcL, platelets less than 20,000/mcL, reticulocytes less than 1%, and bone marrow cellularity less than 20%. The treatment of choice for young adults (under age 40 years) who have an HLA-matched sibling is allogeneic bone marrow transplantation. Children or young adults may also benefit from allogeneic bone marrow transplantation using an unrelated donor. Because of the increased risks associated with unrelated donor allogeneic bone marrow transplantation compared to sibling donors, this treatment is usually reserved for patients who have not responded to immunosuppressive therapy.
For adults over age 40 years or those without HLA-matched hematopoietic stem cell donors, the treatment of choice for severe aplastic anemia is immunosuppression with equine antithymocyte globulin (ATG) plus cyclosporine. Equine ATG is given in the hospital in conjunction with transfusion and antibiotic support. A proven regimen is equine ATG 40 mg/kg/day intravenously for 4 days in combination with cyclosporine, 6 mg/kg orally twice daily. Equine ATG is superior to rabbit ATG, resulting in a higher response rate and better survival. Eltrombopag, a thrombopoietin mimetic, is now being added to ATG plus cyclosporine with tri-lineage hematologic responses as high as 90%. ATG should be used in combination with corticosteroids (prednisone or methylprednisolone 1–2 mg/kg/day orally for 1 week, followed by a taper over 2 weeks) to avoid ATG infusion reactions and serum sickness. Responses usually occur in 1–3 months and are usually only partial, but the blood counts rise high enough to give patients a safe and transfusion-free life. The full benefit of immunosuppression is generally assessed at 4 months post-equine ATG. Cyclosporine and eltrombopag are maintained at full doses for 6 months and then stopped in responding patients. Androgens (such as fluoxymesterone 10–20 mg/day orally in divided doses) have been widely used in the past, with a low response rate, and may be considered in mild cases.
Course & Prognosis
Patients with severe aplastic anemia have a rapidly fatal illness if left untreated. Allogeneic bone marrow transplant from an HLA-matched sibling donor produces survival rates of over 80% in recipients under 20 years old and of about 65–70% in those 20 to 50 years old. Respective survival rates drop 10–15% when the donor is HLA-matched but unrelated. Equine ATG-cyclosporine immunosuppressive treatment leads to a response in approximately 70% of patients (including those with hepatitis virus–associated aplastic anemia) and in up to 90% of patients with the addition of eltrombopag. Up to one-third of patients will relapse with aplastic anemia after ATG-based therapy. Clonal hematologic disorders, such as PNH, AML, or myelodysplasia, may develop in one-quarter of patients treated with immunosuppressive therapy after 10 years of follow-up. Factors that predict response to ATG-cyclosporine therapy are patient’s age, reticulocyte count, lymphocyte count, and age-adjusted telomere length of leukocytes at the time of diagnosis.
When to Refer
All patients should be referred to a hematologist.
When to Admit
Admission is necessary for treatment of neutropenic infection, the administration of ATG, or allogeneic bone marrow transplantation.
Bacigalupo A. How I treat acquired aplastic anemia. Blood. 2017 Mar 16;129(11):1428–36. [PMID: 28096088]
Clucas DB et al; Australian Aplastic Anaemia Registry Steering Committee. Revisiting acquired aplastic anaemia: current concepts in diagnosis and management. Intern Med J. 2019 Feb;49(2):152–9. [PMID: 30324755]
Kumar R et al. Hematopoietic cell transplantation for aplastic anemia. Curr Opin Hematol. 2017 Nov;24(6):509–14. [PMID: 28877042]
Marsh JCW et al. The case for upfront HLA-matched unrelated donor hematopoietic stem cell transplantation as a curative option for adult acquired severe aplastic anemia. Biol Blood Marrow Transplant. 2019 Sep;25(9):e277–84. [PMID: 31129354]
Shallis RM et al. Aplastic anemia: etiology, molecular pathogenesis, and emerging concepts. Eur J Haematol. 2018 Dec;101(6):711–20. [PMID: 30055055]
Townsley DM et al. Eltrombopag added to standard immunosuppression for aplastic anemia. N Engl J Med. 2017 Apr 20;376(16):1540–50. [PMID: 28423296]
NEUTROPENIA
ESSENTIALS OF DIAGNOSIS
Neutrophils less than 1800/mcL (1.8 × 109/L).
Severe neutropenia if neutrophils below 500/mcL (0.5 × 109/L).
General Considerations
Neutropenia is present when the absolute neutrophil count is less than 1800/mcL (1.8 × 109/L), although blacks, Asians, and other specific ethnic groups may have normal neutrophil counts as low as 1200/mcL (1.2 × 109/L) or even less. The neutropenic patient is increasingly vulnerable to infection by gram-positive and gram-negative bacteria and by fungi. The risk of infection is related to the severity of neutropenia. The risk of serious infection rises sharply with neutrophil counts below 500/mcL (0.5 × 109/L), and a high risk of infection within days occurs with neutrophil counts below 100/mcL (0.1 × 109/L) (“profound neutropenia”). The classification of neutropenic syndromes is unsatisfactory as the pathophysiology and natural history of different syndromes overlap. Patients with “chronic benign neutropenia” are free of infection despite very low stable neutrophil counts; they seem to physiologically respond adequately to infections and inflammatory stimuli with an appropriate neutrophil release from the bone marrow. In contrast, the neutrophil count of patients with cyclic neutropenia periodically oscillates (usually in 21-day cycles) between normal and low, with infections occurring during the nadirs. Congenital neutropenia is lifelong neutropenia punctuated with infection.
A variety of bone marrow disorders and nonmarrow conditions may cause neutropenia (Table 13–12). All of the causes of aplastic anemia (Table 13–10) and pancytopenia (Table 13–11) may cause neutropenia. The new onset of an isolated neutropenia is most often due to an idiosyncratic reaction to a medication, and agranulocytosis (complete absence of neutrophils in the peripheral blood) is almost always due to a drug reaction. In these cases, examination of the bone marrow shows an almost complete absence of granulocyte precursors with other cell lines undisturbed. Neutropenia in the presence of a normal bone marrow may be due to immunologic peripheral destruction (autoimmune neutropenia), sepsis, or hypersplenism. The presence in the serum of antineutrophil antibodies supports the diagnosis of autoimmune neutropenia but does not prove this as the pathophysiologic reason for neutropenia. Felty syndrome is an immune neutropenia associated with seropositive nodular rheumatoid arthritis and splenomegaly. Severe neutropenia may be associated with clonal disorders of T lymphocytes, often with the morphology of large granular lymphocytes, referred to as CD3-positive T-cell large granular lymphoproliferative disorder. Isolated neutropenia is an uncommon presentation of hairy cell leukemia or MDS. By its nature, myelosuppressive cytotoxic chemotherapy causes neutropenia in a predictable manner.
Table 13–12. Causes of neutropenia.
Bone marrow disorders
Congenital
Dyskeratosis congenita
Fanconi anemia
Cyclic neutropenia
Congenital neutropenia
Hairy cell leukemia
Large granular lymphoproliferative disorder
Myelodysplasia
Non–bone marrow disorders
Medications: antiretroviral medications, cephalosporins, chlorpromazine, chlorpropamide, cimetidine, methimazole, myelosuppressive cytotoxic chemotherapy, penicillin, phenytoin, procainamide, rituximab, sulfonamides
Aplastic anemia
Benign chronic neutropenia
Pure white cell aplasia
Hypersplenism
Sepsis
Other immune
Autoimmune (idiopathic)
Felty syndrome
Systemic lupus erythematosus
HIV infection
Clinical Findings
Neutropenia results in stomatitis and in infections due to gram-positive or gram-negative aerobic bacteria or to fungi such as Candida or Aspergillus. The most common infectious syndromes are septicemia, cellulitis, pneumonia, and neutropenic fever of unknown origin. Fever in neutropenic patients should always be initially assumed to be of infectious origin until proven otherwise (Chapter 30).
Treatment
Treatment of neutropenia depends on its cause. Potential causative medications should be discontinued. Myeloid growth factors (filgrastim or sargramostim or biosimilar myeloid growth factors) help facilitate neutrophil recovery after offending medications are stopped. Chronic myeloid growth factor administration (daily or every other day) is effective at dampening the neutropenia seen in cyclic or congenital neutropenia. When Felty syndrome leads to repeated bacterial infections, splenectomy has been the treatment of choice, but sustained use of myeloid growth factors is effective and provides a nonsurgical alternative. Patients with autoimmune neutropenia often respond briefly to immunosuppression with corticosteroids and are best managed with intermittent doses of myeloid growth factors. The neutropenia associated with large granular lymphoproliferative disorder may respond to therapy with oral methotrexate, cyclophosphamide, or cyclosporine.
Fevers during neutropenia should be considered as infectious until proven otherwise. Febrile neutropenia is a life-threatening circumstance. Enteric gram-negative bacteria are of primary concern and often empirically treated with fluoroquinolones or third- or fourth-generation cephalosporins (see Infections in the Immunocompromised Patient, Chapter 30). For protracted neutropenia, fungal infections are problematic and empiric coverage with azoles (fluconazole for yeast and voriconazole, itraconazole, posaconazole, or isavuconazole for molds) or echinocandins is recommended. The neutropenia following myelosuppressive chemotherapy is predictable and is partially ameliorated by the use of myeloid growth factors. For patients with acute leukemia undergoing intense chemotherapy or patients with solid cancer undergoing high-dose chemotherapy, the prophylactic use of antimicrobial agents and myeloid growth factors is recommended.
When to Refer
Refer to a hematologist if neutrophils are persistently and unexplainably less than 1000/mcL (1.0 × 109/L).
When to Admit
Neutropenia by itself is not an indication for hospitalization. However, many patients with severe neutropenia have a serious underlying disease that may require inpatient treatment. Most patients with febrile neutropenia require hospitalization to treat infection.
Abdel-Azim H et al. Strategies to generate functionally normal neutrophils to reduce infection and infection-related mortality in cancer chemotherapy. Pharmacol Ther. 2019 Dec;204:107403. [PMID: 31470030]
Barcellini W et al. Autoimmune hemolytic anemia, autoimmune neutropenia and aplastic anemia in the elderly. Eur J Intern Med. 2018 Dec;58:77–83. [PMID: 30527923]
Dale DC et al. An update on the diagnosis and treatment of chronic idiopathic neutropenia. Curr Opin Hematol. 2017 Jan;24(1):46–53. [PMID: 27841775]
LEUKEMIAS & OTHER MYELOPROLIFERATIVE NEOPLASMS
Myeloproliferative disorders are due to acquired clonal abnormalities of the hematopoietic stem cell. Since the stem cell gives rise to myeloid, erythroid, and platelet cells, qualitative and quantitative changes are seen in all of these cell lines. Classically, the myeloproliferative disorders produce characteristic syndromes with well-defined clinical and laboratory features (Tables 13–13 and 13–14). However, these disorders are grouped together because they may evolve from one into another and because hybrid disorders are commonly seen. All of the myeloproliferative disorders may progress to AML.
Table 13–13. World Health Organization classification of myeloproliferative disorders (modified).
Myeloproliferative neoplasms
Chronic myeloid leukemia, BCR-ABL1–positive
Chronic neutrophilic leukemia
Polycythemia vera
Primary myelofibrosis (PMF)
Essential thrombocythemia
Chronic eosinophilic leukemia, not otherwise specified (NOS)
Myeloproliferative neoplasm, unclassifiable
Mastocytosis
Myelodysplastic/myeloproliferative neoplasms (MDS/MPN)
Myelodysplastic syndromes
Acute myeloid leukemia and related neoplasms
Acute myeloid leukemia with recurrent genetic abnormalities
Acute myeloid leukemia with myelodysplasia-related changes
Therapy-related myeloid neoplasms
Acute myeloid leukemia, NOS
Myeloid sarcoma
Myeloid proliferations related to Down syndrome
Acute leukemias of ambiguous lineage
B lymphoblastic leukemia/lymphoma
T lymphoblastic leukemia/lymphoma
Table 13–14. Laboratory features of myeloproliferative neoplasms.
The Philadelphia chromosome seen in chronic myeloid leukemia (CML) was the first recurrent cytogenetic abnormality to be described in a human malignancy. Since that time, there has been tremendous progress in elucidating the genetic nature of these disorders, with identification of mutations in JAK2, MPL, CALR, CSF3R, and other genes.
Masarova L et al. The rationale for immunotherapy in myeloproliferative neoplasms. Curr Hematol Malig Rep. 2019 Aug;14(4):310–27. [PMID: 31228096]
Rumi E et al. Diagnosis, risk stratification, and response evaluation in classical myeloproliferative neoplasms. Blood. 2017 Feb 9;129(6):680–92. [PMID: 28028026]
Rumi E et al. Myeloproliferative and lymphoproliferative disorders: state of the art. Hematol Oncol. 2020 Apr;38(2):121–8. [Epub ahead of print] [PMID: 31833567]
POLYCYTHEMIA VERA
ESSENTIALS OF DIAGNOSIS
JAK2 (V617F) mutation.
Splenomegaly.
Normal arterial oxygen saturation.
Usually elevated white blood count and platelet count.
General Considerations
Polycythemia vera is an acquired myeloproliferative disorder that causes overproduction of all three hematopoietic cell lines, most prominently the red blood cells. Erythroid production is independent of erythropoietin, and the serum erythropoietin level is low. True erythrocytosis, with an elevated red blood cell mass, should be distinguished from spurious erythrocytosis caused by a constricted plasma volume.
A mutation in exon 14 of JAK2 (V617F), a signaling molecule, has been demonstrated in 95% of cases. Additional JAK2 mutations have been identified (exon 12) and suggest that JAK2 is involved in the pathogenesis of this disease and is a potential therapeutic target.
Clinical Findings
A. Symptoms and Signs
Headache, dizziness, tinnitus, blurred vision, and fatigue are common complaints related to expanded blood volume and increased blood viscosity. Generalized pruritus, especially following a warm shower or bath, is related to histamine release from the basophilia. Epistaxis is probably related to engorgement of mucosal blood vessels in combination with abnormal hemostasis. Sixty percent of patients are men, and the median age at presentation is 60 years. Polycythemia rarely occurs in persons under age 40 years.
Physical examination reveals plethora and engorged retinal veins. The spleen is palpable in 75% of cases but is nearly always enlarged when imaged. Thrombosis is the most common complication of polycythemia vera and the major cause of morbidity and death in this disorder. Thrombosis appears to be related both to increased blood viscosity and abnormal platelet function. Uncontrolled polycythemia leads to a very high incidence of thrombotic complications of surgery, and elective surgery should be deferred until the condition has been treated. Paradoxically, in addition to thrombosis, increased bleeding can occur. There is also a high incidence of peptic ulcer disease.
B. Laboratory Findings
According to the WHO 2016 criteria, the hallmark of polycythemia vera is a hematocrit (at sea level) that exceeds 49% in males or 48% in females. Red blood cell morphology is normal (Table 13–14). The white blood count is usually elevated to 10,000–20,000/mcL and the platelet count is variably increased, sometimes to counts exceeding 1,000,000/mcL. Platelet morphology is usually normal. White blood cells are usually normal, but basophilia and eosinophilia are frequently present. Erythropoietin is suppressed and serum levels, usually low. The diagnosis should be confirmed with JAK2 mutation screening. The absence of a mutation in either exon 14 (most common) or 12 should lead the clinician to question the diagnosis.
The bone marrow is hypercellular, with panhyperplasia of all hematopoietic elements, but bone marrow examination is not necessary to establish the diagnosis. Iron stores are usually absent from the bone marrow, having been transferred to the increased circulating red blood cell mass. Iron deficiency may also result from chronic gastrointestinal blood loss. Bleeding may lower the hematocrit to the normal range (or lower), creating diagnostic confusion, and may lead to a situation with significant microcytosis yet a normal hematocrit.
Vitamin B12 levels are strikingly elevated because of increased levels of transcobalamin III (secreted by white blood cells). Overproduction of uric acid may lead to hyperuricemia.
Although red blood cell morphology is usually normal at presentation, microcytosis, hypochromia, and poikilocytosis may result from iron deficiency following treatment by phlebotomy. Progressive hypersplenism may also lead to elliptocytosis.
Differential Diagnosis
Spurious polycythemia, in which an elevated hematocrit is due to contracted plasma volume rather than increased red cell mass, may be related to diuretic use or may occur without obvious cause.
A secondary cause of polycythemia should be suspected if splenomegaly is absent and the high hematocrit is not accompanied by increases in other cell lines. Secondary causes of polycythemia include hypoxia and smoking; carboxyhemoglobin levels may be elevated in smokers (Table 13–15). A renal CT scan or sonogram may be considered to look for an erythropoietin-secreting cyst or tumor. A positive family history should lead to investigation for a congenital high-oxygen-affinity hemoglobin. An absence of a mutation in JAK2 suggests a different diagnosis. However, JAK2 mutations are also commonly found in other myeloproliferative disorders, essential thrombocytosis, and myelofibrosis.
Table 13–15. Causes of polycythemia.
Spurious polycythemia
Secondary polycythemia
Hypoxia: cardiac disease, pulmonary disease, high altitude
Carboxyhemoglobin: smoking
Erythropoietin-secreting tumors, eg, kidney lesions (rare)
Abnormal hemoglobins (rare)
Polycythemia vera
Polycythemia vera should be differentiated from other myeloproliferative disorders (Table 13–14). Marked elevation of the white blood count (above 30,000/mcL) suggests CML. Abnormal red blood cell morphology and nucleated red blood cells in the peripheral blood are seen in myelofibrosis. Essential thrombocytosis is suggested when the platelet count is strikingly elevated.
Treatment
The treatment of choice is phlebotomy. One unit of blood (approximately 500 mL) is removed weekly until the hematocrit is less than 45%; the hematocrit is maintained at less than 45% by repeated phlebotomy as necessary. Patients for whom phlebotomy is problematic (because of poor venous access or logistical reasons) may be managed primarily with hydroxyurea. Because repeated phlebotomy intentionally produces iron deficiency, the requirement for phlebotomy should gradually decrease. It is important to avoid medicinal iron supplementation, as this can thwart the goals of a phlebotomy program. A diet low in iron is not necessary but will increase the intervals between phlebotomies. Maintaining the hematocrit at normal levels has been shown to decrease the incidence of thrombotic complications.
Occasionally, myelosuppressive therapy is indicated. Indications include a high phlebotomy requirement, thrombocytosis, and intractable pruritus. There is evidence that reduction of the platelet count to less than 600,000/mcL will reduce the risk of thrombotic complications. Hydroxyurea is widely used when myelosuppressive therapy is indicated. The usual dose is 500–1500 mg/day orally, adjusted to keep platelets less than 500,000/mcL without reducing the neutrophil count to less than 2000/mcL. The JAK2 inhibitor ruxolitinib is FDA-approved for patients resistant or intolerant to hydroxyurea. In a randomized study comparing best available therapy to ruxolitinib, treatment with ruxolitinib was associated with greater benefit for both hematocrit control without phlebotomy (60%) and splenic volume reduction (38%). Symptom burden improved by greater than 50% in 49% of patients.
Studies of pegylated alfa-2 interferon have demonstrated considerable efficacy, with hematologic responses in greater than 80%, and molecular responses in 20% (as measured by JAK2 mutations). Patients in whom molecular responses were not achieved had a higher frequency of mutations outside the JAK2 pathway and were more likely to acquire new mutations during therapy. Side effects were generally acceptable and much less significant than with nonpegylated forms of interferon. A randomized phase 3 trial comparing PEG-alpha-2 interferon to hydroxyurea demonstrated similar complete remission rates and modifications of disease features (spleen size, karyotype, and histopathologic abnormalities) but with PEG-alpha-2 interferon causing more grade 3/4 adverse events. Alkylating agents have been shown to increase the risk of conversion of this disease to acute leukemia and should be avoided. Lastly, a new and promising therapeutic strategy is induction of apoptosis via the p53 pathway through pharmacologic inhibition of human double minute 2 (mdm2).
Low-dose aspirin (75–81 mg/day orally) has been shown to reduce the risk of thrombosis without excessive bleeding, and should be part of therapy for all patients without contraindications to aspirin. Allopurinol 300 mg orally daily may be indicated for hyperuricemia. Antihistamine therapy with diphenhydramine or other H1-blockers and, rarely, selective serotonin reuptake inhibitors are used to manage pruritus.
Prognosis
Polycythemia is an indolent disease with median survival of over 15 years. The major cause of morbidity and mortality is arterial thrombosis. Over time, polycythemia vera may convert to myelofibrosis or to CML. In approximately 5% of cases, the disorder progresses to AML, which is usually refractory to therapy.
When to Refer
Patients with polycythemia vera should be referred to a hematologist.
When to Admit
Inpatient care is rarely required.
Barbui T et al. The 2016 revision of WHO classification of myeloproliferative neoplasms: clinical and molecular advances. Blood Rev. 2016 Nov;30(6):453–9. [PMID: 27341755]
Gerds AT. Beyond JAK-STAT: novel therapeutic targets in Ph-negative MPN. Hematology Am Soc Hematol Educ Program. 2019 Dec 6;2019(1):407–14. [PMID: 31808852]
Tefferi A et al. Polycythemia vera and essential thrombocythemia: 2019 update on diagnosis, risk-stratification and management. Am J Hematol. 2019;94:133–43. [PMID: 30281843]
ESSENTIAL THROMBOCYTOSIS
ESSENTIALS OF DIAGNOSIS
Elevated platelet count in absence of other causes.
Normal red blood cell mass.
Absence of bcr/abl gene (Philadelphia chromosome).
General Considerations
Essential thrombocytosis is an uncommon myeloproliferative disorder of unknown cause in which marked proliferation of the megakaryocytes in the bone marrow leads to elevation of the platelet count. As with polycythemia vera, the finding of a high frequency of mutations of JAK2 and others in these patients has advanced the understanding of this disorder.
Clinical Findings
A. Symptoms and Signs
The median age at presentation is 50–60 years, and there is a slightly increased incidence in women. The disorder is often suspected when an elevated platelet count is found. Less frequently, the first sign is thrombosis, which is the most common clinical problem. The risk of thrombosis rises with age. Venous thromboses may occur in unusual sites such as the mesenteric, hepatic, or portal vein. Some patients experience erythromelalgia, painful burning of the hands accompanied by erythema; this symptom is reliably relieved by aspirin. Bleeding, typically mucosal, is less common and is related to a concomitant qualitative platelet defect. Splenomegaly is present in at least 25% of patients.
B. Laboratory Findings
An elevated platelet count is the hallmark of this disorder, and may be over 2,000,000/mcL (2000 × 109/L) (Table 13–14). The white blood cell count is often mildly elevated, usually not above 30,000/mcL (30 × 109/L), but with some immature myeloid forms. The hematocrit is normal. The peripheral blood smear reveals large platelets, but giant degranulated forms seen in myelofibrosis are not observed. Red blood cell morphology is normal.
The bone marrow shows increased numbers of megakaryocytes but no other morphologic abnormalities. The peripheral blood should be tested for the bcr/abl fusion gene (Philadelphia chromosome) since it can differentiate CML, where it is present, from essential thrombocytosis, where it is absent.
Differential Diagnosis
Essential thrombocytosis must be distinguished from secondary causes of an elevated platelet count. In reactive thrombocytosis, the platelet count seldom exceeds 1,000,000/mcL (1000 × 109/L). Inflammatory disorders such as rheumatoid arthritis and ulcerative colitis cause significant elevations of the platelet count, as may chronic infection. The thrombocytosis of iron deficiency is observed only when anemia is significant. The platelet count is temporarily elevated after a splenectomy. JAK2 mutations are found in over 50% of cases. MPL and CALR mutations frequently occur in patients with JAK2-negative essential thrombocytosis.
Regarding other myeloproliferative disorders, the lack of erythrocytosis distinguishes it from polycythemia vera. Unlike myelofibrosis, red blood cell morphology is normal, nucleated red blood cells are absent, and giant degranulated platelets are not seen. In CML, the Philadelphia chromosome (or bcr/abl by molecular testing) establishes the diagnosis.
Treatment
Patients are considered at high risk for thrombosis if they are older than 60 years, have a leukocyte count of 11,000/mcL (11 × 109/L) or higher, or have a previous history of thrombosis. They also have a higher risk for bleeding. The risk of thrombosis can be reduced by control of the platelet count, which should be kept under 500,000/mcL (500 × 109/L). The treatment of choice is oral hydroxyurea in a dose of 500–1000 mg/day. In rare cases in which hydroxyurea is not well tolerated because of anemia, low doses of anagrelide, 1–2 mg/day orally, may be added. Higher doses of anagrelide can be complicated by headache, peripheral edema, and heart failure. Pegylated interferon alfa-2 can induce significant hematologic responses and can potentially target the malignant clone in CALR-mutant cases. Strict control of coexistent cardiovascular risk factors is mandatory for all patients.
Vasomotor symptoms such as erythromelalgia and paresthesias respond rapidly to aspirin, and its long-term low-dose use (81 mg/day orally) may reduce the risk of thrombotic complications in low-risk patients. In the unusual event of severe bleeding, the platelet count can be lowered rapidly with plateletpheresis. In cases of marked thrombocytosis (greater than or equal to 1,000,000/mcL [1000 × 109/L]) or of any evidence of bleeding, acquired von Willebrand syndrome must be excluded before starting low-dose aspirin.
Course & Prognosis
Essential thrombocytosis is an indolent disorder that allows long-term survival. Average survival is longer than 15 years from diagnosis, and the survival of patients younger than age 50 years does not appear different from matched controls. The major source of morbidity—thrombosis—can be reduced by appropriate platelet control. Late in the disease course, the bone marrow may become fibrotic, and massive splenomegaly may occur, sometimes with splenic infarction. There is a 10–15% risk of progression to myelofibrosis after 15 years, and a 1–5% risk of transformation to acute leukemia over 20 years.
When to Refer
Patients with essential thrombocytosis should be referred to a hematologist.
Bose P et al. Updates in the management of polycythemia vera and essential thrombocythemia. Ther Adv Hematol. 2019 Aug 30;10:2040620719870052. [PMID: 31516686]
Sankar K et al. Thrombosis in the Philadelphia chromosome-negative myeloproliferative neoplasms. Cancer Treat Res. 2019;179:159–78. [PMID: 31317487]
PRIMARY MYELOFIBROSIS
ESSENTIALS OF DIAGNOSIS
Striking splenomegaly.
Teardrop poikilocytosis on peripheral smear.
Leukoerythroblastic blood picture; giant abnormal platelets.
Initially hypercellular, then hypocellular bone marrow with reticulin or collagen fibrosis.
General Considerations
Primary myelofibrosis is a myeloproliferative disorder characterized by clonal hematopoiesis that is often but not always accompanied by JAK2, CALR, or MPL mutations; bone marrow fibrosis; anemia; splenomegaly; and a leukoerythroblastic peripheral blood picture with teardrop poikilocytosis. Myelofibrosis can also occur as a secondary process following the other myeloproliferative disorders (eg, polycythemia vera, essential thrombocytosis). It is believed that fibrosis occurs in response to increased secretion of platelet-derived growth factor (PDGF) and possibly other cytokines. In response to bone marrow fibrosis, extramedullary hematopoiesis takes place in the liver, spleen, and lymph nodes. In these sites, mesenchymal cells responsible for fetal hematopoiesis can be reactivated. According to the 2016 WHO classification, “prefibrotic” primary myelofibrosis is distinguished from “overtly fibrotic” primary myelofibrosis; the former might mimic essential thrombocytosis in its presentation and it is prognostically relevant to distinguish the two.
Clinical Findings
A. Symptoms and Signs
Primary myelofibrosis develops in adults over age 50 years and is usually insidious in onset. Patients most commonly present with fatigue due to anemia or abdominal fullness related to splenomegaly. Uncommon presentations include bleeding and bone pain. On examination, splenomegaly is almost invariably present and is commonly massive. The liver is enlarged in more than 50% of cases.
Later in the course of the disease, progressive bone marrow failure takes place as it becomes increasingly more fibrotic. Progressive thrombocytopenia leads to bleeding. The spleen continues to enlarge, which leads to early satiety. Painful episodes of splenic infarction may occur. The patient becomes cachectic and may experience severe bone pain, especially in the upper legs. Hematopoiesis in the liver leads to portal hypertension with ascites, esophageal varices, and occasionally transverse myelitis caused by myelopoiesis in the epidural space.
B. Laboratory Findings
Patients are almost invariably anemic at presentation. The white blood count is variable—either low, normal, or elevated—and may be increased to 50,000/mcL (50 × 109/L). The platelet count is variable. The peripheral blood smear is dramatic, with significant poikilocytosis and numerous teardrop forms in the red cell line. Nucleated red blood cells are present and the myeloid series is shifted, with immature forms including a small percentage of promyelocytes or myeloblasts. Platelet morphology may be bizarre, and giant degranulated platelet forms (megakaryocyte fragments) may be seen. The triad of teardrop poikilocytosis, leukoerythroblastic blood, and giant abnormal platelets is highly suggestive of myelofibrosis.
The bone marrow usually cannot be aspirated (dry tap), though early in the course of the disease it is hypercellular, with a marked increase in megakaryocytes. Fibrosis at this stage is detected by a silver stain demonstrating increased reticulin fibers. Later, biopsy reveals more severe fibrosis, with eventual replacement of hematopoietic precursors by collagen. There is no characteristic chromosomal abnormality. JAK2 is mutated in ∼65% of cases, and MPL and CALR are mutated in the majority of the remaining cases; 10% of cases are “triple-negative.”
Differential Diagnosis
A leukoerythroblastic blood picture from other causes may be seen in response to severe infection, inflammation, or infiltrative bone marrow processes. However, teardrop poikilocytosis and giant abnormal platelet forms will not be present. Bone marrow fibrosis may be seen in metastatic carcinoma, Hodgkin lymphoma, and hairy cell leukemia. These disorders are diagnosed by characteristic morphology of involved tissues.
Of the other myeloproliferative disorders, CML is diagnosed when there is marked leukocytosis, normal red blood cell morphology, and the presence of the bcr/abl fusion gene. Polycythemia vera is characterized by an elevated hematocrit. Essential thrombocytosis shows predominant platelet count elevations.
Treatment
Observation with supportive care is a reasonable treatment strategy for asymptomatic patients with low or intermediate-1 Dynamic International Prognostic Scoring system (DIPSS)-plus risk disease, especially in the absence of high-risk mutations. Anemic patients are supported with transfusion. Anemia can also be controlled with androgens, prednisone, thalidomide, or lenalidomide. First-line therapy for myelofibrosis-associated splenomegaly is hydroxyurea 500–1000 mg/day orally, which is effective in reducing spleen size by half in approximately 40% of patients. Both thalidomide and lenalidomide may improve splenomegaly and thrombocytopenia in some patients. Splenectomy is not routinely performed but is indicated for medication-refractory splenic enlargement causing recurrent painful episodes, severe thrombocytopenia, or an unacceptable transfusion requirement. Perioperative complications can occur in 28% of patients and include infections, abdominal vein thrombosis, and bleeding. Radiation therapy has a role for painful sites of extramedullary hematopoiesis, pulmonary hypertension, or severe bone pain. Transjugular intrahepatic portosystemic shunt might also be considered to alleviate symptoms of portal hypertension.
Patients with DIPSS-plus high or intermediate-2 risk disease, or those patients harboring high-risk mutations such as ASXL1 or SRSF2, should be considered for allogeneic stem cell transplant, which is currently the only potentially curative treatment modality in this disease. Nontransplant candidates may be treated with JAK2 inhibitors or immunomodulatory agents for symptom control. Ruxolitinib, the first JAK2 inhibitor to be FDA approved, results in reduction of spleen size and improvement of constitutional symptoms but does not induce complete clinical or cytogenetic remissions or significantly affect the JAK2/CALR/MPL mutant allele burden. Moreover, ruxolitinib can exacerbate cytopenias. The newer selective JAK2 inhibitor fedratinib was approved in 2019. It can lead to sustained reduction in spleen size and improvement in disease-associated symptoms for patients with advanced stage myelofibrosis. However, it carries a significant risk of serious and fatal encephalopathy, including Wernicke encephalopathy, and providers should regularly assess thiamine levels in all patients. The immunomodulatory medications lenalidomide and pomalidomide result in control of anemia in 25% and thrombocytopenia in ∼58% of cases, without significant reduction in splenic size.
Course & Prognosis
The median survival from time of diagnosis is approximately 5 years. Therapies with biologic agents and the application of reduced-intensity allogeneic stem cell transplantation appear to offer the possibility of improving the outcome for many patients. End-stage myelofibrosis is characterized by generalized asthenia, liver failure, and bleeding from thrombocytopenia, with some cases terminating in AML. The DIPSS-plus incorporates clinical and genetic risk variables and is associated with overall survival. Most recently, DIPSS-plus-independent adverse prognostic relevance has been demonstrated for certain mutations including ASXL1 and SRSF2, whereas patients with type 1/like CALR mutations, compared to their counterparts with other driver mutations, displayed significantly better survival.
When to Refer
Patients in whom myelofibrosis is suspected should be referred to a hematologist.
When to Admit
Admission is not usually necessary.
Finazzi G et al. Prefibrotic myelofibrosis: treatment algorithm 2018. Blood Cancer J. 2018 Nov 7;8(11):104. [PMID: 30405096]
Zimran E et al. Novel treatments to tackle myelofibrosis. Expert Rev Hematol. 2018 Nov;11(11):889–902. [PMID: 30324817]
CHRONIC MYELOID LEUKEMIA
ESSENTIALS OF DIAGNOSIS
Elevated white blood cell count.
Markedly left-shifted myeloid series but with a low percentage of promyelocytes and blasts.
Presence of bcr/abl gene (Philadelphia chromosome).
General Considerations
CML is a myeloproliferative disorder characterized by overproduction of myeloid cells. These myeloid cells continue to differentiate and circulate in increased numbers in the peripheral blood.
CML is characterized by a specific chromosomal abnormality and specific molecular abnormality. The Philadelphia chromosome is a reciprocal translocation between the long arms of chromosomes 9 and 22. The portion of 9q that is translocated contains abl, a protooncogene that is received at a specific site on 22q, the break point cluster (bcr). The fusion gene bcr/abl produces a novel protein that possesses tyrosine kinase activity. This disorder is the first recognized example of tyrosine kinase “addiction” by cancer cells.
Early CML (“chronic phase”) does not behave like a malignant disease. Normal bone marrow function is retained, white blood cells differentiate and, despite some qualitative abnormalities, the neutrophils combat infection normally. However, untreated CML is inherently unstable, and without treatment the disease progresses to an accelerated and then acute blast phase, which is morphologically indistinguishable from acute leukemia.
Clinical Findings
A. Symptoms and Signs
CML is a disorder of middle age (median age at presentation is 55 years). Patients usually complain of fatigue, night sweats, and low-grade fevers related to the hypermetabolic state caused by overproduction of white blood cells. Patients may also complain of abdominal fullness related to splenomegaly. In some cases, an elevated white blood count is discovered incidentally. Rarely, the patient will present with a clinical syndrome related to leukostasis with blurred vision, respiratory distress, or priapism. The white blood count in these cases is usually greater than 100,000/mcL (100 × 109/L) but less than 500,000/mcL (500 × 109/L). On examination, the spleen is enlarged (often markedly so), and sternal tenderness may be present as a sign of marrow overexpansion. In cases discovered during routine laboratory monitoring, these findings are often absent. Acceleration of the disease is often associated with fever (in the absence of infection), bone pain, and splenomegaly.
B. Laboratory Findings
CML is characterized by an elevated white blood cell count; the median white blood count at diagnosis is 150,000/mcL (150 × 109/L), although in some cases the white blood cell count is only modestly increased (Table 13–14). The peripheral blood is characteristic. The myeloid series is left shifted, with mature forms dominating and with cells usually present in proportion to their degree of maturation. Blasts are usually less than 5%. Basophilia and eosinophilia may be present. At presentation, the patient is usually not anemic. Red blood cell morphology is normal, and nucleated red blood cells are rarely seen. The platelet count may be normal or elevated (sometimes to strikingly high levels). A bone marrow biopsy is essential to ensure sufficient material for a complete karyotype and for morphologic evaluation to confirm the phase of disease. The bone marrow is hypercellular, with left-shifted myelopoiesis. Myeloblasts compose less than 5% of marrow cells. The hallmark of the disease is the bcr/abl gene that is detected by the polymerase chain reaction (PCR) test in the peripheral blood and bone marrow.
With progression to the accelerated and blast phases, progressive anemia and thrombocytopenia occur, and the percentage of blasts in the blood and bone marrow increases. Blast-phase CML is diagnosed when blasts comprise more than 20% of bone marrow cells.
Differential Diagnosis
Early CML must be differentiated from the reactive leukocytosis associated with infection. In such cases, the white blood count is usually less than 50,000/mcL (50 × 109/L), splenomegaly is absent, and the bcr/abl gene is not present.
CML must be distinguished from other myeloproliferative disease (Table 13–14). The hematocrit should not be elevated, the red blood cell morphology is normal, and nucleated red blood cells are rare or absent. Definitive diagnosis is made by finding the bcr/abl gene.
Treatment
Treatment is usually not emergent even with white blood counts over 200,000/mcL (200 × 109/L), since the majority of circulating cells are mature myeloid cells that are smaller and more deformable than primitive leukemic blasts. In the rare instances in which symptoms result from extreme hyperleukocytosis (priapism, respiratory distress, visual blurring, altered mental status), emergent leukapheresis is performed in conjunction with myelosuppressive therapy.
In chronic-phase CML, the goal of therapy is normalization of the hematologic abnormalities and suppression of the malignant bcr/abl-expressing clone. The treatment of choice consists of a tyrosine kinase inhibitor (eg, imatinib, nilotinib, dasatinib) targeting the aberrantly active abl kinase. It is expected that a hematologic complete remission, with normalization of blood counts and splenomegaly will occur within 3 months of treatment initiation. Second, a major cytogenetic response should be achieved, ideally within 3 months but certainly within 6 months. A major cytogenetic response is identified when less than 35% of metaphases contain the Philadelphia chromosome. Lastly, a major molecular response is desired within 12 months and is defined as a 3-log reduction of the bcr/abl transcript as measured by quantitative PCR. This roughly corresponds to a bcr/abl ratio (compared to abl) of less than 0.01. Patients who achieve this level of molecular response have an excellent prognosis, with 100% of them remaining progression-free at 8 years. On the other hand, patients have a worse prognosis if these targets are not achieved, cytogenetic or molecular response is subsequently lost, or new mutations or cytogenetic abnormalities develop.
Imatinib mesylate was the first tyrosine kinase inhibitor to be approved and it results in nearly universal (98%) hematologic control of chronic-phase disease at a dose of 400 mg/day. The rate of a major molecular response with imatinib in chronic-phase disease is ∼30% at 1 year. The second-generation tyrosine kinase inhibitors, nilotinib and dasatinib, are also used as front-line therapy and can significantly increase the rate of a major molecular response compared to imatinib (71% for nilotinib at 300–400 mg twice daily by 2 years, 64% for dasatinib at 100 mg/day by 2 years) and result in a lower rate of progression to advanced-stage disease. However, these agents can also salvage 90% of patients who do not respond to treatment with imatinib and may therefore be reserved for use in that setting. A dual bcr/abl tyrosine kinase inhibitor, bosutinib, is used for patients who are resistant or intolerant to the other tyrosine kinase inhibitors. The complete cytogenetic response rate to bosutinib is 25%, but it is not active against the T315I mutation.
Patients taking tyrosine kinase inhibitors should be monitored with a quantitative PCR assay. Those with a consistent increase in bcr/abl transcript or those with a suboptimal molecular response as defined above should undergo abl mutation testing and then be switched to an alternative tyrosine kinase inhibitor. The T315I mutation in abl is specifically resistant to therapy with imatinib, dasatinib, nilotinib, and bosutinib but appears to be sensitive to the third-generation agent ponatinib. However, ponatinib is associated with a high rate of vascular thrombotic complications. For patients with the T315I mutation as well as patients who have not responded to multiple tyrosine kinase inhibitors, including ponatinib, the novel allosteric inhibitor asciminib can be tried. It has shown a 54% complete hematologic response rate and a 48% sustained major molecular response in heavily pretreated patients. Dose-limiting toxic effects include asymptomatic elevations in the lipase level and clinical pancreatitis. Lastly, omacetaxine—a non–tyrosine kinase inhibitor therapy approved for patients with CML who are resistant to at least two tyrosine kinase inhibitors—can produce major cytogenetic responses in 18% of patients. Patients in whom a good molecular response to any of these agents cannot be achieved or in whom disease progresses despite therapy should be considered for allogeneic stem cell transplantation.
Patients with advanced-stage disease (accelerated phase or myeloid/lymphoid blast crisis) should be treated with a tyrosine kinase inhibitor alone or in combination with myelosuppressive chemotherapy. The doses of tyrosine kinase inhibitors in that setting are usually higher than those appropriate for chronic-phase disease. Since the duration of response to tyrosine kinase inhibitors in this setting is limited, patients who have accelerated or blast- phase disease should ultimately be considered for allogeneic stem cell transplantation.
Course & Prognosis
Since the introduction of imatinib therapy in 2001, and with the development of molecular-targeted agents, more than 80% of patients remain alive and without disease progression at 9 years. Patients with good molecular responses to tyrosine kinase inhibitor therapy have an excellent prognosis, with essentially 100% survival at 9 years, and it is likely that some fraction of these patients will be cured. Current studies suggest that tyrosine kinase inhibitor therapy may be safely discontinued after 2 years in patients who achieve a sustained major molecular response, with ∼50–60% of patients remaining in molecular remission at least 1 year.
When to Refer
All patients with CML should be referred to a hematologist.
When to Admit
Hospitalization is rarely necessary and should be reserved for symptoms of leukostasis at diagnosis or for transformation to acute leukemia.
Craddock CF. We do still transplant CML, don’t we? Hematology Am Soc Hematol Educ Program. 2018 Nov 30;2018(1):177–84. [PMID: 30504307]
Molica M et al. Insights into the optimal use of ponatinib in patients with chronic phase chronic myeloid leukaemia. Ther Adv Hematol. 2019 Mar 1;10:2040620719826444. [PMID: 30854182]
Westerweel PE et al. New approaches and treatment combinations for the management of chronic myeloid leukemia. Front Oncol. 2019 Aug 6;9:665. [PMID: 31448223]
MYELODYSPLASTIC SYNDROMES
ESSENTIALS OF DIAGNOSIS
Cytopenias with a hypercellular bone marrow.
Morphologic abnormalities in one or more hematopoietic cell lines.
General Considerations
The myelodysplastic syndromes are a group of acquired clonal disorders of the hematopoietic stem cell. They are characterized by the constellation of cytopenias, a usually hypercellular marrow, morphologic dysplasia, and genetic abnormalities. The disorders are usually idiopathic but may be caused by prior exposure to cytotoxic chemotherapy, radiation or both. In addition to cytogenetics, sequencing can detect genetic mutations in 80–90% of MDS patients. Importantly, acquired clonal mutations identical to those seen in MDS can occur in the hematopoietic cells of ∼10% of apparently healthy older individuals, defining the disorder of clonal hematopoiesis of indeterminate potential (CHIP).
Myelodysplasia encompasses several heterogeneous syndromes. A key distinction is whether there is an increase in bone marrow blasts (greater than 5% of marrow elements). The category of MDS with excess blasts represents a more aggressive form of the disease, often leading to AML. Those without excess blasts are characterized by the degree of dysplasia, eg, MDS with single lineage dysplasia and MDS with multilineage dysplasia. The morphologic finding of ringed sideroblasts is used to define a subcategory of the lower-risk MDS syndromes. Patients with isolated 5q loss, which is characterized by the cytogenetic finding of loss of part of the long arm of chromosome 5, comprise an important subgroup of patients with a different natural history. Lastly, a proliferative syndrome including sustained peripheral blood monocytosis more than 1000/mcL (1.0 × 109/L) is termed chronic myelomonocytic leukemia (CMML), a disorder that shares features of myelodysplastic and myeloproliferative disorders. An International Prognostic Scoring System (IPSS) classifies patients by risk status based on the percentage of bone marrow blasts, cytogenetics, and severity of cytopenias. The IPSS is associated with the rate of progression to AML and with overall survival, which can range from a median of 6 years for the low-risk group to 5 months for the high-risk patients.
Clinical Findings
A. Symptoms and Signs
Patients are usually over age 60 years. Many patients are asymptomatic when the diagnosis is made because of the finding of abnormal blood counts. Fatigue, infection, or bleeding related to bone marrow failure are usually the presenting symptoms and signs. The course may be indolent, and the disease may present as a wasting illness with fever, weight loss, and general debility. On examination, splenomegaly may be present in combination with pallor, bleeding, and various signs of infection. Myelodysplastic syndromes can also be accompanied by a variety of paraneoplastic syndromes prior to or following this diagnosis.
B. Laboratory Findings
Anemia may be marked with the MCV normal or increased, and transfusion support may be required. On the peripheral blood smear, macro-ovalocytes may be seen. The white blood cell count is usually normal or reduced, and neutropenia is common. The neutrophils may exhibit morphologic abnormalities, including deficient numbers of granules or deficient segmentation of the nucleus, especially a bilobed nucleus (Pelger-Huet abnormality). The myeloid series may be left shifted, and small numbers of promyelocytes or blasts may be seen. The platelet count is normal or reduced, and hypogranular platelets may be present.
The bone marrow is characteristically hypercellular but occasionally may be hypocellular. Erythroid hyperplasia is common, and signs of abnormal erythropoiesis include megaloblastic features, nuclear budding, or multinucleated erythroid precursors. The Prussian blue stain may demonstrate ringed sideroblasts. In the marrow, too, the myeloid series is often left shifted, with variable increases in blasts. Deficient or abnormal granules may be seen. A characteristic abnormality is the presence of dwarf megakaryocytes with a unilobed nucleus. Genetic abnormalities define MDS; there are frequent cytogenetic abnormalities involving the long arm of chromosome 5 as well as deletions of chromosomes 5 and 7. Some patients with an indolent form of the disease have an isolated partial deletion of chromosome 5 (MDS with isolated del[5q]). Aside from cytogenetic abnormalities, the most commonly mutated genes are SF3B1, TET2, SRSF2, ASXL1, DNMT3A, RUNX1, U2AF1, TP53, and EZH2.
Differential Diagnosis
Myelodysplastic syndromes should be distinguished from megaloblastic anemia, aplastic anemia, myelofibrosis, HIV-associated cytopenias, and acute or chronic drug effect. In subtle cases, cytogenetic evaluation of the bone marrow may help distinguish this clonal disorder from other causes of cytopenias. As the number of blasts increases in the bone marrow, myelodysplasia is arbitrarily separated from AML by the presence of less than 20% blasts.
Treatment
Myelodysplasia is a heterogeneous disease, and the appropriate treatment depends on a number of factors. For patients with anemia who have a low serum erythropoietin level (500 milliunits/mL or less), erythropoiesis-stimulating agents may raise the hematocrit and reduce the red cell transfusion requirement in 40%. Addition of intermittent granulocyte colony-stimulating factor (G-CSF) therapy may augment the erythroid response to epoetin. Unfortunately, the patients with the highest transfusion requirements are the least likely to respond. Patients who remain dependent on red blood cell transfusion and who do not have immediately life-threatening disease should receive iron chelation in order to prevent serious iron overload; the dose of oral agent deferasirox is 20 mg/kg/day in divided dosing. Patients affected primarily with severe neutropenia may benefit from the use of myeloid growth factors such as filgrastim. Oral thrombopoietin analogs, such as romiplostim and eltrombopag, have shown effectiveness in raising the platelet count in myelodysplasia. Finally, occasional patients can benefit from immunosuppressive therapy including ATG. Predictors of response to ATG include age younger than 60 years, absence of 5q–, and presence of HLA DR15.
For patients who do not respond to these interventions, there are several therapeutic options available. Lenalidomide is approved for the treatment of transfusion-dependent anemia due to myelodysplasia. It is the treatment of choice in patients with MDS with isolated del(5q) with significant responses in 70% of patients, and responses typically lasting longer than 2 years. In addition, nearly half of these patients enter a cytogenetic remission with clearing of the abnormal 5q– clone. The recommended initial dose is 10 mg/day orally. The most common side effects are neutropenia and thrombocytopenia, but venous thrombosis occurs and warrants prophylaxis with aspirin, 325 mg/day orally. For patients with high-risk myelodysplasia, azacitidine is the treatment of choice. It can improve both symptoms and blood counts and prolong both overall survival and the time to conversion to acute leukemia. It is used at a dose of 75 mg/m2 daily for 5–7 days every 28 days and up to six cycles of therapy may be required to achieve a response. Decitabine, a related hypomethylating agent, given at 20 mg/m2 daily for 5 days every 28 days can produce similar hematologic responses but has not demonstrated a benefit in overall survival compared to supportive care alone. Unfortunately, the progress that has been made over the past decade in understanding the complex molecular mechanisms underlying MDS has not yet translated into new therapeutic options.
Allogeneic stem cell transplantation is the only curative therapy for myelodysplasia, but its role is limited by the advanced age of many patients and the variably indolent course of the disease. The optimal use and timing of allogeneic transplantation are controversial, but the use of reduced-intensity preparative regimens and alternative donor sources (cord blood, haplotype-matched) has expanded the role of this therapy.
Course & Prognosis
Myelodysplasia is an ultimately fatal disease, and allogeneic transplantation is the only curative therapy, with cure rates of 30–60% depending primarily on the risk status of the disease. Patients most commonly die of infections or bleeding. Patients with MDS with isolated del(5q) have a favorable prognosis, with 5-year survival over 90%. Other patients with low-risk disease (with absence of both excess blasts and adverse cytogenetics) may also do well, with similar survival. Those with excess blasts or CMML have a higher (30–50%) risk of developing acute leukemia, and short survival (less than 2 years) without allogeneic transplantation.
When to Refer
All patients with myelodysplasia should be referred to a hematologist.
When to Admit
Hospitalization is needed only for specific complications, such as severe infection.
Gil-Perez A et al. Management of myelodysplastic syndromes after failure of response to hypomethylating agents. Ther Adv Hematol. 2019 May 9;10:2040620719847059. [PMID: 31156799]
Ogawa S. Genetics of MDS. Blood. 2019 Mar 7;133(10):1049–59. [PMID: 30670442]
Park S et al. Clinical effectiveness and safety of erythropoietin-stimulating agents for the treatment of low- and intermediate-risk myelodysplastic syndrome: a systematic literature review. Br J Haematol. 2019 Jan;184(2):134–60. [PMID: 30549002]
ACUTE LEUKEMIA
ESSENTIALS OF DIAGNOSIS
Short duration of symptoms, including fatigue, fever, and bleeding.
Cytopenias or pancytopenia.
Blasts in peripheral blood in 90% of patients.
More than 20% blasts in the bone marrow.
General Considerations
Acute leukemia is a malignancy of the hematopoietic progenitor cell. Malignant immature cells proliferate in an uncontrolled fashion and replace normal bone marrow elements. Most cases arise with no clear cause. However, radiation and some toxins (benzene) are leukemogenic. In addition, a number of chemotherapeutic agents (especially cyclophosphamide, melphalan, other alkylating agents, and etoposide) may cause leukemia. The leukemias seen after toxin or chemotherapy exposure often develop from a myelodysplastic prodrome and are often associated with abnormalities in chromosomes 5 and 7. Those related to etoposide or anthracyclines may have abnormalities in chromosome 11q23 (MLL locus).
Most of the clinical findings in acute leukemia are due to replacement of normal bone marrow elements by the malignant cells. Less common manifestations result from organ infiltration (skin, gastrointestinal tract, meninges). Acute leukemia is potentially curable with combination chemotherapy.
The myeloblastic subtype, AML, is primarily an adult disease with a median age at presentation of 60 years and an increasing incidence with advanced age. Acute promyelocytic leukemia (APL) is characterized by the chromosomal translocation t(15;17), which produces the fusion gene PML-RAR-alpha, leading to a block in differentiation that can be overcome with pharmacologic doses of retinoic acid. The lymphoblastic subtype of acute leukemia, ALL, comprises 80% of the acute leukemias of childhood. The peak incidence is between 3 and 7 years of age. It is also seen in adults, causing approximately 20% of adult acute leukemias.
Classification of the Leukemias
A. Acute Myeloid Leukemia (AML)
AML is primarily categorized based on recurrent structural chromosomal and molecular abnormalities. The cytogenetic abnormalities can be identified on traditional karyotyping or metaphase fluorescence in situ hybridization (FISH) and the molecular abnormalities are identified by either targeted or genome-wide sequencing of tumor DNA. Favorable cytogenetics such as t(8;21) producing a chimeric RUNX1/RUNX1T1 protein and inv(16)(p13;q22) are seen in 15% of cases and are termed the “core-binding factor” leukemias. These patients have a higher chance of achieving both short- and long-term disease control. Unfavorable cytogenetics confer a very poor prognosis. These consist of chromosomal translocations [t(6;9), t(3;3) or inv (3), t(v;11q23)], isolated monosomy 5 or 7, the presence of two or more other monosomies, or three or more separate cytogenetic abnormalities and account for 25% of the cases. The majority of cases of AML are of intermediate risk by traditional cytogenetics and have either a normal karyotype or chromosomal abnormalities that do not confer strong prognostic significance. However, there are several recurrent gene mutations with prognostic significance in this subgroup. On the one hand, internal tandem duplication in the gene FLT3 occurs in ∼30% of AML and is conditionally associated with a very poor prognosis in the setting of wild type NPM1. Other mutations conferring a poor prognosis occur in RUNX1, ASXL1, and TP53. On the other hand, a relatively favorable group of patients has been identified that lacks FLT3-ITD mutations and includes mutations of nucleophosmin 1 (NPM1) or carries CEBPA biallelic mutations.
B. Acute Promyelocytic Leukemia (APL)
In considering the various types of AML, APL is discussed separately because of its unique biologic features and response to non-chemotherapy treatments. APL is characterized by the cytogenetic finding of t(15;17) and the fusion gene PML-RAR-alpha. It is a highly curable form of leukemia (over 90%) with integration of all-trans-retinoic acid (ATRA) and arsenic trioxide (ATO) in induction, consolidation, and maintenance regimens.
C. Acute Lymphoblastic Leukemia (ALL)
ALL is most usefully classified by immunologic phenotype as follows: common, early B lineage, and T cell. Hyperdiploidy (with more than 50 chromosomes), especially of chromosomes 4, 10, and 17, and translocation t(12;21) (TEL-AML1), is associated with a better prognosis. Unfavorable cytogenetics are hypodiploidy (less than 44 chromosomes), the Philadelphia chromosome t(9;22), the t(4;11) translocation (which has fusion genes involving the MLL gene at 11q23), and a complex karyotype with more than five chromosomal abnormalities.
D. Mixed Phenotype Acute Leukemias
These leukemias consist of blasts that lack differentiation along the lymphoid or myeloid lineage or blasts that express both myeloid and lymphoid lineage-specific antigens. This group is considered very high risk and has a poor prognosis. The limited available data suggest that an “acute lymphoblastic leukemia–like” regimen followed by allogeneic stem cell transplant may be advisable; addition of a tyrosine kinase inhibitor in patients with t(9;22) translocation is recommended.
Clinical Findings
A. Symptoms and Signs
Most patients have been ill only for days or weeks. Bleeding (usually due to thrombocytopenia) occurs in the skin and mucosal surfaces, with gingival bleeding, epistaxis, or menorrhagia. Less commonly, widespread bleeding is seen in patients with disseminated intravascular coagulation (DIC) (in APL and monocytic leukemia). Infection is due to neutropenia, with the risk of infection rising as the neutrophil count falls below 500/mcL (0.5 × 109/L). Common presentations include cellulitis, pneumonia, and perirectal infections; death within a few hours may occur if treatment with appropriate antibiotics is delayed. Fungal infections are also commonly seen.
Patients may also seek medical attention because of gum hypertrophy and bone and joint pain. The most dramatic presentation is hyperleukocytosis, in which a markedly elevated circulating blast count (total white blood count greater than 100,000/mcL) leads to impaired circulation, presenting as headache, confusion, and dyspnea. Such patients require emergent chemotherapy with adjunctive leukapheresis since mortality approaches 40% in the first 48 hours.
On examination, patients appear pale and have purpura and petechiae; signs of infection may not be present. Stomatitis and gum hypertrophy may be seen in patients with monocytic leukemia, as may rectal fissures. There is variable enlargement of the liver, spleen, and lymph nodes. Bone tenderness may be present, particularly in the sternum, tibia, and femur.
B. Laboratory Findings
The hallmark of acute leukemia is the combination of pancytopenia with circulating blasts. However, blasts may be absent from the peripheral smear in as many as 10% of cases (“aleukemic leukemia”). The bone marrow is usually hypercellular and dominated by blasts (greater than 20%).
Hyperuricemia may be seen. If DIC is present, the fibrinogen level will be reduced, the prothrombin time prolonged, and fibrin degradation products or fibrin D-dimers present. Patients with ALL (especially T cell) may have a mediastinal mass visible on chest radiograph. Meningeal leukemia will have blasts present in the spinal fluid, seen in approximately 5% of cases at diagnosis; it is more common in monocytic types of AML and can be seen with ALL.
The Auer rod, an eosinophilic needle-like inclusion in the cytoplasm, is pathognomonic of AML and, if seen, secures the diagnosis. The phenotype of leukemia cells is usually demonstrated by flow cytometry or immunohistochemistry. AML cells usually express myeloid antigens such as CD13 or CD33 and myeloperoxidase. ALL cells of B lineage will express CD19, and most cases will express CD10, formerly known as the “common ALL antigen.” ALL cells of T lineage will usually not express mature T-cell markers, such as CD3, CD4, or CD8, but will express some combination of CD2, CD5, and CD7 and will not express surface immunoglobulin. Almost all ALL cells express terminal deoxynucleotidyl transferase (TdT).
Differential Diagnosis
AML must be distinguished from other myeloproliferative disorders, CML, and myelodysplastic syndromes. Acute leukemia may also resemble a left-shifted bone marrow recovering from a previous toxic insult. If the diagnosis is in doubt, a bone marrow study should be repeated in several days to see if maturation has taken place. ALL must be separated from other lymphoproliferative disease such as CLL, lymphomas, and hairy cell leukemia. It may also be confused with the atypical lymphocytosis of mononucleosis and pertussis.
Treatment
Most patients up to age 60 with acute leukemia are treated with the objective of cure. The first step in treatment is to obtain complete remission, defined as normal peripheral blood with resolution of cytopenias, normal bone marrow with no excess blasts, and normal clinical status. The type of initial chemotherapy depends on the subtype of leukemia.
1. AML—Most patients with AML are treated with a combination of an anthracycline (daunorubicin or idarubicin) plus cytarabine, either alone or in combination with other agents. This therapy will produce complete remissions in 80–90% of patients under age 60 years and in 50–60% of older patients (see Table 39–2). Older patients with AML who are not candidates for intensive chemotherapy may be given 5-azacitidine or decitabine, in combination with the bcl2 inhibitor venetoclax, producing nearly equivalent response rates with lower toxicity.
Once a patient has entered remission, post-remission therapy should be given with curative intent whenever possible. Options include continuation of chemotherapy and allogeneic stem cell transplantation. The optimal treatment strategy depends on the patient’s age, clinical status, and the genetic risk profile of the leukemia. Patients with a favorable genetic profile can be treated with chemotherapy alone or with autologous transplant with cure rates of 60–80%. Patients who do not enter remission (primary induction failure) or those with high-risk genetics have cure rates of less than 10% with chemotherapy alone and are referred for allogeneic stem cell transplantation. For intermediate-risk patients with AML, cure rates are 35–40% with chemotherapy and 40–60% with allogeneic transplantation. Addition of the FLT3 kinase inhibitor midostaurin to the induction, consolidation, and maintenance therapy of AML patients with FLT3 mutations has been shown to prolong event-free and overall survival.
Patients over age 60 have had a poor prognosis, even in first remission, when treated with standard chemotherapy approaches, and only 10–20% become long-term survivors. The use of reduced-intensity allogeneic transplant has improved the outcome for such patients, with studies suggesting that up to 40% of selected patients may be cured.
Once leukemia has recurred after initial chemotherapy, the prognosis is poor. For patients in second remission, transplantation offers a 20–30% chance of cure. Targeted therapies directed at recurrent genetic mutations (FLT3, IDH1/IDH2) are available for these patients.
2. ALL—Adults with ALL are treated with combination chemotherapy, including daunorubicin, vincristine, prednisone, and asparaginase. This treatment produces complete remissions in 90% of patients. Those patients with Philadelphia chromosome-positive ALL (or bcr-abl-positive ALL) should have a tyrosine kinase inhibitor, such as dasatinib or ponatinib, added to their initial chemotherapy. Remission induction therapy for ALL is less myelosuppressive than treatment for AML and does not necessarily produce prolonged marrow aplasia. Patients should also receive central nervous system prophylaxis so that meningeal sequestration of leukemic cells does not develop.
After achieving complete remission, patients may be treated with either additional cycles of chemotherapy or high-dose chemotherapy and stem cell transplantation. Treatment decisions are made based on patient age and disease risk factors. Adults younger than 39 years have uniformly better outcomes when treated under pediatric protocols. For older patients, minimal residual disease testing early on can identify high-risk patients who will not be cured with chemotherapy alone and who will do better with allogeneic transplantation. For patients with relapsed disease, the bispecific antibody blinatumomab targeting CD19 and the antibody-drug conjugate inotuzumab ozogamicin targeting CD22 have shown remarkable activity and are considered superior to traditional chemotherapy options. Tisagenlecleucel is a therapy utilizing autologous T cells engineered to express an anti-CD-19 antigen receptor (CART-19) and is FDA-approved for the treatment of children and young adults with relapsed/refractory B-ALL.
Prognosis
Approximately 70–80% of adults with AML under age 60 years achieve complete remission and ∼50% are cured using risk-adapted post-remission therapy. Older adults with AML achieve complete remission in up to 50% of instances. The cure rates for older patients with AML have been very low (approximately 10–20%) even if they achieve remission and are able to receive post-remission chemotherapy. Reduced-intensity allogeneic transplantation is increasingly being utilized in order to improve on these outcomes.
Patients younger than 39 years with ALL have excellent outcomes after undergoing chemotherapy followed by risk-adapted intensification and transplantation (cure rates of 60–80%). Patients with adverse cytogenetics, poor response to chemotherapy, or older age have a much lower chance of cure (cure rates of 20–40%).
When to Refer
All patients should be referred to a hematologist.
When to Admit
Most patients with acute leukemia will be admitted for treatment.
Marks DI et al. Management of adults with T-cell lymphoblastic leukemia. Blood. 2017 Mar 2;129(9):1134–42. Erratum in: Blood. 2017 Apr 13;129(15):2204. [PMID: 28115371]
Osman AEG et al. Treatment of acute promyelocytic leukemia in adults. J Oncol Pract. 2018 Nov;14(11):649–57. [PMID: 30423270]
Pollyea DA. New drugs for acute myeloid leukemia inspired by genomics and when to use them. Hematology Am Soc Hematol Educ Program. 2018 Nov 30;2018(1):45–50. [PMID: 30504290]
Rytting ME et al. Acute lymphoblastic leukemia in adolescents and young adults. Cancer. 2017 Jul 1;123(13):2398–403. [PMID: 28328172]
Smith CC. The growing landscape of FLT3 inhibition in AML. Hematology Am Soc Hematol Educ Program. 2019 Dec 6;2019(1):539–47. [PMID: 31808872]
CHRONIC LYMPHOCYTIC LEUKEMIA
ESSENTIALS OF DIAGNOSIS
B-cell lymphocytosis with CD19 expression greater than 5000/mcL.
Coexpression of CD19, CD5 on lymphocytes.
General Considerations
CLL is a clonal malignancy of B lymphocytes. The disease is usually indolent, with slowly progressive accumulation of long-lived small lymphocytes. These cells are immune-incompetent and respond poorly to antigenic stimulation.
CLL is manifested clinically by immunosuppression, bone marrow failure, and organ infiltration with lymphocytes. Immunodeficiency is also related to inadequate antibody production by the abnormal B cells. With advanced disease, CLL may cause damage by direct tissue infiltration.
CLL usually pursues an indolent course, but some subtypes behave more aggressively; a variant, prolymphocytic leukemia, is more aggressive. The morphology of the latter is different, characterized by larger and more immature cells. In 5–10% of cases, CLL may be complicated by autoimmune hemolytic anemia or autoimmune thrombocytopenia. In approximately 5% of cases, while the systemic disease remains stable, an isolated lymph node transforms into an aggressive large-cell lymphoma (Richter syndrome).
Clinical Findings
A. Symptoms and Signs
CLL is a disease of older patients, with 90% of cases occurring after age 50 years and a median age at presentation of 70 years. Many patients will be incidentally discovered to have lymphocytosis. Others present with fatigue or lymphadenopathy. On examination, 80% of patients will have diffuse lymphadenopathy and 50% will have enlargement of the liver or spleen.
The long-standing Rai classification system remains prognostically useful: stage 0, lymphocytosis only; stage I, lymphocytosis plus lymphadenopathy; stage II, organomegaly (spleen, liver); stage III, anemia; stage IV, thrombocytopenia. These stages can be collapsed into low risk (stages 0–I), intermediate risk (stage II), and high risk (stages III–IV).
B. Laboratory Findings
The hallmark of CLL is isolated lymphocytosis. The white blood cell count is usually greater than 20,000/mcL (20 × 109/L) and may be markedly elevated to several hundred thousand. Usually 75–98% of the circulating cells are lymphocytes. Lymphocytes appear small and mature, with condensed nuclear chromatin, and are morphologically indistinguishable from normal small lymphocytes, but smaller numbers of larger and activated lymphocytes may be seen. The hematocrit and platelet count are usually normal at presentation. The bone marrow is variably infiltrated with small lymphocytes. The immunophenotype of CLL demonstrates coexpression of the B lymphocyte lineage marker CD19 with the T lymphocyte marker CD5; this finding is commonly observed only in CLL and mantle cell lymphoma. CLL is distinguished from mantle cell lymphoma by the expression of CD23, low expression of surface immunoglobulin and CD20, and the absence of a translocation or overexpression of cyclin D1. Patients whose CLL cells have mutated forms of the immunoglobulin gene (IgVH somatic mutation) have a more indolent form of disease; these cells typically express low levels of the surface antigen CD38 and do not express the zeta-associated protein (ZAP-70). Conversely, patients whose cells have unmutated IgVH genes and high levels of ZAP-70 expression do less well and require treatment sooner. The assessment of genomic changes by FISH provides important prognostic information. The finding of deletion of chromosome 17p (TP53) confers the worst prognosis, while deletion of 11q (ATM) confers an inferior prognosis to the average genotype, and isolated deletion of 13q has a more favorable outcome.
Hypogammaglobulinemia is present in 50% of patients and becomes more common with advanced disease. In some, a small amount of IgM paraprotein is present in the serum.
Differential Diagnosis
Few syndromes can be confused with CLL. Viral infections producing lymphocytosis should be obvious from the presence of fever and other clinical findings; however, fever may occur in CLL from concomitant bacterial infection. Pertussis may cause a particularly high total lymphocyte count. Other lymphoproliferative diseases such as Waldenström macroglobulinemia, hairy cell leukemia, or lymphoma (especially mantle cell) in the leukemic phase are distinguished on the basis of the morphology and immunophenotype of circulating lymphocytes and bone marrow. Monoclonal B-cell lymphocytosis is a disorder characterized by fewer than 5000/mcL B cells and is considered a precursor to B-CLL.
Treatment
The treatment of CLL is evolving as several active targeted agents have emerged. Most cases of early indolent CLL require no specific therapy, and the standard of care for early-stage disease has been observation. Indications for treatment include progressive fatigue, symptomatic lymphadenopathy, anemia, or thrombocytopenia. These patients have either symptomatic and progressive Rai stage II disease or stage III/IV disease. Initial treatment for patients with CLL consists of targeted biologic therapy in most cases. Options include ibrutinib (a Bruton tyrosine kinase inhibitor targeting B-cell receptor signaling) or venetoclax (a bcl2 inhibitor resulting in apoptosis) in combination with anti-CD20 antibody therapy. Choice between these agents is based on toxicity as well as preference. Ibrutinib is a well-tolerated, oral agent given at 420 mg daily; it can be associated with hypertension, cardiac arrhythmias, rash, and increased infections. Caution should be exercised when this agent is used in conjunction with CYP3A inhibitors or inducers. In addition, there is a potential for serious bleeding when it is used in patients taking warfarin. Venetoclax (slowly titrated up to 400 mg daily) is usually given for a shorter course of therapy and is associated with tumor lysis syndrome and neutropenia; some patients may require hospitalization for initial therapy. Venetoclax has to be combined with a monoclonal CD20 antibody, usually obinutuzumab, which can result in infusion reactions. Traditional combination chemotherapy is used only in selected cases (see Table 39–3). For older patients, chlorambucil, 0.6–1 mg/kg orally every 4 weeks, in combination with obinutuzumab is another therapy option.
For patients with relapsed or refractory disease, both venetoclax and ibrutinib or another BTK inhibitor, acalabrutinib, demonstrate significant activity, even for patients with high-risk genetics. Other options include idelalisib and duvelisib (inhibitors of PI3 kinase delta), which are associated with higher toxicity. The dosage for idelalisib is 150 mg orally twice a day, and the dosage for duvelisib is 25 mg orally twice a day. There are risks for colitis, liver injury, and fatal infectious complications in patients treated with PI3k inhibitors. Patients should be given antimicrobial prophylaxis and monitored closely while taking these agents.
Of note, BTK and PI3k inhibitors can be initially associated with marked lymphocytosis due to release of tumor cells from the lymph nodes into the peripheral blood. This results in a significant early reduction in lymphadenopathy but a potentially misleading, more delayed clearance of lymphocytes from peripheral blood and bone marrow.
Associated autoimmune hemolytic anemia or immune thrombocytopenia may require treatment with rituximab, prednisone, or splenectomy. Fludarabine should be avoided in patients with autoimmune hemolytic anemia since it may exacerbate it. Rituximab should be used with caution in patients with past hepatitis B virus (HBV) infection since HBV reactivation, fulminant hepatitis, and, rarely, death can occur without anti-HBV agent prophylaxis. Patients with recurrent bacterial infections and hypogammaglobulinemia benefit from prophylactic infusions of gamma globulin (0.4 g/kg/month), but this treatment is cumbersome and expensive, justified only when these infections are severe. Patients undergoing therapy with a nucleoside analog (fludarabine, pentostatin) should receive anti-infective prophylaxis for Pneumocystis jirovecii pneumonia, herpes viruses, and invasive fungal infections until there is evidence of T-cell recovery.
Allogeneic transplantation offers potentially curative treatment for patients with CLL, but it should be used only in patients whose disease cannot be controlled by the available therapies. Nonmyeloablative allogeneic transplant can result in over 40% long-term disease control in CLL but with risk of moderate toxicity.
Prognosis
Therapies have changed the prognosis of CLL. Patients with stage 0 or stage I disease have a median survival of 10–15 years, and these patients may be reassured that they can live a normal life. Patients with stage III or stage IV disease had a median survival of less than 2 years in the past, but with current therapies, 2-year survival is more than 90% and the long-term outlook appears to be substantially changed. For patients with high-risk and resistant forms of CLL, there is evidence that allogeneic transplantation can overcome risk factors and lead to long-term disease control.
When to Refer
All patients with CLL should be referred to a hematologist.
When to Admit
Hospitalization is rarely needed.
Aitken MJL et al. Emerging treatment options for patients with p53-pathway-deficient CLL. Ther Adv Hematol. 2019 Dec 5;10:2040620719891356. [PMID: 31839919]
Burger JA et al. Evolution of CLL treatment—from chemoimmunotherapy to targeted and individualized therapy. Nat Rev Clin Oncol. 2018 Aug;15(8):510–27. [PMID: 29777163]
Eichhorst B et al. Relapsed disease and aspects of undetectable MRD and treatment discontinuation. Hematology Am Soc Hematol Educ Program. 2019 Dec 6;2019(1):482–9. [PMID: 31808867]
von Keudell G et al. The role of PI3K inhibition in lymphoid malignancies. Curr Hematol Malig Rep. 2019 Oct;14(5):405–13. [PMID: 31359259]
HAIRY CELL LEUKEMIA
ESSENTIALS OF DIAGNOSIS
Pancytopenia.
Splenomegaly, often massive.
Hairy cells present on blood smear and especially in bone marrow biopsy.
General Considerations
Hairy cell leukemia is a rare malignancy of hematopoietic stem cells differentiated as mature B-lymphocytes with hairy cytoplasmic projections. The V600E mutation in the BRAF gene is recognized as the causal genetic event of hairy cell leukemia, since it is detectable in almost all cases at diagnosis and is present at relapse.
Clinical Findings
A. Symptoms and Signs
The disease characteristically presents in middle-aged men. The median age at presentation is 55 years, and there is a striking 5:1 male predominance. Most patients present with gradual onset of fatigue, others complain of symptoms related to markedly enlarged spleen, and some come to attention because of infection.
Splenomegaly is almost invariably present and may be massive. The liver is enlarged in 50% of cases; lymphadenopathy is uncommon.
Hairy cell leukemia is usually an indolent disorder whose course is dominated by pancytopenia and recurrent infections, including mycobacterial infections.
B. Laboratory Findings
The hallmark of hairy cell leukemia is pancytopenia. Anemia is nearly universal, and 75% of patients have thrombocytopenia and neutropenia. The “hairy cells” are usually present in small numbers on the peripheral blood smear and have a characteristic appearance with numerous cytoplasmic projections. The bone marrow is usually inaspirable (dry tap), and the diagnosis is made by characteristic morphology on bone marrow biopsy. The hairy cells have a characteristic histochemical staining pattern with tartrate-resistant acid phosphatase (TRAP). On immunophenotyping, the cells coexpress the antigens CD11c, CD20, CD22, CD25, CD103, and CD123. Pathologic examination of the spleen shows marked infiltration of the red pulp with hairy cells. This is in contrast to the usual predilection of lymphomas to involve the white pulp of the spleen.
Differential Diagnosis
Hairy cell leukemia should be distinguished from other lymphoproliferative diseases such as Waldenström macroglobulinemia and non-Hodgkin lymphomas. It also may be confused with other causes of pancytopenia, including hypersplenism due to any cause, aplastic anemia, and paroxysmal nocturnal hemoglobinuria.
Treatment
Treatment is indicated for symptomatic disease, ie, splenic discomfort, recurrent infections, or significant cytopenias. The treatment of choice is a nucleoside analog, specifically pentostatin or cladribine for a single course, producing a complete remission in 70–95% of patients. Treatment is associated with infectious complications, and patients should be closely monitored. The median duration of response is over 8 years and patients who relapse a year or more after initial therapy can be treated again with one of these agents. Rituximab can be used in the relapsed setting either as a single agent or in combination with a nucleoside analog. The BRAF inhibitor vemurafenib exhibits ∼100% overall response rate in patients with refractory/relapsed hairy cell leukemia, with 35–40% complete remissions. The median relapse-free survival is ∼19 months in patients who achieved complete remission and 6 months in those who obtained a partial response. Moxetumomab pasudotox is a recombinant CD22-targeting immunotoxin recently approved for patients with refractory disease. It has shown a durable complete response rate of 31% in the pivotal trial. However, it can be associated with capillary leak and hemolytic-uremic syndrome attributable to the diphtheria toxin moiety.
Course & Prognosis
More than 95% of patients with hairy cell leukemia live longer than 10 years.
Dhillon S. Moxetumomab pasudotox: first global approval. Drugs. 2018 Nov;78(16):1763–7. Erratum in: Drugs. 2019 Jan;79(1):105. [PMID: 30357593]
Falini B et al. New treatment options in hairy cell leukemia with focus on BRAF inhibitors. Hematol Oncol. 2019 Jun;37(Suppl 1):30–7. [PMID: 31187521]
Grever MR et al. Consensus guidelines for the diagnosis and management of patients with classic hairy cell leukemia. Blood. 2017 Feb 2;129(5):553–60. [PMID: 27903528]
LYMPHOMAS
NON-HODGKIN LYMPHOMAS
ESSENTIALS OF DIAGNOSIS
Often present with painless lymphadenopathy.
Diagnosis is made by tissue biopsy.
General Considerations
The non-Hodgkin lymphomas are a heterogeneous group of cancers of lymphocytes usually presenting as enlarged lymph nodes. The disorders vary in clinical presentation and course from indolent to rapidly progressive.
Molecular biology has provided clues to the pathogenesis of these disorders, often a matter of balanced chromosomal translocations whereby an oncogene becomes juxtaposed next to either an immunoglobulin gene (B-cell lymphoma) or the T-cell receptor gene or related gene (T-cell lymphoma). The net result is oncogene overexpression and development of lymphoma. The best-studied example is Burkitt lymphoma, in which a characteristic cytogenetic abnormality of translocation between the long arms of chromosomes 8 and 14 has been identified. The protooncogene c-myc is translocated from its normal position on chromosome 8 to the immunoglobulin heavy chain locus on chromosome 14. Overexpression of c-myc is related to malignant transformation through excess B-cell proliferation. In the follicular lymphomas, the t(14;18) translocation is characteristic and bcl-2 is overexpressed, resulting in protection against apoptosis, the usual mechanism of B-cell death.
Classification of the lymphomas is a dynamic area still undergoing evolution. The 2017 grouping (Table 13–16) separates diseases based on both clinical and pathologic features. Eighty-five percent of non-Hodgkin lymphomas are B-cell and 15% are T-cell or NK-cell in origin. Even though non-Hodgkin lymphomas represent a diverse group of diseases, they are historically divided in two categories based on clinical behavior and pathology: the indolent (low-grade) and the aggressive (intermediate- or high-grade).
Table 13–16. World Health Organization classification of lymphomas.
Precursor B-cell lymphoblastic lymphoma
Mature B-cell lymphomas
Chronic lymphocytic leukemia/small lymphocytic lymphoma
Monoclonal B-cell lymphocytosis
Hairy cell leukemia
Plasma cell myeloma
Diffuse large B-cell lymphoma
Primary diffuse large B-cell lymphoma of the CNS
High-grade B-cell lymphoma, with MYC and BCL2 and/or BCL6 rearrangements
Mediastinal large B-cell lymphoma
Follicular lymphoma
Small lymphocytic lymphoma
Lymphoplasmacytic lymphoma (Waldenström macroglobulinemia)
Mantle cell lymphoma
Burkitt lymphoma
Marginal zone lymphoma
MALT type
Nodal type
Splenic type
Mature T (and NK cell) lymphomas
Anaplastic large-cell lymphoma
Angioimmunoblastic T-cell lymphoma
Peripheral T-cell lymphoma, NOS
Cutaneous T-cell lymphoma (mycosis fungoides, Sézary syndrome)
Extranodal NK/T-cell lymphoma, nasal type
Adult T-cell leukemia/lymphoma
T-cell large granular lymphocytic leukemia
Hodgkin lymphoma
Nodular lymphocyte predominant Hodgkin lymphoma
Classic Hodgkin lymphoma
Posttransplant lymphoproliferative disorders
Histiocytic and dendritic cell neoplasms
CNS, central nervous system; MALT, mucosa-associated lymphoid tissue; NOS, not otherwise specified.
Clinical Findings
A. Symptoms and Signs
Patients with non-Hodgkin lymphomas usually present with lymphadenopathy. Involved lymph nodes may be present peripherally or centrally (in the retroperitoneum, mesentery, and pelvis). The indolent lymphomas are usually disseminated at the time of diagnosis, and bone marrow involvement is frequent. Many patients with lymphoma have constitutional symptoms such as fever, drenching night sweats, and weight loss of greater than 10% of prior body weight (referred to as “B symptoms”).
On examination, lymphadenopathy may be isolated or diffuse, and extranodal sites of disease (such as the skin, gastrointestinal tract, liver, and bone marrow) may be found. Patients with Burkitt lymphoma are noted to have abdominal pain or abdominal fullness because of the predilection of the disease for the abdomen.
Once a pathologic diagnosis is established, staging is done using a whole-body positron emission tomography (PET)/CT scan, a bone marrow biopsy, and, in patients with high-grade lymphoma or intermediate-grade lymphoma with high-risk features, a lumbar puncture.
B. Laboratory Findings
The peripheral blood is usually normal even with extensive bone marrow involvement by lymphoma. Circulating lymphoma cells in the blood are not commonly seen.
Bone marrow involvement is manifested as paratrabecular monoclonal lymphoid aggregates. In some high-grade lymphomas, the meninges are involved and malignant cells are found with cerebrospinal fluid cytology. The serum LD, a useful prognostic marker, is incorporated in risk stratification of treatment.
The diagnosis of lymphoma is made by tissue biopsy. Needle aspiration may yield evidence for non-Hodgkin lymphoma, but a lymph node biopsy (or biopsy of involved extranodal tissue) is required for accurate diagnosis and classification.
Treatment
A. Indolent Lymphomas
The most common lymphomas in this group are follicular lymphoma, marginal zone lymphomas, and small lymphocytic lymphoma (SLL). The treatment of indolent lymphomas depends on the stage of disease and the clinical status of the patient. A small number of patients have limited disease with only one or two contiguous abnormal lymph node groups and may be treated with localized irradiation with curative intent. However, most patients (85%) with indolent lymphoma have disseminated disease at the time of diagnosis and are not considered curable. Historically, treatment of these patients has not affected overall survival; therefore, treatment is offered only when symptoms develop or for high tumor bulk. Following each treatment response, patients will experience a relapse at traditionally shorter intervals. Some patients will have temporary spontaneous remissions (8%). There are an increasing number of reasonable treatment options for indolent lymphomas, but no consensus exists on the best strategy. Treatment with rituximab (375 mg/m2 intravenously weekly for 4 weeks) is commonly used either alone or in combination with chemotherapy and may be the only agent to affect overall survival in these disorders. Patients should be screened for hepatitis B because rare cases of fatal fulminant hepatitis have been described with the use of anti-CD20 monoclonal therapies without anti-HBV agent prophylaxis. Rituximab is added to chemotherapy regimens including bendamustine; cyclophosphamide, vincristine, and prednisone (R-CVP); and cyclophosphamide, doxorubicin, vincristine, and prednisone (R-CHOP) (see Table 39–3). The immunomodulatory agent lenalidomide in combination with anti-CD20 therapy is an alternative option with similar outcomes to chemotherapy. Some patients with clinically aggressive low-grade lymphomas may be appropriate candidates for allogeneic stem cell transplantation with curative intent. The role of autologous hematopoietic stem cell transplantation remains uncertain, but some patients with recurrent disease appear to have prolonged remissions without the expectation of cure.
Patients with mucosa-associated lymphoid tissue tumors of the stomach may be appropriately treated with combination antibiotics directed against H pylori and with acid blockade but require frequent endoscopic monitoring. Alternatively, mucosa-associated lymphoid tissue tumors confined to the stomach can also be cured with whole-stomach radiotherapy.
B. Aggressive Lymphomas
Patients with diffuse large B-cell lymphoma are treated with curative intent. Most patients are treated with six cycles of immunochemotherapy such as R-CHOP (see Table 39–3). Involved nodal radiotherapy (INRT) may be added for patients with bulky or extranodal disease. About 25% of patients with diffuse large B-cell lymphoma have been identified as “double-protein expressors” with overexpression of MYC and BCL2 proteins by immunohistochemistry. While the outcomes with R-CHOP are inferior, no definitive alternative treatment recommendations can be made at this time. High-grade lymphoma with chromosomal translocations affecting MYC, such as t(8;14), and translocations affecting BCL2, such as t(14;18), also called “double-hit lymphoma,” has a very aggressive course. Patients with this disease may do better with dose-adjusted R-EPOCH as front-line therapy.
Patients with diffuse large B-cell lymphoma or high-grade lymphoma who relapse after initial chemotherapy can still be cured by autologous hematopoietic stem cell transplantation if their disease remains responsive to chemotherapy. For patients who do not respond to second-line chemotherapy, the treatment of choice is chimeric antigen receptor T-cell therapy targeting CD19 with either axicabtagene ciloleucel or tisagenlecleucel, which produces durable complete response rates of ∼40%.
Mantle cell lymphoma is not effectively treated with standard immunochemotherapy regimens. Intensive initial immunochemotherapy including autologous hematopoietic stem cell transplantation has been shown to improve outcomes. Reduced-intensity allogeneic stem cell transplantation offers curative potential for selected patients. The BTK inhibitors ibrutinib and acalabrutinib are active in relapsed or refractory patients with mantle cell lymphoma. For primary central nervous system lymphoma, repetitive cycles of high-dose intravenous methotrexate with rituximab early in the treatment course produce better results than whole-brain radiotherapy and with less cognitive impairment.
Patients with highly aggressive lymphomas (Burkitt or lymphoblastic) require urgent, intense, cyclic chemotherapy in the hospital similar to that given for ALL, and they also require intrathecal chemotherapy as central nervous system prophylaxis.
Patients with peripheral T-cell lymphomas usually have advanced stage nodal and extranodal disease and typically have inferior response rates to therapy compared to patients with aggressive B-cell disease. Autologous stem cell transplantation is often incorporated in first-line therapy. The antibody-drug conjugate brentuximab vedotin has significant activity in patients with CD30 positive peripheral T-cell lymphomas, such as anaplastic large-cell lymphoma.
Prognosis
The median survival of patients with indolent lymphomas is 10–15 years. These diseases ultimately become refractory to chemotherapy. This often occurs at the time of histologic progression of the disease to a more aggressive form of lymphoma.
The International Prognostic Index is widely used to categorize patients with aggressive lymphoma into risk groups. Factors that confer adverse prognosis are age over 60 years, elevated serum LD, stage III or stage IV disease, more than one extranodal site of disease, and poor performance status. Cure rates range from more than 80% for low-risk patients (zero risk factors) to less than 50% for high-risk patients (four or more risk factors).
For patients who relapse after initial chemotherapy, autologous hematopoietic stem cell transplantation and chimeric antigen receptor T-cell therapy offer a 40–50% chance of long-term lymphoma-free survival.
The treatment of older patients with lymphoma has been difficult because of poorer tolerance of aggressive chemotherapy. The use of myeloid growth factors and prophylactic antibiotics to reduce neutropenic complications may improve outcomes.
Molecular profiling techniques using gene array technology and immunophenotyping have defined subsets of lymphomas with different biologic features and prognoses. Those subsets are being studied in clinical trials to determine choice of therapy.
When to Refer
All patients with lymphoma should be referred to a hematologist or an oncologist.
When to Admit
Admission is necessary only for specific complications of lymphoma or its treatment and for the treatment of all high-grade lymphomas.
Chiappella A et al. Diffuse large B-cell lymphoma in the elderly: standard treatment and new perspectives. Expert Rev Hematol. 2017 Apr;10(4):289–97. [PMID: 28290728]
Ferreri AJM et al. Evolving treatments for primary central nervous system lymphoma. Am Soc Clin Oncol Educ Book. 2019 Jan;39:454–66. [PMID: 31099614]
Mehta-Shah N. Emerging strategies in peripheral T-cell lymphoma. Hematology Am Soc Hematol Educ Program. 2019 Dec 6;2019(1):41–6. [PMID: 31808829]
Munshi PN et al. The acceleration of CAR-T therapy in non-Hodgkin lymphoma. Hematol Oncol. 2019 Aug;37(3):233–9. [PMID: 30427551]
Ruan J. Molecular profiling and management of mantle cell lymphoma. Hematology Am Soc Hematol Educ Program. 2019 Dec 6;2019(1):30–40. [PMID: 31808882]
HODGKIN LYMPHOMA
ESSENTIALS OF DIAGNOSIS
Often painless lymphadenopathy.
Constitutional symptoms may or may not be present.
Pathologic diagnosis by lymph node biopsy.
General Considerations
Hodgkin lymphoma is characterized by lymph node biopsy showing Reed-Sternberg cells in an appropriate reactive cellular background. The malignant cell is derived from B lymphocytes of germinal center origin.
Clinical Findings
There is a bimodal age distribution, with one peak in the 20s and a second over age 50 years. Most patients seek medical attention because of a painless mass, commonly in the neck. Others may seek medical attention because of constitutional symptoms such as fever, weight loss, or drenching night sweats, or because of generalized pruritus. An unusual symptom of Hodgkin lymphoma is pain in an involved lymph node following alcohol ingestion.
An important feature of Hodgkin lymphoma is its tendency to arise within single lymph node areas and spread in an orderly fashion to contiguous areas of lymph nodes. Late in the course of the disease, vascular invasion leads to widespread hematogenous dissemination.
Hodgkin lymphoma is divided into two subtypes: classic Hodgkin (nodular sclerosis, mixed cellularity, lymphocyte rich, and lymphocyte depleted) and non-classic Hodgkin (nodular lymphocyte predominant). Hodgkin lymphoma should be distinguished pathologically from other malignant lymphomas and may occasionally be confused with reactive lymph nodes seen in infectious mononucleosis, cat-scratch disease, or drug reactions (eg, phenytoin).
Patients undergo a staging evaluation to determine the extent of disease, including serum chemistries, whole-body PET/CT scan, and bone marrow biopsy.
Treatment
Chemotherapy is the mainstay of treatment for Hodgkin lymphoma and ABVD (doxorubicin, bleomycin, vinblastine, dacarbazine) remains the standard first-line regimen. Even though other regimens such as escalated BEACOPP (bleomycin, etoposide, doxorubicin, cyclophosphamide, vincristine, procarbazine, prednisone) may improve response rates and reduce the need for consolidative radiotherapy, they are usually associated with increased toxicity and lack a definitive overall survival advantage. Low-risk patients are those with stage I or II disease without bulky lymphadenopathy or evidence of systemic inflammation. They traditionally receive a combination of short-course chemotherapy with INRT, but INRT can be eliminated for those with an early negative PET/CT scan without a significant change in outcomes (see Table 39–3). High-risk patients are those with stage III or IV disease or with stage II disease and a large mediastinal or other bulky mass or systemic inflammation. These patients are treated with a full course of ABVD for six cycles. Pulmonary toxicity can unfortunately occur following either chemotherapy (bleomycin) or radiation and should be treated aggressively in these patients, since it can lead to permanent fibrosis and death. A negative interim PET/CT scan after two cycles of chemotherapy can be used to identify patients with an excellent progression-free survival who can have bleomycin eliminated from their treatment. Conversely, an abnormal interim PET/CT scan is associated with a worse prognosis and should prompt early intensification of treatment to achieve a complete response (CR).
Classic Hodgkin lymphoma relapsing after initial treatment is treatable with high-dose chemotherapy and autologous hematopoietic stem cell transplantation. This offers a 35–50% chance of cure when disease is still chemotherapy responsive. The antibody-drug conjugate brentuximab vedotin has shown impressive activity in patients relapsing after autologous stem cell transplantation (overall response rate [ORR] of 75%; CR of 34%) and is FDA-approved for this indication. Last, immune checkpoint inhibition by PD1 blockade with nivolumab or pembrolizumab has shown remarkable activity (ORR of 65%) and is another option for patients with relapsed or refractory disease.
Prognosis
All patients should be treated with curative intent. Prognosis in advanced stage Hodgkin lymphoma is influenced by seven features: stage, age, gender, hemoglobin, albumin, white blood cell count, and lymphocyte count. The cure rate is 75% if zero to two risk features are present and 55% when three or more risk features are present. The prognosis of patients with stage IA or IIA disease is excellent, with 10-year survival rates in excess of 90%. Patients with advanced disease (stage III or IV) have 10-year survival rates of 50–60%. Inferior results are seen in patients who are older, those who have bulky disease, and those with lymphocyte depletion or mixed cellularity on histologic examination. Non-classic Hodgkin lymphoma (nodular lymphocyte predominant) is highly curable with radiotherapy alone for early-stage disease; however, for high-stage disease, it is characterized by long survival with repetitive relapses after chemotherapy or monoclonal anti-CD20 antibody therapy.
When to Refer
• All patients should be sent to an oncologist or hematologist.
• Secondary referral to a radiation oncologist might be appropriate.
When to Admit
Patients should be admitted for complications of the disease or its treatment.
Hu B et al. Checkpoint inhibitors Hodgkin lymphoma and non-Hodgkin lymphoma. Curr Hematol Malig Rep. 2018 Dec;13(6):543–54. [PMID: 30338457]
LaCasce AS. Treating Hodgkin lymphoma in the new millennium: relapsed and refractory disease. Hematol Oncol. 2019 Jun;37(Suppl 1):87–91. [PMID: 31187532]
Spinner MA et al. Risk-adapted therapy for advanced-stage Hodgkin lymphoma. Hematology Am Soc Hematol Educ Program. 2018 Nov 30;2018(1):200–6. [PMID: 30504311]
Straus DJ. Limited-stage Hodgkin lymphoma: minimizing toxicity. Cancer J. 2018 Sep/Oct;24(5):223–9. Erratum in Cancer J. 2019 Mar/Apr;25(2):164. [PMID: 30247257]
PLASMA CELL MYELOMA
ESSENTIALS OF DIAGNOSIS
Bone pain, often in the spine, ribs, or proximal long bones.
Monoclonal immunoglobulin (ie, paraprotein) in the serum or urine.
Clonal plasma cells in the bone marrow or in a tissue biopsy, or both.
Organ damage due to plasma cells (eg, bones, kidneys, hypercalcemia, anemia) or other defined criteria.
General Considerations
Plasma cell myeloma (previously called multiple myeloma) is a malignancy of hematopoietic stem cells terminally differentiated as plasma cells. It is characterized by infiltration of the bone marrow, bone destruction, and paraprotein formation. The diagnosis is established when monoclonal plasma cells (either kappa or lambda light chain restricted) in the bone marrow (any percentage) or as a tumor (plasmacytoma), or both, are associated with end-organ damage (such as bone disease [lytic lesions seen on bone radiographs, magnetic resonance imaging {MRI}, or PET/CT scan], anemia [hemoglobin less than 10 g/dL {100 g/L}], hypercalcemia [calcium greater than 11.5 mg/dL {2.9 mmol/L}], or kidney injury [creatinine greater than 2 mg/dL {176.8 mcmol/L}]) with or without paraprotein elaboration. Sixty percent or more clonal plasma cells in the bone marrow, or a serum free kappa to lambda ratio of greater than 100 or less than 0.01 (both criteria regardless of end-organ damage), are also diagnostic of plasma cell myeloma. Smoldering myeloma is defined as 10–59% clonal plasma cells in the bone marrow, a serum paraprotein level of 3 g/dL (30 g/L) or higher, or both, without plasma cell–related end-organ damage.
Malignant plasma cells can form tumors (plasmacytomas) that may cause spinal cord compression or other soft-tissue–related problems. Bone disease is common and due to excessive osteoclast activation mediated largely by the interaction of the receptor activator of NF-kappa-B (RANK) with its ligand (RANKL). In plasma cell myeloma, osteoprotegerin (a decoy receptor for RANKL) is underproduced, thus promoting the binding of RANK with RANKL with consequent excessive bone resorption.
The paraproteins (monoclonal immunoglobulins) secreted by the malignant plasma cells may cause problems in their own right. Very high paraprotein levels (either IgG or IgA) may cause hyperviscosity, although this is more common with the IgM paraprotein in Waldenström macroglobulinemia. The light chain component of the immunoglobulin, when produced in excess, often leads to kidney injury (frequently aggravated by hypercalcemia or hyperuricemia, or both). Light chain components may be deposited in tissues as amyloid, resulting in kidney failure with albuminuria and a vast array of systemic symptoms.
Myeloma patients are prone to recurrent infections for a number of reasons, including neutropenia, the underproduction of normal immunoglobulins (so-called immunoparesis) and the immunosuppressive effects of chemotherapy. Myeloma patients are especially prone to infections with encapsulated organisms such as Streptococcus pneumoniae and Haemophilus influenzae.
Clinical Findings
A. Symptoms and Signs
Myeloma is a disease of older adults (median age 65 years). The most common presenting complaints are those related to anemia, bone pain, kidney disease, and infection. Bone pain is most common in the back, hips, or ribs or may present as a pathologic fracture, especially of the femoral neck or vertebrae. Patients may also come to medical attention because of spinal cord compression from a plasmacytoma or the hyperviscosity syndrome (mucosal bleeding, vertigo, nausea, visual disturbances, alterations in mental status, hypoxia). Many patients are diagnosed because of laboratory findings of elevated total protein, hypercalcemia, proteinuria, elevated sedimentation rate, or abnormalities on serum protein electrophoresis obtained for symptoms or in routine screening studies. A few patients come to medical attention because of organ dysfunction due to amyloidosis.
Examination may reveal pallor, bone tenderness, or soft tissue masses. Patients may have neurologic signs related to neuropathy or spinal cord compression. Fever occurs mainly with infection. Acute oliguric or nonoliguric kidney injury may be present due to hypercalcemia, hyperuricemia, light chain cast injury, or primary amyloidosis.
B. Laboratory Findings
Anemia is nearly universal. Red blood cell morphology is normal, but rouleaux formation is common and may be marked. The absence of rouleaux formation, however, excludes neither plasma cell myeloma nor the presence of a serum paraprotein. The neutrophil and platelet counts are usually normal at presentation. Only rarely will plasma cells be visible on peripheral blood smear (plasma cell leukemia).
The hallmark of myeloma is the finding of a paraprotein on serum or urine protein electrophoresis (PEP) or immunofixation electrophoresis (IFE). The majority of patients will have a monoclonal spike visible in the gamma- or beta-globulin region of the PEP. The semi-quantification of the paraprotein on the PEP is referred to as the M-protein, and IFE will reveal this to be a monoclonal immunoglobulin. Approximately 15% of patients will have no demonstrable paraprotein in the serum on PEP because their myeloma cells produce only light chains and not intact immunoglobulin (but often seen on serum IFE), and the light chains pass rapidly through the glomerulus into the urine. Urine PEP and IFE usually demonstrate the light chain paraprotein in this setting. The free light chain assay will sometimes demonstrate excess monoclonal light chains in serum and urine, and in a small proportion of patients, will be the only means to identify and quantify the paraprotein being produced. Overall, the paraprotein is IgG (60%), IgA (20%), or light chain only (15%) in plasma cell myeloma, with the remainder being rare cases of IgD, IgM, or biclonal gammopathy. In sporadic cases, no paraprotein is present (“nonsecretory myeloma”); these patients have particularly aggressive disease.
The bone marrow will be infiltrated by variable numbers of monoclonal plasma cells. The plasma cells may be morphologically abnormal often demonstrating multi-nucleation and vacuolization. The plasma cells will display marked skewing of the normal kappa-to-lambda light chain ratio, which will indicate their clonality. Many benign inflammatory processes can result in bone marrow plasmacytosis, but with the absence of clonality and morphologic atypia.
C. Imaging
Bone radiographs are important in establishing the diagnosis of myeloma. Lytic lesions are most commonly seen in the axial skeleton: skull, spine, proximal long bones, and ribs. At other times, only generalized osteoporosis is seen. The radionuclide bone scan is not useful in detecting bone lesions in myeloma, since there is little osteoblastic component. In the evaluation of patients with known or suspected plasma cell myeloma, MRI and PET/CT scans are more sensitive to detect bone disease than plain radiographs and are preferred.
Differential Diagnosis
When a patient is discovered to have a paraprotein, the distinction between plasma cell myeloma or another lymphoproliferative malignancy with a paraprotein (CLL/SLL, Waldenström macroglobulinemia, non-Hodgkin lymphoma, primary amyloid, cryoglobulinemia) or monoclonal gammopathy of undetermined significance (MGUS) must be made. Plasma cell myeloma, smoldering plasma cell myeloma, and MGUS must be distinguished from reactive (benign) polyclonal hypergammaglobulinemia (which is commonly seen in cirrhosis or chronic inflammation).
Treatment
Patients with low-risk smoldering myeloma are observed. Those with high-risk smoldering disease may be treated with lenalidomide (an immunomodulatory agent) and dexamethasone since this therapy prolongs the time to symptomatic myeloma and may prolong survival compared to no treatment though at the expense of treatment-related side effects.
Most patients with plasma cell myeloma require treatment at diagnosis because of bone pain or other symptoms and complications related to the disease. The initial treatment generally involves triple therapy: an immunomodulatory agent, such as lenalidomide; a proteasome inhibitor, such as bortezomib or carfilzomib; and moderate- or high-dose dexamethasone. An immunomodulatory agent is sometimes replaced with an alkylating agent, cyclophosphamide, in the setting of kidney injury. The major side effects of lenalidomide are neutropenia and thrombocytopenia, skin rash, venous thromboembolism, peripheral neuropathy, and possibly birth defects. Bortezomib and carfilzomib have the advantages of producing rapid responses and of being effective in poor-prognosis myeloma. The major side effect of bortezomib is neuropathy (both peripheral and autonomic), which is largely ameliorated when given subcutaneously rather than intravenously. Carfilzomib rarely causes neuropathy but sometimes causes acute pulmonary hypertension or cardiac systolic dysfunction that is usually reversible. A fixed-dose combination of daratumumab (an anti-CD38 monoclonal antibody) plus hyaluronidase-fihj has now received FDA approval for treatment of patients with plasma cell myeloma, including newly diagnosed, transplant-ineligible patients as well as relapsed or refractory patients.
An oral proteasome inhibitor, ixazomib, is available for relapsed disease. Pomalidomide, an immunomodulatory agent, is effective as salvage therapy after relapse. Other salvage agents include daratumumab, elotuzumab (an anti-SLAMF7 monoclonal antibody), panobinostat (a histone deacetylase inhibitor), and selinexor (causes cell cycle arrest and apoptosis).
After initial therapy, many patients under age 80 years are consolidated with autologous hematopoietic stem cell transplantation following high-dose melphalan (an alkylating chemotherapeutic agent). Autologous stem cell transplantation prolongs both duration of remission and overall survival. Lenalidomide or thalidomide prolong remission and survival when given as posttransplant maintenance therapy but at the expense of an elevated rate of second malignancies. Proteasome inhibitors prolong remissions in high-risk patients after autologous stem cell transplantation.
Localized radiotherapy may be useful for palliation of bone pain or for eradicating tumor at the site of pathologic fracture. Vertebral collapse with its attendant pain and mechanical disturbance can be treated with vertebroplasty or kyphoplasty. Hypercalcemia and hyperuricemia should be treated aggressively and immobilization and dehydration avoided. The bisphosphonates (pamidronate 90 mg or zoledronic acid 4 mg intravenously monthly) reduce pathologic fractures in patients with bone disease and are an important adjunct in this subset of patients. The bisphosphonates are also used to treat myeloma-related hypercalcemia. However, long-term bisphosphonates have been associated with a risk of osteonecrosis of the jaw and other bony areas, so the use of bisphosphonates is limited to 1–2 years after definitive initial therapy in most patients. Myeloma patients with oliguric or anuric kidney disease at diagnosis should be treated aggressively with chemotherapy and considered for therapeutic plasma exchange (to reduce the paraprotein burden) because return of kidney function can commonly occur.
Prognosis
The outlook for patients with myeloma has been steadily improving for the past decade. The median survival of patients is more than 7 years. Patients with low-stage disease who lack high-risk genomic changes respond very well to treatment and derive significant benefit from autologous hematopoietic stem cell transplantation and have survivals approaching a decade. The International Staging System for myeloma relies on two factors: beta-2-microglobulin and albumin. Stage 1 patients have both beta-2-microglobulin less than 3.5 mg/L and albumin greater than 3.5 g/dL (survival more than 5 years). Stage 3 is established when beta-2-microglobulin is greater than 5.5 mg/L (survival less than 2 years). Stage 2 is established with values in between stage 1 and 3. Other adverse prognostic findings are an elevated serum LD or bone marrow genetic abnormalities established by FISH involving the immunoglobulin heavy chain locus at chromosome 14q32, multiple copies of the 1q21-23 locus, or 17p chromosome abnormalities (causing the loss or mutation of TP53).
When to Refer
All patients with plasma cell myeloma should be referred to a hematologist or an oncologist.
When to Admit
Hospitalization is indicated for treatment of acute kidney injury, hypercalcemia, or suspicion of spinal cord compression, for certain chemotherapy regimens, or for hematopoietic stem cell transplantation.
Chehab S et al. Daratumumab and its use in the treatment of relapsed and/or refractory multiple myeloma. Future Oncol. 2018 Dec;14(30):3111–21. [PMID: 30136602]
Dingli D et al. Therapy for relapsed multiple myeloma: guidelines from the Mayo stratification for myeloma and risk-adapted therapy. Mayo Clin Proc. 2017 Apr;92(4):578–98. [PMID: 28291589]
Goldschmidt H et al. Navigating the treatment landscape in multiple myeloma: which combinations to use and when? Ann Hematol. 2019 Jan;98(1):1–18. [PMID: 30470875]
Laubach JP et al. Daratumumab, elotuzumab, and the development of therapeutic monoclonal antibodies in multiple myeloma. Clin Pharmacol Ther. 2017 Jan;101(1):81–8. [PMID: 27806428]
Richter J et al. Society of Hematologic Oncology state of the art update and next questions: multiple myeloma. Clin Lymphoma Myeloma Leuk. 2018 Nov;18(11):693–702. [PMID: 30287199]
MONOCLONAL GAMMOPATHY OF UNDETERMINED SIGNIFICANCE
ESSENTIALS OF DIAGNOSIS
Monoclonal immunoglobulin (ie, paraprotein) in the serum (less than 3 g/dL [less than 30 g/L]) or urine.
Clonal plasma cells in the bone marrow less than 10% (if performed).
No symptoms and no organ damage from the paraprotein.
General Considerations
MGUS is present in 1% of all adults (3% of those over age 50 years and more than 5% of those over age 70 years). Among all patients with paraproteins, MGUS is far more common than plasma cell myeloma. MGUS is defined as bone marrow monoclonal plasma cells less than 10% in the setting of a paraprotein (serum M-protein less than 3 g/dL [30 g/L]) and the absence of plasma cell–related end-organ damage. If an excess of serum free light chains (kappa or lambda) is established, the kappa to lambda ratio is 100 or less or 0.01 or greater. In approximately one-quarter of cases, MGUS progresses to overt malignant disease in a median of one decade. The transformation of MGUS to plasma cell myeloma is approximately 1% per year. Two adverse risk factors for progression of MGUS to a plasma cell or lymphoid malignancy are an abnormal serum kappa to lambda free light chain ratio and a serum monoclonal protein (M-protein) level 1.5 g/dL or greater. Patients with MGUS have shortened survival (median 8.1 years vs 12.4 years for age- and sex-matched controls). In addition, 12% of patients with MGUS will convert to primary amyloidosis in a median of 9 years. Plasma cell myeloma, smoldering plasma cell myeloma, and MGUS must be distinguished from reactive (benign) polyclonal hypergammaglobulinemia (common in cirrhosis or chronic inflammation).
Laboratory Findings
To establish the diagnosis, serum and urine should be sent for PEP and IFE to search for a monoclonal protein; serum should be sent for free light chain analysis and quantitative immunoglobulins. Additional tests include a hemoglobin and serum albumin, calcium, and creatinine. If these additional tests are normal (or if abnormal but otherwise explained), then a bone marrow biopsy is usually deferred provided the serum M-protein is less than 3 g/dL (less than 30 g/L). In asymptomatic individuals, a skeletal survey (radiographs) is performed, but if there are some bone complaints or a question regarding bone disease, MRI or PET/CT imaging is preferred. MGUS is diagnosed if patients do not meet the criteria for smoldering plasma cell myeloma or plasma cell myeloma.
Treatment
Patients with MGUS are observed without treatment.
Atkin C et al. What is the significance of monoclonal gammopathy of undetermined significance? Clin Med (Lond). 2018 Oct;18(5):391–6. [PMID: 30287433]
Kyle RA et al. Long-term follow up of monoclonal gammopathy of undetermined significance. N Engl J Med. 2018 Jan 18;378(3):241–9. [PMID: 29342381]
Willrich MAV et al. Laboratory testing for monoclonal gammopathies: focus on monoclonal gammopathy of undetermined significance and smoldering multiple myeloma. Clin Biochem. 2018 Jan;51:38–47. [PMID: 28479151]
WALDENSTRÖM MACROGLOBULINEMIA
ESSENTIALS OF DIAGNOSIS
Monoclonal IgM paraprotein.
Infiltration of bone marrow by plasmacytic lymphocytes.
Absence of lytic bone disease.
General Considerations
Waldenström macroglobulinemia is a syndrome of IgM hypergammaglobulinemia that occurs in the setting of a low-grade non-Hodgkin lymphoma characterized by B cells that are morphologically a hybrid of lymphocytes and plasma cells. These cells characteristically secrete the IgM paraprotein, and many clinical manifestations of the disease are related to this macroglobulin.
Clinical Findings
A. Symptoms and Signs
This disease characteristically develops insidiously in patients in their 60s or 70s. Patients usually present with fatigue related to anemia. Hyperviscosity of serum may be manifested in a number of ways. Mucosal and gastrointestinal bleeding is related to engorged blood vessels and platelet dysfunction. Other complaints include nausea, vertigo, and visual disturbances. Alterations in consciousness vary from mild lethargy to stupor and coma. The IgM paraprotein may also cause symptoms of cold agglutinin disease (hemolysis) or chronic demyelinating peripheral neuropathy.
On examination, there may be hepatosplenomegaly or lymphadenopathy. The retinal veins are engorged. Purpura may be present. There should be no bone tenderness.
B. Laboratory Findings
Anemia is nearly universal, and rouleaux formation is common, although the red blood cells are agglutinated when the blood smear is prepared at room temperature. The anemia is related in part to expansion of the plasma volume by 50–100% due to the presence of the paraprotein. Other blood counts are usually normal. The abnormal plasmacytic lymphocytes may appear in small numbers on the peripheral blood smear. The bone marrow is characteristically infiltrated by the plasmacytic lymphocytes.
The hallmark of macroglobulinemia is the presence of a monoclonal IgM spike seen on serum PEP in the beta-globulin region. The serum viscosity is usually increased above the normal of 1.4–1.8 times that of water. Symptoms of hyperviscosity usually develop when the serum viscosity is over four times that of water, and marked symptoms usually arise when the viscosity is over six times that of water. Because paraproteins vary in their physicochemical properties, there is no strict correlation between the concentration of paraprotein and serum viscosity.
The IgM paraprotein may cause a positive antiglobulin (Coombs) test for complement and have cold agglutinin or cryoglobulin properties. If macroglobulinemia is suspected but the serum PEP shows only hypogammaglobulinemia, the test should be repeated while taking special measures to maintain the blood at 37°C, since the paraprotein may precipitate out at room temperature. Bone radiographs are normal, and there is no evidence of kidney injury.
Differential Diagnosis
Waldenström macroglobulinemia is differentiated from MGUS by the finding of bone marrow infiltration with monoclonal malignant cells. It is distinguished from CLL by bone marrow morphology, the absence of CD5 expression, and the absence of lymphocytosis, and it is distinguished from plasma cell myeloma by bone marrow morphology, the finding of the characteristic IgM paraprotein, and the absence of lytic bone disease.
Treatment
Patients with marked hyperviscosity syndrome (stupor, coma, pulmonary edema) should be treated on an emergency basis with plasmapheresis. On a chronic basis, some patients can be managed with periodic plasmapheresis alone. As with other indolent malignant lymphoid diseases, rituximab (375 mg/m2 intravenously weekly for 4–8 weeks) has significant activity. However, a word of caution: the IgM often rises first after rituximab therapy before it falls. Combination therapy is recommended for advanced disease (see Table 39–3). MYD88 is commonly mutated in Waldenström macroglobulinemia, and in patients with relapsed or refractory disease, the BTK inhibitor ibrutinib, at a dose of 420 mg daily, has shown significant activity with a 90% response rate and a 73% major response rate that can result in durable remissions. Bortezomib, lenalidomide, and bendamustine have also been shown to have activity in this disease. Autologous hematopoietic stem cell transplantation is reserved for relapsed or refractory patients.
Prognosis
Waldenström macroglobulinemia is an indolent disease with a median survival rate of 5 years, and 10% of patients are alive at 15 years.
When to Refer
All patients should be referred to a hematologist or an oncologist.
When to Admit
Patients should be admitted for treatment of hyperviscosity syndrome.
Advani P et al. Updates in prognostication and treatment of Waldenström’s macroglobulinemia. Hematol Oncol Stem Cell Ther. 2019 Dec;12(4):179–88. [PMID: 31158330]
Benevolo G et al. Current options to manage Waldenström’s macroglobulinemia. Expert Rev Hematol. 2017 Jul;10(7):637–47. [PMID: 28592170]
Castillo JJ et al. What is new in the treatment of Waldenström macroglobulinemia? Leukemia. 2019 Nov;33(11):2555–62. [PMID: 31591468]
Dimopoulos MA et al; INNOVATE Study Group and the European Consortium for Waldenström’s Macroglobulinemia. Phase 3 trial of ibrutinib plus rituximab in Waldenström’s macroglobulinemia. N Engl J Med. 2018 Jun 21;378(25):2399–410. [PMID: 29856685]
AMYLOIDOSIS
ESSENTIALS OF DIAGNOSIS
Congo red positive amyloid protein on tissue biopsy.
Primary amyloid protein is kappa or lambda immunoglobulin light chain.
Serum or urine (or both) light chain paraprotein.
General Considerations
Amyloidosis is a rare condition whereby a protein abnormally deposits in tissue resulting in organ dysfunction. The propensity of a protein to be amyloidogenic is a consequence of disturbed translational or posttranslational protein folding and lack of consequential water solubility. The input of amyloid protein into tissues far exceeds its output, so amyloid build up inexorably proceeds to organ dysfunction and ultimately organ failure and premature death.
Amyloidosis is classified according to the type of amyloid protein deposited. The six main categories are primary (immunoglobulin light chain [AL]), secondary (serum protein A, produced in inflammatory conditions [AA]), hereditary (mutated transthyretin [TTR]; many others), senile (wild-type TTR; atrial natriuretic peptide; others), dialysis-related (beta-2-microglobulin, not filtered out by dialysis membranes [Abeta-2M]), and LECT2 (associated with Latino ethnicity). Amyloidosis is further classified as localized (amyloid deposits only in a single tissue type or organ) or, most common, systemic (widespread amyloid deposition).
Clinical Findings
A. Symptoms and Signs
Patients with localized amyloidosis have symptoms and signs related to the affected single organ, such as hoarseness (vocal cords) or proptosis and visual disturbance (orbits). Patients with systemic amyloidosis have symptoms and signs of unexplained medical syndromes, including heart failure (infiltrative/restrictive cardiomyopathy), nephrotic syndrome, malabsorption and weight loss, hepatic dysfunction, autonomic insufficiency, carpal tunnel syndrome (often bilateral), and sensorimotor peripheral neuropathy. Other symptoms and signs include an enlarged tongue; waxy, rough plaques on skin; contusions (including the periorbital areas); cough or dyspnea; and disturbed deglutition. These symptoms and signs arise insidiously, and the diagnosis of amyloidosis is generally made late in the disease process.
B. Laboratory Findings
The diagnosis of amyloid protein requires a tissue biopsy that demonstrates deposition of a pink interstitial substance in the tissue with the hematoxylin and eosin stain. This protein stains red with Congo red and becomes an apple-green color when the light is polarized. Amyloid is a triple-stranded fibril composed of the amyloid protein, amyloid protein P, and glycosaminoglycan. The amyloid fibrils form beta-pleated sheets as demonstrated by electron microscopy. In primary amyloidosis, the amyloid protein is either the kappa or lambda immunoglobulin light chain.
When systemic amyloidosis is suspected, a blind aspiration of the abdominal fat pad will reveal amyloid two-thirds of the time. If the fat pad aspiration is unrevealing, then the affected organ needs biopsy. In 90% of patients with primary amyloidosis, analysis of the serum and urine will reveal a kappa or lambda light chain paraprotein by PEP, IFE, or free light chain assay; in the remainder, mass spectroscopy demonstrates light chain in the tissue biopsy. Lambda amyloid is more common than kappa amyloid, a relative proportion opposite from normal B-cell stoichiometry. Most patients with primary amyloidosis have a small excess of kappa- or lambda-restricted plasma cells in the bone marrow (but less than 10%). The bone marrow may or may not demonstrate interstitial amyloid deposition or amyloid in the blood vessels.
Patients with primary cardiac amyloidosis have an infiltrative cardiomyopathy with thick ventricular walls on echocardiogram that sometimes shows a specific speckling pattern. Paradoxically, QRS voltages are low on ECG. With renal amyloid, albuminuria is present, which can be in the nephrotic range. Late in renal involvement, kidney function decreases.
Differential Diagnosis
Amyloidosis must be distinguished from MGUS and plasma cell myeloma or other malignant lymphoproliferative disorders with an associated paraprotein. Of note, 12% of patients with MGUS will convert to primary amyloidosis in a median of 9 years. One-fifth of patients who have primary amyloidosis will meet the diagnostic criteria for plasma cell myeloma; conversely, 5% of patients with plasma cell myeloma will have amyloid deposition of their paraprotein at diagnosis.
Treatment
The treatment approach to primary amyloidosis closely resembles that of plasma cell myeloma. Prospective, randomized trials of plasma cell myeloma chemotherapy versus colchicine have demonstrated a survival benefit to chemotherapy. The goal is reduction of light chain production and deposition as a means to arrest progressive end-organ dysfunction. Active agents in primary amyloidosis include melphalan, cyclophosphamide, dexamethasone, lenalidomide, and bortezomib (see Table 39–3). The anti-CD38 monoclonal antibody daratumumab may have a role in treating this disorder. New antibodies are being developed that bind the deposited light chain and facilitate its breakage and dissolution. As in plasma cell myeloma, autologous hematopoietic stem cell transplantation after high-dose melphalan is used in patients with reasonable organ function and a good performance status. The treatment-related mortality, however, is higher in patients with primary amyloidosis than in plasma cell myeloma (6% vs 1%). Some patients will demonstrate end-organ improvement after therapy. Agents are being developed that facilitate amyloid dissolution or correct protein folding abnormalities in the amyloid protein.
Prognosis
Untreated primary amyloidosis is associated with progressive end-organ failure and premature death. There is no known cure for primary amyloidosis. Although virtually every tissue examined at autopsy will contain amyloid, patients with primary amyloidosis usually have one or two primary organs failing that clinically drive the presentation and prognosis. The cardiac biomarkers B-type natriuretic peptide (BNP), N-terminal pro-BNP, and troponins T and I are prognostic in this disease regardless of overt clinical cardiac involvement. Historically, patients with predominantly cardiac or autonomic nerve presentations had survivals of 3–9 months, those with carpal tunnel syndrome or nephrosis had survivals of 1.5–3 years, and those with peripheral neuropathy had survivals of 5 years. These survivals are roughly doubled with plasma cell myeloma-like treatment. In those patients able to undergo autologous hematopoietic stem cell transplantation, the median survival approaches 5 years (and approaches 10 years for those achieving a complete hematologic remission).
When to Refer
• All patients who have primary amyloidosis or in whom it is suspected should be referred to a hematologist or oncologist.
• All patients with hereditary amyloidosis should be referred to a hepatologist for consideration of liver transplantation.
When to Admit
• Patients with systemic amyloidosis require hospitalization to treat exacerbations of end-organ failure, including heart, liver, or kidney.
• Patients with primary amyloidosis require hospitalization to undergo autologous hematopoietic stem cell transplantation.
Badar T et al. Recent advances in understanding and treating immunoglobulin light chain amyloidosis. F1000Res. 2018 Aug 29;7. [PMID: 30228867]
Gertz MA. Immunoglobulin light chain amyloidosis: 2018 update on diagnosis, prognosis, and treatment. Am J Hematol. 2018 Sep;93(9):1169–80. [PMID: 30040145]
Vaxman I et al. Recent advances in the diagnosis, risk stratification, and management of systemic light-chain amyloidosis. Acta Haematol. [PMID: 30650422]
Zumbo G et al. New and developing therapies for AL amyloidosis. Expert Opin Pharmacother. 2017 Feb;18(2):139–49. [PMID: 28002971]
BLOOD TRANSFUSIONS
Most blood products are leukoreduced in-line during acquisition and are thus prospectively leukocyte-poor. Leukoreduced blood products reduce the incidence of leukoagglutination reactions, platelet alloimmunization, transfusion-related acute lung injury, and CMV exposure.
RED BLOOD CELL TRANSFUSIONS
Red blood cell transfusions are given to raise the hemoglobin levels in patients with anemia or to replace losses after acute bleeding episodes.
Preparations of Red Cells for Transfusion
Several types of preparations containing red blood cells are available (whole blood, packed red blood cells, frozen red blood cells, or autologous non-frozen red blood cells).
A. Fresh Whole Blood
The advantage of whole blood for transfusion is the simultaneous presence of red blood cells, plasma, and fresh platelets. Fresh whole blood is not absolutely necessary, since all the above components are available separately. The major indications for use of whole blood are cardiac surgery or massive hemorrhage when more than 10 units of blood is required in a 24-hour period.
B. Packed Red Blood Cells
Packed red cells are the component most commonly used to raise the hemoglobin. Each unit has a volume of about 300 mL, of which approximately 200 mL consists of red blood cells. One unit of packed red cells will usually raise the hemoglobin by approximately 1 g/dL. Current guidelines recommend a transfusion “trigger” hemoglobin threshold of 7–8 g/dL (70–80 g/L) for hospitalized critically ill patients, those undergoing cardiothoracic surgery or repair of a hip fracture, those with upper gastrointestinal bleeding, and those with hematologic malignancy undergoing chemotherapy or hematopoietic cell transplant.
C. Autologous Packed Red Blood Cells
Patients scheduled for elective surgery may donate blood for autologous transfusion. These units may be stored for up to 35 days before freezing is necessary.
Compatibility Testing
Before transfusion, the recipient’s and the donor’s blood are typed and cross-matched to avoid hemolytic transfusion reactions. Although many antigen systems are present on red blood cells, only the ABO and Rh systems are specifically tested prior to all transfusions. The A and B antigens are the most important, because everyone who lacks one or both red cell antigens has IgM isoantibodies (called isoagglutinins) in his or her plasma against the missing antigen(s). The isoagglutinins activate complement and can cause rapid intravascular lysis of the incompatible red blood cells. In emergencies, type O/Rh-negative blood can be given to any recipient, but usually packed cells are given to minimize transfusion of donor plasma containing anti-A and anti-B antibodies with the use of whole blood.
The other important antigen routinely tested for is the D antigen of the Rh system. Approximately 15% of the population lacks this antigen. In patients lacking the antigen, anti-D antibodies are not naturally present, but the antigen is highly immunogenic. A recipient whose red cells lack D and who receives D-positive blood may develop anti-D antibodies that can cause severe lysis of subsequent transfusions of D-positive red cells or reject a D-positive fetus.
Blood typing includes a cross-match assay of recipient serum for unusual alloantibodies directed against donor red blood cells by mixing recipient serum with panels of red blood cells representing commonly occurring minor red cell antigens. The screening is particularly important if the recipient has had previous transfusions or pregnancy.
Hemolytic Transfusion Reactions
The most severe hemolytic transfusion reactions are acute (temporally related to the transfusion), involving incompatible mismatches in the ABO system that are isoagglutinin-mediated. Most of these cases are due to clerical errors and mislabeled specimens. With current compatibility testing and double-check clerical systems, the risk of an acute hemolytic reaction is 1 in 76,000 transfused units of red blood cells. Death from acute hemolytic reaction occurs in 1 in 1.8 million transfused units. When hemolysis occurs, it is rapid and intravascular, releasing free hemoglobin into the plasma. The severity of these reactions depends on the dose of red blood cells given. The most severe reactions are those seen in surgical patients under anesthesia.
Delayed hemolytic transfusion reactions are caused by minor red blood cell antigen discrepancies and are typically less severe. The hemolysis usually takes place at a slower rate and is mediated by IgG alloantibodies causing extravascular red blood cell destruction. These transfusion reactions may be delayed for 5–10 days after transfusion. In such cases, the recipient has received red blood cells containing an immunogenic antigen, and in the time since transfusion, a new alloantibody has formed. The most common antigens involved in such reactions are Duffy, Kidd, Kell, and C and E loci of the Rh system. The current risk of a delayed hemolytic transfusion reaction is 1 in 6000 transfused units of red blood cells.
A. Symptoms and Signs
Major acute hemolytic transfusion reactions cause fever and chills, with backache and headache. In severe cases, there may be apprehension, dyspnea, hypotension, and cardiovascular collapse. Patients under general anesthesia will not manifest such symptoms, and the first indication may be tachycardia, generalized bleeding, or oliguria. The transfusion must be stopped immediately. In severe cases, acute DIC, acute kidney injury from tubular necrosis, or both can occur. Death occurs in 4% of acute hemolytic reactions due to ABO incompatibility. Delayed hemolytic transfusion reactions are usually without any or only mild symptoms or signs.
B. Laboratory Findings
When an acute hemolytic transfusion episode is suspected, the identification of the recipient and of the transfusion product bag label should be rechecked. The transfusion product bag with its pilot tube must be returned to the blood bank, and a fresh sample of the recipient’s blood must accompany the bag for retyping and re–cross-matching of donor and recipient blood samples. The hemoglobin will fail to rise by the expected amount. Coagulation studies may reveal evidence of acute kidney injury or acute DIC. The plasma-free hemoglobin in the recipient will be elevated resulting in hemoglobinuria.
In cases of delayed hemolytic reactions, there will be an unexpected drop in hemoglobin and an increase in the total and indirect bilirubins. The new offending alloantibody is easily detected in the patient’s serum.
C. Treatment
If an acute hemolytic transfusion reaction is suspected, the transfusion should be stopped at once. The patient should be vigorously hydrated to prevent acute tubular necrosis. Forced diuresis with mannitol may help prevent or minimize acute kidney injury.
Leukoagglutinin Reactions
Most transfusion reactions are not hemolytic but represent reactions to antigens present on transfused passenger leukocytes in patients who have been sensitized to leukocyte antigens through previous transfusions or pregnancy. Transfusion products relatively rich in leukocyte-rich plasma, especially platelets, are most likely to cause this. Moderate to severe leukoagglutinin reactions occur in 1% of red blood cell transfusions and 2% of platelet transfusions. The risk of a leukoagglutination reaction is minimal if the transfused blood product is leukoreduced in-line upon collection. Most commonly, fever and chills develop in patients within 12 hours after transfusion. In severe cases, cough and dyspnea may occur and the chest radiograph may show transient pulmonary infiltrates. Because no hemolysis is involved, the hemoglobin rises by the expected amount despite the reaction.
Leukoagglutinin reactions may respond to acetaminophen (500–650 mg orally) and diphenhydramine (25 mg orally or intravenously); corticosteroids, such as hydrocortisone (1 mg/kg intravenously), are also of value. Overall, leukoagglutination reactions are diminishing through the routine use of in-line leukotrapping during blood donation (ie, leukoreduced blood). Patients experiencing severe leukoagglutination episodes despite receiving leukoreduced blood transfusions should receive leukopoor or washed blood products.
Hypersensitivity Reactions
Urticaria or bronchospasm may develop during or soon after a transfusion. These reactions are almost always due to exposure to allogeneic plasma proteins rather than to leukocytes. The risk is low enough that the routine use of antihistamine premedications has been eliminated before packed red blood cell transfusions. However, a hypersensitivity reaction, including anaphylactic shock, may develop in patients who are IgA deficient because of antibodies to IgA in the patient’s plasma directed against the IgA in the transfused blood product. Patients with such reactions may require transfusion of washed or even frozen red blood cells to avoid future severe reactions.
Contaminated Blood
Blood products can be contaminated with bacteria. Platelets are especially prone to bacterial contamination because they cannot be refrigerated. Bacterial contamination occurs in 1 of every 30,000 red blood cell donations and 1 of every 5000 platelet donations. Receipt of a blood product contaminated with gram-positive bacteria will cause fever and bacteremia, but rarely causes a sepsis syndrome. Receipt of a blood product contaminated with gram-negative bacteria often causes septic shock, acute DIC, and acute kidney injury due to the transfused endotoxin and is usually fatal. Strategies to reduce bacterial contamination include enhanced venipuncture site skin cleansing, diverting of the first few milliliters of donated blood, use of single-donor blood products (as opposed to pooled-donor products), and point-of-care rapid bacterial screening in order to discard questionable units. Blood products infused with psoralen and then exposed to UVA light will have no living organisms in them, but add cost to acquisition of the blood product. The current risk of a septic transfusion reaction from a culture-negative unit of single-donor platelets (not psoralen treated) is 1 in 60,000. In any patient who may have received contaminated blood, the recipient and the donor blood bag should both be cultured, and antibiotics should be given immediately to the recipient.
Infectious Diseases Transmitted Through Transfusion
Despite the use of only volunteer blood donors and the routine screening of blood, transfusion-associated viral diseases remain a problem. All blood products (red blood cells, platelets, plasma, cryoprecipitate) can transmit viral diseases. All blood donors are screened with questionnaires designed to detect (and therefore reject) donors at high risk for transmitting infectious diseases. For example, the American Red Cross does not accept blood donation from persons with a diagnosis of COVID-19 or from contacts of persons who have or are suspected to have the causal SARS-CoV-2 virus. All blood is screened for hepatitis B surface antigen, antibody to hepatitis B core antigen and syphilis, antibodies to HIV-1 and HIV-2 and NAT (nucleic acid amplification) for HIV, antibody to hepatitis C virus (HCV) and NAT for hepatitis C, antibody to human T-cell lymphotropic/leukemia virus (HTLV), and NAT for West Nile virus. Zika virus contamination is screened for by donor questionnaire but the routine use of an FDA-approved detection test has not been uniformally adopted to screen donated blood. It is recommended that blood donors get screened once for antibodies against Trypanosoma cruzi, the infectious agent that causes Chagas disease (and if negative, no further screening for additional blood donations).
With improved screening, the risk of posttransfusion hepatitis has steadily decreased after the receipt of screened “negative” blood products. The risk of acquiring hepatitis B is about 1 in 200,000 transfused units in the United States (vs about 1 in 21,000 to 1 in 600 transfused units in Asia). The risk of hepatitis C acquisition is 1 in 1.5 to 2 million transfused units in the United States. The risk of HIV acquisition is 1 in 2 million transfused units. Unscreened but leukoreduced blood products appear to be equivalent to CMV screened-negative blood products in terms of the risk of CMV transmission to a CMV-seronegative recipient.
Transfusion Graft-Versus-Host Disease
Allogeneic passenger lymphocytes in transfused blood products will engraft in some recipients and mount an alloimmune attack against tissues expressing discrepant HLA antigens causing graft-versus-host disease (GVHD). The symptoms and signs of transfusion-associated GVHD include fever, rash, diarrhea, hepatitis, lymphadenopathy, and severe pancytopenia. The outcome is usually fatal. Transfusion-associated GVHD occurs most often in recipients with immune defects, malignant lymphoproliferative disorders, solid tumors being treated with chemotherapy or immunotherapy, treatment with immunosuppressive medications (especially purine analogs such as fludarabine), or older patients undergoing cardiac surgery. HIV infection alone does not increase the risk. The use of leukoreduced blood products is inadequate to prevent transfusion-associated GVHD. This complication can be avoided by irradiating blood products (25 Gy or more) to prevent lymphocyte proliferation in blood products given to recipients at high risk for transfusion-associated GVHD.
Transfusion-Related Acute Lung Injury
Transfusion-related acute lung injury (TRALI) occurs in 1 in every 5000 transfused units of blood products. TRALI is clinically defined as noncardiogenic pulmonary edema after a blood product transfusion without other explanation. Transfused surgical and critically ill patients seem most susceptible. It has been associated with allogeneic antibodies in the donor plasma component that bind to recipient leukocyte antigens, including HLA antigens and other granulocyte- and monocyte-specific antigens (such as HNA-1a, -1b, -2a, and -3a). In 20% of cases, no antileukocyte antibodies are identified raising the concern that bioactive lipids or other substances that accumulate while the blood product is in storage can also mediate TRALI in susceptible recipients. Ten to 20% of female blood donors and 1–5% of male blood donors have antileukocyte antibodies in their serum. The risk of TRALI is reduced through the use of male-only plasma donors, when possible. There is no specific treatment for TRALI, only supportive care.
PLATELET TRANSFUSIONS
Platelet transfusions are indicated in cases of thrombocytopenia due to decreased platelet production. They are of some use in immune thrombocytopenia when active bleeding is evident, but the clearance of transfused platelets is rapid as they are exposed to the same pathophysiologic forces as the recipient’s endogenous platelets. The risk of spontaneous bleeding rises when the platelet count falls to less than 80,000/mcL (80 × 109/L), and the risk of life-threatening spontaneous bleeding increases when the platelet count is less than 5000/mcL (5 × 109/L). Because of this, prophylactic platelet transfusions are often given at these very low levels, usually when less than 10,000/mcL (10 × 109/L). Platelet transfusions are also given prior to invasive procedures or surgery in thrombocytopenic patients, and the goal is often to raise the platelet count to 50,000/mcL (50 × 109/L) or more.
Platelets for transfusion are most commonly derived from single-donor apheresis collections (roughly the equivalent to the platelets recovered from six donations of whole blood). A single donor unit of platelets should raise the platelet count by 50,000 to 60,000 platelets per mcL (50–60 × 109/L) in a transfusion-naïve recipient without hypersplenism or ongoing platelet consumptive disorder. Transfused platelets typically last for 2 or 3 days. Platelet transfusion responses may be suboptimal with poor platelet increments and short platelet survival times. This may be due to one of several causes, including fever, sepsis, hypersplenism, DIC, large body habitus, low platelet dose in the transfusion, or platelet alloimmunization (from prior transfusions, prior pregnancy or prior organ transplantation). Many, but not all, alloantibodies causing platelet destruction are directed at HLA antigens. Patients requiring long periods of platelet transfusion support should be monitored to document adequate responses to transfusions so that the most appropriate product can be used. If random platelet transfusions prove inadequate, then the patient should be cross-matched with potential donors who might prove better able to provide adequate platelet-transfusion increments and platelet survival. Patients requiring ongoing platelet transfusions who become alloimmunized may benefit from HLA-matched platelets derived from either volunteer donors or family members.
TRANSFUSION OF PLASMA COMPONENTS
Fresh frozen plasma (FFP) is available in units of approximately 200 mL. FFP contains normal levels of all coagulation factors (about 1 unit/mL of each factor). FFP is used to correct coagulation factor deficiencies and to treat thrombotic thrombocytopenia purpura or other thrombotic microangiopathies. FFP is also used to correct or prevent coagulopathy in trauma patients receiving massive transfusion of packed red blood cell (PRBC). An FFP:PRBC ratio of 1:2 or more is associated with improved survival in trauma patients receiving massive transfusions, regardless of the presence of a coagulopathy.
Cryoprecipitate is made from fresh plasma by cooling the plasma to 4°C and collecting the precipitate. One unit of cryoprecipitate has a volume of approximately 15–20 mL and contains approximately 250 mg of fibrinogen and between 80 and 100 units of factor VIII and von Willebrand factor. Cryoprecipitate is most commonly used to supplement fibrinogen in cases of acquired hypofibrinogenemia (eg, acute DIC) or in rare instances of congenital hypofibrinogenemia. One unit of cryoprecipitate will raise the fibrinogen level by about 8 mg/dL (0.24 mcmol/L). Cryoprecipitate is sometimes used to temporarily correct the acquired qualitative platelet dysfunction associated with kidney disease.
Bryan AW Jr et al. Plasma transfusion demystified: a review of the key factors influencing the response to plasma transfusion. Lab Med. 2017 May 1;48(2):108–12. [PMID: 28444398]
Goodnough LT et al. Blood transfusion therapy. Med Clin North Am. 2017 Mar;101(2):431–47. [PMID: 28189180]
Hoeks MPA et al. Impact of red blood cell transfusion strategies in haemato-oncological patients: a systematic review and meta-analysis. Br J Haematol. 2017 Jul;178(1):137–51. [PMID: 28589623]
Mueller MM; ICC PBM Frankfurt 2018 Group. Patient blood management: recommendations from the 2018 Frankfurt Consensus Conference. JAMA. 2019 Mar 12;321(10):983–97. [PMID: 30860564]
Roubinian N. TACO and TRALI: biology, risk factors, and prevention strategies. Hematology Am Soc Hematol Educ Program. 2018 Nov 30;2018(1):585–94. [PMID: 30570487]