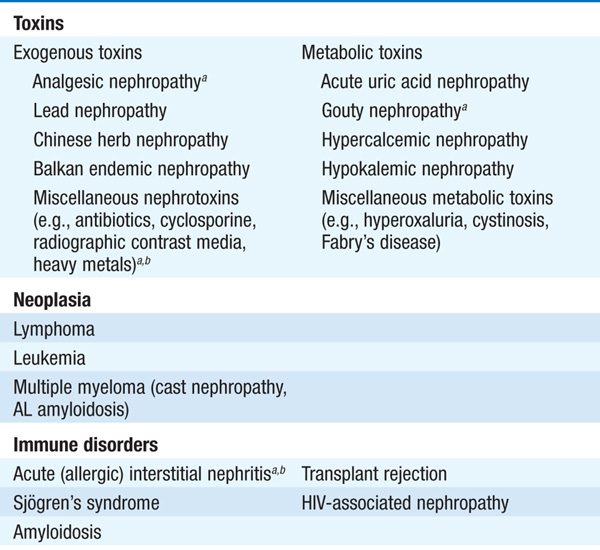
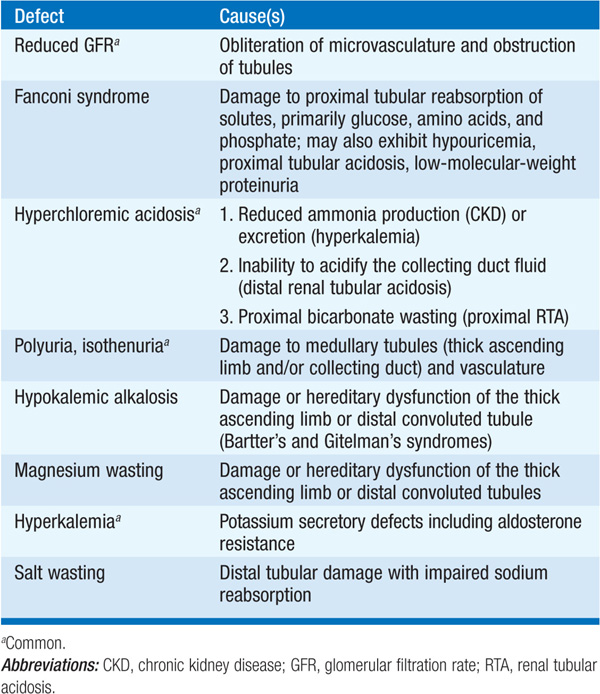
Tubulointerstitial diseases constitute a diverse group of acute and chronic, hereditary and acquired disorders involving the renal tubules and supporting structures (Table 153-1). Functionally, they may result in a wide variety of physiologic phenotypes, including nephrogenic diabetes insipidus (DI) with polyuria, non-anion-gap metabolic acidosis, salt wasting, and hypo-or hyperkalemia. Azotemia is common, owing to associated glomerular fibrosis and/or ischemia. Compared with glomerulopathies, proteinuria and hematuria are less dramatic, and hypertension is less common. The functional consequences of tubular dysfunction are outlined in Table 153-2.
TABLE 153-1 PRINCIPAL CAUSES OF TUBULOINTERSTITIAL DISEASE OF THE KIDNEY
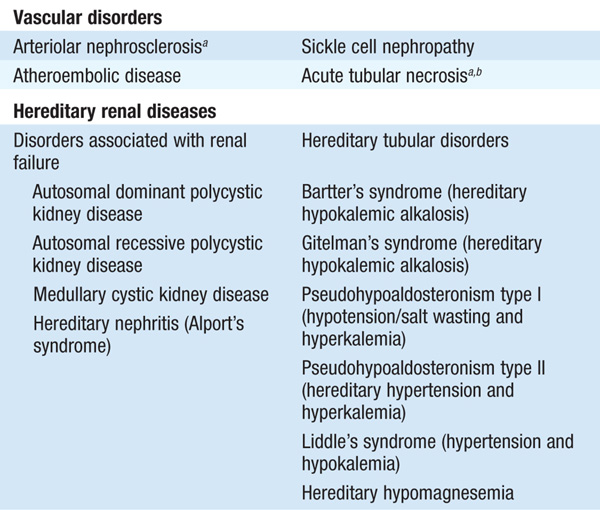
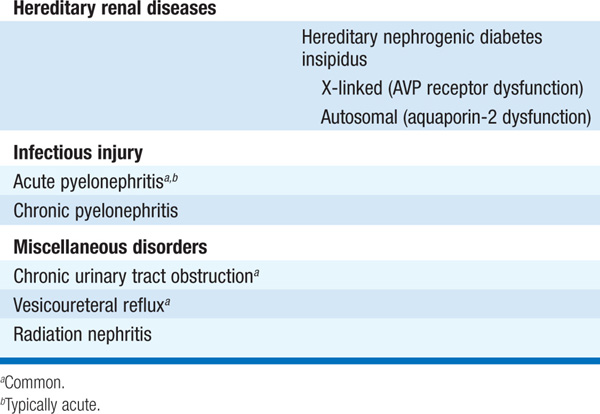
TABLE 153-2 TRANSPORT DYSFUNCTION IN TUBULOINTERSTITIAL DISEASE
Drugs are a leading cause of this type of renal failure, usually identified by a gradual rise in the serum creatinine at least several days after the institution of therapy, occasionally accompanied by fever, eosinophilia, rash, and arthralgias. The onset of renal dysfunction may be very rapid in pts who have previously been sensitized to the offending agent; this is particularly true for rifampin, for which intermittent or interrupted therapy appears to be associated with the development of AIN. In addition to azotemia, there may be evidence of tubular dysfunction (e.g., hyperkalemia, metabolic acidosis). Urinalysis may show hematuria, pyuria, white cell casts, and eosinophiluria on Hansel’s or Wright’s stain; notably, however, eosinophiluria is not specific for AIN, occurring in other causes of acute kidney injury (AKI), including atheroemboli.
Drugs that commonly cause AIN are listed in Table 153-3. Some drugs have a particular predilection for causing AIN, e.g., nafcillin; however, less frequent causes may be apparent only from case reports, such that a detailed history and literature review may be required to make the association with AIN. Many drugs, nonsteroidal anti-inflammatory drugs (NSAIDs) in particular, may elicit a glomerular lesion with similarity to minimal change disease, in addition to AIN; these pts typically have nephrotic-range proteinuria, versus the modest proteinuria typically associated with tubulointerstitial disease.
TABLE 153-3 CAUSES OF ACUTE INTERSTITIAL NEPHRITIS

Renal dysfunction in drug-associated AIN usually improves after withdrawal of the offending drug, but complete recovery may be delayed and incomplete. In uncontrolled studies, glucocorticoids have been shown to promote earlier recovery of renal function and reduce fibrosis; this therapy is generally reserved to avoid or reduce the duration of dialytic therapy in pts who fail to respond to medication withdrawal.
AIN may also occur in the context of systemic infections, classically leptospirosis, Legionella infection, and streptococcal bacterial infection. Interstitial nephritis characterized by a dense infiltrate of IgG4-expressing plasma cells can occur as part of IgG4-related systemic disease; pancreatitis, retroperitoneal fibrosis, and a chronic sclerosing sialadenitis may variably be present. Finally, the tubulointerstitial nephritis and uveitis syndrome (TINU) is another increasingly recognized form of AIN. In addition to uveitis, which may precede or follow the AIN in pts with TINU, systemic symptoms and signs are common, e.g., weight loss, fever, malaise, arthralgias, and an elevated erythrocyte sedimentation rate. The renal disease is typically self-limited; those with progressive disease are often treated with prednisone.
Analgesic nephropathy is an important cause of chronic kidney disease that results from the cumulative (in quantity and duration) effects of combination analgesic agents, usually phenacetin and aspirin. It is thought to be a more common cause of end-stage renal disease (ESRD) in Australia/New Zealand than elsewhere owing to the larger per capita ingestion of analgesic agents in that region of the world. Transitional cell carcinoma may develop. Analgesic nephropathy should be suspected in pts with a history of chronic headache or back pain with chronic kidney disease (CKD) that is otherwise unexplained. Manifestations include papillary necrosis, calculi, sterile pyuria, and azotemia.
A severe form of chronic tubulointerstitial fibrosis has been associated with the ingestion of Chinese herbal medicines, typically employed as part of a dieting regimen; Balkan endemic nephropathy (BEN), geographically restricted to pts from this region of southeastern Europe, shares many similarities with Chinese herbal nephropathy. These disorders are thought to be caused by exposure to aristolochic acid and/or other plant, endemic (in BEN), and medical toxins (the appetite suppressants fenfluramine and diethylpropion, in Chinese herbal nephropathy). Like analgesic nephropathies, these syndromes are both characterized by a high incidence of genitourinary malignancy.
Chronic therapy with lithium can also cause a chronic tubulointerstitial nephritis, often accompanied by nephrogenic DI that persists following discontinuation of the medication. If at all feasible, lithium-treated pts with evolving CKD should be transitioned to alternative medications for their psychiatric disease (e.g., valproic acid).
Metabolic causes of chronic IN include hypercalcemia (with nephrocalcinosis), oxalosis (primary or secondary, e.g., with intestinal disease and hyperabsorption of dietary oxalate), hypokalemia, and hyperuricemia or hyperuricosuria. The renal pathology associated with chronic hypokalemia includes a relatively specific proximal tubular vacuolization, interstitial nephritis, and renal cysts; both chronic and acute renal failure have been described. Chronic IN can occur in association with several systemic diseases, including sarcoidosis, Sjögren’s syndrome, and following radiation or chemotherapy exposure (e.g., ifosfamide, cisplatin).
Monoclonal immunoglobulins are associated with a wide variety of renal manifestations (Table 153-4), of which myeloma-associated cast nephropathy is the most common. The physiochemical characteristics of the monoclonal immunoglobulin light or heavy chains determine the clinical phenotype in individual pts, most commonly cast nephropathy, light chain deposition disease, and AL amyloidosis. In cast nephropathy, filtered light chains aggregate and cause tubular obstruction, tubular damage, and interstitial inflammation. Pts can present with CKD or with AKI; important predisposing factors in acute cast nephropathy include hypercalcemia and volume depletion.
TABLE 153-4 RENAL DISEASES ASSOCIATED WITH MONOCLONAL IMMUNOGLOBULINS
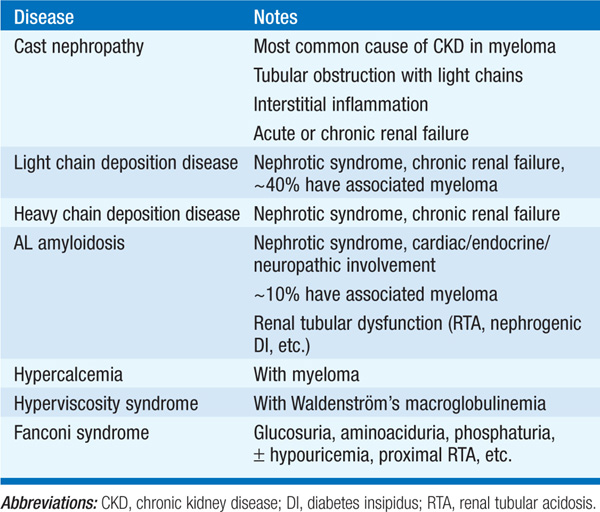
Diagnosis of cast nephropathy relies on the detection of monoclonal light chains in serum and/or urine, typically by protein electrophoresis and immunofixation. Dipstick analysis of the urine for protein is classically negative in cast nephropathy, despite the excretion of up to several grams a day of light chain protein; light chains are not detected by this screening test, which tests only for albuminuria. In contrast, the glomerular deposition of light chains in light chain deposition disease or AL amyloidosis can result in nephrotic-range proteinuria (Table 153-4), with strongly positive urine dipstick for protein.
Management of cast nephropathy encompasses aggressive hydration, treatment of hypercalcemia if present, and chemotherapy for the associated multiple myeloma. Some experts advocate the use of plasmapheresis for pts with severe AKI, high levels of serum monoclonal light chains, and a renal biopsy demonstrating cast nephropathy.
Filtered light chains and multiple other low-molecular-weight proteins are also endocytosed and metabolized by the proximal tubule. Rarely, specific light chains generate crystalline depositions within proximal tubule cells, causing a Fanconi syndrome; again, this property appears to be caused by the specific physicochemical characteristics of the associated light chains. Fanconi syndrome or dysfunction of the distal nephron (hyperkalemic acidosis or nephrogenic DI) may also complicate renal amyloidosis.
Autosomal dominant polycystic kidney disease (ADPKD) is the most common life-threatening monogenic genetic disorder, caused by autosomal dominant mutations in the PKD1 and PKD2 genes; it is a quantitatively important cause of ESRD. Autosomal recessive polycystic disease is a less much common cause of renal failure, typically presenting in infancy; hepatic involvement is much more prominent. The massive renal cysts in ADPKD can lead to progressive CKD, episodic flank pain, hematuria (often gross), hypertension, and/or urinary tract infection. The kidneys are often palpable and occasionally of very large size. Hepatic cysts and intracranial aneurysms may also be present; pts with ADPKD and a family history of ruptured intracranial aneurysms should undergo presymptomatic screening. Other common extrarenal features include diverticulosis and mitral valve prolapse.
The expression of ADPKD is highly variable, with the age of onset of ESRD ranging from childhood to old age. The renal phenotype is much milder in pts with mutations in PKD1, who on average develop ESRD approximately 15 years earlier than those with PKD2 mutations. Indeed, some pts with ADPKD discover the disease incidentally in late adult life, having had mild to moderate hypertension earlier.
The diagnosis is usually made by ultrasonography. In a 15- to 29-year-old at-risk individual from a family with ADPKD, the presence of at least two renal cysts (unilateral or bilateral) is sufficient for diagnosis. Notably, however, renal cysts are a common ultrasound finding in older pts without ADPKD, particularly those with CKD. Therefore, in at-risk individuals 30–59 years of age, the presence of at least two cysts in each kidney is required for the diagnosis; this increases to four cysts in each kidney for those older than 60. Conversely, the absence of at least two cysts in each kidney excludes the diagnosis of ADPKD in at-risk individuals between the ages of 30 and 59.
Hypertension is common in ADPKD, often in the absence of an apparent reduction in glomerular filtration rate. Activation of the renin-angiotensin system appears to play a dominant role; angiotensin-converting enzyme inhibitors or angiotensin receptor blockers are the recommended antihypertensive agents, with a target blood pressure of 120/80 mmHg. Promising treatment modalities for halting progression of CKD in ADPKD include vasopressin antagonists, which dramatically reduce cyst enlargement and renal progression in animal models.
Urinary tract infections are also common in ADPKD. In particular, pts may develop cyst infections, often with negative urine cultures and an absence of pyuria. Pts with an infected cyst may have a discrete area of tenderness, as opposed to the more diffuse discomfort of pyelonephritis; however, clinical distinction between these two possibilities can be problematic. Many commonly used antibiotics, including penicillins and aminoglycosides, fail to penetrate cysts and are ineffective; therapy of kidney infections in ADPKD should use an antibiotic that is known to penetrate cysts (e.g., quinolones), guided initially by local antimicrobial susceptibility patterns.
This describes a number of pathophysiologically distinct entities of tubular function whose common feature is the presence of a non-anion-gap metabolic acidosis. Diarrhea, CKD, and RTA together constitute the vast majority of cases of non-anion-gap metabolic acidosis. Pts with earlier stages of CKD (Table 52-1) typically develop a non-anion-gap acidosis, with a superimposed increase in the anion gap at later stages (Chap. 2). Acidosis may develop at an earlier stage of CKD in those with prominent injury to the distal nephron, as for example in reflux nephropathy.
Pts are unable to acidify the urine despite systemic acidosis; the urinary anion gap is positive, reflective of a decrease in ammonium excretion (Chap. 2). Distal hypokalemic RTA may be inherited (both autosomal dominant and autosomal recessive) or acquired due to autoimmune and inflammatory diseases (e.g., Sjögren’s syndrome, sarcoidosis), urinary tract obstruction, or amphotericin B therapy. Chronic type I RTA is typically associated with hypercalciuria and osteomalacia, a consequence of the long-term buffering of acidosis by bone.
There is a defect in bicarbonate reabsorption, usually associated with features of Fanconi syndrome, including glycosuria, aminoaciduria, phosphaturia, and uricosuria (indicating proximal tubular dysfunction). Isolated proximal RTA is caused by hereditary dysfunction of the basolateral sodium-bicarbonate cotransporter. Fanconi syndrome may be inherited or acquired due to myeloma, chronic IN (e.g., Chinese herbal nephropathy), or drugs (e.g., ifosfamide, tenofovir). Treatment requires large doses of bicarbonate (5–15 mmol/kg per day), which may aggravate hypokalemia.
This may be due to hyporeninemic hypoaldosteronism or to resistance of the distal nephron to aldosterone. Hyporeninemic hypoaldosteronism is typically associated with volume expansion and most commonly seen in elderly and/or diabetic pts with CKD. The hyperkalemia associated with NSAIDs and cyclosporine is at least partially due to hyporeninemic hypoaldosteronism. Pts with hyporeninemic hypoaldosteronism are typically hyperkalemic; they may also exhibit a mild non-anion-gap acidosis, with urine pH <5.5 and a positive urinary anion gap. Acidosis often improves with reduction in serum [K+]; hyperkalemia appears to interfere with medullary concentration of ammonium by the renal countercurrent mechanism. Should reduction in serum [K+] not improve acidosis, pts should be treated with oral bicarbonate or citrate. Finally, various forms of distal tubular injury and tubulointerstitial disease, e.g., interstitial nephritis, are associated with distal insensitivity to aldosterone; urine pH is classically >5.5, again with a positive urinary anion gap.

For a more detailed discussion, see Beck LH, Salant DJ: Tubulointerstitial Diseases of the Kidney, Chap. 285, p. 2367, in HPIM-18.