Chapter 47
Learning and Memory
Basic Mechanisms
Significant advances have been made in understanding the ways in which the nervous system encodes and retrieves information. One focus of current research is on the understanding of learning and memory at the cellular level, where the information-encoding process can be traced to changes in neuronal properties such as membrane excitability and synaptic strength. One emerging principle is that no single universal mechanism for learning and memory exists. Instead, different memory systems (see Chapter 48) can use different mechanisms. Therefore, a comprehensive understanding of memory mechanisms requires an understanding of the general ways in which neurons are changed by learning and the ways in which those changes are maintained and expressed at the cellular level.
Research during the past several decades on several vertebrate and invertebrate model systems has led to the development of several general principles. A list of these principles (Byrne, 1987) might include the following:
1. Multiple memory systems are present in the brain (see also Chapter 48).
2. Short-term and long-term forms of learning and memory involve changes in existing neural circuits.
3. These changes may involve multiple cellular mechanisms within individual neurons.
4. Second-messenger systems play a role in mediating cellular changes.
5. Changes in membrane channels are often correlated with learning and memory.
6. Long-term memory requires new protein synthesis, whereas short-term memory does not.
This chapter describes several types of neural and molecular mechanisms implicated in learning and memory. The chapter first considers some of the paradigms that have been used to study simple forms of nonassociative and associative learning and provides an example of mechanistic analyses that have been performed in a selected invertebrate model system. The later sections of the chapter describe the mechanisms of two phenomena, which are known as long-term potentiation (LTP) and long-term depression (LTD). Both LTP and LTD occur in forebrain structures, and LTP and LTD are thought to be mechanisms for memory storage in the central nervous system.
Paradigms have been Developed to Study Associative and Nonassociative Learning
Associative Learning
Associative learning is a broad category that includes many of our daily learning activities that involve the formation of associations among stimuli and/or responses. It is usually subdivided into classical conditioning and instrumental conditioning. Classical (or Pavlovian) conditioning is induced by a procedure in which a generally neutral stimulus, termed a conditioned stimulus (CS), is paired with a stimulus that generally elicits a response, termed an unconditioned stimulus (US). Two examples of unconditioned stimuli are food, which elicits salivation, or a shock to the foot, which elicits limb withdrawal. Instrumental (or operant) conditioning is a process by which an organism learns to associate consequences with its own behavior. The delivery of a reinforcing stimulus is contingent upon the expression of a designated behavior. The probability that this behavior will be expressed is then altered. This chapter focuses on classical conditioning, as it is mechanistically the best understood type of associative conditioning.
An astute observation by Ivan Pavlov, a Russian physiologist who had been studying digestion in dogs, led to his discovery of classical conditioning. He first noticed that the mere sight of the food dish caused dogs to salivate. He continued the experiments to see if dogs would also salivate in response to a bell rung at feeding time. Pavlov trained dogs to stand in a harness and, after the sound of a bell, fed them meat powder (Fig. 47.1). At first, the bell by itself did not elicit any response, but the meat powder elicited reflex salivation, which was termed the unconditioned response (UR). He noted that after a few pairings of the bell and meat powder the dogs began to salivate when the bell rang, before they received the meat powder. This response is termed the conditioned response (CR). This type of conditioning came to be called reward or appetitive classical conditioning. If the bell or another stimulus was followed by an unpleasant event, such as an electric shock, then a variety of autonomic responses became conditioned. This type of conditioning is termed aversive conditioning.
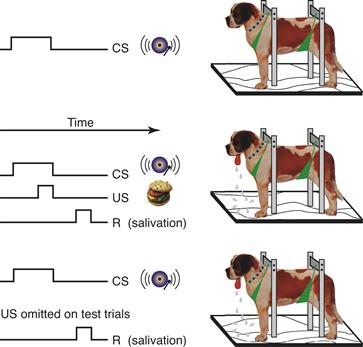
Figure 47.1 Classical conditioning. In the procedure introduced by Pavlov, the production of saliva is monitored continuously. Presentation of meat powder (US) reliably leads to salivation, whereas some “neutral” stimulus such as a bell (CS) initially does not. With repeated pairings of the bell and meat powder, the animal learns that the bell predicts the food and salivates in response to the bell alone.
Modified from Rachlin (1991).
Nonassociative Learning
Three examples of nonassociative learning have received the most experimental attention: habituation, dishabituation, and sensitization. Habituation is defined as a reduction in the response to a stimulus that is delivered repeatedly. Dishabituation refers to the restoration or recovery of a habituated response due to the presentation of another, typically strong, stimulus to the animal. Sensitization is an enhancement or augmentation of a response produced by the presentation of a strong stimulus. The following sections focus on the neural and molecular mechanisms of sensitization.
Invertebrate Studies: Key Insights from Aplysia into Basic Mechanisms of Learning
Since the mid-1960s, the marine mollusc Aplysia has proven to be an extremely useful model system to gain insights into the neural and molecular mechanisms of simple forms of memory. Indeed, the pioneering discoveries of Eric Kandel using this animal were recognized by his receipt of the Nobel Prize in Physiology or Medicine in 2000. A number of characteristics make Aplysia well suited for the examination of the molecular, cellular, morphological, and network mechanisms underlying neuronal modifications (plasticity) and learning and memory. The animal has a relatively simple nervous system with large, individually identifiable neurons that are accessible for detailed analyses. Indeed, the neurons and neural circuits that mediate many behaviors in Aplysia have been identified. In several cases, these behaviors have been shown to be modifiable by learning. Moreover, specific loci within neural circuits at which modifications occur during learning have been identified, and aspects of the cellular mechanisms underlying these modifications have been analyzed (For reviews see Bailey & Kandel, 2008; Byrne & Kandel, 1996; Fioravante, Antzoulatos, & Byrne, 2008; Kandel, 2001).
Sensitization of withdrawal reflexes has been particularly well studied. A single sensitizing stimulus, such as a brief several second-duration electric shock, can produce a reflex enhancement that lasts minutes (short-term sensitization), whereas prolonged training (e.g., multiple stimuli over an hour or more) produces an enhancement that lasts from days to weeks (long-term sensitization).
Multiple Cellular Processes Mediate Short- and Long-Term Sensitization in Aplysia
A discussion of memory mechanisms can be divided into three parts: induction, expression, and maintenance. Induction refers to the initial events that trigger or initiate the modification process; expression concerns how the modification process is ultimately expressed; and maintenance addresses the manner in which the modification is made to endure over time.
Short-Term Sensitization
Short-term (minutes) sensitization is induced when a single, brief train of shocks is delivered to the body wall of the animal. This process results in the release of modulatory transmitters, such as serotonin (5-HT), from a distinct class of interneurons (IN) referred to as facilitatory neurons (Fig. 47.2A1). These facilitatory neurons regulate the properties of the sensory neurons (SN) and the strength of their connections with postsynaptic interneurons and motor neurons (MN) through a process called heterosynaptic facilitation (Byrne & Kandel, 1996; Figs. 47.2A2 and 47.2A3). The molecular mechanisms contributing to short-term heterosynaptic facilitation are illustrated in Figure 47.2B. Serotonin binds to multiple types of G-protein-coupled receptors localized in the membrane of SNs and activation of these receptors recruits at least two predominant signaling cascades in the SNs: the cyclic adenosine monophosphate/protein kinase A (cAMP/PKA) and the diacylglycerol/protein kinase C (DAG/PKC) cascades (Byrne & Kandel, 1996). Calcium/calmodulin-dependent protein kinase (CaM kinase II) appears to be engaged as well. PKA and PKC have multiple targets, including membrane ion channels, thus affecting the spike waveform and the excitability of the SN. For example, activation of the PKA and PKC cascades modifies various K+ channels, leading to an enhanced spike activity (i.e., enhanced excitability), and broadening of the action potential. Reflections of enhanced excitability include an increase in the number of action potentials elicited in a sensory neuron by a fixed extrinsic current injected into the cell or by a fixed stimulus to the skin (Fig. 47.2A3). Spike broadening leads to enhanced Ca2+ influx in presynaptic terminals and facilitates transmitter release. In addition to spike broadening, spike-duration-independent mechanisms of enhanced transmitter release have been implicated in 5-HT-induced heterosynaptic facilitation. These mechanisms involve direct modulation of a presynaptic calcium conductance independently of spike broadening (Leal & Klein, 2009) as well as an increase in the number of transmitter-containing synaptic vesicles that become available for release, possibly through synapsin-regulated vesicle mobilization (Fig. 47.2; Byrne & Kandel, 1996; Fioravante, Liu, Netek, Cleary, & Byrne, 2007; Zhao & Klein, 2002). This process is represented in Figure 47.2B (large open arrow) as the translocation or mobilization of transmitter vesicles from a reserve pool to a releasable pool results in an increase in the number of transmitter-containing vesicles available for release, with subsequent action potentials in the sensory neuron. The maintenance of short-term sensitization is dependent on the persistence of the PKA- and PKC-induced phosphorylations of the various substrate proteins. Prolonged treatments of 5-HT (1.5 h) also activate mitogen-activated protein kinase (MAPK) (for review, see Sharma & Carew, 2004). However, this pathway appears to be important only for the induction of longer-term processes (see section on Long-term Sensitization).
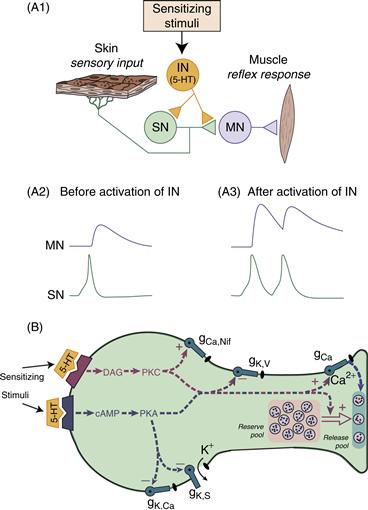
Figure 47.2 Model of short-term heterosynaptic facilitation of the sensorimotor connection that contributes to short-term sensitization in Aplysia. (A1) Sensitizing stimuli activate facilitatory interneurons (IN) that release modulatory transmitters, one of which is 5-HT. The modulator leads to an alteration of the properties of the sensory neuron (SN). (A2 and A3) The enhanced synaptic input to the motor neuron (MN) (A3) results from enhanced sensory input, partly due to two mechanisms. First, the same peripheral stimulus can evoke a greater number of action potentials in the presynaptic SN (i.e., enhanced excitability). Second, each action potential fired by an SN produces a stronger synaptic response in the MN (i.e., synaptic facilitation). (B) Model of a SN that depicts the multiple processes for short-term facilitation and changes in excitability that contribute to short-term sensitization. 5-HT released from facilitatory neurons binds to at least two distinct classes of receptors on the outer surface of the membrane and leads to the transient activation of two intracellular second messengers, DAG and cAMP, and their respective kinases (PKC and PKA). These kinases affect multiple cellular processes, the combined effects of which lead to changes in excitability and enhanced transmitter release when subsequent action potentials are fired in the sensory neuron (see text for additional details).
Modified from Byrne and Kandel (1996).
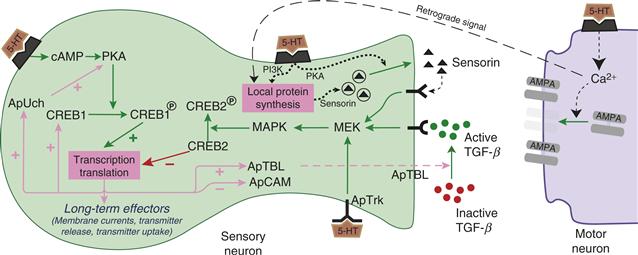
Figure 47.3 Simplified scheme of the mechanisms in SNs that contribute to long-term sensitization. Sensitization training leads to cAMP-dependent regulation of CREB1. Serotonin also leads to activation of MAPK, which regulates CREB2. Whereas CREB1 acts as an initiator of gene transcription, CREB2 acts as a repressor of gene transcription. The combined effects of activation of CREB1 and suppression of CREB2 lead to regulation of the synthesis of at least 10 proteins, only some of which are shown. ApTBL is believed to activate latent forms of TGF-β, which can then bind to receptors on the SN. TGF-β activates MAPK, which may act by initiating a second round of gene regulation by affecting CREB2-dependent pathways. Serotonin can also increase the release of the peptide sensorin, which binds to autoreceptors leading to further activation of MAPK. Increased local synthesis and subsequent release of sensorin can also be evoked by 5-HT, through the PI3 kinase and type II PKA, respectively. Because increased synthesis of sensorin requires elevation of postsynaptic calcium, a retrograde signal is also postulated. In addition to the retrograde signal, 5-HT-induced postsynaptic signaling also leads to an increased number of glutamate receptors.
The consequences of activating these multiple second-messenger systems and modulating these various cellular processes are expressed when test stimuli elicit action potentials in the sensory neuron at various times after the presentation of the sensitizing stimuli (Fig. 47.2A3). More transmitter is available for release as a result of the mobilization process and each action potential results in a larger influx of Ca2+ to trigger release of the available transmitter. This facilitation of transmitter release from the sensory neuron leads to a larger postsynaptic potential in the motor neuron. Larger postsynaptic potentials lead to enhanced activation of interneurons and motor neurons and thus to an enhanced behavioral response.
Of general significance is the observation that a single modulatory transmitter (i.e., 5-HT) activates multiple kinase systems. The involvement of multiple second messenger systems in synaptic plasticity also appears to be a theme emerging from mammalian studies. For example, as discussed in a later section of the chapter, the induction of LTP in the CA1 area of the hippocampus appears to involve MAPK, PKC, CaM kinase II, and tyrosine kinase (reviewed in Dineley et al., 2001; Miyamoto, 2006).
Long-Term Sensitization
Sensitization also exists in a long-term form, which persists for at least 24 h. Whereas short-term sensitization can be produced by a single brief stimulus, the induction of long-term sensitization requires a more extensive training period over an hour or more.
Both short- and long-term sensitization share some common cellular pathways during their induction. For example, both forms activate the cAMP/PKA cascade (Fig. 47.3). However, in the long-term form, unlike the short-term form, activation of the cAMP/PKA cascade induces gene transcription and new protein synthesis (Kandel, 2001; Fioravante et al., 2008). Repeated training leads to a translocation of PKA to the nucleus, where it phosphorylates the transcriptional activator CREB1 (cAMP responsive element binding protein1). CREB1 binds to a regulatory region of genes known as a CRE (cAMP responsive element). Whereas CREB1 acts as an activator of gene transcription, a related transcription factor, CREB2, acts constitutively as a repressor of gene transcription. This repression of transcription is in turn suppressed by MAPK phosphorylation of CREB2 (Guan et al., 2002). MAPK is activated by multiple pathways including a 5-HT sensitive tyrosine receptor kinase-like molecule (ApTrk), and through feedback pathways involving the peptide sensorin and transforming growth factor β (TGF-β) (see following).
The role of transcription factors in long-term memory formation is not limited to the induction phase but may also extend to the consolidation phase, where consolidation is defined as the time window during which RNA and protein synthesis are required for converting short- to long-term memory. For example, treatment of ganglia with 5 pulses of 5-HT over a 1.5 h period to mimic sensitization training leads to the binding of CREB1 to the promoter of its own gene and induces CREB1 synthesis. The necessity of prolonged transcription and translation for long-term facilitation (LTF) observed 24 h after treatment persists for at least 10 h after induction. These results suggest that CREB1 can regulate its own level of expression, giving rise to a CREB1 positive feedback loop that supports memory consolidation (Liu, Cleary, & Byrne, 2011).
The combined effects of activation of CREB1 and removal of CREB2’s repression of transcription lead to changes in the synthesis of specific proteins. So far, more than 10 gene products that are regulated by sensitization training have been identified, and others are likely to be found in the future. These results indicate that there is not a single memory gene or protein, but that multiple genes are regulated, and they act in a coordinated way to alter neuronal properties and synaptic strength. The following section discusses several regulated proteins of particular significance.
The downregulation of a homologue of a neuronal cell adhesion molecule (NCAM), denoted ApCAM, plays a key role in long-term facilitation. This downregulation has two components. First, the synthesis of ApCAM is reduced. Second, preexisting ApCAM is internalized via increased endocytosis. The internalization and degradation of ApCAM allow for the restructuring of the axon arbor (Bailey & Kandel, 2008). The SN can now form additional connections with the same postsynaptic target or make new connections with other cells. In addition, the presynaptic cell-adhesion protein neurexin, along with its postsynaptic counterpart neuroligin and their transsynaptic interaction, are required for 5-HT-induced LTF and the associated presynaptic structural changes (Choi et al., 2011). Furthermore, the stabilization of new structures depends on a translation-regulating protein, the Aplysia homolog of cytoplasmic polyadenylation element-binding protein (ApCPEB; Miniaci et al., 2008). ApCPEB appears to have prion-like properties in that it can appear in one of two or more conformations, one of which dominates and allows ApCPEB to self-perpetuate. Conversion of ApCPEB to the self-perpetuating state is enhanced by 5-HT and required for the persistence of LTF (Si, Kandel, Majumdar, Grindley, & Choi, 2010). Finally, it has been suggested that persistence of long-term facilitation and sensitization can be disrupted by inhibition of an Aplysia homolog of the protein kinase M (PKM Apl III; Cai, Pearce, Chen, & Glanzman, 2011). PKM (PKMζ) is a constitutively active PKC isoform that phosphorylates vertebrate AMPA-type glutamate receptors and enhances their responses, thus supporting the persistence of long-term potentiation of synapses (Sacktor, 2011).
Another protein whose synthesis is regulated by long-term facilitation is Aplysia tolloid/BMP-like protein (ApTBL-1). Tolloid and the related molecule BMP-1 appear to function as secreted Zn2+ proteases. In some systems, they activate members of the transforming growth factor β (TGF-β) family. Indeed, in SNs, TGF-β mimics the effects of 5-HT in that it produces long-term increases in the synaptic strength of the SNs (Zhang, Endo, Cleary, Eskin, & Byrne, 1997). Interestingly, TGF-β activates MAPK in the SNSs and induces its translocation to the nucleus. Thus, TGF-β could be part of an extracellular positive feedback loop, possibly leading to another round of protein synthesis (Fig. 47.3) to further consolidate the memory (Zhang, Endo, Cleary, Eskin, & Byrne, 1997). Another extracellular positive feedback loop involves the 5-HT-induced regulation of the release of the SN-specific neuropeptide sensorin (Fig. 47.3). Synthesis of sensorin is stimulated by 5-HT in a PI3 kinase (Phosphatidylinositol 3-kinase)-dependent manner and somewhat surprisingly, requires elevation of postsynaptic calcium (Cai, Chen, & Glanzman, 2008; Hu, Wu, & Schacher, 2006). The mechanism through which postsynaptic calcium regulates presynaptic local protein synthesis of sensorin remains unclear, but the release of a retrograde signal has been postulated (Cai et al., 2008). Interestingly, although the existence of a postsynaptic neuron seems to be required for long-term facilitation, it is not required for another correlate of long-term sensitization, increased SN excitability (Cleary, Lee, & Byrne, 1998; Liu et al., 2011). Sensorin binding to presynaptic autoreceptors activates MAPK (Hu et al., 2006), which phosphorylates CREB2 and contributes to transcriptional regulation during the consolidation of LTF.
Another important protein, Aplysia ubiquitin hydrolase (ApUch), appears to be involved in an intracellular positive feedback loop. During the induction of long-term facilitation, ApUch levels in SNs are increased, possibly via CREB phosphorylation and a consequent increase in ApUch transcription. The increased levels of ApUch increase the rate of degradation of proteins, via the ubiquitin-proteosome pathway, including the regulatory subunit of PKA (Chain et al., 1999). The catalytic subunit of PKA, when freed from the regulatory subunit, is highly active. Thus, increased ApUch will lead to an increase in PKA activity and a more protracted phosphorylation of CREB1. This phosphorylated CREB1 may act to further prolong ApUch expression, thus closing a positive feedback loop. Protein degradation, in general, and the role of ubiquitination in particular, is an emerging theme in recent studies on the neural basis of long-term memory (Fioravante & Byrne, 2011).
One simplifying hypothesis is that the mechanisms underlying the expression of short- and long-term sensitization are the same, but extended in time for long-term sensitization. Some evidence supports this hypothesis. For example, long-term sensitization, like short-term sensitization, is associated with an enhancement of sensorimotor connections. In addition, K+ currents and the excitability of sensory neurons are modified by long-term sensitization (Cleary et al., 1998). However, the mechanisms underlying the expression of short- and long-term sensitization differ in four fundamental ways. First, long-term sensitization training leads to a decrease in the duration of the SNs action potential (Antzoulatos & Byrne, 2007). Second, structural changes such as neurite outgrowth and active zone remodeling are associated with the expression of long-term but not with short-term sensitization (Bailey & Kandel, 2008). Interestingly, the structural changes take time to develop. Long-term synaptic facilitation measured one day after four days of training is associated with structural changes, whereas long-term facilitation measured one day after a single training session is not (Wainwright, Byrne, & Cleary, 2004). Third, long-term sensitization is associated with an increase in high-affinity glutamate uptake (Levenson et al., 2000). A change in glutamate uptake could potentially exert a significant effect on synaptic efficacy by regulating the amount of transmitter available for release, the rate of clearance from the cleft, and thereby the duration of the excitatory postsynaptic potential (EPSP) and the degree of receptor desensitization. Finally, long-term sensitization has been correlated with changes in the postsynaptic cell (i.e., the motor neuron; Cleary et al., 1998). Thus, as with other examples of memory, multiple sites of plasticity exist even within this simple reflex system.
Other Temporal Domains for the Memory of Sensitization
Historically, memory has been divided into two temporal domains, short term and long term. It has become increasing clear from studies of a number of memory systems that this distinction is overly simplistic. For example, in Aplysia, Carew and colleagues (Sutton, Masters, Bagnall, & Carew, 2001) and Kandel and colleagues (Ghirardi, Montarolo, & Kandel, 1995) have discovered an intermediate phase of memory that has distinctive temporal characteristics and a unique molecular signature. The intermediate-phase memory for sensitization is expressed at times approximately 5 min to 3 h after the beginning of training. Like long-term sensitization, its induction requires protein synthesis. Intermediate-term sensitization relies on both presynaptic and postsynaptic translation (protein synthesis; Antonov, Kandel, & Hawkins, 2010; Jin, Kandel, & Hawkins, 2011; Villareal, Li, Cai, & Glanzman, 2007), but not on transcription (mRNA synthesis). Intermediate-term synaptic facilitation, induced by 5-HT, requires activity of presynaptic PKC and postsynaptic CaM kinase II (Jin et al., 2011). In contrast to the well-established role of presynaptic PKA in short-term facilitation, its role in intermediate-term facilitation appears to be more variable, depending on the training protocol (Antonov et al., 2010; Ghirardi et al., 1995; Jin et al., 2011). Finally, intermediate-term facilitation has been correlated with activation of previously “silent” release sites by the recruitment of synaptic vesicles to preexisting varicosities (Kim et al., 2003).
In addition to intermediate-term memory, it is likely that Aplysia has different phases of long-term memory. For example, increased synthesis of a number of proteins occurs at 24 h after sensitization training and some of these proteins are different from those whose synthesis is increased during and immediately after training. These results suggest that the memory for sensitization that persists for times greater than 24 h may be dependent on the synthesis of proteins occurring at 24 h and may have a different molecular signature than the 24-h memory.
Mechanisms Underlying Associative Learning of Withdrawal Reflexes in Aplysia
In addition to sensitization, the withdrawal reflexes of Aplysia are subject to classical conditioning. The short-term classical conditioning observed at the behavioral level reflects, at least in part, a cellular mechanism called activity-dependent neuromodulation (Hawkins, Abrams, Carew, & Kandel, 1983; Walters & Byrne, 1983). A diagram of the general scheme is presented in Figure 47.4. The US pathway is activated by a shock to the animal, which elicits a withdrawal response (the UR). When a CS is paired consistently with the US, the animal will develop a withdrawal response (CR) to the CS. Activity-dependent neuromodulation is proposed as the mechanism for this pairing-specific effect. The US activates both a motor neuron (UR) and a modulatory system. The modulatory system delivers 5-HT to all the SNs (parts of the various CS pathways), which leads to a nonspecific enhancement of transmitter release from the SNs. This nonspecific enhancement contributes to short-term sensitization (see prior discussion). SNs whose activity is temporally contiguous with the US-mediated reinforcement are additionally modulated. Spiking in a SN during the presence of 5-HT leads to changes in that cell relative to other SNs whose activity was not paired with the US. Thus, a subsequent CS will lead to an enhanced activation of the reflex (Fig. 47.4B). Figure 47.5 illustrates a more detailed model of the proposed cellular mechanisms responsible for this example of classical conditioning. The modulator (US) acts by increasing the activity of adenylyl cyclase (AC), which in turn increases the levels of cAMP. Paired spiking in the SNs (CS) leads to a simultaneous increase in the level of intracellular calcium, which acting through calmodulin (CaM) greatly enhances the action of the modulator to increase AC activity and cAMP production. Thus, this system determines CS–US contiguity by a method of coincidence detection at the presynaptic terminal.
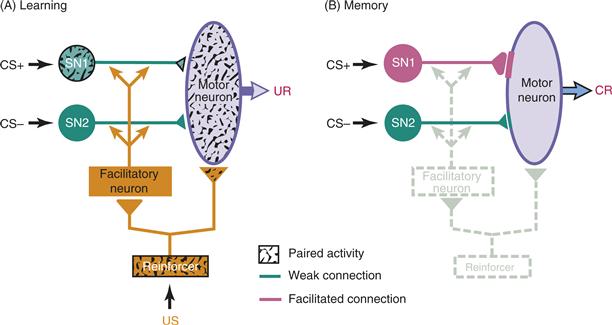
Figure 47.4 Model of classical conditioning of a withdrawal reflex in Aplysia. (A) Activity in a sensory neuron (SN1) along the CS+ (paired) pathway is coincident with activity in neurons along the reinforcement pathway (US). However, activity in the sensory neuron (SN2) along the CS– (unpaired) pathway is not coincident with activity in neurons along the US pathway. The US directly activates the motor neuron, producing the UR. The US also activates a modulatory system in the form of the facilitatory neuron, resulting in the delivery of a neuromodulatory transmitter to the two sensory neurons. The pairing of activity in SN1 with the delivery of the neuromodulator yields the associative modifications. (B) After the paired activity in A, the synapse from SN1 to the motor neuron is selectively enhanced. Thus, it is more likely to activate the motor neuron and produce the conditioned response (CR) in the absence of US input.
Modified from Lechner and Byrne (1998).
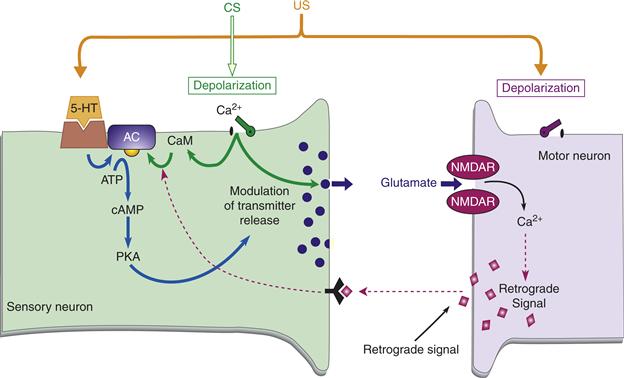
Figure 47.5 Model of associative facilitation at the Aplysia sensorimotor synapse. This model has both a presynaptic and a postsynaptic detector for the coincidence of the CS and the US. Furthermore, a putative retrograde signal allows for the integration of these two detection systems at the presynaptic level. The CS leads to activity in the sensory neuron, yielding presynaptic calcium influx, which enhances the US-induced cAMP cascade. The CS also induces glutamate release, which results in postsynaptic calcium influx through NMDA receptors if the glutamate release is paired with the US-induced depolarization of the postsynaptic neuron. The postsynaptic calcium influx putatively induces a retrograde signal, which further enhances the presynaptic cAMP cascade. The end result of the cAMP cascade is to modulate transmitter release and enhance the strength of the synapse.
Modified from Lechner and Byrne (1998).
Now, consider the postsynaptic side of the synapse. The postsynaptic region contains NMDA-type receptors (see Chapters 8 and 10). These receptors need concurrent delivery of glutamate and depolarization in order to allow calcium to enter. The glutamate is provided by the activated SN (CS), and the depolarization is provided by the US (Antonov, Antonova, Kandel, & Hawkins, 2003; Roberts & Glanzman, 2003). Thus, the postsynaptic neuron provides another example of coincidence detection. The increase in intracellular calcium putatively causes a retrograde signal to be released from the postsynaptic cell to the presynaptic terminal, ultimately acting to further enhance the cAMP cascade in the SN. The overall amplification of the cAMP cascade acts to raise the level of PKA, which in turn leads to the modulation of transmitter release. These activity-dependent changes enhance synaptic efficacy between the specific SN of the CS pathway and the motor neuron. Thus, the SN along the CS pathway will be better able to activate the motor neuron and produce the CR.
Summary
Certain invertebrates display an enormous capacity for learning and offer particular experimental advantages for analyzing the cellular and molecular mechanisms of learning. For example, behaviors in Aplysia are mediated by relatively simple neural circuits, which can be analyzed with conventional anatomical and electrophysiological approaches. Once the circuit is specified, the neural locus for the particular example of learning can be found, and biophysical, biochemical, and molecular approaches can then be used to identify mechanisms underlying the change. The relatively large size of some of these cells allows these analyses to take place at the level of individually identified neurons. Individual neurons can be removed surgically and assayed for changes in the levels of second messengers, protein phosphorylation, RNA, and protein syntheses. Moreover, peptides and nucleotides can be injected into individual neurons. This chapter has focused exclusively on Aplysia, but many other invertebrates have proven to be valuable model systems for the cellular and molecular analysis of learning and memory. Each has its own unique advantages.
Other invertebrate model systems such as Drosophila, although not ideal for cell biological approaches because of their small neurons, offer tremendous advantages for obtaining insights into mechanisms of learning and memory through the application of genetic approaches. A frequently used protocol in Drosophila employs a two-stage differential odor–shock avoidance procedure, in which animals learn to avoid odors paired (CS+) with shock but not odors explicitly unpaired (CS–). This learning is typically retained for 4–6 h, but retention for 24 h to 1 week can be produced by a spaced training procedure. Several mutants deficient in learning have been identified. Analysis of the affected genes has revealed elements of the cAMP signaling pathway as key in learning and memory. It is now known that in Drosophila as in Aplysia, the formation of long-term memory requires activation of the cAMP signaling pathway, which, in turn, activates members of the CREB transcription family. These proteins appear to be the key molecular switch that enables expression of genes necessary for the formation of long-term memories. These transcription factors are also important for long-term memory in vertebrates (see following). See Byrne (1987) and Carew (2000) for a review of other selected invertebrate model systems that have contributed importantly to the understanding of memory mechanisms.
Vertebrate Studies: Long-Term Potentiation
In contrast to the studies on invertebrates like Aplysia described earlier, it has been more difficult to link synaptic plasticity with specific examples of learning in vertebrates. However, one exciting and extensively studied candidate memory mechanism is the synaptic phenomenon termed long-term potentiation (LTP). This phenomenon is defined as a persistent increase in synaptic strength (as measured by the amplitude of the EPSP in a follower neuron) that can be induced rapidly by a brief burst of spike activity in the presynaptic afferents. The intense experimental interest in LTP is driven by the working hypothesis that this form of synaptic plasticity may participate in information storage in several brain regions. This section describes the properties of LTP and how it is studied, reviews its underlying mechanisms, and explores the possibility of linkages between LTP and learning and memory.
Long-Term Potentiation Occurs in a Variety of Neural Synapses
The first evidence that long-term modification of mammalian synapses could be induced by experimental means appeared in 1973, when Timothy Bliss and Terje Lomo demonstrated LTP in the hippocampus of the anesthetized rabbit. Brief, high-frequency stimulation (HFS) of the perforant-pathway input to the dentate gyrus produced a long-lasting enhancement of the extracellularly recorded field potential. Subsequent studies of nonanesthetized animals have shown that LTP can last for weeks or months. Originally thought to be unique to the mammalian hippocampal formation, LTP is now known to occur in the cerebellum, neocortical regions, subcortical regions such as the amygdala, mammalian peripheral nervous system, the arthropod neuromuscular junction, and the Aplysia sensorimotor synapse. It is important to bear in mind that no universal mechanism exists for inducing LTP. Indeed, different mechanisms may be used at the same synapse, depending on the experimental conditions. This chapter focuses primarily on the LTP at the synapse made by a pyramidal neuron in the CA3 region of the hippocampus to a pyramidal neuron in the CA1 region of the hippocampus.
Long-Term Potentiation at the CA3–CA1 Synapse
Within the hippocampus proper, by far the best studied synapse is that from the Schaffer collateral/commissural (Sch/com) fibers of the CA3 pyramidal cells to the CA1 pyramidal cells (Fig. 47.6). In fact, this is probably the most commonly studied synapse in the mammalian brain, due in part to its relatively simple circuitry and its laminar organization. These features make it possible to extract useful data from extracellular recordings, which are easier to perform than intracellular recordings and preferable for some purposes. Examples of LTP induction are illustrated in Figure 47.7B. The lower waveforms are extracellularly recorded field EPSPs recorded in the CA1 region in response to a single weak stimulation of the Sch/com pathway. Brief electric stimuli delivered to this pathway lead to the initiation of action potentials in the individual axons in the pathway. These action potentials then propagate to the synaptic terminals. The release of transmitter from the multiple afferent terminals produces a summated EPSP in the postsynaptic cell, which can be detected with an extracellular electrode. Test stimuli are delivered repeatedly at a low rate that produces stable EPSPs in the postsynaptic cell (Figs. 47.7A and B1). After a baseline period, a brief high-frequency tetanus is delivered. Subsequent test stimuli produce enhanced EPSPs (Fig. 47.7B2). The enhancement persists for many hours. Although the synaptic enhancement is stable after the tetanus, LTP, like heterosynaptic facilitation of the sensorimotor synapse in Aplysia, has multiple temporal domains. One domain is associated with an enhancement of the EPSP that persists for about 90 min. This form of LTP is referred to as early LTP (E-LTP). A second domain referred to as late LTP (L-LTP) is associated with synaptic enhancement that persists for periods of time greater than about 90 min. As described later, different mechanisms underlie the induction and maintenance of E- and L-LTP.
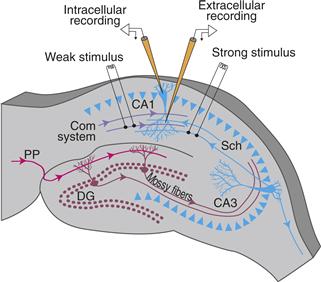
Figure 47.6 Schematic of a transverse hippocampal brain slice preparation from the rat. Two extracellular stimulating electrodes are used to activate two nonoverlapping inputs to pyramidal neurons of the CA1 region of the hippocampus. By suitably adjusting the current intensity delivered to the stimulating electrodes, different numbers of Schaffer collateral/commissural (Sch/com) axons can be activated. In this way, one stimulating electrode was made to produce a weak postsynaptic response and the other to produce a strong postsynaptic response. Also illustrated is an extracellular recording electrode placed in the stratum radiatum (the projection zone of the Sch/com inputs) and an intracellular recording electrode in the stratum pyramidale (the cell body layer). Also indicated is the mossy fiber projection from granule cells of the dentate gyrus (DG) to the pyramidal neurons of the CA3 region.
Adapted from Barrionuevo and Brown (1983).
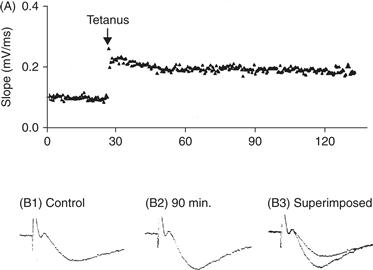
Figure 47.7 LTP at the CA3–CA1 synapse in the hippocampus. (A) Test stimuli are delivered repeatedly once every 10 s while the strength of the synaptic connection is monitored. Strength can be assessed by the amplitude of the extracellularly recorded EPSP or, as was done in this example, as the slope of the rising phase of the EPSP, which provides an accurate reflection of its strength. To induce LTP, two 1 s, 100 Hz tetani were delivered with a 20-s interval. Subsequent test stimuli produce enhanced EPSPs. The enhancement is stable and persists for at least 2 h. Examples of extracellulary recorded field EPSPs before (B1) and 90 min after the induction of LTP (B2). In B3 the traces from B1 and B2 are superimposed.
Modified from Nicoll et al. (1988).
Properties of Long-Term Potentiation at the CA3–CA1 Synapse Include Cooperativity, Associativity, and Input Specificity
The CA3–CA1 synapses exhibit a form of LTP characterized by “classical” properties that have been termed “cooperativity,” “associativity,” and “input specificity” (Fig. 47.8) (Bliss & Collingridge, 1993; Brown, Ganong, Kairiss, & Keenan, 1990). These “classical properties” are actually different manifestations of the same underlying mechanism that is responsible for this type of LTP. Other less commonly studied types of LTP have different signatures.
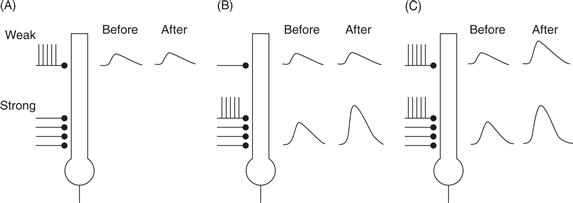
Figure 47.8 Features of LTP at CA3–CA1 synapses in the hippocampus. A single hippocampal pyramidal cell is shown receiving a weak and strong synaptic input. (A) Tetanic stimulation of the weak input alone does not cause LTP in that pathway (compare the EPSP before and after the tetanus). (B) Tetanic stimulus of the strong input alone causes LTP in the strong pathway, but not in the weak pathway. (C) Tetanic stimulation of both the weak and the strong pathway together causes LTP in both the weak and the strong pathway.
Modified from Nicoll et al. (1988).
Cooperativity refers to the fact that the probability of inducing LTP, or the magnitude of the resulting change, increases with the number of stimulated afferents. Weak HFS, which activates fewer afferents, often fails to induce LTP (Fig. 47.8A). In contrast, strong stimulation, which activates more afferents, produces LTP more reliably (Fig. 47.8B). Thus, the additional axons recruited by higher stimulation intensities “cooperate” to trigger LTP.
Associativity was shown in preparations in which two distinct axonal inputs converged onto the same post-synaptic target. Consider the interactions between two stimulus pathways, one termed the weak pathway with a small number of stimulated afferents, and the other termed the strong pathway with a large number of stimulated afferents (Fig. 47.8C). HFS of the weak input by itself failed to produce LTP in that pathway unless this stimulation was paired with tetanic stimulation of the strong input. Thus, LTP was induced in a weak input only when its activity was associated with activity in the strong input.
Input specificity means that LTP is restricted to only the inputs that received the tetanic stimulation. An unstimulated weak pathway was not facilitated after the tetanus to the strong pathway (Fig. 47.8B).
A Hebbian Mechanism Explains the Properties of Long-Term Potentiation at the CA3–CA1 Synapse in the Hippocampus
How can these classical properties of LTP in the CA1 region of the hippocampus be explained? In the late 1940s, the Canadian psychologist Donald Hebb (1949) formulated a postulate regarding the conditions that cause synapses to change. His thinking proved to be influential and guided later experiments that probed the mechanisms behind LTP. According to Hebb’s postulate:
When an axon of cell A is near enough to excite a cell B and repeatedly or persistently takes part in firing it, some growth process or metabolic change takes place in one or both cells such that A’s efficiency, as one of the cells firing B, is increased. (p. 62)
In short, coincident activity in two synaptically coupled neurons was proposed to cause increases in the synaptic strength between them. Numerous modern interpretations of Hebb’s postulate exist, but most are captured by the mnemonic: “Cells that fire together, wire together.”
Could the classical properties of LTP all be consequences of synapses that obey a Hebbian rule? Possibly so if a critical amount of postsynaptic depolarization were a necessary condition for inducing LTP in active synapses. In this case, cooperativity would result when enough input fibers were stimulated to produce the critical amount of postsynaptic depolarization. Associativity would emerge from the fact that the strong input caused sufficient depolarization of the postsynaptic membrane during the presynaptic activity in the weak input. Input specificity would occur because LTP was induced only in those inputs to a neuron that were active at the same time that the cell was sufficiently depolarized by the strong input to that neuron. In other words, these classical phenomena could all be manifestations of a single underlying Hebbian mechanism at the CA3–CA1 synapse.
Not all forms of LTP are Hebbian, however. Examples of non-Hebbian LTP can be found at the mossy fiber–CA3 synapse in the hippocampus and at the parallel fiber–Purkinje cell synapse in the cerebellum. These results indicate that the classical properties of cooperativity, associativity, and input specificity are not universal.
Mechanisms for Induction, Expression, and Maintenance of Long-Term Potentiation
LTP Induction
It is currently thought that there are multiple mechanisms or at least multiple second-messenger pathways that can lead to persistent synaptic enhancement. Multiple mechanisms may also contribute to expression and maintenance.
Calcium Ions And LTP
It is generally agreed that the induction of LTP depends on an increase in the intracellular concentration of calcium ions ([Ca2+]i) in some key compartments of pre- and/or postsynaptic cells (Bliss & Collingridge, 1993; Johnston, Williams, Jaffe, & Gray, 1992; Nicoll & Malenka, 1995). The exact role of calcium in the induction process depends on the particular form of LTP and the synaptic system. In the CA1 region of the hippocampus, LTP induction in the Sch/com synapse depends on changes in postsynaptic [Ca2+]i (Fig. 47.9). Two major pathways that have been studied extensively are implicated in some aspect of LTP induction: calcium influx through ionotropic GluRs, especially the N-methyl-D-aspartate receptor (NMDAR); and calcium influx through voltage-gated calcium channels (VGCCs).
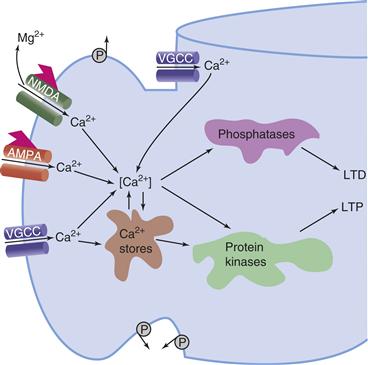
Figure 47.9 Events leading to some forms of LTP and LTD. The schematic depicts a postsynaptic spine with various sources of Ca2+. The NMDA receptor channel complex admits Ca2+ only after depolarization removes the Mg2+ block in the presence of bound glutamate. Calcium may also enter through the ligand-gated AMPA receptor channel or voltage-gated calcium channels (VGCC), which may be located on the spine head or dendritic shaft. Calcium pumps (P), located on the spine head, neck, and dendritic shaft, are hypothesized to help isolate Ca2+ concentration changes in the spine head from those in the dendritic shaft.
Nmdar-Dependent LTP
Recall that the classical form of LTP in the CA1 region of the hippocampus has properties that can be explained in terms of a Hebbian mechanism. For this form of LTP, considerable evidence shows a role for the NMDAR (Bliss & Collingridge, 1993). Numerous pharmacological studies have shown that competitive antagonists of NMDA, such as D-2-amino-5-phosphonopentenoic acid (D-AP5, also termed AP5 or APV) or NMDA ion channel blockers, such as the noncompetitive antagonist (+)-5-methyl-10, 11-dihydro-5H-dibenzo[a,d] cyclohepten-5,10-imine (MK-801), can prevent the induction of LTP.
The NMDAR has two properties that immediately suggest the nature of its role in LTP induction at Hebbian synapses (Bliss & Collingridge, 1993; Brown et al., 1990; Gustafsson & Wigstrom, 1988). First, NMDARs are permeable to Ca2+ (in addition to Na+ and K+). This property is significant because postsynaptic [Ca2+]i plays a critical role in inducing NMDAR-dependent LTP. Second, the channel permeability is a function of both pre- and postsynaptic factors. Channel opening requires the neurotransmitter glutamate (or some related agonist) to bind to the NMDA site. This in turn requires presynaptic activity for glutamate release. At the usual resting membrane potential, the ionic channels of NMDARs are normally blocked by magnesium ions (Mg2+), but this channel block is relieved by sufficient depolarization of the postsynaptic membrane. Thus, the NMDAR-mediated conductance is voltage dependent, allowing Ca2+ entry only when presynaptic release is combined with post-synaptic depolarization.
At this point, you should recall the distinction between the properties of the NMDAR and those of the AMPAR (α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid receptor) (see Chapters 8 and 10 for details), which are also found in the postsynaptic membrane. The AMPAR does not exhibit voltage-dependent Mg2+ block, has relatively lower Ca2+ permeability, and the AMPAR-mediated conductance is essentially voltage independent. Released glutamate can potentially act on both the AMPARs and the NMDARs associated with the membrane on the dendritic spine (Fig. 47.9). With this knowledge, one can easily envision a possible role for the AMPAR and NMDAR in a Hebbian modification. Nearly concurrent presynaptic activity (producing glutamate release and binding) and postsynaptic depolarization (relieving the Mg2+ block) allow Ca2+ influx into the dendritic spine of the post-synaptic neuron. The increased [Ca2+]i in some critical region of the dendritic spine, presumably very close to the NMDAR, is thought to activate Ca2+-dependent enzymes, such as CaM kinase II, which play a key role in LTP induction (Fig. 47.9).
In qualitative terms, one can understand how these molecular events could help account for the properties of cooperativity, associativity, and spatiotemporal specificity. Active synapses release glutamate, which can bind to the NMDAR, causing Ca2+ influx into dendritic spines on the postsynaptic cell. This Ca2+ influx acts locally and results in input-specific LTP. However, the Ca2+ influx occurs only when the synaptic input is strong enough to depolarize the postsynaptic membrane sufficiently to relieve the Mg2+ block, giving rise to cooperativity (Fig. 47.8). The depolarization itself is mediated in large part by the (voltage-independent) AMPARs, which are also colocalized on the dendritic spine (Fig. 47.9). Note that activity in a weak input by itself would not depolarize the postsynaptic cell sufficiently to relieve the Mg2+ block unless this activity were properly timed in relationship to activity in a strong input to the same cell. The combined depolarization of the two inputs gives rise to associativity and input specificity (Fig. 47.8).
Nmdar-Independent LTP
Most of the preceding accounts of the Hebbian, NMDAR-dependent form of LTP apply only to certain synapses under some conditions, and even then it may be only one aspect of the story (Johnston et al., 1992; Nicoll & Malenka, 1995). In many synapses, LTP induction does not appear to require the NMDAR (Johnston et al., 1992; Teyler et al., 1994). Even within the hippocampus, some synapses exhibit NMDAR-independent forms of LTP. For example, in the presence of the competitive antagonist APV, even the Sch/com inputs to CA1 pyramidal neurons, which are known to exhibit the classical Hebbian form of LTP that relies on the NMDAR, can exhibit an NMDAR-independent type of LTP. The onset of NMDAR-independent LTP is relatively slow (20–30 min), exhibits input specificity, and is prevented by a blocker of L-type voltage-gated calcium channels (VGCCs). Thus, a distinction exists between “NMDA LTP” and “VGCC LTP.” Other work has suggested that VGCCs are likely to be responsible for certain types of LTP in the CA3 region of the hippocampus and in the visual cortex.
The general case may be that both NMDAR-dependent and NMDAR-independent forms of LTP may coexist in the same brain region, among different classes of synaptic inputs onto the same postsynaptic neuron, and even among the same class of synaptic inputs to the same postsynaptic neuron.
Induction of Late LTP
A common requirement for the induction of both early LTP and late LTP at the CA3–CA1 synapse is the elevation of levels of intracellular calcium in the postsynaptic (i.e., CA1) neuron. Additional steps are involved in the induction of L-LTP, however. As was the case for the induction of long-term facilitation of the sensorimotor synapse in Aplysia, activation of PKA appears to be necessary for the induction of L-LTP (Huang, Nguyen, Abel, & Kandel, 1996). However, in the CA1 neuron the cAMP pathway is engaged directly by the activation of a calcium-sensitive adenylyl cyclase rather than being activated by 5-HT as in Aplysia. (Transmitters that regulate cAMP in CA1 neurons can profoundly modulate LTP, however.) Thus, at the CA1 neuron elevated levels of calcium lead to activation of adenylyl cyclase, increased synthesis of cAMP, and activation of PKA. Also, like long-term facilitation in Aplysia, MAPK is necessary for the induction of L-LTP (Dineley et al., 2001). The mechanisms of activation of MAPK in CA1 neurons have not been elucidated in detail, but may involve PKA, PKC, or both acting together. The final steps in the induction of L-LTP involve a PKA- and MAPK-dependent phosphorylation of CREB and induction of CREB responsive genes. These genes encode the proteins underlying changes at the synapse (but see Box 47.1).
Box 47.1 Is Memory More than Changes in Synaptic Strength?
The search for the biological basis of learning and memory has led many of the leading neuroscientists of the twentieth century to direct their efforts to investigating the synapse. The focus of most recent work on LTP (and indeed the bulk of this chapter) has been to elucidate the mechanisms underlying changes in synaptic strength. Although changes in synaptic strength are certainly ubiquitous, they are not the exclusive means for the expression of neuronal plasticity associated with learning and memory. Both short-term and long-term sensitization and classical conditioning of defensive reflexes in Aplysia are associated with an enhancement of excitability of the sensory neurons in addition to changes in synaptic strength. Classical and operant conditioning of feeding behavior in Aplysia also leads to changes in excitability of neuron B51. Interestingly, in this case there is a bidirectional control. The excitability of B51 is increased by operant conditioning, whereas it is decreased by classical conditioning. Changes in excitability of sensory neurons in the mollusc Hermissenda are produced by classical conditioning. In vertebrates, classical conditioning of eye-blink reflexes produces changes in the excitability of cortical neurons. Eye-blink conditioning also produces changes in the spike afterpotential of hippocampal pyramidal neurons. Finally, as described in their original report on LTP, Bliss and Lomo found that the expression of LTP was also associated with an apparent enhanced excitability. For recent review see Mozzachoidi and Byrne (2010).
John H. Byrne
The mechanisms underlying the induction of long-term neuronal plasticity and memory seem to be highly conserved across species. NMDAR-dependent LTP has been characterized at the sensorimotor synapse of Aplysia (Lin & Glanzman, 1994). Formation of some long-term memories in Drosophila depends on both an increase in the activity of a transcriptional activator of the bZip family, dCREB2a, and a decrease in the activity of a related repressor, dCREB2b. Establishment of LTM in Drosophila therefore appears analogous to induction of Aplysia LTF, involving transcriptional activation by one bZip family member (CREB1 in Aplysia) and relief of repression by another member (CREB2 in Aplysia). CREB activation also correlates with mammalian long-term memories. Interference with CREB function can inhibit long-term memories whereas overexpression of CREB can facilitate them.
LTP Expression
Up to this point, evidence related to early events in the causal chain that triggers the induction process has been emphasized. Another question follows naturally: What biochemical and biophysical changes incorporate this modification once it has been triggered? Most of the ideas about enhanced synaptic transmission concern either increased transmitter release or increased receptivity to released transmitter. The former entails presynaptic changes the latter, postsynaptic. Although some of the induction mechanisms discussed previously implicated a postsynaptic increase in [Ca2+]i, this does not necessarily imply that expression must also be postsynaptic. Ample evidence is available for ongoing two-way communication across the synaptic cleft, so a postsynaptic trigger could, in principle, give rise to a pre- and/or postsynaptic modification (see also Fig. 47.5). Extensive and seemingly conflicting accounts can be found in the literature regarding the nature of the changes responsible for the observed increase in synaptic efficacy following LTP induction. Some of the possibilities are illustrated schematically in Figure 47.10. However, regarding LTP at the CA3–CA1 synapse, the expression of LTP is associated with an increase in the number of functional AMPA receptors in the postsynaptic neuron (mechanism No. 5 in Fig. 47.10; for review, see Malinow & Malenka, 2002). LTP at this synapse also involves phosphorylation of the AMPA receptor, which leads to an increased conductance that contributes to enhanced synaptic efficacy (mechanism No. 4 in Fig. 47.10). Recent results indicate that a family of small transmembrane AMPA receptor regulatory proteins (TARPs) controls both AMPA receptor trafficking and channel gating. Finally, LTP at hippocampal and cortical excitatory synapses correlates with increases in the size of dendritic spines on the postsynaptic neurons (reviewed in Kasai, Fukuda, Watanabe, Hayashi-Takagi, & Noguchi, 2010).
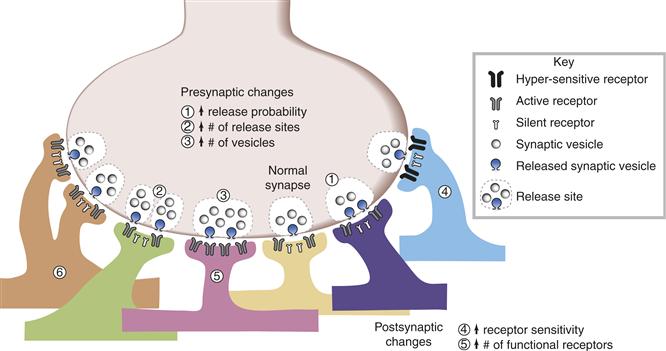
Figure 47.10 Schematic representation of possible loci for cellular changes involved in the enhancement of synaptic efficacy. The efficacy of a synapse can be potentiated through at least six mechanisms. First, there could be an increase in the fraction (release probability) of available presynaptic vesicles that undergo exocytosis. For example, in mechanism 1, two out of four available vesicles are released (i.e., 50% release probability), in contrast to the normal synapse, where only one out of four vesicles is released (i.e., 25% release probability). Second, there could be an increase in the number of release sites at the presynaptic neuron (mechanism 2). Third, the synapse could be potentiated through an increase in the number of vesicles available for release. For example, at a release site with eight vesicles, two of them will be exocytosed (instead of one) (mechanism 3), even if the release probability (25%) is the same as at the normal synapse. Fourth, there could be an increase in the sensitivity of the preexisting receptors to presynaptically released neurotransmitter or a greater conductance of the channel (mechanism 4). Fifth, there could be an increase in the number of functional receptors (illustrated as an increase from two active receptors at the normal synapse to four at the potentiated synapse; mechanism 5). Finally, the synapse could also be potentiated through coordinated presynaptic and postsynaptic morphological changes, such as the growth of new synaptic contacts between the same pair of neurons (mechanism 6).
Adapted from Wang, Ko, and Kelly (1997).
In contrast to the predominant evidence for postsynaptic expression of LTP at the CA3–CA1 synapse (see, however, Enoki, Hu, Hamilton, & Fine, 2009), LTP at the mossy fiber–CA3 synapse appears to involve an increase in transmitter release.
LTP Maintenance
Regardless of the ultimate nature and locus of the modification that gives rise to LTP expression, the more general problem remains of how a synaptic change can endure over long periods of time in the face of constant molecular turnover.
Maintenance of Early LTP
The maintenance of E-LTP is due to the persistence of phosphorylation of substrate proteins involved in expression mechanisms (see earlier discussion). This phosphorylation, in turn, is regulated by the engagement of protein kinases and phosphatases. For E-LTP, autonomously active forms of PKC and CaM kinase II appear to be particularly important.
Maintenance of Late LTP
The maintenance of persistent forms of LTP ultimately involves alterations in gene expression and changes in protein synthesis. High-frequency electrical stimulation in the rat hippocampus raises levels of specific mRNAs that encode transcription factors (e.g., Fos, zif268), cyto-skeletal proteins (e.g., Arc), and signal transduction molecules such as CaM kinase II. Moreover, protein synthesis inhibitors block the late phase of LTP but not earlier phases. One interesting observation is that brain slices that received protein synthesis inhibitors just 2 h after HFS showed no decline in late LTP, indicating that there is a critical time window during which protein synthesis might be necessary to maintain long-term plasticity. One protein PKMζ appears both necessary and sufficient for maintenance of a least some forms of L-LTP and seems necessary for maintaining the increase in postsynaptic AMPA receptors (Sacktor, 2011).
Although the involvement of new protein synthesis is consistent with the data, it immediately raises the problem of synaptic input specificity. If neural activity ultimately affects gene expression in the nucleus, then the mRNAs and proteins produced in the somatic compartment could, in principle, travel to any synapse within the cell. The problem is how to modify only the appropriate synapses and maintain synapse specificity. One solution is for the specific kinase(s) activated locally during a tetanus to phosphorylate one or more substrates and that these substrates act to “tag” the synapse as one that has been potentiated. For example, a stimulus that activates one CA1 synaptic input sufficiently to induce L-LTP can lead to the induction of L-LTP by a weaker stimulus to a second synaptic input even when that second input is by itself sufficient to only induce E-LTP. This effect is limited to a time window where the second weaker stimulus occurs within an hour or so of the stronger one. These results indicate that the weaker stimulus can lead to the transient (~1 h) activation of a synaptic “tag” that can specificially “capture” “plasticity factors” induced by a strong stimulus to a different synapse (Redondo & Morris, 2011). A major goal of current LTP research is to identify the nature of the tag and the plasticity factors.
Support of a more permanent synaptic modification might require a self-perpetuating tag and/or a biochemical positive feedback loop that maintains synaptic strength (Hayer & Bhalla, 2005). One possibility is that CPEB, the prion-like molecule that has been implicated in Aplysia LTF (Miniaci et al., 2008), might help to maintain synthesis of PKMζ (Sacktor, 2011). Positive feedback based on network activity may also be important. Periodic reactivation of strengthened synapses may be necessary to reactivate mechanisms of L-LTP, maintaining synaptic strength for months or longer (Smolen, 2007; Wang & Tsien, 2006).
Links between Long-Term Potentiation and Learning
Long-term potentiation has properties that have long been considered necessary for the encoding and retrieval of information. Hebbian forms of LTP exhibit associativity, which appears to be a desirable property, and all forms of LTP appear to be well suited to rapid learning. One of the more important challenges entails linking LTP (and LTD, see later) to learning and memory. This task has proven rather difficult to achieve, although evidence is accumulating for a causal link between LTP and memory. For example, learning in rats was verified to induce hippocampal LTP (Whitlock, Heynen, Shuler, & Bear, 2006). Also, inhibition of PKMζ in the hippocampus in vivo was demonstrated to disrupt LTP maintenance and eliminate spatial learning. Instrumental and classically conditioned long-term memories are similarly erased by locally inhibiting PKMζ in different brain regions of rats and mice, from days to even weeks and months after training (Saktor, 2011; Serrano et al., 2008). These results have greatly strengthened the link between LTP and learning. However, inhibiting PKMζ does not erase all forms of memory (Serrano et al., 2008).
Additional promising work in this area has come from studies of genetically engineered knockout mice. The first generation of knockouts prevented the expression of some factor that was thought to be necessary for LTP, such as CaM kinase II. Although these studies were intriguing, they suffered from the fact that the consequences of a gene knockout might alter brain development and might not be confined to a specific part of the brain under study. Some of these problems have been overcome in a second generation of knockouts that have temporal as well as spatial specificity. For example, temporal and spatial control of gene expression can now be achieved using binary transgene systems such as tetracycline transactivating systems and Cre/LoxP recombination systems. The NMDAR gene in only CA1 pyramidal cells of the hippocampus has been knocked out using the Cre/LoxP system (Tsien, Huerta, & Tonegawa, 1996). The results provide strong evidence in favor of the notion that NMDA receptor-dependent synaptic plasticity at CA3–CA1 synapses is required for the acquisition of spatial memory. Further spatial specificity can be obtained by using promoters that are specific for subfields of the hippocampus. For example, one of the kainate receptor subunits, KA-1, exhibits high levels of expression in the CA3 pyramidal cell layer. Using the KA-1 promoter and the α-CaM kinase II promoter, Nakashiba, Young, McHugh, Buhl, and Tonegawa (2008) drove expression of tetanus toxin specifically in CA3 neurons. Expression of the toxin inhibited transmitter release from CA3 neurons which was associated with a block of one-trial contextual learning. Similar approaches using cell-specific promoters to drive expression of toxins or their receptors have been used to examine the role of cerebellar circuits that mediate classical conditioning of eye-blink reflexes (for review, see Nakanishi, 2009). The molecular approaches described above hold tremendous promise for testing hypotheses of the functional role of various forms of LTP in specific brain regions as well as the role of specific neural circuits in mediating memory processes.
Long-Term Depression
LTD is believed by many to be the mechanism by which learning is encoded in the cerebellum (Ito, 2001), as well as a process whereby LTP could be reversed in the hippocampus and neocortex (Bear & Malenka, 1994). In the hippocampus, brief HFS (e.g., four trains of 10 shocks at 100 Hz) can induce classical LTP, whereas low-frequency stimulation (LFS) over longer periods (1 Hz for 10 min) can induce LTD.
Some forms of LTD appear to be mediated by the NMDAR and these forms appear to be due to dephosphorylation of AMPAR subunits, which decreases their conductance and hence the synaptic efficacy, as well as endocytosis of AMPARs, which decreases their surface number and hence the synaptic efficacy. In addition, NMDAR-independent forms of LTD exist. These often depend on activation of metabotropic glutamate receptors. For example, NMDAR-independent LTD in the parallel fiber input to Purkinje cells in the cerebellum is induced by the combined increase in intracellular levels of Ca2+ (by activity) in postsynaptic Purkinje cells and activation of metabotropic glutamate receptors produced by the release of glutamate from parallel fibers.
The form of LTD may vary in different brain regions and sometimes among different inputs to the same brain region. Even within the CA1 region, the Sch/com input may exhibit both NMDAR-dependent and NMDAR-independent forms of LTD.
Calcium levels in the dendritic spines appear to be a common locus for the induction of NMDAR-dependent LTP and LTD. For example, at the Sch/com input to CA1, both LTP and LTD can be blocked by injecting Ca2+ chelators into the postsynaptic cell. If both LTP and LTD are triggered by Ca2+ entry, then how are their induction processes different? Presumably, more Ca2+ influx occurs during an LTP-inducing HFS than during an LTD-inducing LFS. One formal molecular model developed by John Lisman incorporates this Ca2+-dependent, bidirectional control of synaptic strength. In this model, high [Ca2+]i activates a protein kinase that phosphorylates a protein causing LTP induction, whereas intermediate [Ca2+]i activates a protein phosphatase that dephosphorylates this protein and causes LTD. The synaptic strength thus depends on which of these competing processes is most active, which in turn will be a function of the pattern of activity experienced by the cell (Fig. 47.9).
LTP and LTD can be readily reversed. If LFS is applied after a synapse is potentiated by HFS, the synaptic strength decreases. This phenomenon is called depotentiation. In contrast, if HFS is applied after a synapse is depressed by LFS, the synaptic strength increases. This phenomenon is called dedepression. Accumulating evidence indicates that the mechanisms of depotentiation differ from that of LTD, and the mechanisms of dedepression differ from that of LTP.
Summary
The understanding of LTP and LTD is evolving rapidly (Bliss, Collingridge, & Morris, 2007). For example, whereas the N-methyl-D-aspartate receptor was once the pivotal focus of long-term potentiation research, it is now clear that other mechanisms should be considered. It has also become evident that HFS is but one end of a spectrum of stimulations that can induce synaptic changes. Lower stimulation frequencies can induce long-term depression, which may share some common molecular mechanisms with LTP. Finally, several stages in the maintenance of LTP have been identified, and probably more will be found. A remaining challenge is to clarify the varieties of LTP and LTD mechanisms and to demonstrate their functional significance by establishing convincing links to the encoding and retrieval of information.
How Does a Change in Synaptic Strength Store a Complex Memory?
The relationship between the synaptic changes and the behavior of conditioned reflexes can be straightforward because the locus for the plastic change is part of the mediating circuit. Thus, the change in the strength of the sensorimotor synapse in Aplysia can be related to the memory for sensitization (e.g., Fig. 47.2A1). However, the idea that an increase in synaptic strength leads to an enhanced behavioral response, and a decrease in synaptic strength leads to a decreased behavioral response, can be misleading. For example, a decrease in synaptic strength in a postsynaptic neuron that exerts an inhibitory action can be translated into an enhanced behavioral response. Indeed, in the parallel fiber-to-Purkinje cell connection in the cerebellum, such a disinhibition is precisely the mechanism that has been proposed to contribute to classical conditioning of the eye-blink reflex (see Chapter 48 for a discussion of this form of conditioning). Nevertheless, for relatively simple reflex systems in which the circuit is well understood, it is possible to directly relate a change in synaptic strength to learning. However, in most other examples of memory, it is considerably less clear how the synaptic changes are induced and, once induced, how the information is retrieved. This is especially true in memory systems that involve the storage of information for patterns, facts, and events. Neurobiologists have turned to artificial neural circuits to gain insights into these issues.
A simple network that can store and “recognize” patterns is illustrated in Figure 47.11. The network is artificial, but nevertheless is inspired by actual circuitry in the CA3 region of the hippocampus. In this example, six different input projections make synaptic connections with the dendrites of each of six postsynaptic neurons (Fig. 47.11A). The postsynaptic neurons serve as the output of the network. Input projections can carry multiple types of patterned information, and these patterns can be complex. In order to simplify the present discussion, consider that the particular input pathway in Figure 47.11A carries information regarding the pattern of neural activity induced by a single brief flash of a spatial pattern of light. For example, activity in the top pathway (line a) might represent light falling on the temporal region of the retina, whereas activity in the pathway on the bottom (line f) might represent light falling on the nasal region of the retina. Thus, the spatial pattern of an image falling upon the retina could be reconstructed from the pattern of neuronal activity over the n (in this case 6) input projections to the network.
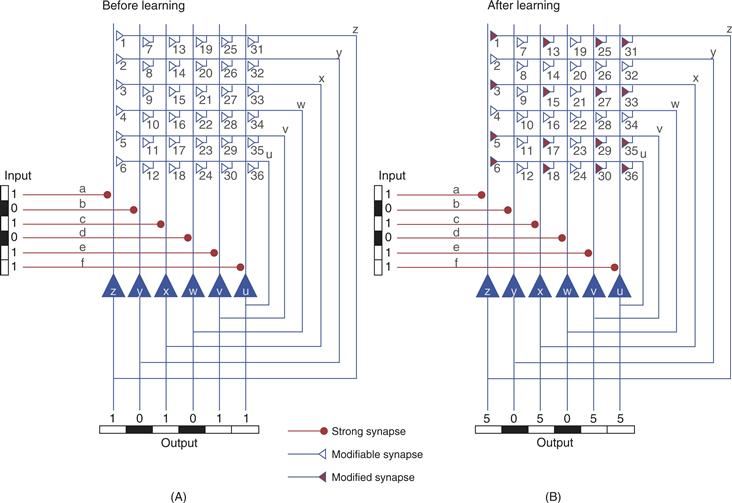
Figure 47.11 Autoassociation network for recognition memory. The artificial circuit consists of six input pathways that make strong connections to each of six output neurons. The output neurons have axon collaterals that make synaptic connections (numbered 1–36) with each of the output cells. (A) A pattern represented by activity in the input lines or axons (a, b, c, d, e, f) is presented to the network. A 1 represents an active axon (e.g., a spike), whereas a 0 represents an inactive axon. The input pathways make strong synapses (•) with the postsynaptic output cells. Thus, output cells (u, v, w, x, y, z) generate a pattern that is a replica of the input pattern. The collateral synapses were initially weak and do not contribute to the output. Nevertheless, the activity in the collaterals that occurred in conjunction (assume minimal delays within the circuit) with the input pattern led to a strengthening of a subset of the 36 synapses. (B) A second presentation of the input produces an output pattern that is an amplified, but an otherwise intact, replica of the input. An incomplete input pattern can be used as a cue to retrieve the complete pattern.
Three aspects of the circuit endow it with the ability to store and retrieve patterns. First, each of the input lines makes a sufficiently strong connection with its corresponding postsynaptic cell to activate it reliably. Second, each output cell (z to u) sends an axon collateral that makes an excitatory connection with itself as well as the other five output cells. This pattern of synaptic connectivity leads to a network of 36 synapses (42 including the 6 input synapses). Third, each of the 36 synaptic connections is modifiable through an LTP-like mechanism (see earlier discussion). Specifically, the strength of a particular synaptic connection is initially weak, but it will increase if the presynaptic and postsynaptic neurons are active at the same time. The circuit configuration with the embedded synaptic “learning rule” leads to an autoassociation or autocorrelation matrix. The autoassociation is derived from the fact that the output is fed back to the input where it associates with itself.
Now consider the consequences of presenting the patterned input to the network of Figure 47.11A. The input pattern will activate the six postsynaptic cells in such a way as to produce an output pattern that will be a replica of the input pattern. In addition, the pattern will induce changes in the synaptic strength of the active synapses in the network. For example, synapse 3 will be strengthened because the postsynaptic cell, cell z, and the presynaptic cell, cell x, will be active at the same time. Note also that synapses 1, 5, and 6 will be strengthened as well. This occurs because these input pathways to cell z are also active. Thus, all synapses that are active at the same time as cell z will be strengthened. When the pattern is presented again as in Figure 47.11B, the output of the cell will not only be governed by the input, but also by the feedback connections, a subset of which were strengthened (Fig. 47.11B, filled synapses) by the initial presentation of the stimulus. Thus, for output cell z, a component of its activity will be derived from input a, but components will also come from synapses 1, 3, 5, and 6. If each of the initially strong and newly modified synapses is assumed to contribute equally to the firing of output cell z, the activity would be five times greater than the activity produced by input a before the learning. After learning, the output is an amplified version of the input but the basic features of the pattern are preserved.
Note that the “memory” for the pattern does not reside in any one synapse or in any one cell. Rather, it is distributed throughout the network at multiple sites. The properties of these types of autoassociation networks have been examined by James Anderson, Teuvo Kohonen, David Marr, Edmond Rolls, David Wilshaw, and their colleagues and found to exhibit a number of phenomena that would be desirable for a biological recognition memory system. For example, such networks exhibit pattern completion. If a partial input pattern is presented, the autoassociation network can complete the pattern in the sense that it can produce an output that is approximately what is expected for the full input pattern. Thus, any part of the stored pattern can be used as a cue to retrieve the complete pattern. For the example of Figure 47.11, the input pattern was {101011}. This pattern led to an output pattern of {505055}. If the input pattern is degraded to {101000}, then neurons z and x each receive two less recurrent inputs, and neurons v and u each receive two less recurrent inputs and lose their direct input. Nevertheless neurons z, x, v, and u each still receive two recurrent inputs and neurons z and x receive direct inputs. Therefore, these four neurons fire, and the output pattern is {303022}. Compared to the original output pattern {505055}, some decrease in the strength of firing occurs, but the basic pattern is preserved. This preservation is a simple example of the phenomenon known as graceful degradation, which is exhibited by autoassociative networks. With graceful degradation, the network can still function if some of the input connections or postsynaptic cells are lost. This property arises from the distributed representation of the memory within the circuit.
Summary
The concept of distributed representation of memory crosses multiple levels of organization of memory systems. Multiple brain systems are involved in memory, and memory is distributed among synapses in a particular memory circuit (Fig. 47.11). Also, memory at any one synapse is represented by multiple cellular changes (Figs. 47.2, 47.3, and 47.9). The reductionist approaches described in this chapter have provided key insights into cellular memory mechanisms. In the near future, a major experimental question to be answered is the extent to which the mechanisms for learning are common both within any one animal and between different species. Although many common features are emerging, there seem to be some differences. Thus, it will be important to understand the extent to which specific mechanisms are used selectively for one type of learning and not another. Irrespective of the particular example of learning and memory that is analyzed, whether it be simple or complex, it will be important to pay attention to three major details: details of the circuit interactions, details of the learning rule, and details of the intrinsic biophysical properties of the neurons within the circuit.
References
1. Antonov I, Kandel ER, Hawkins RD. Presynaptic and postsynaptic mechanisms of synaptic plasticity and metaplasticity during intermediate-term memory formation in Aplysia. Journal of Neuroscience. 2010;30:5781–5791.
2. Antonov I, Antonova I, Kandel ER, Hawkins RD. Activity-dependent presynaptic facilitation and Hebbian LTP are both required and interact during classical conditioning in Aplysia. Neuron. 2003;37:135–147.
3. Antzoulatos EG, Byrne JH. Long-term sensitization training produces spike narrowing in Aplysia sensory neurons. Journal of Neuroscience. 2007;27:676–683.
4. Bailey CH, Kandel ER. Synaptic remodeling, synaptic growth and the storage of long-term memory in Aplysia. Progress in Brain Research. 2008;169:179–198.
5. Barrionuevo G, Brown TH. Associative long-term potentiation in hippocampal slices. Proceedings of the National Academy of Sciences of the United States of America. 1983;80:7347–7351.
6. Bear MF, Malenka RC. Synaptic plasticity: LTP and LTD. Current Opinion in Neurobiology. 1994;4:389–399.
7. Bliss TVP, Collingridge GL. A synaptic model of memory: Long-term potentiation in the hippocampus. Nature (Lond.). 1993;361:31–39.
8. Bliss TVP, Collingridge GL, Morris R. Synaptic plasticity in the hippocampus. In: Anderson P, Morris R, Amaral D, Bliss T, O’Keefe J, eds. The hippocampus book. New York: Oxford University Press; 2007:343–474.
9. Brown TH, Ganong AH, Kairiss EW, Keenan CL. Hebbian synapses: Biophysical mechanisms and algorithms. Annual Review of Neuroscience. 1990;13:475–512.
10. Byrne JH. Cellular analysis of associative learning. Physiological Reviews. 1987;67:329–439.
11. Byrne JH, Kandel ER. Presynaptic facilitation revisited: State- and time-dependence. Journal of Neuroscience. 1996;16:425–435.
12. Cai D, Chen S, Glanzman DL. Postsynaptic regulation of long-term facilitation in Aplysia. Current Biology. 2008;18:920–925.
13. Cai D, Pearce K, Chen S, Glanzman DL. Protein kinase M maintains long-term sensitization and long-term facilitation in Aplysia. Journal of Neuroscience. 2011;31:6421–6431.
14. Carew TJ. Behavioral neurobiology Sunderland, MA: Sinauer; 2000.
15. Chain DG, Casadio A, Schacher S, et al. Mechanisms for generating the autonomous cAMP-dependent protein kinase required for long-term facilitation in Aplysia. Neuron. 1999;22:147–156.
16. Choi YB, Li HL, Kassabov SR, et al. Neurexin-neuroligin transsynaptic interaction mediates learning-related synaptic remodeling and long-term facilitation in Aplysia. Neuron. 2011;70:468–481.
17. Cleary LJ, Lee WL, Byrne JH. Cellular correlates of long-term sensitization in Aplysia. Journal of Neuroscience. 1998;18:5988–5998.
18. Dineley KT, Weeber EJ, Atkins C, Adams JP, Anderson AE, Sweatt JD. Leitmotifs in the biochemistry of LTP induction: Amplification, integration and coordination. Journal of Neurochemistry. 2001;77:961–971.
19. Enoki R, Hu YL, Hamilton D, Fine A. Expression of long-term plasticity at individual synapses in hippocampus is graded, bidirectional, and mainly presynaptic: Optical quantal analysis. Neuron. 2009;62:242–253.
20. Fioravante D, Byrne JH. Protein degradation and memory formation. Brain Research Bulletin. 2011;85:14–20.
21. Fioravante D, Antzoulatos EG, Byrne JH. Sensitization and habituation: Invertebrate. In: Sweatt JD, Byrne JH, eds. Oxford: Elsevier Science Limited; 2008;31–51. Volume 4 of Learning and memory: A comprehensive reference. Vols. 4.
22. Fioravante D, Liu RY, Netek AK, Cleary LJ, Byrne JH. Synapsin regulates basal synaptic strength, synaptic depression and serotonin-induced facilitation of sensorimotor synapses in Aplysia. Journal of Neurophysiology. 2007;98:3568–3580.
23. Ghirardi M, Montarolo PG, Kandel ER. A novel intermediate stage in the transition between short- and long-term facilitation in the sensory to motor neuron synapse of Aplysia. Neuron. 1995;14:413–420.
24. Guan Z, Giustetto M, Lomvardas S, et al. Integration of long-term-memory-related synaptic plasticity involves bidirectional regulation of gene expression and chromatin structure. Cell. 2002;111:483–493.
25. Gustafsson B, Wigstrom H. Physiological mechanisms of long-term potentiation. Trends in Neuroscience. 1988;11:156–162.
26. Hawkins RD, Abrams TW, Carew TJ, Kandel ER. A cellular mechanism of classical conditioning in Aplysia: Activity-dependent amplification of presynaptic facilitation. Science. 1983;219:400–405.
27. Hayer A, Bhalla US. Molecular switches at the synapse emerge from receptor and kinase traffic. PLoS Computational Biology. 2005;1:137–154.
28. Hebb DO. The organization of behavior New York: Wiley (Interscience); 1949.
29. Hu JY, Wu F, Schacher S. Two signaling pathways regulate the expression and secretion of a neuropeptide required for long-term facilitation in Aplysia. Journal of Neuroscience. 2006;26:1026–1035.
30. Huang Y-Y, Nguyen PV, Abel T, Kandel ER. Long-lasting forms of synaptic potentiation in the mammalian hippocampus. Learning & Memory. 1996;3:74–85.
31. Ito M. Cerebellar long-term depression: Characterization, signal transduction, and functional roles. Physiological Reviews. 2001;81:1143–1195.
32. Jin I, Kandel ER, Hawkins RD. Whereas short-term facilitation is presynaptic, intermediate-term facilitation involves both presynaptic and postsynaptic protein kinases and protein synthesis. Learning & Memory. 2011;18:96–102.
33. Johnston D, Williams D, Jaffe D, Gray R. NMDA-receptor-independent long-term potentiation. Annual Review of Physiology. 1992;54:489–505.
34. Kandel ER. The molecular biology of memory storage: A dialogue between genes and synapses. Science. 2001;294:1030–1038.
35. Kasai H, Fukuda M, Watanabe S, Hayashi-Takagi A, Noguchi J. Structural dynamics of dendritic spines in memory and cognition. Trends in Neurosciences. 2010;33:121–129.
36. Kim JH, Udo H, Li HL, et al. Presynaptic activation of silent synapses and growth of new synapses contribute to intermediate and long-term facilitation in Aplysia. Neuron. 2003;40:151–165.
37. Leal K, Klein M. Direct enhancement of presynaptic calcium influx in presynaptic facilitation at Aplysia sensorimotor synapses. Molecular and Cellular Neurosciences. 2009;41:247–257.
38. Lechner HA, Byrne JH. New perspectives on classical conditioning: A synthesis of Hebbian and Non-Hebbian mechanisms. Neuron. 1998;20:355–385.
39. Levenson J, Endo S, Kategaya LS, et al. Long-term regulation of neuronal high-affinity glutamate and glutamate uptake in Aplysia. Proceedings of the National Academy of Sciences. 2000;97:12858–12863.
40. Lin XY, Glanzman DL. Hebbian induction of long-term potentiation of Aplysia sensorimotor synapses: Partial requirement for activation of an NMDA-related receptor. Proceedings Biological Sciences. 1994;22:215–221.
41. Liu RY, Cleary LJ, Byrne JH. The requirement for enhanced CREB1 expression in consolidation of long-term synaptic facilitation and long-term excitability in sensory neurons of Aplysia. Journal of Neuroscience. 2011;31:6871–6879.
42. Malinow R, Malenka RC. AMPA receptor trafficking and synaptic plasticity. Annual Review of Neuroscience. 2002;25:103–126.
43. Miniaci MC, Kim JH, Puthanveettil SV, et al. Sustained CPEB-dependent local protein synthesis is required to stabilize synaptic growth for persistence of long-term facilitation in Aplysia. Neuron. 2008;59:1024–1036.
44. Miyamoto E. Molecular mechanism of neuronal plasticity: induction and maintenance of long-term potentiation in the hippocampus. Journal of Pharmacological Sciences. 2006;100:433–442.
45. Mozzachiodi R, Byrne JH. More than synaptic plasticity: Role of nonsynaptic plasticity in learning and memory. Trends in Neurosciences. 2010;33:17–26.
46. Nakanishi S. Genetic manipulation study of information processing in the cerebellum. Neuroscience. 2009;162:723–731.
47. Nakashiba T, Young JZ, McHugh TJ, Buhl DL, Tonegawa S. Transgenic inhibition of synaptic transmission reveals role of CA3 output in hippocampal learning. Science. 2008;319:1260–1264.
48. Nicoll RA, Kaur JA, Malenka RC. The current excitement of long-term potentiation. Neuron. 1988;1:97–103.
49. Nicoll RA, Malenka RC. Contrasting properties of two forms of long-term potentiation in the hippocampus. Nature (Lond.). 1995;377:115–118.
50. Rachlin H. Introduction to modern behaviourism 3rd ed. New York: Freesman; 1991.
51. Redondo RL, Morris RGM. Making memories last: the synaptic tagging and capture hypothesis. Nature Reviews Neuroscience. 2011;12:17–30.
52. Roberts AC, Glanzman DL. Learning in Aplysia: looking at synaptic plasticity from both sides. Trends in Neuroscience. 2003;26:662–670.
53. Sacktor TC. How does PKMzeta maintain long-term memory?. Nature Reviews Neuroscience. 2011;12:9–15.
54. Serrano P, Friedman EL, Kenney J, et al. PKMzeta maintains spatial, instrumental, and classically conditioned long-term memories. PLoS Biology. 2008;6:2698–2706.
55. Sharma SK, Carew TJ. The roles of MAPK cascades in synaptic plasticity and memory in Aplysia: Facilitatory effects and inhibitory constraints. Learning & Memory. 2004;11:373–378.
56. Si K, Choi YB, White-Grindley E, Majumdar A, Kandel ER. Aplysia CPEB can form prion-like multimers in sensory neurons that contribute to long-term facilitation. Cell. 2010;140:421–435.
57. Smolen P. A model of late long-term potentiation simulates aspects of memory maintenance. PLoS ONE. 2007;2:e445.
58. Sutton MA, Masters SE, Bagnall MW, Carew TJ. Molecular mechanisms underlying a unique intermediate phase of memory in Aplysia. Neuron. 2001;31:143–154.
59. Teyler TJ, Cavus I, Coussens C, et al. Multideterminant role of calcium in hippocampal synaptic plasticity. Hippocampus. 1994;4:623–634.
60. Tsien JZ, Huerta PT, Tonegawa S. The essential role of hippocampal CA1 NMDA receptor-dependent synaptic plasticity in spatial memory. Cell. 1996;87:1317–1326.
61. Villareal G, Li Q, Cai D, Glanzman DL. The role of rapid, local, postsynaptic protein synthesis in learning-related synaptic facilitation in Aplysia. Current Biology. 2007;17:2073–2080.
62. Wainwright ML, Byrne JH, Cleary LJ. Dissociation of morphological and physiological changes associated with long-term memory in Aplysia. Journal of Neurophysiology. 2004;92:2628–2632.
63. Walters ET, Byrne JH. Associative conditioning of single sensory neurons suggests a cellular mechanism for learning. Science. 1983;219:405–408.
64. Wang JH, Ko GY, Kelly PT. Cellular and molecular basis of memory: Synaptic and neuronal plasticity. Journal of Clinical Neurophysiology. 1997;14:264–293.
65. Wang H, Hu Y, Tsien JZ. Molecular and systems mechanisms of memory consolidation and storage. Progress in Neurobiology. 2006;79:123–135.
66. Whitlock JR, Heynen AJ, Shuler MG, Bear MF. Learning induces long-term potentiation in the hippocampus. Science. 2006;313:1093–1097.
67. Zhang F, Endo S, Cleary LJ, Eskin A, Byrne JH. Role of transforming growth factor-b in long-term facilitation in Aplysia. Science. 1997;275:1318–1320.
68. Zhao Y, Klein M. Modulation of the readily releasable pool of transmitter and of excitation-secretion coupling by activity and by serotonin at Aplysia sensorimotor synapses in culture. Journal of Neuroscience. 2002;22:10671–10679.
Suggested Readings
1. Anderson P, Morris R, Amaral D, Bliss T, O’Keefe J, eds. The hippocampus book. New York: Oxford University Press; 2007.
2. Byrne JH. Learning and memory. In: Byrne JH, ed. Neuroscience online: An electronic textbook for the neurosciences. 1997; http://nba.uth.tmc.edu/neuroscience/ Department of Neurobiology and Anatomy, The University of Texas Medical School at Houston (UTHealth) © 1997.
3. Byrne JH, ed. Concise learning and memory—the editor’s selection. Oxford: Elsevier; 2009.
4. Dudai Y. Memory from a to z: Keywords, concepts, and beyond Oxford: Oxford University Press; 2002.
5. Kandel ER. In search of memory: The emergence of a new science of mind New York: W. W. Norton & Company; 2007.
6. Squire LR, Kandel ER. Memory: From mind to molecules New York: Henry Holt and Company; 1999.
7. Sweatt JD. Mechanisms of memory Amsterdam: Academic Press; 2010.