Chapter 5
Pharmaceuticals and medicinal chemistry
One of the most important applications of organic chemistry involves the design and synthesis of pharmaceutical agents—a topic that is defined as medicinal chemistry. This is a relatively new scientific discipline. Before the 1960s, the discovery of pharmaceutical agents was very much a hit-and-miss affair. Thousands of organic compounds were made in the laboratory, or were extracted from natural sources, in the hope that they would have pharmacological activity. Success was more down to luck than design. From the 1960s on, there has been much greater understanding of how drugs work, and of the targets with which they interact. Advances in biology, genetics, chemistry, and computing have now made it possible to design new drugs, rather than to rely on trial and error. Medicinal chemists are key players in the pharmaceutical industry because they are experts at both designing and synthesizing drugs. The two skills go hand in hand. For example, there is no point in designing a drug that cannot be synthesized. Similarly, it is wasteful to generate thousands of novel compounds if they have little chance of being active drugs.
The pathfinder years
In the past, societies depended on herbs and extracts from natural sources to treat illness. No doubt there was a powerful placebo effect involved, as most ancient therapies have little beneficial effect. However, some of these concoctions are effective. Examples include the sedative effects of various opium preparations, and the physical and psychological effects obtained from chewing coca leaves—a habit which is still common in some South American communities. Extracts of willow bark have been known for centuries to reduce fever, pain, and inflammation.
In the 19th century, chemists isolated chemical components from known herbs and extracts. Their aim was to identify a single chemical that was responsible for the extract’s pharmacological effects—the active principle. For example, morphine is the active principle responsible for the sedative properties of opium, while cocaine is the active principle present in coca leaves. The active principle in willow bark is salicyclic acid. Other active principles isolated in the 19th century included quinine, caffeine, atropine, physostigmine, and theophylline. Quinine was of particular importance since it is effective in treating malaria. Caffeine and theophylline are stimulants found in beverages. Atropine has been used in cardiovascular medicine and as an antidote to pesticide poisoning, while physostigmine can be used to treat glaucoma.
It was not long before chemists synthesized analogues of active principles. Analogues are structures which have been modified slightly from the original active principle. Such modifications can often improve activity or reduce side effects. This led to the concept of the lead compound—a compound with a useful pharmacological activity that could act as the starting point for further research. Fully synthetic compounds were also investigated for pharmacological activity leading to the discovery of general anaesthetics, local anaesthetics, and barbiturates during the late 19th and early 20th centuries.
The first half of the 20th century culminated in the discovery of effective antimicrobial agents. At the beginning of the century, Paul Ehrlich developed arsenic-containing drugs which proved effective against syphilis, while early antimalarial agents were discovered in the 1920s. The sulphonamides were discovered in the 1930s, but the most important advance was penicillin, which was introduced in the 1940s. The original penicillin was isolated from a fungus, and this sparked a massive worldwide study of fungal cultures in the post-war years, which led to the identification of many of the antibiotics used in medicine today. The middle part of the 20th century was a golden age of antibacterial research, and marked one of the most important advances in medicine. Before the antibiotic revolution, even simple wounds could prove life-threatening, and many of the surgical operations carried out routinely today were totally impractical.
The development of rational drug design
The 1960s can be viewed as the birth of rational drug design. During that period there were important advances in the design of effective anti-ulcer agents, anti-asthmatics, and beta-blockers for the treatment of high blood pressure. Much of this was based on trying to understand how drugs work at the molecular level and proposing theories about why some compounds were active and some were not.
However, rational drug design was boosted enormously towards the end of the century by advances in both biology and chemistry. The sequencing of the human genome led to the identification of previously unknown proteins that could serve as potential drug targets. For example, kinase enzymes have proved important targets for novel anti-cancer agents in recent years. These enzymes catalyse phosphorylation reactions and play a key role in controlling cell growth and division. Similarly, the sequencing of genomes from viruses led to the identification of viral-specific proteins that could serve as novel targets for new antiviral agents. Advances in automated, small-scale testing procedures (high-throughput screening) also allowed the rapid testing of potential drugs.
In chemistry, advances were made in X-ray crystallography and NMR spectroscopy, allowing scientists to study the structure of drugs and their mechanisms of action. Powerful molecular modelling software packages were developed that allowed researchers to study how a drug binds to a protein binding site. Novel synthetic methods have boosted the ability of chemists to create new compounds. In addition, the development of automated synthetic methods has vastly increased the number of compounds that can be synthesized in a given time period. Companies can now produce thousands of compounds that can be stored and tested for pharmacological activity. Such stores have been called chemical libraries and are routinely tested to identify compounds capable of binding with a specific protein target. These advances have boosted medicinal chemistry research over the last twenty years in virtually every area of medicine.
There has also been a significant change in the way pharmaceutical research is tackled. For most of the 20th century, drug research depended on the discovery of a lead compound with a useful pharmacological activity. Thousands of analogues were then synthesized in an effort to find an improved compound. Years later, the molecular target might be discovered, allowing a better understanding of the biological mechanisms affected by these agents. In this approach, progress was dictated by whatever lead compound was discovered.
Nowadays, most research projects are initiated by choosing a potential drug target, such as an enzyme or a receptor. A lead compound that interacts with that protein target is then sought. Of course, there are still research projects that are determined by the chance discovery of a pharmacologically active compound, but the scientific approach towards the design of novel drugs now follows the pathway shown in Figure 54. Those stages requiring knowledge of organic chemistry are highlighted in bold.
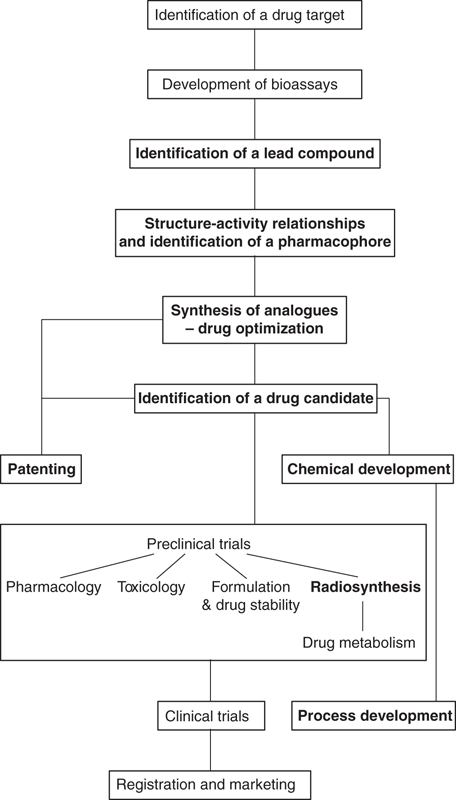
54. A typical approach to the development of a drug.
Identification of a drug target
Drugs interact with molecular targets in the body such as proteins and nucleic acids. However, the vast majority of clinically useful drugs interact with proteins, especially receptors, enzymes, and transport proteins (Chapter 4).
Drugs can be designed to activate receptors in the same way as the natural messenger. Such drugs are called agonists. Alternatively, drugs can be designed to block a receptor without activating it. Such drugs are known as antagonists. Examples of receptor antagonists include the beta-blocker propranolol, and the anti-ulcer agents cimetidine and ranitidine. Examples of receptor agonists include the anti-asthmatic drug salbutamol and the analgesic morphine.
Enzymes are also important drug targets. Drugs that bind to the active site and prevent the enzyme acting as a catalyst are known as enzyme inhibitors. Examples of enzyme inhibitors include the anti-HIV drug saquinavir, and the anti-hypertensive agent captopril. Enzymes are located inside cells, and so enzyme inhibitors have to cross cell membranes in order to reach them—an important consideration in drug design. There is no point designing a potent enzyme inhibitor if it fails to cross the cell membrane.
Transport proteins are targets for a number of therapeutically important drugs. For example, a group of antidepressants known as selective serotonin reuptake inhibitors prevent serotonin being transported into neurons by transport proteins. As a result, serotonin levels increase and produce the observed antidepressant effect.
Drug testing and bioassays
Bioassays are tests that are used to identify whether a drug interacts with a protein target. In vitro tests are carried out on target molecules or cell cultures. For example, enzyme inhibitors can be tested on a purified enzyme in a test tube to see whether they prevent the enzyme catalysing a particular reaction. In vitro tests can be automated, allowing the rapid testing of thousands of compounds in a very small time period. This is known as high-throughput screening. These tests are ideal for identifying whether drugs interact with a molecular target to produce a particular pharmacological effect. The ability of a drug to bind to its target and produce such an effect is known as pharmacodynamics.
In vivo bioassays are carried out on living organisms, and are complimentary to in vitro tests. In vivo tests establish whether a drug produces a physiological effect, such as analgesia or the lowering of blood pressure. In vivo tests also establish whether a drug reaches its molecular target when it is administered to an organism. The range of factors affecting a drug’s ability to reach its target is known as pharmacokinetics.
The main pharmacokinetic factors are absorption, distribution, metabolism, and excretion. Absorption relates to how much of an orally administered drug survives the digestive enzymes and crosses the gut wall to reach the bloodstream. Once there, the drug is carried to the liver where a certain percentage of it is metabolized by metabolic enzymes. This is known as the first-pass effect. The ‘survivors’ are then distributed round the body by the blood supply, but this is an uneven process. The tissues and organs with the richest supply of blood vessels receive the greatest proportion of the drug. Some drugs may get ‘trapped’ or sidetracked. For example fatty drugs tend to get absorbed in fat tissue and fail to reach their target. The kidneys are chiefly responsible for the excretion of drugs and their metabolites. They are particularly efficient at excreting polar molecules.
In vivo tests can sometimes identify unexpected activity that would not be picked up by in vitro tests. For example, the dye prontosil was shown to have antibacterial activity in vivo, but proved inactive in vitro. This is because prontosil itself is inactive, and is metabolized to an active sulphonamide within the body. A compound that acts in this way is known as a prodrug.
Finally, in vivo tests can detect side effects which would not be observed by in vitro tests. Sometimes, this suggests unexpected applications for the drug. For example, the anti-impotence drug sildenafil was originally tested as an anti-hypertensive drug, and its anti-impotence effects were only identified during early clinical trials.
Identification of lead compounds
A lead compound is a chemical structure that can bind to a desired molecular target. It may not bind particularly strongly and it may not be particularly active, but the fact that it binds to the desired target means that it can serve as a starting point for further research. The medicinal chemist can then ‘tweak’ the structure to find analogues that bind more strongly and have better activity and selectivity.
Lead compounds are obtained from both the natural world and the laboratory. Historically, the natural world has been a rich source of novel lead compounds and remains so today. However, finding them is usually a slow process. Moreover, there is no guarantee of success. Nowadays, there is much greater emphasis on generating lead compounds by synthesis or rational design.
Structure–activity relationships and pharmacophores
Having identified a lead compound, it is important to establish which features of the compound are important for activity. This, in turn, can give a better understanding of how the compound binds to its molecular target.
Most drugs are significantly smaller than molecular targets such as proteins. This means that the drug binds to quite a small region of the protein—a region known as the binding site (Figure 49). Within this binding site, there are binding regions that can form different types of intermolecular interactions such as van der Waals interactions, hydrogen bonds, and ionic interactions. If a drug has functional groups and substituents capable of interacting with those binding regions, then binding can take place.
A lead compound may have several groups that are capable of forming intermolecular interactions, but not all of them are necessarily needed. One way of identifying the important binding groups is to crystallize the target protein with the drug bound to the binding site. X-ray crystallography then produces a picture of the complex which allows identification of binding interactions. However, it is not always possible to crystallize target proteins and so a different approach is needed. This involves synthesizing analogues of the lead compound where groups are modified or removed. Comparing the activity of each analogue with the lead compound can then determine whether a particular group is important or not. This is known as an SAR study, where SAR stands for structure–activity relationships.
Once the important binding groups have been identified, the pharmacophore for the lead compound can be defined. This specifies the important binding groups and their relative position in the molecule. The pharmacophore can be represented by highlighting the binding groups on the lead compound. For example, the pharmacophore for estradiol consists of three functional groups—the phenol group, the aromatic ring, and the alcohol group (Figure 55). The remainder of the tetracyclic structure serves as a rigid scaffold to hold the important binding groups in the correct positions, such that they interact simultaneously with the target binding site.
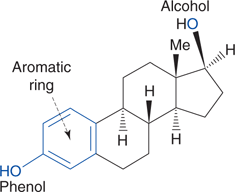
55. The pharmacophore for estradiol.
Identifying the pharmacophore of a rigid molecule such as estradiol is relatively straightforward, but with flexible compounds it is difficult to determine the relative position of important binding groups since the drug can exist in different shapes (conformations). For example, the binding groups for dopamine (an important neurotransmitter in the brain) are the two phenol groups, the aromatic ring, and the charged amine group (Figure 56). The relative positions of the phenol groups and the aromatic ring are easy to define as this is a rigid part of the molecule, but the position of the charged amine cannot be defined. This is because the bonds in the side chain can rotate, producing a large number of possible conformations. The conformation that binds most effectively to the binding site will have the charged amine group positioned in a particular way, and this is known as the active conformation. Other conformations will bind less effectively.
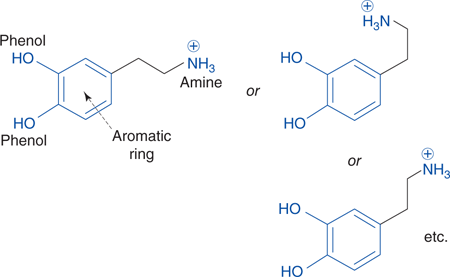
56. Different conformations of dopamine.
One way of identifying the active conformation of a flexible lead compound is to synthesize rigid analogues where the binding groups are locked into defined positions. This is known as rigidification or conformational restriction. The pharmacophore will then be represented by the most active analogue. For example, the structures shown in Figure 57 are rigid analogues of dopamine which have particular conformations of dopamine trapped within their structure. The bold bonds highlight the three-carbon chain that is present in dopamine. If one of these analogues proved significantly more active than the others, it can be used to define the active conformation and the pharmacophore.
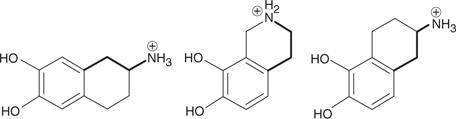
57. Rigid analogues of dopamine.
A large number of rotatable bonds is likely to have an adverse effect on drug activity. This is because a flexible molecule can adopt a large number of conformations, and only one of these shapes corresponds to the active conformation. If the molecule enters the binding site in an inactive conformation, it will depart again without binding. Indeed, a flexible molecule may have to enter and depart a binding site several times before it adopts the correct active conformation for binding. In contrast, a totally rigid molecule containing the required pharmacophore will bind the first time it enters the binding site, resulting in greater activity.
Drug design and drug optimization
Drug optimization involves designing and synthesizing analogues of the lead compound in order to find structures with improved activity, selectivity, and pharmacokinetics. A crystal structure of the lead compound bound to the target protein will help enormously in this quest—a process known as structure-based drug design—but it is not always possible to crystallize the target protein. Fortunately, there are a number of well-established design strategies that can aid the medicinal chemist in deciding which analogues are worth synthesizing. The strategy of designing a rigid analogue that mimics the active conformation of the lead compound has already been mentioned. Another strategy is to add extra groups to the structure, which would allow additional binding interactions to take place with parts of the binding site not occupied by the lead compound.
It is also important to optimize a drug’s pharmacokinetic properties such that it can reach its target in the body. Strategies include altering the drug’s hydrophilic/hydrophobic properties to improve absorption, and the addition of substituents that block metabolism at specific parts of the molecule.
Drug candidates and patenting
The drug optimization process produces a large number of compounds, several of which could be considered as a drug candidate for preclinical tests and clinical trials. Several factors are involved in deciding which one goes forward. The drug candidate must have useful activity and selectivity, with minimal side effects. It must have good pharmacokinetic properties, lack toxicity, and preferably have no interactions with other drugs that might be taken by a patient. Finally, it is important that it can be synthesized as cheaply as possible in order to make the maximum profit. Therefore, if there is a choice between two compounds of similar activity, then the choice may well be determined by identifying which one is cheaper to synthesize.
Once a promising-looking drug has been identified, it needs to be patented such that the company has exclusive rights to marketing. Since patenting takes place relatively early on in the drug development process, several years of the patent are lost due to the time taken to carry out preclinical and clinical trials.
Patenting drugs can lead to ethical dilemmas since the majority of people in the developing world cannot afford them. To address this, the World Trade Organization’s TRIPS (trade-related aspects of intellectual property rights) agreement allows governments in the developing world to grant compulsory licences for the manufacture of potentially life-saving drugs. This allows a country to bypass patent regulations and produce urgently needed medicines for its own citizens. Unfortunately, some countries have stretched the definition of life-threatening conditions. For example, in 2012, India imposed a compulsory licence on sorafenib—an anti-cancer agent which is considered to be life extending rather than life saving. This has created concerns that pharmaceutical companies might stop developing pharmaceuticals in therapeutic areas such as cancer or tropical diseases.
Chemical and process development
Once a candidate drug has been identified, work starts on developing a large-scale synthesis that will provide sufficient quantities of the drug for preclinical and clinical trials. This is known as chemical development. The development chemist has a demanding role as it is necessary to produce large quantities of the drug as quickly as possible, while maintaining the quality of each batch produced. This is crucial since the preclinical tests and clinical trials must be carried out on batches having a consistent purity. Otherwise, the tests are not comparing like with like. The chemical development process is more than just scaling up the original synthesis. Reactions may need to be modified, or altered altogether, in order to optimize yields. Indeed, the final production synthesis may be totally different from the original research synthesis.
Preclinical trials and formulation
Preclinical trials involve testing the drug candidate for selectivity, toxicity, and possible side effects. Most of this work is carried out by toxicologists, pharmacologists, and biochemists. However, organic chemists are needed to synthesize samples of the drug containing a radioisotope such as 14C. Such radiolabelled compounds are used to monitor the distribution and metabolism of the drug during in vivo tests.
Formulation involves pharmacists and pharmaceutical chemists who identify how best to store and administer the drug, for example as a pill or capsule.
Clinical trials and regulatory affairs
Clinical trials are the province of the clinician. There are four phases of clinical trials. Phase 1 involves a small group of healthy volunteers, whereas the later phases involve patients. The clinical trials are the most expensive and time-consuming part of the process in getting a drug to market and many drugs fail the process. This can be because they are not sufficiently effective, or they produce unacceptable side effects.
Regulatory bodies such as the US Food and Drug Administration (FDA) and the European Agency for the Evaluation of Medicinal Products monitor the process, and have to give their approval before the drug is finally marketed.
The future
Since the 1980s, there has been significant progress in treating diseases that were once considered untreatable. For example, there has been remarkable progress in designing effective antiviral drugs, inspired by the need to treat HIV. There has also been significant progress in treating various cancers. The development of a class of drugs called kinase inhibitors has been particularly important in this respect. However, there are some diseases that are still proving to be problematic. There are no cures for Alzheimer’s or Parkinson’s disease, and finding treatments for these diseases provides major challenges for the future. Most drugs that have reached clinical trials for the treatment of Alzheimer’s disease have failed. Between 2002 and 2012, 244 novel compounds were tested in 414 clinical trials, but only one drug gained approval. This represents a failure rate of 99.6 per cent as against a failure rate of 81 per cent for anti-cancer drugs.
The increase in drug-resistant bacterial strains is another concern. Several bacterial strains (such as Staphylococcus aureus) gain resistance to antibacterial agents due to their relatively high rates of mutation. For example, in the 1960s, S. aureus strains emerged that had gained resistance to the early penicillins. This crisis was averted by designing a new penicillin called methicillin that could combat these strains, but further strains have now developed that are resistant to methicillin (MRSA, or methicillin-resistant S. aureus). Other problem infections include multidrug-resistant tuberculosis and E. faecalis. Therefore, it is important to continue the search for novel antibacterial agents.
Several approaches to finding new antibacterial agents are being investigated. For example, the pharmaceutical company GlaxoSmithKline is currently investigating compounds that inhibit a bacterial enzyme called polypeptide deformylase. It has also been suggested that research should move away from finding new broad-spectrum antibacterial drugs that treat a wide variety of infections, to designing drugs that target specific infections. This is because broad-spectrum drugs have proved more susceptible to drug resistance. A therapy that combines a number of ‘narrow-spectrum’ drugs that act on different targets could be effective because there is little chance of drug resistance arising to all the drugs present in the combination.
Unfortunately, the trend in recent decades has been to cut back on antibacterial research due to the relatively low rates of success in obtaining new agents. Moreover, any novel agents that are discovered are likely to be placed on a reserve list to limit the chances of resistance arising. Consequently, pharmaceutical industries are unlikely to gain a significant financial return for the huge research investment required to design new drugs. Various national and world organizations have recognized the dangers and are now warning that more research is needed. In April 2014, the World Health Organisation declared that urgent, coordinated action was needed on a global scale to stop the world entering a post-antibiotic era where bacterial infections could once again become untreatable and result in even the most minor of injuries being potentially fatal.
Antimicrobial resistance is now seen as a ticking time bomb of global proportions, which poses as serious a threat to civilization as climate change. Tackling that threat is likely to require coordinated and funded collaborations between governments, pharmaceutical companies, and academic institutions. Fortunately, governments have now recognized the threat and have introduced new initiatives to encourage collaborative research.
Veterinary drugs
The same strategies used to design human medicines are applied to the development of veterinary drugs. Veterinary drugs are often different from human drugs because animals have different biochemical and metabolic systems. A compound that is safe for humans may be toxic to an animal. For example, theobromine is a constituent of chocolate that is toxic to dogs, but not to humans. ‘Doggy chocolates’ have to be specially formulated to lack theobromine.
Different animal species may also require different medicines for a specific disease. A further complication involves medicines for farm animals, since traces of the medication could end up in the food we eat. Therefore, there are regulations determining how long farmers should wait before slaughtering or milking animals that have received drug treatment. The discovery of horse meat in some European meat products during 2013 was a concern because it raised the possibility that traces of veterinary drugs might be present in the meat. For example, phenylbutazone is an anti-inflammatory agent used on horses, but causes adverse effects in humans.
The use of antibacterial agents in veterinary medicine is another concern since this could increase the prevalence of antibacterial resistance. For that reason, it is best to use agents that are not used in human medicine. Controversially, antibiotics such as penicillins and cephalosporins have been used to promote animal growth, but legislation is being passed in various countries to prevent this practice.
Each drug that is used in veterinary practice is approved for specific species, which means that a drug approved for use on a dog is not necessarily approved for use on a cat. To date, there are 634 drugs approved for dogs and 313 for cats. A complication in the treatment of dogs relates to different breeds. Some breeds are more susceptible to certain diseases than others, and there can be differences in drug metabolism. For example, the antiparasitic drug ivermectin is licensed for dogs, but can prove toxic to collies. As far as livestock is concerned, there are 688 products available for cattle in the USA, most of which are used to treat infections or inflammation. Veterinary medicine even includes medicines for bees. The varroa mite is one of the factors thought to be responsible for colony collapse disorder, where worker bees suddenly disappear from the hive. The mite can be treated with pyrethroid and organophosphate pesticides (Chapter 6), but mild cases of infection can be treated with antibiotics such as oxytetracycline.
Drugs of abuse
Several drugs that were once lauded as medical breakthroughs are now classed as drugs of abuse. For example, heroin was marketed at the end of the 19th century and was hailed as the ‘heroic’ drug that would eliminate pain. Unfortunately, nobody anticipated its addictive properties. Similarly, Sigmund Freud advocated the use of cocaine as an antidepressant until its addictive properties became clear. The psychoactive drug LSD was originally introduced as a medicine, and, in the 1970s, a psychoactive compound called MDMA was investigated as an aid to psychotherapy. However, the drug’s euphoric effects means that it is now taken as a ‘social drug’. This is the drug now known as ‘ecstasy’.
These are examples of drugs that were originally developed with the best of intentions, but a number of unscrupulous chemical companies are now deliberately designing drugs of abuse. These include stimulants that act in the same way as amphetamines. Because they are novel structures, they are not illegal and it is legitimate to sell them, as long as they are not advertised for human consumption. Instead, they are advertised as bath salts, plant food, or window cleaner.
Such designer drugs have been labelled ‘legal highs’, which might mislead consumers into thinking that they are legally approved. However, none of these drugs have undergone the preclinical and clinical trials required for medicines. Anyone taking them is gambling with their health, if not their life. When the UK government banned the legal high ‘serotoni’, it had already been responsible for thirty-seven deaths in the UK alone. Another forty-two people died taking the stimulant mephedrone.
Unfortunately, it takes time to identify novel legal highs, and even longer to make them illegal. By the time a ‘legal high’ has been banned, the company that produces it has usually modified the structure and introduced a new stimulant. For example, the product called Ivory Wave was sold as bath salts, and contained a psychoactive compound called methylenedioxyprovalerone. When this compound was banned, it was replaced with a similar structure called naphthylpyrovalerone. When this was banned, desoxypipradol was added instead. Desoxypipradol is more potent than the previous two compounds, and many regular users of Ivory Wave overdosed on the product.
The problem has been increasing in recent years. In 2009, there were twenty-four ‘legal highs’ sold across Europe, but this had risen to eighty-one by 2013. Chinese labs are thought to be producing most of the legal highs that are currently available. There has also been a rise in synthetic cannabinoids such as ‘annihilation’ which was responsible for nine people being hospitalized in 2012.
The UK government has now passed a law that bans any substance capable of producing a psychoactive effect, rather than banning each structure as it appears. However, this may only serve to drive the market underground. Moreover, genuine research on psychoactive drugs may be hindered by the need for government licences. There could be even wider ramifications if chemical suppliers feel obliged to stop selling chemicals used in the synthesis of legal highs, as that would adversely affect many legitimate research projects.