KEY CONCEPTS
An increasing risk of schizophrenia with advancing paternal age has been demonstrated through epidemiologic studies during the past decade. Before 2001, the results of research studying paternal age and schizophrenia were inconsistent with regard to risk of schizophrenia and advancing paternal age (Granville-Grossman, 1966; Hare & Moran, 1979; Kinnell, 1983; Malama et al., 1988; Bertranpetit & Fananas, 1993). However, in a large prospective cohort study in Jerusalem, Malaspina et al. (2001) reported that the risk of schizophrenia doubled with each 10-year increment of paternal age. Other studies confirmed that older paternal age is a risk factor for schizophrenia (Brown et al., 2002; Dalman & Allebeck, 2002; Byrne et al., 2003; Zammit et al., 2003; El-Saadi et al., 2004; Sipos et al., 2004; Tsuchiya et al., 2005), although two others did not find an association (El-Saadi et al., 2004; Pulver et al., 2004). Collectively, these studies strengthened the evidence that paternal age at birth is an important risk factor for schizophrenia. In this chapter we will outline (1) the epidemiologic evidence supporting the relationship between advanced paternal age and schizophrenia; (2) animal and human models pointing to a role for paternal age in learning and cognition; and (3) genetic and epigenetic mechanisms that may underlie the relationship between advanced paternal age and schizophrenia.
Seminal Findings
The maintenance of schizophrenia in the population despite the reduced fecundity of affected individuals has been one of the enigmatic features of the disease. One possible explanation could be the replenishment of disease genes through new mutations. Paternal age is reported to be the major source of de novo mutations in humans and other mammals, likely due to the constant cell replication cycles that occur in spermatogenesis. Following puberty, spermatogonia undergo some 23 divisions per year. At ages 20 and 40, a man’s germ cell precursors will have undergone about 200 and 660 such divisions, respectively. During a man’s life, the spermatogonia are vulnerable to DNA damage, and mutations may accumulate in clones of spermatogonia as men age (Crow, 1999). We hypothesize, therefore, that advanced paternal age might increase the risk of schizophrenia in the offspring through de novo genetic changes.
The Jerusalem Perinatal Study (JPS) is a population-based cohort that contains information on all 92,408 individuals born in West Jerusalem between 1964 and 1976. Information on maternal conditions, obstetric complications and interventions during labor and delivery, parental ages, immigration status, ethnicity, and occupation was recorded at birth. Postpartum and antepartum interviews of the mothers occurred from 1965 to 1968 and 1974 to 1976, respectively. Staff from the Israeli Ministry of the Interior linked the JPS data to the Israeli Psychiatric Case Registry in order to identify subjects with psychiatric diagnoses and maintain full confidentiality. In 2001, Malaspina and colleagues reported a monotonic increase in the risk of schizophrenia as paternal age increased (Malaspina et al., 2001).
Epidemiologic Evidence
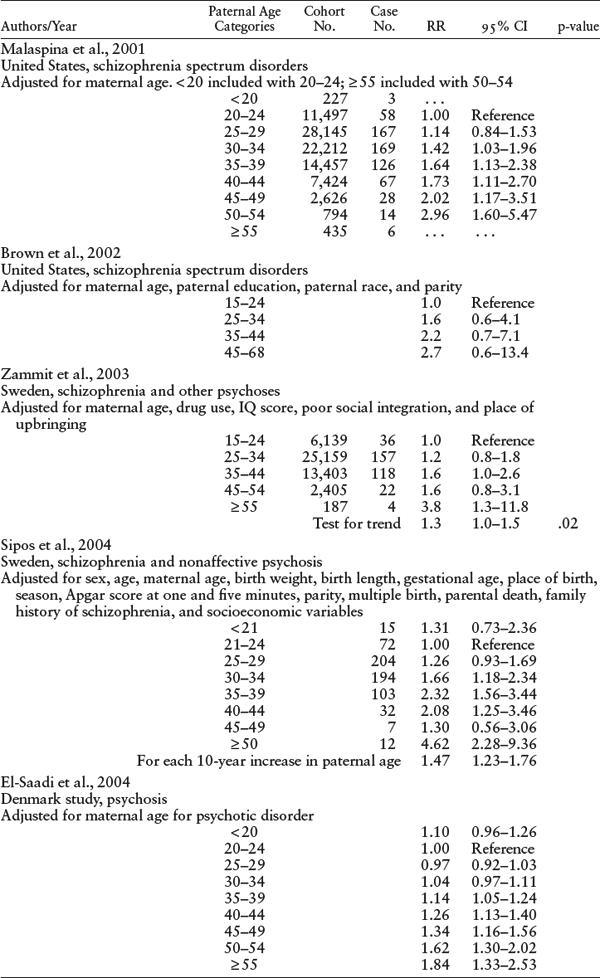
Malaspina’s finding was replicated in numerous studies in the United States (Malaspina et al., 2001; Brown et al., 2002), Europe (Dalman & Allebeck, 2002; Byrne et al., 2003; Zammit et al., 2003; El-Saadi et al., 2004; Sipos et al., 2004), Australia (El-Saadi et al., 2004), and Japan (Tsuchiya et al., 2005). Many studies used a cohort design (Malaspina et al., 2001; Brown et al., 2002; Zammit et al., 2003; El-Saadi et al., 2004; Sipos et al., 2004), but others employed a case-control design in a large population data set (Dalman & Allebeck, 2002; Byrne et al., 2003; El-Saadi et al., 2004; Pulver et al., 2004; Tsuchiya et al., 2005; St Clair, 2009). Details of these studies are provided in tables 5.1 and 5.2.
Collectively, the studies showed a tripling of risk for schizophrenia in the offspring of the oldest group of fathers, in comparison with the risk from younger fathers. Furthermore, the research demonstrated that the paternal age effect is not explained by other factors, including family history of psychosis, maternal age, parental education and social ability, family social integration, social class, birth order, birth weight, or birth complications.
Biological Plausibility
Several approaches have been employed to examine the biological plausibility of paternal age as a risk factor for schizophrenia. First, animal models were used to examine whether paternal age is related to specific outcomes that are relevant to schizophrenia. A translational approach using an animal model offers the opportunity to identify candidate genes, epigenetic mechanisms, or both that may explain the association of cognitive functioning with advancing paternal age. Second, epidemiologic studies have examined whether advanced paternal age is related to specific cognitive or social deficits in offspring.
Animal Models of Paternal Age Effects
Auroux was the first scientist to report that paternal age could negatively affect the learning capacity of Wistar rats sired by aged males (Auroux, 1983). Between 10 and 13 weeks of age, rat offspring were evaluated for learning capacity with an avoidance conditioning test. The results demonstrated that offspring of aged fathers had less spontaneous activity and worse learning capacity than those of mature rodents, despite having no noticeable physical anomalies.
In the laboratory of Jay Gingrich, this effect was replicated in inbred mice: the behavioral performance of progeny of 18- to 24-month-old sires was inferior to the performance of progeny of 4-month-old sires (Bradley-Moore et al., 2002). This study demonstrated significantly decreased learning in an active avoidance test, less exploration in the open field, and a number of other behavioral decrements in the offspring of older sires. Other investigations showed that offspring of 10-month-old sires had significantly fewer social and exploratory behaviors than offspring of two-month-old sires (Smith et al., 2009) and that aged rats sired offspring with a reduced capacity for passive-avoidance learning (Garcia-Palomares et al., 2009). Male progeny of older rats are also reported to have alterations in cortical growth, which are perhaps relevant to the behavioral findings (Foldi et al., 2010). These studies strongly indicate that advancing paternal age is related to cognitive, social, and anatomic changes in offspring.
CI = confidence interval; RR = relative risk.
SOURCE: Authors.
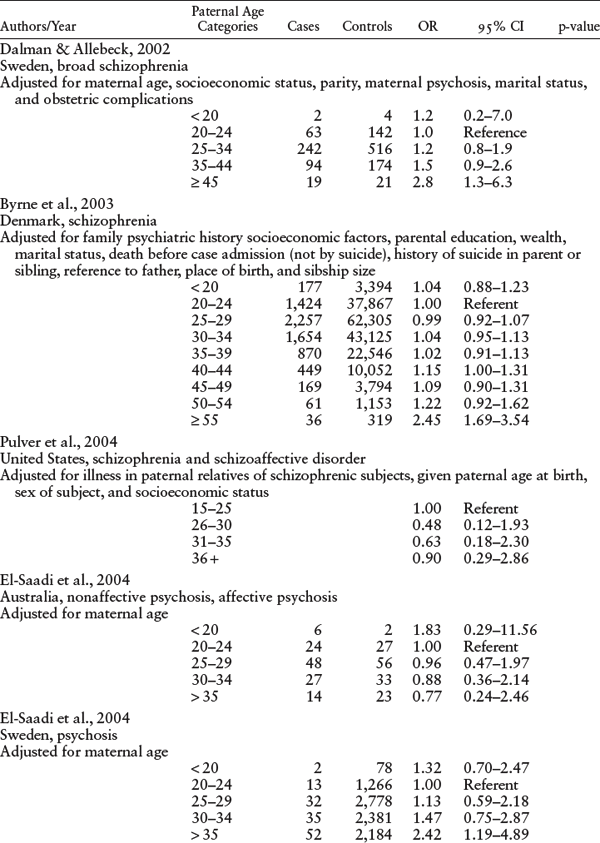
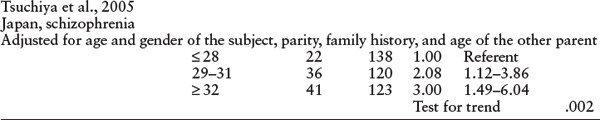
CI = confidence interval; OR = odds ratio.
SOURCE: Authors.
Paternal Age and Intelligence
A relationship between cognitive function and later paternal age in humans was explored by Auroux and colleagues, following their discovery of paternal age effects in rodents. The authors demonstrated a “U shaped” relationship between paternal age and cognition in a study of 1,700 male military recruits in Nancy, France, in 1985, wherein sons of very young fathers and of much older fathers did less well on psychometric tests without significant maternal age effects (Auroux et al., 1989).
Another study showed that higher paternal age (35 years or older) was associated with decreased reading ability in girls, although not in boys (St Sauver et al., 2001). A study that included more than 44,000 of the offspring from the JPS population-based cohort showed that maternal and paternal age exerted independent effects on intelligence in late adolescence (Malaspina et al., 2005). These results were adjusted for parental education, social class, sex, birth order, birth weight, and birth complications. They showed reductions in performance intelligence only for the offspring of the oldest fathers, whereas later maternal age comparably lowered verbal and nonverbal intelligence.
An effect of later paternal age on cognition in children was demonstrated in a sample of 33,437 offspring from the U.S. Collaborative Perinatal Project (CPP). In models that adjusted for potential confounders (including maternal age), they reported that children of older fathers were more likely to be cognitively impaired. Later paternal age was associated with worse performance in five out of six cognitive measures, whereas later maternal age was linked with better performance on intelligence tests (Saha et al., 2009).
The pathophysiology linking paternal age to intelligence is yet to be elucidated. Numerous genes participate in determining intelligence, and any of these might undergo mutations. As described here, paternal age is the most robust factor in determining the human spontaneous mutation rate (Crow, 1993). Both schizophrenia and autism entail reduced learning ability, so these findings may be relevant to the mechanism linking later paternal age to these conditions (Reichenberg et al., 2006; Corbett et al., 2009; Koenen et al., 2009; Mesholam-Gately et al., 2009).
This work is still nascent, and many explanations remain to be proposed and tested to explain the linkage of paternal age to intelligence and learning. For example, cigarette smoking can damage DNA in germ cells (Zenzes, 2000), and there may be higher rates of smoking in older cohorts. As detailed later in this chapter, changes may occur in the epigenetic processes in sperm cells with male aging. Epigenetic mechanisms are heritable regulators of gene expression that are independent of DNA sequence (Oakes et al., 2003) (see chapter 9 in this book). Socioeconomic status is also related to cognition in offspring (Turkheimer et al., 2003; Lawlor et al., 2006). In the study of paternal age and cognition in the CPP that was mentioned here, adjustment for socioeconomic status reduced the difference in intelligence scores for offspring of older and younger fathers, although it did not account for the paternal age effect (Saha et al., 2009). Additional large studies are needed to elucidate better the relationship between advanced paternal age and cognition. As childbearing is increasingly delayed, this association, if confirmed, could have important public health implications.
A Variant of Schizophrenia
A persistent question is whether the association of paternal age and schizophrenia could be explained by psychiatric problems in the parents—problems that both delay childbearing and are heritable by their offspring. If so, then advanced paternal age would be more common among familial cases than sporadic cases; however, this is not so (Sipos et al., 2004). It is also possible that de novo genetic events in the paternal germ line are affecting the same genes that influence the risk in familial cases. Available evidence suggests this is not the case. The few genetic studies that examined familial and sporadic cases separately found that the “at-risk haplotypes” linked to familial schizophrenia were not associated with sporadic cases, including dystrobrevin-binding protein (Van Den Bogaert et al., 2003) and neuregulin (Williams et al., 2003). Segregating sporadic cases from the analyses actually strengthened the magnitude of the genetic association in the familial cases, consistent with etiologic heterogeneity between familial and sporadic groups. Furthermore, sporadic cases have more de novo copy number variants (CNVs) than do familial cases in which CNVs are more likely to be inherited (Xu et al., 2008.).
Individuals with schizophrenia who have no family history of psychosis may have different phenotypes than familial cases. For example, only sporadic subjects showed a significant improvement in negative symptoms after changing from a “medication-free” condition to one where they received antipsychotic treatment (Malaspina et al., 2000). Sporadic subjects also have significantly more disruptions in their smooth pursuit eye movement quality (Malaspina et al., 1998) and have distinct intelligence test profiles (Wolitzky et al., 2006). The sporadic group of subjects had greater hypofrontality, with increased medial temporal lobe activity (frontotemporal imbalance), whereas the familial group had left lateralized temporoparietal hypoperfusion along with widespread regional cerebral blood flow (rCBF) changes in cortico-striato-thalamo-cortical regions (Malaspina et al., 2004). These findings support the hypothesis that familial and sporadic cases of schizophrenia may have distinct neural underpinnings.
Genetic Mechanisms
Several genetic mechanisms might explain the relationship between paternal age and the risk for schizophrenia. Spermatagonial stem cells divide constantly, undergoing hundreds of replications throughout a lifetime. Males have a higher frequency of point mutations, and the frequency increases with age and higher rates of de novo genetic disorders and birth defects in offspring (Crow, 1999).There are other possible explanations for the paternal age effects, as described in the sections that follow.
De Novo Mutations
Approximately 20% of birth defects are attributed to new mutations (Nelson & Holmes, 1989; Crow, 1997). Single base pair changes are the most frequently occurring mutations in paternal gametes. These may be random events or they may occur in certain “hot spots.” Achondroplastic dwarfism is an example of a “hot spot” mutation (Wilkin et al., 1998), wherein 90% of cases have a de novo mutation at the same single codon, leading to a missense mutation in the fibroblast growth factor 3 gene (Orioli et al., 1995; Yu et al., 2000).
Trinucleotide Repeat Expansions
Trinucleotide repeat disorders are caused by multiple insertions of three specific nucleotides in certain genes. An example is the (CAG)n repeat in the androgen receptor gene. Multiple insertions of a repeat sequence in genes may lead to repeat sizes that are above the normal threshold. These larger-than-normal repeat lengths may often result in a defective protein or no protein. These disorders are characterized by an autosomal dominant mode of inheritance, a progressive course, and anticipation—i.e., an earlier age of onset and an increasing severity of disease from one generation to the next. Anticipation has been reported in several neuropsychiatric disorders including myotonic dystrophy, spinocerebellar ataxias, and Huntington’s disease. For a detailed review of trinucleotide repeat disorders, see Orr and Zoghbi (2007).
The sex of the transmitting parent may influence anticipation (Telenius et al., 1993); many disorders show greater trinucleotide repeat expansion with paternal inheritance than with maternal inheritance (Duyao et al., 1993; Lindblad & Schalling, 1999; Schols et al., 2004). Consistent with anticipation, onset of schizophrenia may occur earlier in successive generations of multiply affected pedigrees (Gorwood et al., 1995; Petronis et al., 1995; Heiden et al., 1999).
It is not known if anticipation in schizophrenia is more likely for paternal or maternal transmission of the trinucleotide repeats, and the results of studies have been mixed (Gorwood et al., 1997; Johnson et al., 1997; Husted, Scutt, & Bassett, 1998; Imamura et al., 1998). Not all studies find longer trinucleotide repeats in family members with schizophrenia who have an earlier onset than the preceding generation (Morris et al., 1995; O’Donovan et al., 1996; O’Donovan & Owen, 1999; Bowen et al., 2000; Vincent et al., 2000).
Copy Number Variation
Over the last decade there has been an upsurge of interest in the possible role of CNVs in certain genes or regions of the genome in the etiology of schizophrenia, autism, mental retardation, and other neuropsychiatric disorders (St Clair, 2009). Copy number variation refers to a segment of DNA that can be from 1 kilobase to 1 or more megabases (Cook & Scherer, 2008) and that can include one or more genes, copy duplications, copy deletions, or multiallelic or complex rearrangements. A significant number of CNVs are over 50 kilobases in size and are recurrent. They have an increased mutation rate and are located in unstable parts of the genome. CNVs are discussed in more detail in chapter 8.
A difference between the rare inherited variants within some familial cases and de novo variants in sporadic cases of schizophrenia may be related to paternal age, given that the genetic variation is introduced through the paternal line.
Epigenetics and Advanced Paternal Age
Epigenetic phenomena are defined as heritable changes in the genome that are unrelated to the DNA sequence (Feinberg, 2004). Epigenetic mechanisms regulate the expression of genes and include methylation, acetylation, alterations in chromatin structure, and RNAi. Epigenetic changes occur in somatic cells with aging more frequently than mutations do (Bennett-Baker, Wilkowski, & Burke, 2003; Fraga et al., 2005). Epigenetics is described in detail in chapter 9.
A number of genes are imprinted in that they are only expressed from the paternal or maternal allele. Imprinting errors could increase with advancing paternal age through a number of pathways, including environmental exposures. In a recent study, it was reported that paternal occupation as a dry cleaner significantly increased the risk of schizophrenia in the offspring, an effect possibly due to chronic exposure to perchlorethylene, a common dry-cleaning solvent (Perrin et al., 2007).
X-Chromosome Inactivation
The association between advancing paternal age and schizophrenia among women may result at least in part from imprinting errors in the paternal X-chromosome of older fathers. X-chromosome inactivation is typically a random process in which 50% of cells have a paternal X-chromosome activated and maternal X-chromosome inactivated and 50% of cells have the maternal X-chromosome activated and the paternal X-chromosome inactivated, resulting in dosage compensation between males and females. A significant number of females have skewed X-chromosome inactivation—i.e., deviating from the 50:50 ratio just described. In females, only one allele is expressed on the X-chromosome and, in combination with skewed X-inactivation, results in a functional loss of heterozygosity (LOH) (Buller et al., 1999). This functional LOH may render women more susceptible to cognitive and neuropsychiatric disorders.
The hypothesis that imprinting errors on the X-chromosome may explain some of the risk in women associated with paternal age is supported by studies in women with Turner’s syndrome (45, X). It has been reported in some but not all studies that among women with Turner’s syndrome there are differences between those women whose X-chromosome has been transmitted through the maternal line (XM) and those whose X-chromosome has been transmitted through the paternal line (XP). Some studies have noted differences in social skills and executive function and arithmetic ability between 45, XP and 45, XM women (Skuse et al., 1997; Chong et al., 2000). Others have noted that there is strong correlation with cardiovascular disease in XM women but not XP women (Chu et al., 1994). Another more recent study reported that XM and XP women differed in superior temporal gyrus gray matter but not in white matter (Kesler et al., 2003). On the basis of these and other studies, imprinted genes are expected to be identified on the X-chromosome. Imprinting errors associated with advanced paternal age, coupled with skewed X-chromosome inactivation, could result in partial or complete loss of protein from a paternally expressed allele, increasing the risk of schizophrenia in females.
Failures to Replicate
Among all the epidemiologic evidence examining paternal age in relation to schizophrenia, there have been few failures to replicate either the paternal age effect or its approximate magnitude (El-Saadi et al., 2004; Pulver et al., 2004). The uniformity of the results across different cultures lends further credence to the robust relationship between advanced paternal age and schizophrenia. This relation is likely to reflect an innate human biological phenomenon that progresses over aging in the male germ line, independent of regional environmental risk factors for schizophrenia. The most plausible explanation is that the relation between advanced paternal age and schizophrenia is mediated through de novo genetic mechanisms and alterations in the epigenetic profile of different alleles.
Conclusion
Current findings on the relationship between paternal age and schizophrenia suggest new directions for research into the etiology of the disease. The studies linking advanced paternal age to the risk of schizophrenia indicate that we should expand this event horizon to consider the effects of environmental exposures on the fidelity of the male germ cell line over the life span of the father. The mutational stigmata of an exposure may remain in spermatogonial cells and be manifested in the clones of spermatozoa that it will subsequently generate over a man’s reproductive life.
KEY AREAS FOR FUTURE RESEARCH
Acknowledgments
Dr. Malaspina is supported by the following grants: National Institute of Mental Health (NIMH) 5K24 MH01699-11 and NIMH 5R01 MH59114-09. M. Perrin is supported by the following grants: National Institutes of Health (NIH) 5K07 CA131094-03, NIH 3K07 CA131094-02S1, and a National Alliance for Research on Schizophrenia and Depression Young Investigator Award. K. Kleinhaus is supported by NIH K08 MH085807.
Selected Readings
Harlap, S., Davies, A. M., Deutsch, L., Calderon-Margalit, R., Manor, O., Paltiel, O., Tiram, E., Yanetz, R., Perrin, M. C., Terry, M. B., et al. (2007). The Jerusalem Perinatal Cohort, 1964–2005: Methods and a review of the main results. Paediatric and Perinatal Epidemiology 21(3): 256–273.
Midgeon, B. R. (2007). Females Are Mosaics. New York: Oxford University Press.
Perrin, M., Brown, A. S., & Malaspina, D. (2007). Aberrant epigenetic regulation could explain the relationship of paternal age to schizophrenia. Schizophrenia Bulletin 33(6): 1270–1273.
Roth, T. L. & Lubin, F. D. (2009). Lasting epigenetic influence of early-life adversity on the BDNF gene. Biological Psychiatry 65(9): 760–769.
Rutten, B. P. F. & Mill, J. (2009). Epigenetic mediation of environmental influences in major psychotic disorders. Schizophrenia Bulletin 35(6): 1045–1056.
References
Auroux, M. (1983). Decrease of learning capacity in offspring with increasing paternal age in the rat. Teratology 27(2): 141–148.
Auroux, M. R., Mayaux, M. J., Guihard-Moscato, M. L., Fromantin, M., Barthe, J., & Schwartz, D. (1989). Paternal age and mental functions of progeny in man. Human Reproduction 4(7): 794–797.
Bennett-Baker, P., Wilkowski, J., & Burke, D. (2003). Age-associated activation of epigenetically repressed genes in the mouse. Genetics 165: 2055–2062.
Bertranpetit, J. & Fananas, L. (1993). Parental age in schizophrenia in a case-controlled study. British Journal of Psychiatry 162: 574.
Bowen, T., Guy, C. A., Cardno, A. G., Vincent, J. B., Kennedy, J. L., Jones, L. A., Gray, M., Sanders, R. D., McCarthy, G., Murphy, K. C., et al. (2000). Repeat sizes at CAG/CTG loci CTG18.1, ERDA1 and TGC13-7a in schizophrenia. Psychiatric Genetics 10(1): 33–37.
Bradley-Moore, M., Abner, R., Edwards, T., Lira, J., Lira, A., Mullen, T., Paul, S., Malaspina, D., Brunner, D., & Gingrich, J. A. (2002). Modeling the effect of advanced paternal age on progeny behavior in mice. Developmental Psychobiology 41(3): 230.
Brown, A. S., Schaefer, C. A., Wyatt, R. J., Begg, M. D., Goetz, R., Bresnahan, M. A., Harkavy-Friedman, J., Gorman, J. M., Malaspina, D., & Susser, E. S. (2002). Paternal age and risk of schizophrenia in adult offspring. American Journal of Psychiatry 159(9): 1528–1533.
Buller, R. E., Sood, A. K., Lallas, T., Buekers, T., & Skilling, J. S. (1999). Association between nonrandom X-chromosome inactivation and BRCA1 mutation in germline DNA of patients with ovarian cancer. Journal of the National Cancer Institute 91(4): 339–346.
Byrne, M., Agerbo, E., Ewald, H., Eaton, W. W., & Mortensen, P. B. (2003). Parental age and risk of schizophrenia: A case-control study. Archives of General Psychiatry 60(7): 673–678.
Chong, E. Y. Y., Pang, J. C. S., Ko, C. W., Poon, W. S., & Ng, H. K. (2000). Telomere length and telomerase catalytic subunit expression in non-astrocytic gliomas. Pathology, Research and Practice 196(10): 691–699.
Chu, C. E., Donaldson, M. D., Kelnar, C. J., Smail, P. J., Greene, S. A., Paterson, W. F., & Connor, J. M. (1994). Possible role of imprinting in the Turner phenotype. Journal of Medical Genetics 31(11): 840–842.
Cook, E. & Scherer, S. (2008). Copy-number variations associated with neuropsychiatric conditions. Nature 455: 919–923.
Corbett, B. A., Constantine, L. J., Hendren, R., Rocke, D., & Ozonoff, S. (2009). Examining executive functioning in children with autism spectrum disorder, attention deficit hyperactivity disorder and typical development. Psychiatry Research 166(2–3): 210–222.
Crow, J. F. (1993). How much do we know about spontaneous human mutation rates? Environmental and Molecular Mutagenesis 21(2): 122–129.
Crow, J. F. (1997). The high spontaneous mutation rate: Is it a health risk? Proceedings of the National Academy of Sciences U.S.A. 94(16): 8380–8386.
Crow, J. F. (1999). Spontaneous mutation in man. Mutation Research 437(1): 5–9.
Dalman, C. & Allebeck, P. (2002). Paternal age and schizophrenia: Further support for an association. American Journal of Psychiatry 159(9): 1591–1592.
Duyao, M., Ambrose, C., Myers, R., Novelletto, A., Persichetti, F., Frontali, M., Folstein, S., Ross, C., Franz, M., Abbott, M., et al. (1993). Trinucleotide repeat length instability and age of onset in Huntington’s disease. Nature Genetics 4(4): 387–392.
El-Saadi, O., Pedersen, C. B., McNeil, T. F., Saha, S., Welham, J., O’Callaghan, E., Cantor-Graae, E., Chant, D., Mortensen, P. B., & McGrath, J. (2004). Paternal and maternal age as risk factors for psychosis: Findings from Denmark, Sweden and Australia. Schizophrenia Research 67(2–3): 227–236.
Feinberg, A. P. (2004). The epigenetics of cancer etiology. Seminars in Cancer Biology 14(6): 427–432.
Foldi, C. J., Eyles, D. W., McGrath, J. J., & Burne, T. H. (2010). Advanced paternal age is associated with alterations in discrete behavioural domains and cortical neuroanatomy of C57BL/6J mice. European Journal of Neuroscience 31(3): 556–564.
Fraga, M., Ballestar, E., Paz, M., Ropero, S., Setien, F., Ballestar, M., Heine-Suñer, D., Cigudosa, J., Urioste, M., Benitez, J., et al. (2005). Epigenetic differences arise during the lifetime of monozygotic twins. Proceedings of the National Academy of Sciences U.S.A. 102(30): 10604–10609.
Garcia-Palomares, S., Pertusa, J. F., Minarro, J., Garcia-Perez, M. A., Hermenegildo, C., Rausell, F., Cano, A., & Tarin, J. J. (2009). Long-term effects of delayed fatherhood in mice on postnatal development and behavioral traits of offspring. Biology of Reproduction 80(2): 337–342.
Gorwood, P., Leboyer, M., Falissard, B., Rouillon, F., Jay, M., & Feingold, J. (1997). Further epidemiological evidence for anticipation in schizophrenia. Biomedicine and Pharmacotherapy 51(9): 376–380.
Gorwood, P., Leboyer, M., Jay, M., Payan, C., & Feingold, J. (1995). Gender and age at onset in schizophrenia: Impact of family history. American Journal of Psychiatry 152(2): 208–212.
Granville-Grossman, K. L. (1966). Parental age and schizophrenia. British Journal of Psychiatry 112(490): 899–905.
Hare, E. H. & Moran, P. A. (1979). Parental age and birth order in homosexual patients: A replication of Slater’s study. British Journal of Psychiatry 134: 178–182.
Heiden, A., Willinger, U., Scharfetter, J., Meszaros, K., Kasper, S., & Aschauer, H. N. (1999). Anticipation in schizophrenia. Schizophrenia Research 35(1): 25–32.
Husted, J., Scutt, L. E., & Bassett, A. S. (1998). Paternal transmission and anticipation in schizophrenia. American Journal of Medical Genetics 81(2): 156–162.
Imamura, A., Honda, S., Nakane, Y., & Okazaki, Y. (1998). Anticipation in Japanese families with schizophrenia. Journal of Human Genetics 43(4): 217–223.
Johnson, J. E., Cleary, J., Ahsan, H., Harkavy Friedman, J., Malaspina, D., Cloninger, C. R., Faraone, S. V., Tsuang, M. T., & Kaufmann, C. A. (1997). Anticipation in schizophrenia: Biology or bias? American Journal of Medical Genetics 74(3): 275–280.
Kesler, S. R., Blasey, C., Brown, W. E., Yankowitz, J., Zeng, S. M., Bender, B. G., & Reiss, A. L. (2003). Effects of X-monosomy and X-linked imprinting on superior temporal gyrus morphology in Turner Syndrome. Biological Psychiatry 54(6): 636–646.
Kinnell, H. G. (1983). Parental age in schizophrenia. British Journal of Psychiatry 142: 204.
Koenen, K. C., Moffitt, T. E., Roberts, A. L., Martin, L. T., Kubzansky, L., Harrington, H., Poulton, R., & Caspi, A. (2009). Childhood IQ and adult mental disorders: A test of the cognitive reserve hypothesis. American Journal of Psychiatry 166(1): 50–57.
Lawlor, D. A., Najman, J. M., Batty, G. D., O’Callaghan, M. J., Williams, G. M., & Bor, W. (2006). Early life predictors of childhood intelligence: Findings from the Mater-University study of pregnancy and its outcomes. Paediatric and Perinatal Epidemiology 20(2): 148–162.
Lindblad, K. & Schalling, M. (1999). Expanded repeat sequences and disease. Seminars in Neurology 19(3): 289–299.
Malama, I. M., Papaioannou, D. J., Kaklamani, E. P., Katsouyanni, K. M., Koumantaki, I. G., & Trichopoulos, D. V. (1988). Birth order sibship size and socio-economic factors in risk of schizophrenia in Greece. British Journal of Psychiatry 152: 482–486.
Malaspina, D., Friedman, J. H., Kaufmann, C., Bruder, G., Amador, X., Strauss, D., Clark, S., Yale, S., Lukens, E., Thorning, H., et al. (1998). Psychobiological heterogeneity of familial and sporadic schizophrenia. Biological Psychiatry 43(7): 489–496.
Malaspina, D., Goetz, R. R., Yale, S., Berman, A., Friedman, J. H., Tremeau, F., Printz, D., Amador, X., Johnson, J., Brown, A., et al. (2000). Relation of familial schizophrenia to negative symptoms but not to the deficit syndrome. American Journal of Psychiatry 157(6): 994–1003.
Malaspina, D., Harkavy-Friedman, J., Corcoran, C., Mujica-Parodi, L., Printz, D., Gorman, J. M., & Van Heertum, R. (2004). Resting neural activity distinguishes subgroups of schizophrenia patients. Biological Psychiatry 56(12): 931–937.
Malaspina, D., Harlap, S., Fennig, S., Heiman, D., Nahon, D., Feldman, D., & Susser, E. S. (2001). Advancing paternal age and the risk of schizophrenia. Archives of General Psychiatry 58(4): 361–367.
Malaspina, D., Reichenberg, A., Weiser, M., Fennig, S., Davidson, M., Harlap, S., Wolitzky, R., Rabinowitz, J., Susser, E., & Knobler, H. Y. (2005). Paternal age and intelligence: Implications for age-related genomic changes in male germ cells. Psychiatric Genetics 15(2): 117–125.
Mesholam-Gately, R. I., Giuliano, A. J., Goff, K. P., Faraone, S. V., & Seidman, L. J. (2009). Neurocognition in first-episode schizophrenia: A meta-analytic review. Neuropsychology 23(3): 315–336.
Morris, A. G., Gaitonde, E., McKenna, P. J., Mollon, J. D., & Hunt, D. M. (1995). CAG repeat expansions and schizophrenia: Association with disease in females and with early age-at-onset. Human Molecular Genetics 4(10): 1957–1961.
Nelson, K. & Holmes, L. B. (1989). Malformations due to presumed spontaneous mutations in newborn infants. New England Journal of Medicine 320(1): 19–23.
O’Donovan, M. C., Guy, C., Craddock, N., Bowen, T., McKeon, P., Macedo, A., Maier, W., Wildenauer, D., Aschauer, H. N., Sorbi, S., et al. (1996). Confirmation of association between expanded CAG/CTG repeats and both schizophrenia and bipolar disorder. Psychological Medicine 26(6): 1145–1153.
O’Donovan, M. C. & Owen, M. J. (1999). Candidate-gene association studies of schizophrenia. American Journal of Human Genetics 65(3): 587–592.
Oakes, C. C., Smiraglia, D. J., Plass, C., Trasler, J. M., & Robaire, B. (2003). Aging results in hypermethylation of ribosomal DNA in sperm and liver of male rats. Proceedings of the National Academy of Sciences U.S.A. 100(4): 1775–1780.
Orioli, I. M., Castilla, E. E., Scarano, G., & Mastroiacovo, P. (1995). Effect of paternal age in achondroplasia, thanatophoric dysplasia, and osteogenesis imperfecta. American Journal of Human Genetics 59(2): 209–217.
Orr, H. T. & Zoghbi, H. Y. (2007). Trinucleotide repeat disorders. Annual Review of Neuroscience 30: 575–621.
Perrin, M. C., Opler, M. G., Harlap, S., Harkavy-Friedman, J., Kleinhaus, K., Nahon, D., Fennig, S., Susser, E. S., & Malaspina, D. (2007). Tetrachloroethylene exposure and risk of schizophrenia: Offspring of dry cleaners in a population birth cohort, preliminary findings. Schizophrenia Research 90(1–3): 251–254.
Petronis, A., Sherrington, R. P., Paterson, A. D., & Kennedy, J. L. (1995). Genetic anticipation in schizophrenia: Pro and con. Journal of Clinical Neuroscience 3(2): 76–80.
Pulver, A. E., McGrath, J. A., Liang, K. Y., Lasseter, V. K., Nestadt, G., & Wolyniec, P. S. (2004). An indirect test of the new mutation hypothesis associating advanced paternal age with the etiology of schizophrenia. American Journal of Medical Genetics Part B: Neuropsychiatric Genetics 124(1): 6–9.
Reichenberg, A., Gross, R., Weiser, M., Bresnahan, M., Silverman, J., Harlap, S., Rabinowitz, J., Shulman, C., Malaspina, D., Lubin, G., et al. (2006). Advancing paternal age and autism. Archives of General Psychiatry 63(9): 1026–1032.
Saha, S., Barnett, A. G., Foldi, C., Burne, T. H., Eyles, D. W., Buka, S. L., & McGrath, J. J. (2009). Advanced paternal age is associated with impaired neurocognitive outcomes during infancy and childhood. PLoS Medicine 6(3): e40.
Schols, L., Bauer, P., Schmidt, T., Schulte, T., & Riess, O. (2004). Autosomal dominant cerebellar ataxias: Clinical features, genetics, and pathogenesis. Lancet Neurology 3(5): 291–304.
Sipos, A., Rasmussen, F., Harrison, G., Tynelius, P., Lewis, G., Leon, D. A., & Gunnell, D. (2004). Paternal age and schizophrenia: A population based cohort study. British Medical Journal 329(7474): 1070.
Skuse, D. H., James, R. S., Bishop, D. V., Coppin, B., Dalton, P., Aamodt-Leeper, G., Bacarese-Hamilton, M., Creswell, C., McGurk, R., & Jacobs, P. A. (1997). Evidence from Turner’s syndrome of an imprinted X-linked locus affecting cognitive function. Nature 387(6634): 705–708.
Smith, R. G., Kember, R. L., Mill, J., Fernandes, C., Schalkwyk, L. C., Buxbaum, J. D., & Reichenberg, A. (2009). Advancing paternal age is associated with deficits in social and exploratory behaviors in the offspring: A mouse model. PLoS One 4(12): e8456.
St Clair, D. (2009). Copy number variation and schizophrenia. Schizophrenia Bulletin 35(1): 9–12.
St Sauver, J. L., Katusic, S. K., Barbaresi, W. J., Colligan, R. C., & Jacobsen, S. J. (2001). Boy/girl differences in risk for reading disability: Potential clues? American Journal of Epidemiology 154(9): 787–794.
Telenius, H., Kremer, H. P., Theilmann, J., Andrew, S. E., Almqvist, E., Anvret, M., Greenberg, C., Greenberg, J., Lucotte, G., Squitieri, F., et al. (1993). Molecular analysis of juvenile Huntington disease: The major influence on (CAG)n repeat length is the sex of the affected parent. Human Molecular Genetics 2(10): 1535–1540.
Tsuchiya, K. J., Takagai, S., Kawai, M., Matsumoto, H., Nakamura, K., Minabe, Y., Mori, N., & Takei, N. (2005). Advanced paternal age associated with an elevated risk for schizophrenia in offspring in a Japanese population. Schizophrenia Research 76(2–3): 337–342.
Turkheimer, E., Haley, A., Waldron, M., D’Onofrio, B., & Gottesman, I. I. (2003). Socioeconomic status modifies heritability of IQ in young children. Psychological Science 14(6): 623–628.
Van Den Bogaert, A., Schumacher, J., Schulze, T. G., Otte, A. C., Ohlraun, S., Kovalenko, S., Becker, T., Freudenberg, J., Jonsson, E. G., Mattila-Evenden, M., et al. (2003). The DTNBP1 (dysbindin) gene contributes to schizophrenia, depending on family history of the disease. American Journal of Human Genetics 73(6): 1438–1443.
Vincent, J. B., Paterson, A. D., Strong, E., Petronis, A., & Kennedy, J. L. (2000). The unstable trinucleotide repeat story of major psychosis. American Journal of Medical Genetics 97(1): 77–97.
Wilkin, D. J., Szabo, J. K., Cameron, R., Henderson, S., Bellus, G. A., Mack, M. L., Kaitila, I., Loughlin, J., Munnich, A., Sykes, B., et al. (1998). Mutations in fibroblast growth-factor receptor 3 in sporadic cases of achondroplasia occur exclusively on the paternally derived chromosome. American Journal of Human Genetics 63(3): 711–716.
Williams, N. M., Preece, A., Spurlock, G., Norton, N., Williams, H. J., Zammit, S., O’Donovan, M. C., & Owen, M. J. (2003). Support for genetic variation in neuregulin 1 and susceptibility to schizophrenia. Molecular Psychiatry 8(5): 485–487.
Wolitzky, R., Goudsmit, N., Goetz, R. R., Printz, D., Gil, R., Harkavy-Friedman, J., & Malaspina, D. (2006). Etiological heterogeneity and intelligence test scores in patients with schizophrenia. Journal of Clinical and Experimental Neuropsychology 28(2): 167–177.
Xu, B., Roos, J., Levy, S., van Rensburg, E., Gogos, J., & Karayiorgou, M. (2008). Strong association of de novo copy number mutations with sporadic schizophrenia. Nature Genetics 40(7): 880–885.
Yu, K., Herr, A. B., Waksman, G., & Ornitz, D. M. (2000). Loss of fibroblast growth factor receptor 2 ligand-binding specificity in Apert syndrome. Proceedings of the National Academy of Sciences U.S.A. 97(26): 14536–14541.
Zammit, S., Allebeck, P., Dalman, C., Lundberg, I., Hemmingson, T., Owen, M. J., & Lewis, G. (2003). Paternal age and risk for schizophrenia. British Journal of Psychiatry 183: 405–408.
Zenzes, M. T. (2000). Smoking and reproduction: Gene damage to human gametes and embryos. Human Reproduction Update 6(2): 122–131.