65 | DISEASE-MODIFYING THERAPIES FOR ALZHEIMER’S DISEASE
JOSHUA D. GRILL AND JEFFREY CUMMINGS
Alzheimers disease (AD) is an age-related progressive neurodegenerative disorder. It is the most common cause of dementia, underlying 60–70% of all cases of late-onset cognitive decline. In the United States alone, more than 5 million persons are afflicted with AD and the prevalence is increasing. Whereas modern medicine has seen significant decline in deaths resulting from human immunodeficiency virus/acquired immunodeficiency syndrome, stroke, heart disease, and prostate and breast cancer, AD-related death has sharply risen. Alzheimer’s disease is the sixth leading cause of death in the United States. The association of AD with age is marked; disease risk doubles for every five years of life beginning after age 60. With the recent explosion of the baby boomer generation into retirement age and the expected increases of the world’s elderly population, an epidemic of AD is upon us. More than 30 million patients have dementia worldwide and the global prevalence of AD will double every 20 years (Ferri et al., 2005). The progressive course of AD eventually results in complete dependence and a high burden of care. The cost of this care is substantial, estimated at $180 billion annually in the United States, with worldwide expectations of unsustainable increases in cost. Thus, finding interventions that can reduce the burden associated with AD represents one of the most important scientific challenges in medical research.
The previous chapters in this section have described the substantial increases in understanding AD over the last several decades. The clinical phenomenology of AD is now well characterized and understood to include the slow and unrelenting progression of dementia, as well as a decade-long period of silent and then prodromal disease that eventually leads to clinical dementia. These predementia phases are characterized by accumulation of pathological substrates in the brain and initial neurodegeneration and may represent the ideal time of treatment.
Significant discoveries related to the genetic, molecular, and cellular underpinnings of AD have also been described in the preceding text. These discoveries have led to the identification of a wide array of therapeutic targets. Despite this, few drugs have achieved regulatory approval. Moreover, none of the approved drugs impact the underlying course of AD, instead providing mild improvement related to AD symptoms.
This chapter describes the state of the science of the pursuit of therapies that are capable of slowing the course of disease and, if started early enough, can impact the impending epidemic of AD. Specifically, the chapter:
• Defines disease modification and discuss its demonstration in the setting of AD clinical trials
• Presents the current state of trial design and outcome choices for studies of new potential disease-modifying agents
• Identifies the challenges to disease-modifying trials and how they may differ depending upon the clinical population enrolled
• Reviews findings from AD trials and how they contribute to the current understanding of potential disease modification and trial conduct
• Describes the current understanding of AD pathology and classes of drug candidates for disease-modification
• Presents the identified targets and candidate agents for potential disease-modifying therapies for AD
DEFINING DISEASE MODIFICATION
Many current clinical trials for AD address the pathophysiology of the disorder using pharmacologic targets affecting Aβ, tau-protein related mechanisms, mitochondrial function, neuroprotection, apoptosis, or neuroregenerative strategies (Salloway et al., 2008). These “disease-modifying” interventions aim to prevent, delay the onset, or slow the progression of the disease process. Neuroregenerative strategies or interventions that rescue dysfunctional but not moribund neurons could theoretically produce an improvement in function. These mechanism-based strategies are generally contrasted with symptomatic interventions that aim to compensate for deteriorating neural systems function but do not slow or ameliorate the underlying disease process. Neurotransmitter-related interventions, for example, are viewed as compensatory and not disease-modifying interventions.
No specific consensus definition of disease modification has evolved. Katz (2008) suggested that a strong definition of disease-modification is “a therapy that affects the underlying pathology and structure of the disease.” Cummings (2006) suggested that a disease-modifying agent is a pharmacological treatment that intervenes in the neurobiological processes that lead to cell death and produces a corresponding clinical response. This definition is essentially identical to that of the European Medicines Agency (EMA) (Committee for Medicinal Products for Human Use, 2008) that states that “for regulatory purposes a disease-modifying effect will be considered when the pharmacologic treatment delays the underlying pathological or pathophysiological disease processes and when this is accompanied by an improvement of clinical signs and symptoms of the dementing condition.” The EMA goes on to suggest a two-step approach in which the first step depends on delaying the course of the progression of the disease based on clinical signs and symptoms and leading to the limited claim of delay of disability. If these results are supported by a convincing package of biological and/or neuroimaging data (e.g., slowing, delay in the progression of brain atrophy) a full claim of disease modification can be considered.
The distinction between disease-modifying and symptomatic compounds cannot be drawn sharply. Some agents have dual action—such as ladostigil with cholinesterase inhibition plus monoamine oxidase inhibition (Weinreb et al., 2011)—and agents such as cholinesterase inhibitors that are viewed as largely symptomatic may have modest disease-modifying effects (Sabbagh et al., 2008).
Disease modification from a clinical trialist or regulatory point of view may differ from that of a practicing physician or patient and caregiver. Patients and caregivers would likely see a therapy that slows clinical progression but does not produce improvement of symptoms as less than successful.
The terminology of disease modification is typically not used in regulatory language appearing in the package insert for a medication. Interferon beta-1a, for example, has clinical and biomarker support as a disease-modifying intervention for multiple sclerosis. Its label reads, “to slow the accumulation of physical disability and decrease the frequency of exacerbations.” Similarly, etanercept has clinical and biomarker evidence of disease modification for rheumatoid arthritis. Its label notes its utility for “inhibiting the progression of structural damage and improving physical function”(Cummings, 2009). Regulatory terminology for AD interventions with clinical and biomarker evidence of disease modification will likely have similar trial-specific labeling regarding reduction in loss of cognition and function with supporting evidence from a specified biomarker.
The FDA tends to avoid mechanism-related terminology. The language for the indication for the use of a cholinesterase inhibitor is “for the treatment of mild-to-moderate Alzheimer’s disease” and does not stipulate that this is symptomatic or disease-modifying therapy (Katz, 2008).
The development of therapies targeting the pathophysiology and biology of AD has led many companies to adopt trial outcomes that will support labeling related to disease modification. Two strategies have been discussed in this context. The randomized withdrawal design and the randomized start design can theoretically demonstrate delay of progression in the absence of a biomarker. However, the statistical and ethical issues involved in these designs (Bodick et al., 1997; Rascol et al., 2011) have led to rare use of this approach. These are discussed in more detail in the following.
For practical purposes of constructing clinical trials and demonstrating disease-modifying properties of an intervention, a combination of a clinical trial demonstrating a drug–placebo difference in clinical outcomes and accompanied by a related drug–placebo difference in key biomarker outcomes is typically used. The terminology for the indication will likely reflect the clinical trial outcomes, such as reduction in loss of cognition and activities of daily living and corresponding improvement in a biomarker indicative of the biology of the disease such as atrophy on magnetic resonance imaging (MRI), amyloid burden on amyloid imaging, brain-metabolism on fluorodeoxyglucose positron emission tomography (FDG PET), or spinal fluid measures such as cerebrospinal fluid (CSF) Aβ42, tau, or phospho-tau. There remains substantial uncertainty about exactly how a regulatory agency such as the FDA will view data from biomarkers included in clinical trials of disease-modifying compounds (Katz, 2004). Paradoxical effects have confounded clinical trial interpretation; the observation that patients in the AN 1792 trial (active vaccination comprised of Aβ42) with a more robust antibody response had greater atrophy was unanticipated (Fox et al., 2005). Similarly in a trial of divalproex designed to show disease modification with a kinase inhibitor, there was greater MRI atrophy in the treatment than the placebo group (Fleisher et al., 2011). These observations complicate the planning for optimal use of biomarkers in trials.
DEMONSTRATING DISEASE MODIFICATION IN CLINICAL TRIALS
CLINICAL TRIAL DESIGNS
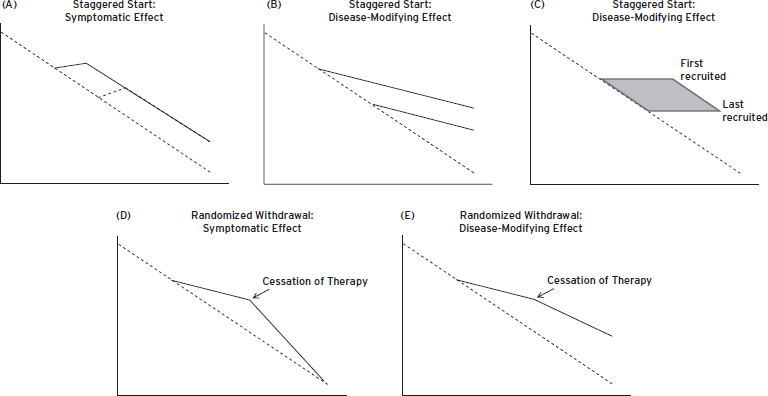
Two clinical trial designs have been suggested as adequate to indicate disease modification: These are the staggered start design and the randomized withdrawal design (Bodick et al., 1997). In the staggered start design, individuals are randomized to drug or placebo at study onset (Fig. 65.1A). After a defined interval, individuals in the placebo arm are switched to active treatment. If the switched group “catches up” with the original treatment group, then no disease modification has been demonstrated and a symptomatic benefit is concluded. On the other hand, if the delayed treatment group fails to “catch up” with the original treatment group, this suggests that the disease has been modified in the group first randomized to therapy (Fig. 65.1B). There are several uncertainties with this design. It is unclear how long of a therapeutic exposure might be required to establish therapeutic benefit, and likewise it is unclear how long of an observation period would be required to determine if the delayed start group has “caught up” with the original therapy group.
A novel interpretation of the delayed start design is the observation that in any recruitment period some patients begin early after the trial is initiated and some may begin as long as twelve months later (e.g., in a twelve-month recruitment period). Patients recruited later in the trial can be compared with patients recruited earlier in the trial. This allows a statistical “staggered start” analysis (Fig. 65.1C) (Hendrix S., personal communication).
The randomized withdrawal design is based on the fact that a group withdrawn from therapy will decline to the level of function expected of a placebo group if no disease modification has occurred (Fig. 65.1D), whereas a withdrawal group that continues to remain above the expected level has experienced disease modification (Fig. 65.1E). The randomized withdrawal design is subject to the same uncertainties as the randomized start design. The duration of treatment and the duration of observation off treatment are both uncertain.
Figure 65.1 (A) Staggered start design with symptomatic effect shown.(B) Staggered start design with disease-modifying effect shown.(C) Natural history staggered start with observations collected over the course of the recruitment period.(D) Randomized withdrawal design with symptomatic effect shown.(E) Randomized withdrawal design with disease-modifying effect shown.
These designs have rarely been implemented in neurodegenerative disorders due to their complexities and uncertainties. When used in a Parkinson’s disease trial, the results did not lead to a clear-cut conclusion, interpretation (Rascol et al., 2011), and/or new labeling.
Delay of disability is consistent with disease modification but can be achieved with symptomatic therapy (Mohs et al., 2001). Delay of milestones is not conclusive evidence of disease modification.
Use of biomarkers in conjunction with clinical outcomes is an alternative for establishing that an agent has changed the underlying biology of the disorder. Most biomarkers are inferential in that they represent imaging findings, electrophysiological observations, or peripheral indicators of a central process. The central process itself is undocumented. Although MRI correlates with nerve cell loss, nerve cell loss does not account for all the variance of MRI atrophy (Burton et al., 2012). This suggests that other as yet unidentified factors account for some aspects of MRI atrophy. Atrophy is also common across neurodegenerative disorders, is not specific to AD, does not identify a mechanism of action of an intervention, and does not indicate a specific change in pathology. Similarly, FDG PET is a measure of metabolism, a non-specific indicator of synaptic activity. A beneficial effect of an intervention on FDG PET compared with placebo would indicate improved neuronal and synaptic function in the treatment group, but would not establish the mechanism for such improvement. Symptomatic agents may improve FDG PET activity in AD (Keller et al., 2011; Mega et al., 2005). Amyloid PET provides specific information on deposited fibrillar amyloid in the cortex, and its removal can be demonstrated by amyloid imaging in clinical trials (Rinne et al., 2010; Ostrowitzki et al., 2012). However, fibrillar amyloid may be a nontoxic species of Aβ and removal of fibrillar amyloid does not necessarily predict cognitive benefit (Holmes et al., 2008). Fibrillar Aβ is a proxy for the amyloidogenic process and possibly the presence of neurotoxic oligomers. It is not necessarily a direct measure of a neurotoxic species or the effect on neuronal and synaptic function. Serum and CSF measures such as Aβ40 and Aβ42 or CSF tau and phospho-tau are measures of cellular products and processes that have migrated from the interstitial space of the brain across the blood-brain or brain–CSF barrier and into the serum or CSF. Measures of these elements provide indirect reflections of the processes leading to cell death and dysfunction (Clark et al., 2003) but are not direct brain measures. Biomarkers are indirect and inferential measures of the basic processes leading to cell death and dementia.
To be supportive of a disease-modifying effect, the response of a biomarker (drug–placebo difference) should be correlated with the clinical outcomes. Biomarkers chosen as key outcomes should correlate with the primary clinical outcomes. For example, hippocampal atrophy may be a good choice to support a diagnosis of AD, but whole brain atrophy or ventricular enlargement may correlate better with trial outcomes such as Alzheimer’s Disease Assessment Scale-Cognitive Subscale (ADAS-Cog) and Clinical Dementia Rating (CDR) (Ridha et al., 2008). The correlation of a biomarker with a clinical outcome is necessary but not sufficient evidence to establish that the biomarker and clinical outcomes reflect a common mechanism. Most drugs have multiple effects, some known and some unknown. Biomarker and clinical effects could be mediated by different pathways or could even have opposite effects. Establishing the relationship between a biomarker and a clinical outcome will take experience across multiple clinical trials and multiple therapeutic mechanisms (Katz, 2004).
To incorporate a biomarker in a clinical trial and be included in the package insert labeling of a therapeutic agent, the biomarker must go through a qualification process with review by the FDA. Decisions about how the biomarker is best included in the trial and how biomarker data will be managed in data analysis plan (Cummings, 2011b; Wagner et al., 2007) must be prespecified.
The use of biomarkers as an outcome for treatment in prevention trials including patients who are without symptoms is under consideration (Aisen et al., 2011; Vellas et al., 2011). A surrogate marker would have predictive value for a drug–placebo difference on a clinical outcome. No biomarker has risen to surrogate status. Although biomarkers have been shown to correlate with clinical measures, none have been shown to predict clinical outcomes. An incremental evolutionary approach is most likely to succeed in developing surrogate measures over time. Showing that a biomarker change predicts a clinical outcome in a mildly affected patient population would be a step towards incorporating the biomarker in a trial of asymptomatic individuals. The biomarker might eventually replace the clinical outcome and serve as a surrogate. This is a multiyear, multitrial process.
Clinical trial outcomes used in conjunction with biomarkers in trials of disease-modifying agents might include a drug–placebo difference at trial conclusion, delay in milestones, increasing drug–placebo difference or time, and change in slope of decline (discussed in more detail in the following).
POPULATIONS CONSIDERED FOR DISEASE-MODIFYING TRIALS
AD is traditionally described as a disease with a progressive clinical course over 8–12 years, ultimately resulting in complete dependence and then death. Fulfillment of the National Institute on Neurological and Communicative Disorders and Stroke-Alzheimer’s Disease and Related Dementias Association (NINCDS-ADRDA) criteria for probable AD has been used as an inclusion criterion in most trials. The NINCDS-ADRDA criteria are purely clinical and incorporate the concept of dementia. A patient must demonstrate memory impairment and impairment to another domain of cognition, and these cognitive impairments must affect the patient’s ability to perform activities of daily living and not be better accounted for by another possible cause. The cognitive impairment must be progressive and present for at least six months.
The course of AD, however, spans 20 years and includes a period of biological onset that precedes dementia. Recent improvements to AD criteria incorporate biomarkers, increase diagnostic accuracy, and facilitate earlier diagnosis—including before criteria for dementia are met. Differing nomenclatures and clinical and biological criteria for the phases of AD have been proposed. Disease-modifying trials can use these criteria as an initial means for defining the population to be studied and can enroll any sample of patients with AD, be they asymptomatic, mildly symptomatic, or demented. The group of patients to be included is decided based on trial goals.
TRIALS ENROLLING ASYMPTOMATIC PATIENTS AT GREATEST GENETIC RISK
Alzheimer’s disease can be predicted with certainty from conception in a fraction of cases. Autosomal dominant AD results from gene mutations to the amyloid precursor protein(APP), presenilin-1, and presenilin-2 genes. Within families that carry these mutations, there is a 50% heritability, 100% penetrance, and substantial consistency in the age of symptom onset. This creates an opportunity for trial designs that initiate therapy at an approximate time frame preceding clinical onset to examine drug potential to delay that onset. Two major international programs are currently underway with the objective of better characterizing this familial form of AD and conducting prevention trials (Bateman et al., 2011; Reiman et al., 2010). The Dominantly Inherited Alzheimer’s Network includes seven US sites, one UK site, and three Australian sites. The AD Prevention Initiative (API) is a collaboration between US and Colombian investigators that follows the largest known kindred of familial AD, a cohort of presenilin-1 mutation carriers in Antioquia, Colombia. Asymptomatic offspring will be enrolled (non-randomly assigning non-carriers to placebo). Crenezumab, a monoclonal antibody targeting Aβ, is the candidate agent to be studied in this cohort.
A second component of the API proposes trials in those at greatest genetic risk for the sporadic form of AD: carriers of the ε4 allele of the apolipoprotein E(APOE) gene. Apolipoprotein E was the first identified and remains the strongest known genetic risk factor for sporadic AD. Three alleles have been identified: ε2, ε3, and ε4. The ε3 allele is most common among the general population. Hetero- and homozygotic carriers of ε4 are at significantly increased risk for sporadic AD. Current studies estimate up to a 12-fold increased risk associated with ε4 homozygosity (see http://www.alzgene.org/). Furthermore some ε4 carriers, by middle age, demonstrate AD-like abnormalities on functional MRI, FDG PET, and amyloid PET. Therefore, enrollment of ε4 carriers is an enrichment strategy.
Takeda and Zinfandel pharmaceuticals are conducting a prevention trial in which patients are selected on the basis of risk associated with APOE and TOMM40 carrier status for specific age strata. Those at highest risk are assigned to pioglitazone or placebo and those at low risk are assigned to placebo.
For each genetic risk strategy, it is unknown what proportion of asymptomatic patients will meet biomarker criteria for trial entry (see the following) and at what time points. The overlap between genetics and biology is substantial, but imperfect. Combined biomarker and genetic inclusion criteria are feasible.
TRIALS ENROLLING ASYMPTOMATIC PATIENTS AT GREATEST BIOLOGICAL RISK
Disease-modifying trials can identify and enroll cognitively normal older persons who demonstrate AD biomarkers. At autopsy, approximately 20–40% of cognitively normal elderly (age 65 or older) meet criteria for neuropathological diagnosis of AD. Amyloid PET imaging reveals a similar proportion of amyloid positive older living individuals who, at the time of scanning, have no cognitive impairment. A growing literature supports the inclusion of such patients in disease-modifying trials, because they are at substantially increased risk to develop cognitive impairment and symptomatic AD. For example, in one study of 159 nondemented participants with follow-up periods ranging from one to five years, amyloid PET positivity at baseline was associated with a hazard ratio of 4.85 [95% CI: 1.2–19.0] for progression to AD dementia (Morris et al., 2009).
The first trial enrolling biomarker positive cognitively normal older participants is being planned. The anti-amyloid in asymptomatic AD (A4) trial will enroll amyloid PET positive volunteers age 70 or older and randomize to placebo or amyloid immunotherapy. These patients have been described as asymptomatic at risk (Dubois et al., 2010) or preclinical AD (Sperling et al., 2011). The length of the asymptomatic phase of AD is unknown, and one challenge for the A4 trial is to determine an optimal study length. Too short of a trial would not permit examination of prevention or delay of onset of cognitive impairment as an outcome. Alternatively, amyloid biomarker positive patients experience greater and more rapid change in other biomarkers, such as brain volume and metabolic rate, but such outcome measures are unlikely to satisfy FDA requirements for approval (see previous). Validation of biomarkers in demented populations may lead to a regulatory pathway for demonstration of disease modification in asymptomatic patients (Aisen et al., 2011). Another potential pitfall of preclinical AD trials is that they will require very large numbers of subjects and are likely to encounter high rates of screen failures for biomarker criteria for entry (Grill, Di et al., 2012a).
TRIALS ENROLLING SYMPTOMATIC PATIENTS
Most disease-modifying trials are conducted in impaired patients. DuBois and colleagues describe the earliest symptomatic phase of disease as “prodromal AD.” Prodromal AD patients demonstrate episodic memory impairment (adjusted for education) and AD biomarkers. Recent trials have applied these criteria for entry. Prodromal AD trials have used Aβ biomarkers (low CSF Aβ42, high ratio of CSF tau/CSF Aβ42, or amyloid PET).
The development of criteria for prodromal AD was in part the result of challenges previously met by trials enrolling patients with mild cognitive impairment (MCI). MCI is based only on clinical criteria for impairment of memory or other cognitive domains (generally 1 to 1.5 standard deviations below age- and education-adjusted norms). MCI trials that examined the rate of conversion to AD dementia as a primary outcome measure frequently met lower than expected rates of conversion, necessitating trial extension in some cases. No trial succeeded in demonstrating a therapeutic effect in MCI patients. MCI is a heterogeneous disorder with several underlying pathologies and diverse clinical outcomes. Many observational studies have shown that MCI patients demonstrating AD biomarkers are at greatest risk for progression to AD dementia and proposed research criteria have incorporated biomarker support to the diagnosis of MCI caused by AD (Albert et al., 2011). The use of prodromal AD criteria obviates the need for the MCI construct.
The majority of AD trials have enrolled patient populations meeting criteria for dementia and mild-to-moderate AD. As discussed, the most commonly applied diagnostic criteria are those developed by the NINCDS-ADRDA. Criteria for mild-to-moderate severity are frequently defined using the Mini-Mental State Examination (MMSE), typically set at MMSE 14–26. Most AD patients will meet these criteria at the time of dementia diagnosis or shortly thereafter and for approximately the next three to five years. By the time of AD dementia diagnosis, most but not all AD patients will be readily identifiable on multiple AD biomarkers. Recent work suggests that some biomarkers such as amyloid PET may reach a plateau (peak level of abnormality) by the time of AD dementia diagnosis (Jack et al., 2010). Such findings suggest that this phase of AD biology may already be too advanced for targeted treatments to be successful. Alternatively, biomarkers such as hippocampal volume, cortical thickness, and FDG PET metabolism appear to decline linearly through the dementia phase of AD. Therapeutic strategies able to arrest this decline may therefore still be able to modify the natural history of disease, rescuing brain tissue or function. Since no trial for disease-modifying agents has succeeded in any AD populations, it is premature to conclude which population are most treatment responsive.
AD trials have been conducted in more impaired patients, including moderately demented (e.g., global CDR = 2) or severely demented patients (e.g., defined as minimum MMSE of 5–7 and maximum MMSE defined as 14–17). The capacity of severely demented patients to sign informed consent or complete cognitive outcome assessments is diminished. For many families, these trials represent a last attempt to find alternative therapy, often out of desperation. Successful disease modification that fails to provide clinical improvement in severely demented patients, however, presents a serious ethical dilemma; the likely outcome is prolongation of a significantly compromised state.
CHALLENGES RELATED TO TRIAL SAMPLES
Trial inclusion and exclusion criteria are designed to ensure a homogeneous sample and facilitate interpretation of trial results. Trials that enroll heterogeneous samples face significant challenges. Mild-to-moderate AD dementia trial populations are often wide-ranging in age, requiring various stratifications as part of randomization strategies (e.g., age, genotype, site, MMSE). These trials may also have enrolled heterogenous populations in terms of disease biology. Most AD dementia trials have applied clinical criteria without biomarker requirements related to disease diagnosis. It is likely that a portion of trial participants do not suffer from AD, but instead some other underlying pathology, potentially affecting trial internal validity. The application of biomarker criteria, even in dementia trials, may facilitate improved sample homogeneity. Even the application of biological criteria, however, is not a guarantee of sample homogeneity. For example, amyloid PET may label some Lewy body dementia patients (and may fail to label some AD patients). Furthermore, a large portion (20–40%) of AD patients suffer vascular comorbidity and a smaller portion (<5%) may suffer comorbid neurodegenerative disease. Only through combined clinical and biological criteria (including MRI exclusion of those with vascular pathology likely to contribute to cognitive impairment) can enrollment of an appropriate and homogeneous sample be achieved. Evolving biomarkers may allow further narrowing of populations to better match biological subtype and mechanism of action of the test compound.
CLINICAL OUTCOMES IN DISEASE-MODIFYING TRIALS
Dependent upon the trial design and the population enrolled, a large number of clinical outcome scenarios may be used to examine if a drug or other treatment is capable of altering the natural history of AD. As yet, there is no clear preferred outcome choice for disease-modifying AD trials. Broadly categorized, these trials either directly examine efficacy or test the ability of a drug to delay a milestone. For each choice, primary comparison is versus placebo. An active comparison is sometimes included as a means of assessing trial quality. A trial with donepezil that did not demonstrate a donepezil-placebo difference would be regarded skeptically.
DRUG–PLACEBO DIFFERENCE AT TRIAL END
The most straightforward outcome metric for assessing drug efficacy is to compare the group randomized to drug to the group randomized to placebo at trial end. This parallel design assumes successful randomization (otherwise the groups might not be equivalent at baseline, complicating analyses and interpretation). In most trials to date, mean group changes from baseline were compared, rather than cross-sectional comparisons at trial completion. The parallel group design has been used in most mild-to-moderate or moderate-to-severe AD dementia trials, including those that resulted in the regulatory approval of the cholinesterase inhibitors and memantine.
DELAY IN ONSET OF MILD COGNITIVE IMPAIRMENT
When enrolling asymptomatic patients, a delay-to-milestone approach can be used, incorporating the mean time to a diagnosis of MCI as the outcome or using a survival-type design. Both primary prevention trials (enrolling community based samples with no clinical or biological sign of disease) and secondary prevention trials (enrolling “asymptomatic at risk” or “preclinical AD”) could use the MCI construct as an outcome. If the mean time to MCI diagnosis is delayed or a significantly lower proportion of subjects randomly assigned to the active medication fulfills MCI criteria at a predetermined time point, the claim of delay of disability would be supported. As with all predementia disease-modifying trials, there is currently little guidance on the choice of trial length. A resulting potential challenge is that insufficient numbers of subjects may develop cognitive impairment to provide adequate statistical power to examine a drug effect. Adaptive designs with variable lengths determined by prespecified outcomes are one solution to this problem. To support a claim of disease modification, the delay in MCI should be supported by delay in progression of biomarkers.
DELAY IN ONSET OF DEMENTIA
Trials enrolling MCI populations can examine a treatment’s ability to delay dementia onset. Several trials have employed this design, including trials of rofecoxib, rivastigmine, and donepezil and vitamin E (Jelic et al., 2006). These trials employed fulfillment of NINCDS-ADRDA criteria for possible or probable AD or fulfillment of NINCDS-ADRDA and DSM-IV-TR criteria for AD as primary outcomes. In most cases, assessment of progression to dementia was performed every three months. In the trial of donepezil and vitamin E, there was a suggestion after one year that fewer participants assigned to donepezil had progressed to dementia than in the placebo group (secondary outcome). By three years (the time point of the primary outcome assessment), however, there was no difference in the proportion of participants who had progressed to dementia between the groups. A primary outcome at one-year (or a one-year trial) might have concluded drug efficacy or inappropriately concluded a disease-modifying effect. No other MCI trial has observed even a trend toward disease modification. In the trials of rofecoxib and rivastigmine, fewer than expected participants progressed to dementia (in both the treatment and the placebo groups). The rofecoxib trial was halted prior to completion because of this low conversion rate and high drop out. The rivastigmine trial was subsequently extended to permit examination of the primary outcome. Symptomatic benefit will delay onset of dementia and by themselves the delay to dementia designs do not establish disease modification. These challenges encountered in the cholinesterase inhibitor dementia progression trials demonstrate the importance of trial planning and adequate data support for trial design decisions.
DELAY IN OTHER MILESTONES
Besides delay to diagnostic outcomes (onset of dementia or onset of MCI), AD trials have examined other milestones and the ability of a drug to delay the onset of those milestones. Two very large trials in MCI examined progression to a global CDR score greater than or equal to 1(mild dementia, see the following) as a primary outcome. The rofecoxib MCI trial discussed earlier incorporated this endpoint as a secondary outcome. Other trials enrolling dementia populations, such as that examining vitamin E and selegiline, have incorporated a time to global CDR score 3(severe dementia), loss of activities of daily living (ADLs), institutionalization, or death. An ADCS study of valproic acid enrolled moderate AD patients with no history of agitation or psychosis and investigated the drug’s ability to reduce (relative to placebo) the time to emergence of such symptoms.
Mohs and colleagues demonstrated in a one-year trial of moderate AD patients that donepezil delayed the time to functional decline, relative to placebo, as measured by the functional assessment and change scale. This trial, especially in combination with the results of the donepezil MCI trial, display the possibility for symptomatic benefit to imitate disease modification. Longer studies, at least 18-months, and combined clinical and biological support are necessary to support a claim of disease modification in AD.
SLOPE CHANGE
The progressive nature of AD presents an opportunity to examine drug effect on disease course. In contrast to endpoint comparisons, slope analyses examine drug effect throughout the course of study, in effect using the entire course of the trial as an outcome. Drug ability to slow the rate of cognitive decline over time serves as the primary outcome. The phase III trial of tarenflurbil included a prespecified slope analysis as a secondary outcome, with the aim of supporting a claim of disease modification, though the drug failed to demonstrate efficacy. Statistical slope analyses are complicated and challenging, due in large part to the nonlinear progression of disease and the influence of outliers (see the following). Placebo response and potential symptomatic effects, especially at trial initiation, can further complicate these analyses. Nevertheless, slope analyses may be sufficient for a disease-modifying label for the European Medicines Agency; the US Food and Drug Administration (FDA) has not, as yet, agreed that successful demonstration of altered disease slope of decline is sufficient for this claim.
CLINICAL OUTCOME MEASURES FOR DISEASE-MODIFYING TRIALS
The registration trials of the available symptomatic AD treatments provided a roadmap to drug approval that has subsequently been embraced by regulatory agencies, investigators, and sponsors. This has been particularly relevant in the choice of clinical outcome measures. To receive marketing approval from the FDA, a new AD drug must demonstrate efficacy on coprimary outcomes, including one cognitive measure and one functional or global measure. The standard approach to trial designs and outcome choices may soon require amending, however. It remains unclear if this approach is optimal in disease-modifying trials that aim at new targets, test medications with unique mechanisms of action, that depend more on biomarkers, and that ultimately incorporate more challenging (and beneficial) objectives, including early symptomatic and presymptomatic intervention.
COGNITIVE OUTCOME MEASURES
Successful registration trials in mild-to-moderate populations have utilized the ADAS-cog. The ADAS-cog is a 70-point instrument that examines memory, orientation, comprehension of language and commands, naming, word finding, and ideational and constructional praxis. The ADAS-cog lacks linearity when used as a longitudinal or cross-sectional measure of disease severity. In early or late dementia, the annual change in the ADAS-cog is minimal, relative to that observed in mild-to-moderate dementia. This issue is particularly critical in trials that move earlier in disease with the goal of intervention at a stage of minimal neuropathological burden. An 80-point version of the ADAS-cog that includes a delayed recall task (ADAS12) is available and multiple lines of evidence suggest increased sensitivity for the ADAS12 in MCI and prodromal AD populations, relative to the standard ADAS-cog. A 13-item version of the ADAS-cog includes executive measures.
Few trials in mild-to-moderate AD have implemented other cognitive outcomes. The neuropsychological test battery (NTB) was developed as alternative to the ADAS-cog, attempting to account for some of its weaknesses (the standard version of the ADAS-cog largely fails to examine attention, working memory, and executive function). Preliminary analyses of psychometric properties and longitudinal change in early AD suggested that the NTB may be more sensitive than the ADAS-cog to cognitive change in early AD. Administrative demands of the NTB are high. In moderate-to-severe dementia, only the severe impairment battery has been used in trials to successfully demonstrate cognitive efficacy.
GLOBAL AND FUNCTIONAL OUTCOMES
The most common choice of second coprimary outcome measure for AD trials has been the ADCS-activities of daily living (ADCS-ADL) scale. Similar scales include the Disability Assessment for Dementia and Functional Assessment Questionnaire. The overriding principle behind each of these informant-based scales is impairment to basic and instrumental ADLs and a sensitivity to detect further impairment, slowing of that decline, or improvement with treatment.
Alternatively, some AD dementia trials have incorporated the CDR as a global outcome. This scale includes assessment of memory and function and may offer greater sensitivity in milder populations, including MCI and prodromal AD. The CDR can be examined as an outcome measure in two capacities. A global staging score can be calculated using a scoring algorithm that weights memory (relative to the other components: orientation, judgment and problem solving, community affairs, home and hobbies, and self-care). Alternatively, the six components or “boxes” can be treated equally and summed (sum of the boxes score, CDR-sob).
Another commonly used global scale is the Clinician Interview Based Impression of Change plus caregiver input (CIBIC+). This tool compares the outcomes at prespecified intervals to baseline global severity. The interview assesses cognition, function, and behavior. The CIBIC+ has generally been used in shorter trials (3 and 6 months) and the CDR-sob in longer-duration trials (12 and 18 months).
OUTCOME CHOICES IN FUTURE DISEASE-MODIFYING TRIALS
With the evolution of AD research and the realization of the need for trials earlier in the disease, use of single outcome measures to demonstrate drug efficacy has been proposed (Aisen et al., 2011). The CDR-sob may represent a logical choice as a single outcome. The CDR includes memory, cognition, and function; it lacks behavioral measures included in other global assessments. In very mild AD, the CDR is internally valid, sensitive to change over time, and requires fewer trial participants than do cognitive outcome measures. In trials of the earliest AD patients, however, which will enroll asymptomatic at risk or preclinical populations, the CDR-sob may not achieve the necessary sensitivity. Ongoing discussions and research aim to identify and develop unique metrics to quantify the earliest cognitive impairments in AD. For example, the planned API prevention trial in autosomal dominant mutation carriers, the proposed ADCS A4 trial in asymptomatic preclinical participants, and the pioglitazone trial are likely to use newly developed composite measures of cognition.
SECONDARY OUTCOMES IN DISEASE-MODIFYING TRIALS
Alzheimer’s disease trials include a large variety of secondary outcome measures. The neuropsychiatric inventory is the most commonly included metric of behavioral symptoms. Behavioral symptoms occur in 75% of AD patients and few safe treatment options adequately manage these symptoms. Importantly, even in the prodromal stages of AD, behavioral symptoms are common and behavior tends both to worsen in severity and change in type over the course of AD. Both earlier and later stage trials therefore generally include the neuropsychiatric inventory.
Global and functional measures such as the CDR, MMSE, and ADCS-ADL, if not used as coprimary outcomes in a given trial, are often included as secondary outcomes. Scales that will support the registration and use of a drug if it is approved also are commonly incorporated into disease-modifying trials. These include scales of quality of life (QoL). In particular, the QoL-AD has been developed for use in AD studies and is now commonly incorporated. Three variants of the QoL-AD exist: the patient’s assessment of patient QoL, the caregiver assessment of patient QoL, and the caregiver assessment of caregiver QoL. A variety of other scales measure the burden of care: these include the Dependence Scale.
Increasingly, sponsors may need to anticipate data required by payers to inform decisions regarding adopting drugs, placing them on formularies, and reimbursing their costs. The Resource Utilization in Dementia scale examines caregiver time spent supervising and assisting the patient with ADLs. This and other pharmacoeconomic tools are needed to provide insight into the clinical meaningfulness of treatment effects.
COMPUTERIZED OUTCOMES
Computerized cognitive assessment batteries may bring additional sensitivity and convenience to outcomes assessment in disease-modifying trials. Sufficient data now exist to support use of computerized measures in elderly populations, including the development of age normative data. These batteries are associated with reduced variance and may afford smaller sample sizes, especially in early phase trials. Although computerized testing affords standardized administration and response/reaction time measurements, the correlations between computerized tests and activities of daily living are less established (Wild et al., 2008). A major strength (and rationale for development) of computerized batteries is to increase sensitivity to very mild changes in cognition. In theory, batteries could use adaptive testing and item response theory to examine cognitive ability more efficiently, though none of the currently available systems uses such technology. Computerized cognitive testing systems include the NeuroTrax Mindstreams system, the Cambridge Neuropsychological Test Automated Battery, and the CogState.
PATIENT-REPORTED OUTCOMES
The FDA has recently provided guidance recommending the use of patient-reported outcomes (PRO) as supportive for the demonstration of drug efficacy. As is implied by their names, PROs require no interpretation from a clinician. The PRO in cognitive impairment (PROCOG) has been developed for possible use in disease-modifying trials. The PROCOG has been validated in MCI and AD and was designed to assess cognition, as well as behavior, function, and quality of life.
BIOMARKERS FOR CLINICAL TRIALS
Neuroimaging and fluid biomarkers are discussed in Chapters 62 and 63, respectively. The characteristics of biomarkers will not be reviewed here. Biomarkers can have multiple roles in clinical trials (Table 65.1). State or trait markers might be used for clinical trial enrollment. APOE ε4 is a trait marker of increased risk for AD. Patients with MCI who are ε4 carriers are more likely to progress to AD than those who are not ε4 carriers. APOE ε4 carrier status could be used to enrich a population of MCI patients likely to progress to AD (Elias-Sonnenschein et al., 2011). State biomarkers can be used for enrollment also. Asymptomatic individuals who have positive amyloid imaging for example, might be considered at risk for progression to symptomatic AD (Dubois et al., 2010; Sperling et al., 2011). Similarly, low CSF Aβ42 or low Aβ42 and high tau or phospho-tau could also be used as biological measures that would enrich an asymptomatic or MCI population likely to progress to AD dementia (Heister et al., 2011). Biomarkers could also be used to further enrich a population in ways that would match the proposed mechanism of action of an intervention. For example, patients with high serum isoprostane levels might be appropriate candidates for antioxidant interventions, or patients with high cytokine levels might be appropriate candidates for anti-inflammatory interventions (Cummings, 2011a).
Biomarkers can be used to demonstrate target engagement and proof of mechanism. Stable isotope labeled kinetics (SILK)(Bateman et al., 2006) can be used to show inhibition of amyloid production by secretase inhibitors (Bateman et al., 2009). SILK is valuable for demonstrating target engagement of agents that affect Aβ reduction or clearance. The long-term relationship and clinical outcomes related to these relatively brief physiologic measures has not yet been established.
TABLE 65.1. Potential roles of biomarkers in clinical trials (examples of biomarker applications)
| Patient selection |
| Amyloid imaging |
| Hippocampal atrophy |
| CSF with low Aβ42 or elevated tau/phospho-tau |
| APOEε4 allele |
| Target engagement |
| Stable isotope labeled kinetics (SILK) |
| Serum Aβ40 or Aβ42 |
| Amyloid imaging |
| Efficacy |
| Whole brain atrophy or ventricular enlargement |
| FDG PET |
| CSF tau/phosphor-tau |
| Amyloid imaging |
| Cytokines |
| Isoprostanes |
| Side-effect monitoring |
| MRI for effusions and microhemorrhages |
| Liver function tests and other safety measures |
An important use of biomarkers is to support disease modification with proposed disease-modifying intervention. Reduced atrophy on MRI, reduced hypometabolism on FDG PET, and reduced CSF tau or phospho-tau compared with placebo groups in a placebo-controlled clinical trial would be evidence of disease modification. Isoprostane and cytokine measures might also reflect disease-modifying activity of therapeutic candidates. Neuronal injury biomarkers promise to be better measures of disease modification than amyloid biomarkers because amyloid measures such as CSF Aβ and amyloid imaging have had limited relationships to cognitive decline and dementia (Engler et al., 2006).
Biomarkers have an important role in detecting adverse events associated with antidementia therapies. Encephalitis associated with AN1792 was evident on MRI scans (Orgogozo et al., 2003) and MRI is used to follow amyloid related imaging abnormalities (ARIA), hemorrhagic type (ARIA-H) and ARIA effusion (ARIA-E), associated with immunotherapy and possibly with other amyloid related interventions (Salloway et al., 2009; Sperling et al., 2012).
A biomarker tightly linked to the mechanism of action of a drug might be approved in combination with the agent as a “theranostic” combination. For example, if an AD population with a specific genotype was identified as responding to a specific intervention, the test for the genotype might be approved together with use of the intervention conditional on the presence of the associated biological abnormality.
CHALLENGES TO DISEASE-MODIFYING TRIALS
Disease-modifying trials for AD face many unique barriers to success. Throughout, this chapter has addressed the many challenges to disease-modifying trial design. This section outlines some of the specific challenges to successful disease-modifying trial conduct.
RECRUITMENT
Essentially all clinical trials struggle to meet their recruitment goals. For most trials, the actual exceeds the planned time to complete study enrollment. In other cases, slow enrollment of sufficient numbers of suitable trial candidates can require study amendment (to add sites or altered inclusion/exclusion criteria), render a trial underpowered, or stop a trial entirely. Some barriers to trial enrollment are ubiquitous. These include low trial awareness, low rates of primary care (and even specialty) referrals to trials, and patient skepticism of research.
Alzheimer’s disease trial enrollment has many specific barriers (Grill and Karlawish, 2010). AD is a disease of the elderly and older patients are less likely to qualify for trials. The average American over the age of 60 takes 6–12 prescription medications and, especially in early phase trials, such medications may be prohibited by protocols. Elderly patients also suffer a variety of comorbidities that may prevent trial participation. These include cancers, diabetes (especially if uncontrolled), hypertension (especially if uncontrolled), and other problems that impact cognition, mobility, or independence. In fact, based on data from the State of California’s Alzheimer’s Disease Diagnostic and Treatment Centers, Schneider and colleagues estimated that less than 10% of AD patients are eligible for AD trials (Schneider et al., 1997). Using data from the National Alzheimer’s Coordinating Center, Grill and colleagues found that approximately 25% of participants in that large natural history study met generalized disease-modifying trial criteria for eligibility (Grill, Monsell et al., 2012c). Of eligible patients, only a portion will be aware of and interested in trials.
Less is known about the barriers to participation in disease-modifying trials in earlier (predementia) disease. In MCI/prodromal trials, a major barrier to enrollment is that few patients have received a diagnosis. Community screens inevitably identify a large number of patients with cognitive impairment, but after identification, there are no guarantees that these patients will be eligible or interested in trial participation. For example, those identified as MCI in community screens would need to undergo testing for AD biomarkers because entry criteria for prodromal AD trials and patient attitudes toward these biomarker testing procedures may be a barrier to participation (Grill et al., 2012b). Little is known about the barriers to disease-modifying trials in asymptomatic at-risk/preclinical populations.
RETENTION
Retention of enrolled participants is as important as their initial recruitment. Ethical guidelines outline the voluntary nature of research participation and ensure participant rights to withdraw consent and cease participation at any time. Therefore, participant retention begins with the recruitment process; investigators should enroll only participants likely to complete the trial. Like slow enrollment, high dropout can render a trial underpowered to examine the primary scientific question. Alternatively, skewed dropout can lead to scientific error. For example, were a symptomatic drug to lead to increased dropout throughout the trial (and the trial analyzed using the last observation carried forward analysis), the results could be misinterpreted as representing a disease-modifying effect.
Alzheimer’s disease dementia trials reported in the literature have demonstrated an approximately 75% retention rate (Grill and Karlawish, 2010). Mild cognitive impairment trials have had a lower mean rate of retention, with dropout ranging from 40% to 50%, rendering these trials far more challenging to interpret (Jelic et al., 2006). MCI trials of cholinesterase inhibitors have been hypothesized to suffer high dropout as a result of drug side effects, since dropout is higher in the group randomized to drug. Trial retention in early (preclinical) disease will be critical, as these trials are likely to require long durations. Though existing primary AD prevention trials have had good retention rates, most have employed benign interventions and infrequent visits (e.g., annual) and may not provide optimal guidance for preclinical AD studies. Preclinical trials studies are likely to employ riskier interventions that require more frequent assessment of safety.
Retrospective analyses have examined factors that predict trial dropout. In the ADCS MCI trial of donepezil and vitamin E, Edland and colleagues found that minority participants were more likely to drop out than non-Hispanic white participants and non-married participants were more likely to drop out than married participants (Edland et al., 2010). In a recent examination of six ADCS dementia trials, Grill, Raman et al.(2012d) observed dropout rates of 25% for patients with spouse partners, 32% for those with adult child partners, and 34% for those with other study partners. Edland and colleagues also observed a significantly higher rate of drop out at commercial trial sites, relative to academic sites. We recently observed a similar (though not statistically significant) difference between academic and non-academic sites in the rate of trial completers in a phase II mild-to-moderate dementia trial.
REPRESENTATIVE SAMPLES
Another major challenge in AD disease-modifying trials is increasing the representativeness of trial samples. Alzheimer’s disease trials enroll predominantly highly educated non-Hispanic White participants who are joined in their participation by spouse caregivers. Faison and colleagues examined minority participation in a sample of trials conducted over a ten-year period and observed less than 10% participation in ADCS trials and less than 3% participation in industry-sponsored trials. It is critical to increase the diversity of AD trial samples, especially because African Americans and Hispanics may be at increased risk for AD. Lower education has also been suggested as a risk factor for AD, though trial samples generally are of high education levels. Similarly, 67% of participants enrolled in a sample of ADCS trials did so with a spouse study partner (Grill, Raman et al., 2012d), despite the observation that in the U.S. most AD patients receive care from non-spouse family members.
GLOBALIZATION
Given the frequent struggles of AD trials to meet enrollment goals, there is an increasing need to conduct trials outside of the United States, where abundant samples of (especially treatment naïve) AD patients may exist. The majority of AD trials now include international (non-US) sites (Cummings et al., 2011). The conduct of clinical trials in a wide variety of global regions is associated with a variety of challenges. Life expectancy and access to health care vary substantially across global regions. Such variance is likely to manifest in differences in survival especially in the setting of longer trials, may affect the probability of adverse events, and may impact the representativeness of trial results. Mean levels of education also are considerably different among global regions that conduct AD trials. In addition to potential implications related to education-associated differences in AD progression, educational differences have substantial impact on the psychometric properties of cognitive outcome measures. Other challenges to interpreting global AD trials include potential differences in genetics among populations under study, including drug metabolism and AD risk genes; differences in attitudes toward AD diagnosis or biomarker procedures; and cultural differences in caregiving (and subsequent implications to trial outcome measures that rely on an informant). Finally, the availability of qualified investigators and nation-specific differences in trial regulations may hinder successful disease-modifying trials conducted globally.
TRIAL ETHICS
A number of ethical issues are raised in AD trials. Whether a patient suffering cognitive impairment is capable of providing truly informed consent and how to assess which patients are and which are not capable remains a challenging issue. Having capacity is not an all-or-none phenomenon. At one time, a patient may have capacity to make one decision (what they would like to eat for dinner) but lack the capacity to make another decision (whether to enroll in a high-risk trial). A variety of capacity scales exist and trials and the institutions in which they are conducted often standardize the metrics for assessing the patients’ understanding of the trial.
For trials that may directly benefit the patients or advance the science of the field, proxies may provide surrogate consent for AD patients that lack it. The use of surrogate consent has substantial research support. Many studies have examined the attitudes toward surrogate consent of patients regarding their future selves, caregivers and families regarding patients, expert investigators, and the general public. The wide consensus is in favor of the use of surrogate consent for the majority of AD trial scenarios and most current trials will enroll patients if there is a spouse, family member, or another person identified in an advanced directive that can provide informed consent on behalf of the patient. This varies only slightly from trial-to-trial; for example, the ongoing ADCS-conducted trial of nerve growth factor (NGF) gene transfer delivery to the basal forebrain will enroll only patients with capacity to provide informed consent. Greater variability exists on a state-by-state basis regarding who can provide informed consent in the patient’s stead and trialists are referred to their local regulatory bodies to determine a decision tree for this issue. Informed consent practices vary among nations and must be carefully monitored in global trials.
With the successful development of the cholinesterase inhibitors, a brief debate over the ethics of placebo controlled AD trials occurred (Kawas et al., 1999). This debate subsided, in large part because of the recognition that the available medications were only symptomatic. Thus, withholding these medications for the sake of a trial is not seen to have long-term ramifications on patient health. Moreover, trials examining agents anticipated to possibly have disease-modifying properties will generally enroll patients on symptomatic AD drugs, provided that they are maintained on stable doses. It is unclear how this landscape may change in the scenario of approval of the first disease-modifying AD drugs. Further, as trials have become more global in their conduct, AD drug development may bring the ethical challenge of developing a drug where it may not be made readily available, if proven effective.
Despite the management of ethical matters in dementia trials, the initiation of prodromal and preclinical AD trials brings new challenges. As discussed, the testing of agents in predementia populations will be critical to disease understanding, testing the amyloid hypothesis, and effectively treating and defraying the cost of AD. Ethical challenges must not derail predementia AD trials. However, a variety of ethical issues remain understudied in the setting of predementia trials. For example, these trials will enroll MCI or asymptomatic patients at highest risk for AD. The language that should be used to describe the criteria for entry (preclinical AD, at risk for AD, and the like) is uncertain; few prospective studies of long-term outcomes related to biomarker positivity are available to guide study conduct, including informed consent; and how enrolled participants (or those deemed ineligible) will interpret risk information is unknown. The strongest guidance for delivering risk information has been provided by the Risk Evaluation and Education for AD study, which randomized cognitively normal participants with a family history of AD to learn their APOE genotype or not. In this study, learning APOE genotype did not cause depression or anxiety, but those who were informed they were ε4 carriers were more likely than those who learned they were non-carriers to begin taking vitamins and supplements. Grill, Raman et al.(2012b) have recently shown in a qualitative study that persons in a hypothetical situation of learning they are at increased risk for AD will approach the decision of whether to participate in AD prevention trials differently—more frequently citing a desire to lower their personal AD risk and less frequently citing logistical barriers to participation. The ethical issues surrounding predementia trials are in need of future research to more adequately address each of these issues.
LESSONS LEARNED FROM TRIALS
There have been no successful phase III trials for any disease-modifying compound of any neurodegenerative disorder. All trials conducted to date have failed to show a compelling, consistent, and repeatable drug–placebo difference. In some cases, trial conduct is impugned and the efficacy of the drug in the clinical trial was inadequately tested. In others, appropriately conducted clinical trials failed to show a drug–placebo difference or there was an emergence of unacceptable side effects (Cummings, 2010). It is important to distinguish between failed trials and failed drugs because failed trials cannot provide evidence for or against a specific pathway or specific intervention. Hallmark features of failed trials include a failure of decline in the placebo group, failure to demonstrate a drug–placebo difference with the active comparator (typically donepezil), and excessive variability in measures (Table 65.2).
A well-conducted trial that fails to show a drug–placebo difference is valuable in demonstrating that the agent is not effective in this particular population, at this specific dose, used for this specific period of time. Trials answer very limited questions. Trials are focused experiments that can answer only limited questions and do not provide evidence of a possible effect in a different population, dose, or exposure duration.
A well-conducted trial that demonstrates excessive and unacceptable side effects also provides valuable information on whether to advance a molecule. The increased cognitive and functional decline and increased emergence of skin cancer associated with semagacestat—the gamma-secretase inhibitor—exemplifies a well-conducted trial in which adverse events were demonstrated suggesting that the agent should not be advanced, at least in patients with mild-to-moderate AD exposed at this dose (Cummings, 2010). More experience with gamma secretase inhibitors is needed before concluding whether these side effects are inevitably associated with gamma secretase inhibition or unique to semagacestat.
TABLE 65.2. Approach to trials that fail to show a drug–placebo difference
| Failed trials |
| No placebo decline or placebo improvement |
| No drug–placebo difference in an active comparator arm (e.g., donepezil) |
| High measurement variability or site-to-site inconsistency |
| Failed drugs |
| Lack of efficacy |
|
• Did not cross the blood–brain barrier |
|
• Dose too low |
|
• Lack of effect on target |
| Unacceptable side effects |
It is important to understand a compound as well as possible before advancing it to phase III pivotal trials. Phase III trials are large, long, and expensive. Agents not likely to succeed in phase III should not be advanced. Phase II trials large enough to show a drug–placebo difference are as large as phase III trials. Drug development programs often combine phases II and III (Cummings, 2008) in a II/III design, anticipating using the data for registration purposes. Alternatives to this approach include greater use of adaptive trial designs in phase II to adjust for duration, doses, and side effects, and greater use of biomarkers in phase II to insure patient selection, target engagement, and disease modification. Biomarkers may show disease modification in shorter times or with smaller populations than required for demonstration of clinical evidence of disease modification (Jack et al., 2003). Positive biomarker effects will de-risk a phase III program but do not guarantee its success since the predictive validity of biomarkers is currently unknown.
DISEASE-MODIFYING TREATMENTS OVERVIEW AND CLASSIFICATION
Many types of treatments are in development as disease- modifying therapies for AD and have advanced to the point of clinical testing (Table 65.3) (Grill and Cummings, 2010). They have various targets, potential mechanisms of action, and modes of administration. As is the case for any therapy targeting the CNS, candidate treatments must be able to penetrate the blood–brain barrier if on-target activity is to be achieved. Therefore, many drug candidates for AD are small molecules and are being developed as oral medications. The unique biology of AD, however, presents additional challenges, and many promising agents in clinical trials utilize alternate modes of administration. These include small peptides, such as davunetide (AL-108), a potentially neuroprotective agent that is administered intranasally. Similarly, a recent study of intranasal insulin administration suggested possible symptomatic efficacy in early AD (Craft et al., 2012). An active vaccination, AN-1792, was tested in phase I and phase II trials and served as a catalyst to development of a variety of immunotherapies, including some that are currently in development as intramuscular injections. Several candidate passive immunotherapies are in development and administered through intravenous infusion or subcutaneous injection. Finally, some treatments such as gene transfer and cell-based therapies are administered directly to the brain.
The broad range of modes of administration of these AD candidate therapies are paralleled by a variety of targets and mechanisms of action. Many small molecules aim to alter protein processing of one of the abnormally folded and accumulated proteins in the brains of AD patients. These include the two abnormally processed proteins responsible for the hallmark pathologies first identified by Alzheimer himself, the amyloid beta (Aβ) protein that accumulates in neuritic plaques within the cortical parenchyma, and tau, the microtubule-associated protein intrinsic to every neuron that becomes hyperphosphorylated and condensed in neurofibrillary tangles. Most small molecules target specific molecular components of the cascade of events that lead to the formation of these brain pathologies (candidates are described further in the following).
TABLE 65.3. Classes and examples of emerging therapies for the treatment of Alzheimer’s disease
| CLASS | MECHANISM | EFFECT OR AGENT |
| Anti-amyloid therapies | Decrease APP expression | Posiphen |
| Increase Aβ metabolism | Neprilysin/IDE stimulators | |
| BACE inhibition | MK-8931; LY-2811376 | |
| Gamma secretase inhibition | BMS708163 | |
| Gamma secretase modulation | EVP-0962 | |
| Alpha-secretase enhancement | M1 agonists | |
| Aggregation inhibitors | AZD-103/ELND-005; PBT2 | |
| Monoclonal antibodies | Bapineuzumab; solanezumab; gantenerumab; crenezumab | |
| Polyclonal antibodies | IVIg | |
| Active vaccination | CAD-106; ACC-001 | |
| BBB transport inhibitors (to brain) | RAGE inhibitors | |
| BBB transport facilitators (to blood) | LRP enhancement | |
| ApoE expression increase | Bexarotene | |
| Anti-tau therapies | Aggregation inhibitors | Methylene blue |
| Kinase inhibitors | GSK-3β inhibitors | |
| Phosphatases | Protein phosphatase 2A (PP2A) enhancers | |
| Microtubule stabilization | AL-108 | |
| Neuroprotective agents | Anti-inflammatory agents | NSAIDs |
| Antioxidants | Resveratrol; curcumin; vitamin E | |
| PPAR gamma agonists | Pioglitazone; rosiglitazone | |
| Trophic factors | NGF and BDNF-like molecules | |
| Regenerative approaches | Stem cell therapies | Cell replacement; generation of support cells |
Alternatively, treatments may target the cellular processes associated with disease, be they downstream or independent of plaques and tangles. Such targets include the known inflammation associated with AD; cellular metabolism and production of reactive oxygen species that may accompany AD and normal aging; mitochondrial functioning or misfunctioning as part of neurodegenerative disease; apoptosis and apoptosis-associated genes and cell cycling molecules; and genes or proteins that may result in neuroprotection. Despite a plethora of possible molecular targets, these therapies can largely be distilled to a single goal: preventing neuron and synapse loss in the AD brain. Once such synapse loss has occurred, which is likely to have begun by the time of prodromal AD or before, regenerative strategies may be necessary if a return to baseline function is to be achieved. Replenishing tissue through stem cell treatment, cell transplants, or treatment with neurotrophic factors that increase endogenous neurogenesis may offer such promise, though successful neuroregeneration remains a distant goal.
Most agents have more than one mechanism of action. Off-target action may be responsible for side effects of some agents. Off-target effects may also lead to repurposing agents for non-AD indications.
DISEASE-MODIFYING TREATMENTS—CLASSES AND AGENTS
Aβ PRODUCTION
Neuritic plaques are pathognomonic to AD, and the Aβ protein that accumulates in the plaques is a primary target for disease- modifying AD drug development. Central to the amyloid hypothesis is the abnormal proteolytic processing of the amyloid precursor protein (APP). Initial cleavage of APP occurs by either α- or β-site enzymes. In either case, the produced C-terminal fragment is subsequently modified by gamma secretase within the transmembrane region. If the α-secretase product is cleaved by gamma secretase, the resulting protein fragment (p3) is believed to be non-toxic. In contrast, the product of sequential beta-site cleavage enzyme (BACE) and gamma secretase cleavages are a variety of Aβ peptide species ranging in size from ~37–43 amino acids in length. Aβ40 is the most abundant species and Aβ42 is less abundant but very important from the disease standpoint. Aβ42 is especially prone to aggregation and concentrated in the core of neuritic plaques. Recent evidence suggests that high molecular weight species of Aβ aggregated together (e.g., consisting of 2 to 20 Aβ molecules), collectively referred to as oligomers, may represent the most synapto- or neurotoxic forms of Aβ.
Several candidate drugs targeting Aβ are now in clinical development. Many of these drugs directly target APP or molecules involved in its proteolytic processing. Posiphen is a positive enantiomer of phenserine, the acetylcholinesterase inhibitor. Though it has no cholinergic activity, Posiphen reduces expression of APP and subsequently lowers Aβ in culture and animal models. Clinical studies of Posiphen are ongoing.
Small molecules that inhibit or modulate the enzymes critical to the amyloidogenic pathway may reduce levels of Aβ and therefore slow disease course. Because it represents the most upstream cleavage step, BACE (also known as β-secretase and memapsin-2) may represent an ideal target to halt production of all pathologic posttranslational APP products. The activity of BACE on APP, however, requires multisite binding and successful inhibition may require large molecules (>500Da) that cross the blood–brain barrier with difficulty. Small lipophilic agents able to access the CNS have been developed and clinical trial results are pending. Alternatively, basic science studies have linked BACE-neutralizing antibodies to transferrin receptor antibodies as a novel mechanism of transporting larger molecules including antibodies into the CNS (Yu et al., 2011).
Gamma secretase inhibitors have been more approachable for small molecule drug development; candidates have reached clinical testing. Gamma secretase is a four-part complex, consisting of the membrane proteins presenilin-1 or -2, nicastrin, Aph-1, and Pen-2. Presenilin serves as the catalytic subunit and is critical to APP proteolysis, but also to function of other transmembrane proteins including the Notch signaling receptor. As a result, presenilin knockouts are lethal and potent nonselective gamma secretase (and Notch) inhibitors lack tolerability sufficient for clinical use. Semagacestat, the non-selective gamma secretase inhibitor in development by Eli Lilly Pharmaceuticals had phase III studies halted because of unacceptable levels of skin cancer and other adverse events, including worsening cognition. Other gamma secretase-targeting drugs remain under clinical study. For these drugs to be successful, activity must sufficiently lower Aβ to have clinical impact (though to what extent is unknown) without causing significant off-target side effects. Alternatively, recent basic studies have identified other targets related to gamma secretase that include a protein critical to this secretase activity (He et al., 2010).
An alternate mechanism by which Aβ production may be lowered without directly preventing APP cleavage by gamma secretase is the modulation of gamma secretase activity, such that the postcleavage product is an alternate-length peptide fragment. The obvious benefit of drugs with such mechanisms of action is that they may have therapeutic effect without altering Notch activity. Several compounds with gamma secretase modulating activity have been identified, including some nonsteroidal anti-inflammatory drugs (NSAIDS). R-flurbiprofen is a non-NSAID gamma secretase modulator that was demonstrated as being no different from placebo on any outcome measure in a large phase III mild-to-moderate clinical trial. Penetration of the CNS by the agent may have been insufficient. Several other gamma secretase-modulating compounds have been identified and may soon enter clinical development.
Another mechanism to reduce Aβ levels in the CNS would be to enhance alternate processing of APP through the non-amyloidogenic pathway by enhancing the activity of α-secretase. Few compounds have been identified that successfully activate α-secretase. Laboratory studies have suggested such activating effects for muscarinic ACh receptor agonists. M1 and M3 receptor agonism may regulate APP processing. In one small uncontrolled trial of 19 AD patients, mean decline in CSF Aβ was observed after treatment with an M1 agonist (Nitsch et al., 2000).
AMYLOID BETA DEGRADATION
Endogenous Aβ degradation occurs through activity of neprilysin (also known as neutral endopeptidase), insulin-degrading enzyme (IDE), endothelin-converting enzyme, angiotensin-converting enzyme, metalloproteinase 9, and as many as 15 other recently identified proteinases. Neprilysin and IDE are thought to be the primary regulators of Aβ degradation and are reduced in AD. Studies in transgenic mouse models suggest that degrading enzyme activity is dose-dependently related to AD pathology and may be a suitable target for new treatments, but that targeting multiple degrading enzymes may be needed to ensure that treatment effect is not washed out by compensatory pathways. For example, APP transgenic mice that overexpress the neprilysin transgene demonstrate increased Aβ degradation but not behavioral improvement (Meilandt et al., 2009). Mice engineered to overexpress neprilysin and IDE, alternatively, have reduced Aβ levels, reduced astrogliosis, microgliosis, and dystrophic neurites, and improved spatial memory.
Targeting molecular regulators of Aβ degrading enzyme levels in vivo may be more readily accomplished. Saito and colleagues identified somatostatin receptors as a potential regulator of neprilysin activity (Saito et al., 2005), and Cabrol and colleagues recently completed a high-throughput screen in which they identified multiple small molecule activators of IDE(Cabrol et al., 2009).
ANTIAGGREGATION AND IMMUNOTHERAPIES
In the CNS, Aβ is prone to aggregation. Dimers, trimers, and other high-molecular-weight combinations of Aβ may provide a compelling treatment target, since transgenic animals with reduced plaque burden but high oligomer levels do not demonstrate behavioral improvement (Cheng et al., 2007). Small molecules may specifically inhibit oligomerization (Necula et al., 2007), fibrillization, or both, and it remains unclear which (if any) is optimal for slowing the course of AD.
The only antiaggregation agent tested in a large phase III trial—tramiprosate—failed to demonstrate efficacy, but a variety of other agents that aim specifically to prevent the oligomerization of Aβ are in development. Cyclohexanehexol (also known as AZD-103 and ELND-005) prevents oligomerization in vitro, improved behavioral performance in a mouse model of AD, and was tolerated in a human phase I trial. A metal protein attenuating compound, PBT2, is believed to prevent Aβ oligomerization by chelating copper and zinc, which have been proposed as critical to this process. The compound demonstrated initial efficacy in AD, lowering CSF levels of Aβ and mitigating cognitive deterioration, relative to placebo (Lannfelt et al., 2008).
Aβ plaques may be deleterious to synaptic function and can be removed from the brain. Several types of immunotherapies have demonstrated an ability to reduce fibrillar amyloid burden, as measured by amyloid PET, and AD immunotherapies represent the most active area of disease-modifying trials.
Active vaccination with a synthetic full-length Aβ peptide (AN1792) in 300 mild AD patients initially suggested a clinical benefit in participants who received therapy and generated antibodies against Aβ but was halted because of a 6% incidence of T-cell mediated meningoencephalitis. Although analysis of the primary outcomes at the time of trial interruption demonstrated no drug–placebo difference, long-term follow up of survivors suggested a benefit in activities of daily living among antibody responders. Long-term follow-up to autopsy of the first subjects to die demonstrated that there was no impact on the rate of progression to severe dementia, but that responders demonstrated substantial and sometimes complete removal of plaque burden. Pathological examination has also suggested an impact on hyperphosphorylated tau and dystrophic neurites in the hippocampus of antibody responders (Serrano-Pozo et al., 2010). These findings have challenged the validity of Aβ plaques as a target in AD. Aβ vaccinations modified to remove T-cell activity, consisting primarily of peptide fragments, are in clinical development.
The initial active vaccination studies sparked initiation of robust passive immunotherapy development programs. Treatment with antibody therapy should avoid the T-cell mediated response that was believed to underlie meningoencephalitis in the AN1792 studies. Furthermore, since only a portion of patients administered the vaccine responded by generating antibodies, passive immunotherapy is hypothesized to increase the proportion of patients who respond to therapy. Several not mutually exclusive hypotheses for the mechanism of action of anti-Aβ antibodies have been proposed and studied in animal models. In such studies, passive immunization has been demonstrated to result in enhanced breakdown of Aβ through activation of microglia and some antibodies appear to serve as a “peripheral sink” binding Aβ in the blood and creating a flow of Aβ out of the brain.
Multiple antibodies in development for AD have demonstrated ability to reduce fibrillar Aβ burden, as measured by amyloid PET (Ostrowitzki et al., 2012), and one has been shown to alter tau levels in the CSF (Blennow et al., 2012). Ongoing studies will determine if these biological drug effects are associated with reduced cognitive decline. In the first phase III studies, the monoclonal antibody bapineuzumab was reported to not meet its primary outcome measures in mild-to-moderate AD. It will be important to know whether the antibody was efficacious in reducing amyloid plaques in this trial. Although no safety issues related to meningoencephalitis have been observed with passive immunotherapies to date, antibody treatments do result in symptomatic and asymptomatic amyloid-related imaging abnormalities (ARIA), which have been categorized as consisting of vasogenic edema and effusion changes (ARIA-E) or the deposition of hemosiderin indicating microhemorrhages (ARIA-H) (Sperling et al., 2012). Ongoing trials will determine the rates of occurrence of ARIA and its subtypes, as well as how often ARIA results in symptoms. It is likely that these rates will differ among candidate therapies and some antibodies are being developed specifically with the goal of improving the profile. Preliminary data suggest that ARIA may be most likely in areas of substantial Aβ removal.
While most antibodies in development for AD are monoclonal and specifically target a particular fragment of the Aβ peptide, polyclonal antibodies may also remove Aβ and provide clinical efficacy. Plaques in the brains of untreated AD patients are decorated with IgG antibodies, which elicit a microglial response, and an inverse relationship exists between IgG level and plaque burden in AD patients. Therapeutic use of naturally produced autoantibodies in the form of intravenous immunoglobulin (IVIg) as treatment for AD is a current area of clinical investigation. Treatment with IVIg increased plasma and decreased CSF levels of Aβ in a study of eight AD patients (Relkin et al., 2008). There is a large multisite clinical trial of IVIg in the United States and a second trial is planned.
One hypothetical mechanism by which immunotherapies reduce Aβ is through transport across the blood–brain barrier (BBB). Apolipoproteins do not cross the BBB, but play a role in Aβ transport and may regulate passage of Aβ between the CNS and the periphery. Beside age, APOE genetic status is the most well established risk factor for AD. The extent to which the different isoforms of ApoE bind Aβ is debated (Fan et al., 2009). ApoE ε4 likely decreases passage of Aβ from brain to blood. Mouse models that overproduce and deposit Aβ, when combined with transgenic animals that lack ApoE, have reduced Aβ deposition (Bales et al., 1997). Synthesized compounds that prevent ApoE binding to Aβ reduce plaque deposition and memory impairment in animal models of AD (Sadowski, Pankiewicz et al. 2006). Alternatively, in another mouse model, the FDA-approved cutaneous T-cell lymphoma drug, bexarotene, demonstrated rapid plaque clearance and behavioral improvement. Bexarotene targets ApoE and liver X receptors and is being considered for clinical development for AD (Cramer et al., 2012).
One proposed mechanism by which ApoE regulates brain Aβ is by facilitating passage across the BBB via the low-density lipoprotein receptor-related protein (LRP). Antibodies against the LRP reduce Aβ efflux from the brain, and peripheral administration of soluble LRP as a mechanism to pull Aβ out of the brain has been proposed as a potential therapy. Alternatively, because Aβ travels bidirectionally, it may be useful to prevent its entry to the CNS from peripheral sources. The receptor for advanced glycation end-products(RAGE) is a multiligand receptor that binds Aβ with high affinity. Preventing ligands from binding RAGE lowers brain levels of Aβ. RAGE-inhibiting compounds are in development, though the first major phase II trial of a RAGE inhibitor failed to demonstrate efficacy and may have resulted in cognitive worsening, relative to placebo.
TAU
The second pathognomonic hallmark of AD is the neurofibrillary tangle (NFT). NFTs can be used to stage AD severity, based on a stereotypical progression that begins in the entorhinal cortex and parahippocampal gyrus and progresses to include neocortical regions. Pathological studies have found the correlation between NFT pathology and disease progression to be stronger than that for neuritic plaque burden.
Neurofibrillary tangles are intracellular, composed of condensed hyperphosphorylated paired helical filaments of the microtubule-associated protein tau (MAPtau or tau). Although phosphorylation of tau is critical to its normal function, hyperphosphorylated tau (phospho-tau) aggregates into paired helical filaments rather than binding to microtubules. The resulting instability disrupts axonal transport and may result in neuronal injury and cell death, though neurons may live a decade or longer with neurofibrillary tangles (Braak and Del Tredici, 2011).
Abnormally high levels of phosphorylated or total tau in the CSF are indicators of neurodegenerative disease or injury. Clinical examinations suggest that abnormalities in Aβ do not predict cognitive decline unless p-tau levels are also abnormal (Desikan et al., 2012). In basic science models, tau is essential to AD and reducing tau can alleviate Aβ-dependent cognitive impairment (Roberson et al., 2007). Given that tau is a constitutive part of the cytoplasm of neurons, removal of cytoplasmic tau is not a realistic target. Staging of tau pathology in postmortem AD brains and a mouse model in which abnormal tau expression is limited to the entorhinal cortex but still develops pathology throughout the limbic structures (Liu et al., 2012) suggest that tau is transynaptically transported, perhaps underlying the anatomic spread of AD pathology. Investigational therapies aim to stabilize tau or to prevent its hyperphosphorylation, aggregation, or spread between cells.
Several classes of agents, including anthraquinones, polyphenols, aminothienopyridazines, and phenothiazines, may prevent tau aggregation. Wischik and colleagues have begun clinical development of the phenothiazine methylene blue as a treatment for AD (Wischik and Staff, 2009) The initial clinical trial of this agent failed to meet its prespecified endpoints but long-term observations and biomarker studies suggested possible benefit. Clinical development in AD continues. Methylene blue may reduce tau levels through autophagy (Congdon et al., 2012) and preventing tau fibril formation.
Reducing hyperphosphorylation might slow AD. Small molecule inhibitors of glycogen synthase kinase 3 beta (GSK-3β) and the cyclin-dependent kinase-5(cdk5), both of which phosphorylate tau at multiple sites, are logical candidates to prevent NFT development (Schneider and Mandelkow, 2008). Lithium and valproate inhibit GSK-3β, but large controlled studies have failed to confirm initial promising results from small open label studies (Hampel et al., 2009; Tariot et al., 2009). Caffeine also inhibits GSK-3β (Arendash et al., 2009).
Drugs that dephosphorylate tau may offer an alternative approach. Protein phosphatase 2A (PP2A) is decreased in AD and inhibition can cause tau hyperphosphorylation, tangle-like pathology, and behavioral impairment in animal models (Kins et al., 2001). Multiple PP2As exist and candidates capable of increasing PP2A activity, potentially through targeting upstream regulators, are logical (Iqbal et al., 2002), though no such candidates have reached clinical testing. NAP is an 8 amino acid peptide and the active component of the glial-derived activity dependent neuroprotective protein (ADNP). NAP stabilizes microtubules and can reduce tau phosphorylation (Divinski et al., 2004). NAP reduced AD pathology in mouse models and has reached clinical investigation in an intranasal form (Gozes et al., 2005).
NEUROPROTECTION
Neuroprotection is defined here as including therapeutic strategies that aim to increase neuronal and synaptic survival in the face of AD pathology. Many neuroprotective strategies are independent of the actual mechanisms of cell and synapse loss. Epidemiological findings suggest AD-preventing properties for the NSAIDs and the cholesterol-lowering drugs (Sano et al., 2008). If these agents indeed prevent AD, the mechanisms by which they do so are unknown. As discussed, NSAIDs may alter proteolytic processing of APP, but AD pathology also includes a significant inflammatory component. Similarly, cholesterol may be involved in Aβ processing and statin therapy may impact AD pathology. Despite this and the epidemiologic results, prospective randomized trials in mild-to-moderate AD have failed to confirm disease-slowing properties for the NSAIDs diclofenac, misoprostol, rofecoxib, or naproxen; or for the HMG-CoA reductase inhibitors atorvastatin and simvastatin. Additionally, rofecoxib was no better than placebo at delaying conversion to dementia in MCI, and primary prevention trials of naproxen and celecoxib were halted for safety reasons (Sano et al., 2008).
Other therapies target the mechanisms by which cells die. Given that age is the strongest risk factor for AD, cellular aging is one logical correlate for study and the production of reactive oxygen species (ROS) in particular may be critical both in normal aging and AD. ROS may penetrate cellular mitochondria, and Aβ-dependent and independent mitochondrial dysfunction has been implicated in AD. The mitochondrial membrane stabilizer, latrepirdine (Dimebon, dimebolin) showed early promise in a small phase II trial, but two larger phase III studies failed to confirm latrepirdine’s efficacy (Cummings, 2010). Other antioxidant compounds remove ROS and demonstrated preclinical efficacy in AD models, but trial results are generally mixed. One study suggested a delay in the time to severe AD (as measured with the CDR) in patients treated with vitamin E, but other trials have suggested no benefit (Sano et al., 2008). Sirtuins are another family of compounds believed to have antioxidant and potential Aβ-lowering properties. A large phase II trial of resveratrol, the sirtuin found in red wine and the skin of red grapes, is currently underway.
Both neuronal survival and apoptotic cell death are critical to proper CNS development and this process is largely dependent upon neurotrophic factor regulation. Because of their potent survival effects in development, neurotrophic factors are logical treatment candidates in neurodegenerative disease. Trophic factor therapies face a variety of challenges, most specifically size that limits penetration of the CNS and intolerability related to potent off-target effects (Eriksdotter Jonhagen et al., 1998). To address both of these challenges, neurosurgical delivery of gene transfer technology can be applied. An ongoing clinical development program uses neurosurgery to deliver adeno-associated virus (AAV)-delivered nerve growth factor DNA to the trk-A receptor-expressing cholinergic neurons of nucleus basalis of Meynert. These cells provide cholinergic supply to the neocortex and hippocampus and are known to degenerate early in the disease. Their enhanced survival may increase cholinergic tone and slow disease progression. Similarly, in a primate model, gene transfer to the entorhinal cortex with brain-derived neurotrophic factor (BDNF) DNA enhances memory performance (Nagahara et al., 2009).
Cell-based therapies for AD present unique challenges. The synapse and cell loss in AD is widespread, making replacement unlikely. Recent animal studies suggest that replacement may not be necessary, however, to achieve therapeutic efficacy. In triple transgenic mice, stem cell therapy resulted in increased hippocampal synaptic density, possibly as a result of increased BDNF signaling (Blurton-Jones et al., 2009).
CONCLUSIONS
Alzheimers disease is one of the most important areas of medical research. Without improvements in treatment options and earlier intervention AD will bankrupt world health care systems. No treatment can slow the course of AD, but a wide range of targets for potential disease-modifying therapies have been identified and many candidate drugs are in development. Clinical trials will be essential to finding disease-modifying drugs, but face a variety of challenges. As trials move earlier in disease, the reliance on standards set in dementia trials may hinder success. Improved outcome measures, surrogate biomarkers, and better means of enrolling samples are critically needed. Despite these challenges, optimism exists for the numerous compounds that have entered or will soon begin clinical study. The field is on the cusp of disease-modifying therapies, which may act as catalysts for continued improvement in therapeutics, implementation of trials to delay cognitive impairment, and amelioration of the human and financial toll associated with AD.
DISCLOSURES
Dr. Grill has received grant/research support from the National Institute on Aging (AG016570), the Alzheimer’s Association (NIRG-12-242511), the John Douglas French Foundation, the Sidell-Kagan Foundation, and as a site investigator for clinical trials sponsored by Elan, Genentech Janssen Alzheimer Immunotherapy, Bristol-Myers Squibb, Medivation, Pfizer, and the Alzheimer’s Disease Cooperative Study (ADCS). He has served as a paid consultant to Avanir Pharmaceuticals and Phloronol Inc.
Dr. Cummings owns the copyright of the Neuropsychiatric Inventory. He has provided consultation to Abbott, Acadia, ADAMAS, Anavex, Astellas, Avanir, Avid, Baxter, Bayer, Bristol-Myers Squibb, Eisai, Elan, EnVivo, Forest, GE, Genentech, GlaxoSmithKline, Janssen, Lilly, Lundbeck, MedAvante, Medivation, Medtronics, Merck, Neurokos, Neurotrax, Novartis, Otsuka, Pfizer, Prana, QR Pharma, Sonexa, Takeda, and UBC. He owns stock in ADAMAS, Prana, Sonexa, MedAvante, Neurotrax, and Neurokos. He has been a speaker for Eisai, Forest, Janssen, Novartis, Pfizer, Lundbeck. Dr. Cummings has provided expert witness consultation regarding olanzapine and ropinirole.
REFERENCES
Aisen, P.S., Andrieu, S., et al.(2011). Report of the task force on designing clinical trials in early (predementia) AD. Neurology 76:280–286.
Albert, M.S., DeKosky, S.T., et al.(2011). The diagnosis of mild cognitive impairment due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 7:270–279.
Arendash, G.W., Mori T., et al.(2009). Caffeine reverses cognitive impairment and decreases brain amyloid-beta levels in aged Alzheimer’s disease mice. J. Alzheimers Dis. 17(3):661–680.
Bales, K.R., Verina T., et al.(1997). Lack of apolipoprotein E dramatically reduces amyloid beta-peptide deposition. Nat. Genet. 17(3):263–264.
Bateman, R.J., Aisen P.S., et al.(2011). Autosomal-dominant Alzheimer’s disease: a review and proposal for the prevention of Alzheimer’s disease. Alzheimers Res. Ther. 2(6):35.
Bateman, R.J., Munsell, L.Y., et al.(2006). Human amyloid-beta synthesis and clearance rates as measured in cerebrospinal fluid in vivo. Nat. Med. 12(7):856–861.
Bateman, R.J., Siemers, E.R., et al.(2009). A gamma-secretase inhibitor decreases amyloid-beta production in the central nervous system. Ann. Neurol. 66(1):48–54.
Blennow, K., Zetterberg, H., et al.(2012). Effect of immunotherapy with bapineuzumab on cerebrospinal fluid biomarker levels in patients with mild to moderate Alzheimer disease. Arch. Neurol. 69(8):1002–1010.
Blurton-Jones, M., Kitazawa M., et al.(2009). Neural stem cells improve cognition via BDNF in a transgenic model of Alzheimer disease. Proc. Natl. Acad. Sci. USA 106(32):13594–13599.
Bodick, N., Forette, F., et al.(1997). Protocols to demonstrate slowing of Alzheimer disease progression. Position paper from the International Working Group on harmonization of dementia drug guidelines. The disease progression sub-group. Alzheimer Dis. Assoc. Disord. 3:50–53.
Braak, H., and Del Tredici, K.(2011). Alzheimer’s pathogenesis: is there neuron-to-neuron propagation? Acta Neuropathol. 121(5):589–595.
Burton, E.J., Mukaetova-Ladinska, E.B., et al.(2012). Quantitative neurodegenerative pathology does not explain the degree of hippocampal atrophy on MRI in degenerative dementia. Int. J. Geriatr. Psychiatry 27(12):1267–1274.
Cabrol, C., Huzarska M.A., et al.(2009). Small-molecule activators of insulin-degrading enzyme discovered through high-throughput compound screening. PLoS One 4(4):e5274.
Cheng, I.H., Scearce-Levie K., et al.(2007). Accelerating amyloid-beta fibrillization reduces oligomer levels and functional deficits in Alzheimer disease mouse models. J. Biol. Chem. 282(33):23818–23828.
Clark, C.M., Xie, S., et al.(2003). Cerebrospinal fluid tau and beta-amyloid: how well do these biomarkers reflect autopsy-confirmed dementia diagnoses? Arch. Neurol. 60(12):1696–1702.
Committee for Medicinal Products for Human Use.(2008). Guideline on medicinal products for the treatment of Alzheimer’s disease and other dementias. European Medicines Agency.
Congdon, E.E., Wu, J.W., et al.(2012). Methylthioninium chloride (methylene blue) induces autophagy and attenuates tauopathy in vitro and in vivo. Autophagy 8(4):609–622.
Craft, S., Baker L.D., et al.(2012). Intranasal insulin therapy for Alzheimer disease and amnestic mild cognitive impairment: a pilot clinical trial. Arch. Neurol. 69(1):29–38.
Cramer, P.E., Cirrito, J.R., et al.(2012). ApoE-directed therapeutics rapidly clear beta-amyloid and reverse deficits in AD mouse models. Science 335(6075):1503–1506.
Cummings, J.L.(2011a). Alzheimer’s disease clinical trials: changing the paradigm. Curr. Psychiatry Rep. 13(6):437–442.
Cummings, J.L.(2011b). Biomarkers in Alzheimer’s disease drug development. Alzheimers Dement. 7(3):e13–44.
Cummings, J.L.(2006). Challenges to demonstrating disease-modifying effects in Alzheimer’s disease clinical trials. Alzheimers Dement. 2:263–271.
Cummings, J.L.(2009). Defining and labeling disease-modifying treatments for Alzheimer’s disease. Alzheimers Dement. 5:406–418.
Cummings, J.L.(2008). Optimizing phase II of drug development for disease-modifying compounds. Alzheimers Dement. 4(Suppl 1):S15–20.
Cummings, J.L.(2010). What can be inferred from the interruption of the semagacestat trial for treatment of Alzheimer’s disease? Biol. Psychiatry 68(10):876–878.
Cummings, J., Reynders R., et al.(2011). Globalization of Alzheimer’s disease clinical trials. Alzheimers Res. Ther. 3(4):24.
Desikan, R.S., McEvoy L.K., et al.(2012). Amyloid-beta-associated clinical decline occurs only in the presence of elevated P-tau. Arch. Neurol. 69(6):709–713.
Divinski, I., Mittelman, L., et al.(2004). A femtomolar acting octapeptide interacts with tubulin and protects astrocytes against zinc intoxication. J. Biol. Chem. 279(27):28531–28538.
Dubois, B., Feldman, H.H., et al.(2010). Revising the definition of alzheimer’s disease: a new lexicon. Lancet Neurol. 9:1118–1127.
Edland, S.D., Emond J.A., et al.(2010). NIA-funded Alzheimer centers are more efficient than commercial clinical recruitment sites for conducting secondary prevention trials of dementia. Alzheimer Dis. Assoc. Disord. 24(2):159–164.
Elias-Sonnenschein, L.S., Viechtbauer, W., et al.(2011). Predictive value of APOE-ε4 allele for progression from MCI to AD-type dementia: a meta-analysis. J. Neurol. Neurosurg. Psychiatry 82(10):1149–1156.
Engler, H., Forsberg, A., et al.(2006). Two-year follow-up of amyloid deposition in patients with Alzheimer’s disease. Brain 129 (Pt 11):2856–2866.
Eriksdotter Jonhagen, M., Nordberg A., et al.(1998). Intracerebroventricular infusion of nerve growth factor in three patients with Alzheimer’s disease. Dement. Geriatr. Cogn. Disord. 9(5):246–257.
Fan, J., Donkin J., et al.(2009). Greasing the wheels of Abeta clearance in Alzheimer’s disease: The role of lipids and apolipoprotein E. Biofactors 35(3):239–248.
Ferri, C.P., Prince, M., et al.(2005). Global prevalence of dementia: A delphi consensus study. Lancet 366:2112–2117.
Fleisher, A.S., Truran, D., et al.(2011). Chronic divalproex sodium use and brain atrophy in Alzheimer disease. Neurology 77(13):1263–1271.
Fox, N.C., Black, R.S., et al.(2005). Effects of Abeta Immunization (AN1792) on MRI measures of cerebral volume in Alzheimer disease. Neurology 64(9):1563–1572.
Gozes, I., Morimoto, B.H., et al.(2005). NAP: research and development of a peptide derived from activity-dependent neuroprotective protein (ADNP). CNS Drug Rev. 11(4):353–368.
Grill, J.D., and Cummings, J.L.(2010). Current therapeutic targets for the treatment of Alzheimer’s disease. Exp. Rev. Neurother. 10(5):711–728.
Grill, J.D., and Karlawish, J.(2010). Addressing the challenges to successful recruitment and retention in Alzheimer’s disease clinical trials. Alzheimers Res. Ther. 2(6):34.
Grill, J.D., Di, L., et al.(2012a). Estimating sample sizes for predementia Alzheimer’s trials based on the Alzheimer’s Disease Neuroimaging Initiative. Neurobiol. Aging 34(1):62–72.
Grill, J.D., Karlawish, J., et al.(2012b). Risk disclosure and preclinical Alzheimer’s disease clinical trial enrollment. Alzheimers Dement.
Grill, J.D., Monsell, S., et al.(2012c). Are patients whose study partners are spouses more likely to be eligible for Alzheimer’s disease clinical trials? Dement. Geriatr. Cogn. Disord. 33:334–340.
Grill, J.D., Raman, R., et al.(2012d). Effect of study partner on the conduct of Alzheimer disease clinical trials. Neurology. [Epub ahead of print].
Hampel, H., Ewers, M., et al.(2009). Lithium trial in Alzheimer’s disease: a randomized, single-blind, placebo-controlled, multicenter 10-week study. J. Clin. Psychiatry 70(6):922–931.
He, G., Luo, W., et al.(2010). Gamma-secretase activating protein is a therapeutic target for Alzheimer’s disease. Nature 467(7311):95–98.
Heister, D., Brewer, J.B., et al.(2011). Predicting MCI outcome with clinically available MRI and CSF biomarkers. Neurology 77(17):1619–1628.
Holmes, C., Boche, D., et al.(2008). Long-term effects of Abeta42 immunisation in Alzheimer’s disease: follow-up of a randomised, placebo-controlled phase I trial. Lancet 372(9634):216–223.
Iqbal, K., Alonso Adel, C., et al.(2002). Significance and mechanism of Alzheimer neurofibrillary degeneration and therapeutic targets to inhibit this lesion. J. Mol. Neurosci. 19(1–2):95–99.
Jack, C.R., Jr., Knopman, D.S., et al.(2010). Hypothetical model of dynamic biomarkers of the Alzheimer’s pathological cascade. Lancet Neurol. 9:119–128.
Jack, C.R., Jr., Slomkowski, M., et al (2003). MRI as a biomarker of disease progression in a therapeutic trial of milameline for AD. Neurology 60(2):253–260.
Jelic, V., Kivipelto, M., et al.(2006). Clinical trials in mild cognitive impairment: lessons for the future. J. Neurol. Neurosurg. Psychiatry 77:429–438.
Katz, R.(2004). Biomarkers and surrogate markers: an FDA perspective. NeuroRx 1:189–195.
Katz, R.(2008). Food and Drug Administration regulation. CNS Spectr. 13:10.
Kawas, C.H., Clark, C.M., et al.(1999). Clinical trials in Alzheimer disease: debate on the use of placebo controls. Alzheimer Dis. Assoc. Disord. 13(3):124–129.
Keller, C., Kadir, A., et al.(2011). Long-term effects of galantamine treatment on brain functional activities as measured by PET in Alzheimer’s disease patients. J. Alzheimers Dis. 24(1):109–123.
Kins, S., Crameri A., et al.(2001). Reduced protein phosphatase 2A activity induces hyperphosphorylation and altered compartmentalization of tau in transgenic mice. J. Biol. Chem. 276(41):38193–38200.
Lannfelt, L., Blennow, K., et al.(2008). Safety, efficacy, and biomarker findings of PBT2 in targeting Abeta as a modifying therapy for Alzheimer’s disease: a phase IIa, double-blind, randomised, placebo-controlled trial. Lancet Neurol. 7(9):779–786.
Liu, L., Drouet, V., et al.(2012). Trans-synaptic spread of tau pathology in vivo. PLoS One 7(2):e31302.
Mega, M.S., Dinov, I.D., et al.(2005). Metabolic patterns associated with the clinical response to galantamine therapy: a fludeoxyglucose f 18 positron emission tomographic study. Arch. Neurol. 62(5):721–728.
Meilandt, W.J., Cisse, M., et al.(2009). Neprilysin overexpression inhibits plaque formation but fails to reduce pathogenic Abeta oligomers and associated cognitive deficits in human amyloid precursor protein transgenic mice. J. Neurosci. 29(7):1977–1986.
Mohs, R.C., Doody, R.S., et al.(2001). A 1-year placebo-controlled preservation of function survival study of donepezil in AD patients. Neurology 57:481–488. Erratum in: Neurology 57:1942.
Morris, J.C., Roe, C.M., et al.(2009). Pittsburgh compound b imaging and prediction of progression from cognitive normality to symptomatic Alzheimer disease. Arch. Neurol. 66:1469–1475.
Nagahara, A.H., Merrill, D.A., et al.(2009). Neuroprotective effects of brain-derived neurotrophic factor in rodent and primate models of Alzheimer’s disease. Nat. Med. 15(3):331–337.
Necula, M., Breydo L., et al.(2007). Methylene blue inhibits amyloid Abeta oligomerization by promoting fibrillization. Biochemistry 46(30):8850–8860.
Nitsch, R.M., Deng, M., et al.(2000). The selective muscarinic M1 agonist AF102B decreases levels of total Abeta in cerebrospinal fluid of patients with Alzheimer’s disease. Ann. Neurol. 48(6):913–918.
Orgogozo, J.M., Gilman, S., et al.(2003). Subacute meningoencephalitis in a subset of patients with AD after Abeta42 immunization. Neurology 61(1):46–54.
Ostrowitzki, S., Deptula, D., et al.(2012). Mechanism of amyloid removal in patients with Alzheimer disease treated with gantenerumab. Arch. Neurol. 69(2):198–207.
Rascol, O., Fitzer-Attas, C.J., et al.(2011). A double-blind, delayed-start trial of rasagiline in Parkinson’s disease (the ADAGIO study): prespecified and post-hoc analyses of the need for additional therapies, changes in UPDRS scores, and non-motor outcomes. Lancet Neurol. 10:415–423.
Reiman, E.M., Langbaum, J.B., et al.(2010). Alzheimer’s prevention initiative: a proposal to evaluate presymptomatic treatments as quickly as possible. Biomarkers Med. 4(1):3–14.
Relkin, N.R., Szabo, P., et al.(2008). 18-month study of intravenous immunoglobulin for treatment of mild Alzheimer disease. Neurobiol. Aging 30(11):1728–1736.
Ridha, B.H., Anderson, V.M., et al.(2008). Volumetric MRI and cognitive measures in Alzheimer disease: comparison of markers of progression. J. Neurol. 255(4):567–574.
Rinne, J.O., Brooks, D.J., et al.(2010). 11C-PiB PET assessment of change in fibrillar amyloid-beta load in patients with Alzheimer’s disease treated with bapineuzumab: a phase 2, double-blind, placebo-controlled, ascending-dose study. Lancet Neurol. 9(4):363–372.
Roberson, E.D., Scearce-Levie, K., et al.(2007). Reducing endogenous tau ameliorates amyloid beta-induced deficits in an Alzheimer’s disease mouse model. Science 316(5825):750–754.
Sabbagh, M.N., Richardson, S., et al.(2008). Disease-modifying approaches to Alzheimer’s disease: challenges and opportunities—lessons from donepezil therapy. Alzheimers Dement. 4:S109–118.
Sadowski, M.J., Pankiewicz, J., et al.(2006). Blocking the apolipoprotein E/amyloid-beta interaction as a potential therapeutic approach for Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 103(49):18787–18792.
Saito, T., Iwata, N., et al.(2005). Somatostatin regulates brain amyloid beta peptide Abeta42 through modulation of proteolytic degradation. Nat. Med. 11(4):434–439.
Salloway, S., Mintzer, J., et al.(2008). Disease-modifying therapies in Alzheimer’s disease. Alzheimers Dement. 4(2):65–79.
Salloway, S., Sperling, R., et al.(2009). A phase 2 multiple ascending dose trial of bapineuzumab in mild to moderate Alzheimer disease. Neurology 73(24):2061–2070.
Sano, M., Grossman, H., et al.(2008). Preventing Alzheimer’s disease: separating fact from fiction. CNS Drugs 22(11):887–902.
Schneider, A., and Mandelkow, E.(2008). Tau-based treatment strategies in neurodegenerative diseases. Neurotherapeutics 5(3):443–457.
Schneider, L.S., Olin, J.T., et al.(1997). Eligibility of Alzheimer’s disease clinic patients for clinical trials. J. Am. Geriatr. Soc. 45(8):923–928.
Serrano-Pozo, A., William, C.M., et al.(2010). Beneficial effect of human anti-amyloid-beta active immunization on neurite morphology and tau pathology. Brain 133(Pt 5):1312–1327.
Sperling, R., Salloway, S., et al.(2012). Amyloid-related imaging abnormalities in patients with Alzheimer’s disease treated with bapineuzumab: a retrospective analysis. Lancet Neurol. 11(3):241–249.
Sperling, R.A., Aisen, P.S., et al (2011). Toward defining the preclinical stages of Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 7:280–292.
Tariot, P.N., Cummings, J., et al.(2009). The ADCS Valproate Neuroprotection Trial: primary efficacy and safety results. Alzheimers Dement. 5(4):P84–P85.
Vellas, B., Aisen, P.S., et al.(2011). Prevention trials in Alzheimer’s disease: an EU-US task force report. Prog. Neurobiol. 95(4):594–600.
Wagner, J.A., Williams, S.A., et al.(2007). Biomarkers and surrogate end points for fit-for-purpose development and regulatory evaluation of new drugs. Clin. Pharmacol. Ther. 81(1):104–107.
Weinreb, O., Amit, T., et al.(2011). A novel anti-Alzheimer’s disease drug, ladostigil neuroprotective, multimodal brain-selective monoamine oxidase and cholinesterase inhibitor. Int. Rev. Neurobiol. 100:191–215.
Wild, K., Howieson, D., et al.(2008). Status of computerized cognitive testing in aging: a systematic review. Alzheimers Dement. 4(6):428–437.
Wischik, C., and Staff, R.(2009). Challenges in the conduct of disease-modifying trials in AD: practical experience from a phase 2 trial of Tau-aggregation inhibitor therapy. J. Nutr. Health Aging 13(4):367–369.
Yu, Y.J., Zhang, Y., et al.(2011). Boosting brain uptake of a therapeutic antibody by reducing its affinity for a transcytosis target. Sci. Transl. Med. 3(84):84ra44.