70 | EPIDEMIOLOGY OF NEUROPSYCHIATRIC AND DEVELOPMENTAL DISORDERS OF CHILDHOOD
ELISE B. ROBINSON, BENJAMIN M. NEALE, AND MARK J. DALY
Psychiatric and developmental disorders of childhood onset have been studied for much of the twentieth century. There is an emerging recognition, however, that neuropsychiatric conditions affecting children are rising in estimated prevalence and place an enormous burden on parents, educators, and the health care system (Charman, 2011a; King and Bearman, 2009). Although this rise in prevalence likely contains elements of diagnostic changes, greater awareness of these disorders, and true changes in incidence, there is little doubt that psychiatric and developmental disorders among children are a major concern to society today. A recent survey of more than 10,000 US adolescents concluded that there is nearly a 50% lifetime childhood prevalence of one or more DSM-IV mood, anxiety, or behavioral disorders (excluding eating and substance abuse disorders) and that more than 20% of children meet the definition of severe impairment (Merikangas et al., 2010).
It has become increasingly common for traditionally “adult” psychiatric disorders, such as bipolar disorder or major depression, to be diagnosed in children. However, the focus of this chapter is classical childhood neuropsychiatric conditions whose diagnosis requires the presence of symptoms before age 18 within the DSM-IV-TR framework (APA, 2000). We consider four major diagnostic categories falling within these specifications and highlight the major diagnosis within each—specifically, intellectual disability (ID; formerly referred to as mental retardation and referred to as learning disability in Europe), pervasive developmental disorders (autism spectrum disorders [ASDs]), hyperactive and inattentive behavior (attention deficit/hyperactivity disorder [ADHD]), and tic disorders (Tourette disorder [TD]). Within this first subsection, we address the epidemiology, heritability, and implied genetic architecture within each diagnostic category. We draw synergies and overlaps where appropriate and articulate the connection between traditional diagnostic categories and common behavioral and cognitive traits.
Some of these connections bear relevance to the fifth edition of the DSM, currently under development. In the DSM-IV, the central approach for disease classification is one of categorical symptoms without an empirical basis for diagnostic thresholds. In construction of the DSM-5, the recognition of a continuum underpinning some psychiatric illnesses is growing, exemplified by the planned adoption of autism spectrum disorder in place of a discrete diagnosis within the Pervasive Developmental Disorders category. This shift recognizes a robust literature on more quantitative approaches to neuropsychiatric phenotypes and may influence estimates of disease prevalence. We discuss several implications of assessing psychiatric traits with a dimensional approach in the following, particularly as they relate to genetic architecture.
DEFINITION AND IMPACT OF CHILDHOOD PSYCHIATRIC AND DEVELOPMENTAL DISORDERS
INTELLECTUAL DISABILITY
Intellectual disability and the formal diagnosis of mental retardation in the DSM-IV are characterized by intellectual functioning that is substantially below average and significant limitations in adaptive behavior with onset before the age of 18 (APA, 2000). Approximately 2% of people worldwide meet criteria for IDs. Most individuals with a diagnosis require some level of assisted services throughout adulthood, although there is considerable inter-individual variability in the degree of impairment.
Intellectual functioning is most often assessed via IQ tests (e.g., the Wechsler Intelligence Scales for Children) (Weschler et al., 1992). Intellectual disability is traditionally defined as a score below 70 (at or below two standard deviations below the mean). Although some people with IDs are not diagnosed until mid- or late-childhood, most are identified early in life subsequent to parental concerns about developmental delays (Leonard and Wen, 2002). The genetic sources of certain syndromic cases of ID have long been recognized (Lejeune et al., 1959), but in most cases the cause of cognitive impairment is unknown. Intellectual disability in general is accepted to have numerous genetic and non-genetic causes, representing the end result of a defect in any number of many possible elements of central nervous system (CNS) development.
AUTISM SPECTRUM DISORDERS
Autism spectrum disorders (ASDs) are a system of neurodevelopmental conditions with onset before age three, characterized by impairments in social interaction and communication, along with significantly restricted interests and repetitive behaviors (APA, 2000). Although ASDs vary widely in phenotypic expression, they are universally impairing and present an enormous burden to affected individuals and their families. Approximately 40% to 60% of children with ASDs have concomitant intellectual disabilities, and it is estimated that an even larger fraction require lifelong external supports (Chakrabarti and Fombonne, 2005; Charman et al., 2011b; Centers for Disease Control and Prevention, 2012). For each patient, the lifetime societal costs associated with an ASD diagnosis are estimated to be $3.2 to $5 million in the United States and £0.8 to £1.2 in the United Kingdom (Ganz, 2007; Knapp et al., 2009).
These costs become staggering in aggregate, especially given the growing frequency of ASD diagnoses. The prevalence of ASDs and their apparent rise in incidence has been a point of enormous contention in recent years and is discussed in the following section. Current estimates place the prevalence of ASDs at approximately 1% today in both the United States and the United Kingdom (Baron-CoShen et al., 2009; CDC, 2012).
ATTENTION DEFICIT/HYPERACTIVITY DISORDER
Excess levels of activity along with difficulties in attention and impulse control have been characterized in children for more than a century (Still, 1902). Three subtypes of ADHD are recognized in the DSM-IV: (1) hyperactive/impulsive, (2) inattentive, and (3) combined type, reflecting endorsed criteria in both categories. Examples of ADHD symptoms include interrupting, always being on the go, fidgeting and difficulty sitting still (which reflect the hyperactive/impulsive dimension), as well as difficulty completing tasks, an inability to maintain focus on an activity, and flipping focus (which reflects the inattention dimension). In developing the DSM-5, the psychiatric community is moving to abandon the distinction between the three subtypes in favor of a single construct. This is motivated in large part by the strong phenotypic correlation between the two dimensions, both among patients and the population at large (Larsson et al., 2011b).
Attention deficit/hyperactivity disorder is one of the most common psychiatric disorders of childhood, affecting an estimated 4% to 12% of children worldwide. Approximately 40% of cases are estimated to persist into adulthood, but method of assessment substantially impacts estimated adult prevalence. Self-report measures tend to be least reliable and yield the most restrictive estimates (Faraone et al., 2003). Attention deficit/hyperactivity disorder predicts a range of behaviors and outcomes in both childhood and adulthood that carry a societal cost. Children and adults with ADHD are more likely to be disruptive in school environments, have lower educational attainment, and higher rates of substance use, sexually transmitted diseases, criminality, incarceration, and incurred health care costs. In 2006, worldwide costs of ADHD were estimated to be between $77.5 and $115.9 billion annually because of the costs of treatment and more significantly lost productivity and indirect social costs (Barbaresi and Olsen, 1998; Leibson et al., 2001).
TOURETTE DISORDER
Tourette disorder (TD) and the related phenotype of tic disorder are characterized by uncontrolled movements, speech, or actions. Tourette was originally proposed in 1885 by Georges Gilles de la Tourette as a phenotype that included multiple motor tics and at least one vocal tic (Gilles de la Gilles de la Tourette et al., 1982). Motor tics span from simple movements such as eye blinking to complex movements including jumping or body contortions. Vocal tics similarly vary from simple tics of throat clearing or sniffing to more complex tics, including syllables, words, coprolalia (undesired vocalization of profanities), echolalia (uncontrolled repetition of what other people say), and palilalia (repetition of one’s own words) (Singer, 2005). In the DSM-IV, a Tourette diagnosis requires both motor and vocal tics persisting for a year or longer. When only vocal or motor tics are present, the patient is considered to have tic disorder, with presentation for less than a year considered transient and longer than a year considered chronic (APA, 2000). Onset of these phenotypes is primarily in childhood and is typically most severe during development.
Tourette Disorder and tic disorder are somewhat unusual as psychiatric phenotypes in that neither deficit nor difficulty resulting from the behaviors is necessary to define the disorder. Nevertheless, patients with these disorders experience increased average levels of anxiety and depression (Coffey and Park, 1997; Sukhodolsky et al., 2003). In addition to greater stress, patients with TD have higher rates of ADHD, obsessive compulsive disorder (OCD), and other neuropsychiatric phenotypes (Cavanna et al., 2009; Grados and Mathews, 2009). These comorbidities are well documented but poorly understood. (A discussion of the genetics of anxiety disorders including OCD is found in Chapter 41.)
TRAJECTORIES INTO ADULTHOOD FOR THOSE DIAGNOSED WITH CHILDHOOD PSYCHIATRIC DISORDERS
INTELLECTUAL DISABILITY
Although individuals with developmental disabilities can benefit from early intervention strategies (Charman, 2011c; Dawson et al., 2010), measured intelligence is, on the whole, stable following early childhood (Plomin et al., 2008). Recent evidence suggests that the common genetic influences on intelligence are highly consistent across the lifespan (Deary et al., 2012). The rare, sporadic genetic events associated with major cognitive impairment—e.g., Down syndrome and Fragile X syndrome—similarly influence cognition across the life course (Plomin et al., 2008). However, because of the great heterogeneity within the ID diagnostic category, there is also likely to be substantial variation in developmental trajectories both within and between etiologically distinct sets of cases.
AUTISM SPECTRUM DISORDERS
Studies in clinical populations have suggested that there is substantial heterogeneity in the developmental trajectory of ASDs (Eaves and Ho, 1996; Fountain et al., 2012; Lord et al., 2006; Starr et al., 2003). Improvements are common; for example, gains are often made in the domain of verbal fluency (Anderson et al., 2007; Shattuck et al., 2007; Szatmari et al., 2000). However, even though some children with ASDs experience growth in specific skills (Fountain et al., 2012), the great majority continue to manifest the core symptoms of the disorder throughout their lives (McGovern and Sigman, 2005; Piven et al., 1996; Szatmari et al., 2009). The population of adults with an ASD diagnosis is growing rapidly, and further study is needed to understand their range of functioning and need for services (Shattuck et al., 2012).
ATTENTION DEFICIT/HYPERACTIVITY DISORDER
Longitudinal studies of ADHD have suggested that approximately 40% of patients continue to meet full criteria for ADHD in early adulthood and approximately 60% manifest residual symptoms (Faraone et al., 2006). Although the disorder as a whole is moderately stable over time, there is evidence for varying stability among ADHD’s phenotypic domains. Multiple studies have noted greater persistence of inattentive as compared with hyperactive/impulsive symptomology (Biederman et al., 2000; Larsson et al., 2011b). However, few studies have assessed persistence beyond the age of 25. Children diagnosed with ADHD have significantly higher rates of arrest for both misdemeanor and felony charges than children without an ADHD diagnosis, although the samples in which this has been investigated are predominantly male (Barkley et al., 2004). In a longitudinal study of girls diagnosed with ADHD, higher rates of all major classes of psychopathology were found (Biederman et al., 2010).
TOURETTE DISORDER
Typically, vocal and motor tic symptoms peak during early puberty and tend to diminish in adulthood. Even though symptoms ease in adulthood, an estimated 90% of patients with Tourette continue to experience tics (Pappert et al., 2003). Tic severity in childhood has been shown to be inversely correlated with health-related quality of life (Cavanna et al., 2012).
HERITABILITY AND PREVALENCE OF CHILDHOOD PSYCHIATRIC DISORDERS
INTELLECTUAL DISABILITY
Unlike the other developmental phenotypes discussed in this chapter, intellectual disability is currently defined by a quantitative measure. Intelligence is assessed on a continuum in the general population, and IQ scores are standardized such that they are normally distributed around an empirically derived population average. The DSM-IV defines intellectual disability as an IQ score more than two standard deviations below the mean—accordingly corresponding with the lowest scoring 2.5% of individuals, an inherent marker of prevalence (APA, 2000).
Although the population rate of ID is stable under the DSM-IV definition, there can be fluctuation in the commonality of specific ID-associated syndromic conditions. For example, Down syndrome remains the most significant single, identified cause of major cognitive impairment (Mefford et al., 2012), but its prevalence varies by time period and geographic region. Like many spontaneous genetic events, the incidence of chromosomal disorders like DS varies significantly with parental age and likely with other sociodemographic and environmental factors (Irving et al., 2008).
It is difficult to estimate the heritability of ID given its limited prevalence and etiologic heterogeneity. IQ itself is moderately to highly heritable (Haworth et al., 2010), and evidence suggests that at least mild ID represents the low end of typical variation (Plomin et al., 2008). However, the etiologic structure of ID is likely to vary across levels of severity (Nichols, 1984). The empirical basis for this model is drawn from family studies showing that the siblings of children with mild cognitive disabilities had, on the whole, below-average IQs themselves. The siblings of children with severe ID, by contrast, were on average IQ-normative. This pattern suggests that the genetic sources of rare and severe cognitive impairments are more likely to be sporadic, and the genetic sources of more common and mild cognitive impairments more likely to be familial.
On the whole, severe ID is considered to be significantly less heritable than most common neuropsychiatric conditions (or indeed nearly all medical and anthropometric traits in general) (Plomin et al., 2008). Although some inherited genetic causes have been identified (Lossie and Driscoll, 1999), there are a variety of clear environmental risk factors that can induce major cognitive impairment (e.g., trauma, infection, poisoning, or malnutrition before, during, or after birth, early childhood diseases). Furthermore, many of the documented genetic influences are large-scale spontaneous chromosomal anomalies, which themselves can be environmentally induced (Mefford et al., 2012). Such a heterogeneous etiology necessitates that inherited factors explain a smaller proportion of variance for ID than many other common medical phenotypes.
AUTISM SPECTRUM DISORDERS
The estimated prevalence of ASDs has increased dramatically over four decades in the United States and the United Kingdom, and is currently approximated at around 1% in both countries (Baird et al., 2006; Baron-CoShen et al., 2009; CDC, 2012; Elsabbagh et al., 2012). The increase in measured prevalence is at least in large part secondary to definitional expansion and greater diagnostic awareness. Neither Asperger syndrome nor pervasive developmental disorder not otherwise specified (PDD-NOS) were included in the DSM description of autism until the text’s fourth edition (1994), and recent studies suggest that the probability of ASD ascertainment continues to grow within certain US populations (Pedersen et al., 2012; “Prevalence of autism spectrum disorders—Autism and Developmental Disabilities Monitoring Network, 14 sites, United States, 2008”). A recent UK study highlighted the probable importance of changes to the definition and study of ASDs in prevalence estimation. Using modern clinical criteria and research methods, Brugha et al. (2011) found that the prevalence of ASDs among UK adults was also approximately 1%. Because ASDs are typically lifelong phenotypes, this suggests that these adults would have met current clinical criteria as children but were not diagnosed because ascertainment had yet to become common (Brugha et al., 2011). Studies such as these do not, however, preclude the possibility of a “true” increase in prevalence, and the degree to which current data may reflect such a trend is unknown.
The estimated prevalence of certain types of intellectual disability, discussed in the preceding, may also change with diagnostic climate. Although ID to ASD diagnostic substitution does not account for the majority of the increase in ASD prevalence, it has clearly made a substantial contribution. Using data from the California Department of Developmental Services, King and Bearman (2009) estimated that one-fourth of the observed increase in ASD prevalence in California between 1992 and 2005 was a direct consequence of diagnostic exchange between ID and ASD (King and Bearman, 2009). These changes are likely to have implications for the etiological structure and pathophysiological heterogeneity of ASDs.
The male to female ratio among individuals with an ASD diagnosis is approximately 4:1. The overall ratio, however, obscures a substantial sex by IQ interaction in the population distribution of the disorder. Among individuals with ASDs who have very low IQ, the male to female ratio is closer to 2:1. In cases with above average IQ, the ratio is much higher, likely 6:1 or above (Fombonne, 2003; Skuse, 2009). This trend has held constant across the period of prevalence increase (Lord and Schopler, 1985). Because much of the recent diagnostic expansion has occurred within the high functioning range (Rutter), several population-based studies have begun to report overall male:female ratios that are higher than 4:1 (Brugha et al., 2011; Williams et al., 2008). Although some of the sex by IQ interaction likely reflects greater ascertainment probabilities in boys (Russell et al., 2010), above average cognitive abilities do appear to confer greater ASD protection to women (at least as the diagnostic category is currently defined). Although the data are strong that some etiological overlap exists between ASDs and ID, ascertainment considerations do preclude a conclusive interpretation of population data. In fact, it has been proposed that the observed correlation between ID and ASD could be in part explained by the fact the presence of both in an individual dramatically increases the chance of the individual being ascertained owing to severe impairment (Skuse, 2007). This thesis is not inconsistent with the high rate of identification of IQ-normative ASD in adults who were not ascertained as children in the Brugha study (Brugha et al., 2011).
Autism spectrum disorders have long been considered among the most heritable of neuropsychiatric conditions. Through the period of diagnostic expansion, most twin studies of ASDs have suggested that the disease category is at least 60% heritable (Ronald and Hoekstra, 2011). However, the largest of those studies, both conducted recently, have noted a trend that reinforces the possibility of both genetic and environmental influence on autistic behavior (Hallmayer et al., 2011; Rosenberg et al., 2009). Rosenberg et al. (2009) and Hallmayer et al. (2011) reported that the dizygotic (DZ) twin of an individual with ASD has approximately a 30% chance of being diagnosed with autism him- or herself. This figure suggests that ASD recurrence in DZ twins may be higher than that estimated for regular siblings (Constantino et al., 2010; Ozonoff et al., 2011). This is notable because DZ twins are no more genetically similar than regular siblings. Some of the estimated difference may be caused by higher ascertainment probabilities among twins (e.g., because twins are the same age, they are more likely to be assessed by the same clinician at the same time). There is also a possibility that the registries employed for these studies are not representative of the population as a whole, and families with multiple affected twins were more likely to participate than families with only one affected child. However, elevated DZ twin risk may also suggest that ASDs, in part, reflect environmental exposures during fetal development (Szatmari, 2011). The nature of such possible exposures, and the manner in which they interact with genetic vulnerability, are important areas of future research (Croen et al., 2011).
ATTENTION DEFICIT/HYPERACTIVITY DISORDER
The estimated prevalence of ADHD varies substantially based on age at assessment, informant (clinician, parent, teacher, self, or some combination), country, and diagnostic criteria. Using DSM-IV criteria, the estimated prevalence ranges from 5.8% in Brazil (Rohde et al., 1999) to 7.5% in Australia (Graetz et al., 2001) and 17% in Germany (Baumgaertel et al., 1995). In Sweden, using the Autism-Tics, ADHD, and Other Comorbidities inventory, prevalence was estimated at 1.8% with a 2.5-fold enrichment in boys compared with girls (Lichtenstein et al., 2010). In the United States, prevalence estimates range between 9.5% and 16% (Hudziak et al., 1998; Gimpel and Kuhn, 2000; Nolan et al., 2001), but can be lower when functional impairment is taken into consideration (Wolraich et al., 1998).
The estimated heritability of ADHD is consistently high. Faraone and colleagues conducted a metaanalysis of diagnostic and dimensional assessments of ADHD determining a mean estimate of 76% for heritability across 20 different twin studies conducted in the United States, the European Union, and Australia with point estimates ranging from approximately 60% to more than 95% (Faraone et al., 2005).
TOURETTE DISORDER
The prevalence of Tourette disorder is estimated between 0.1% and 1% with the expanded phenotype of tic disorder estimated to be between 3% and 12% (Kurlan et al., 2001; Lichtenstein et al., 2010; Singer, 2005). In a large population cohort of twins from Sweden, tic disorder is estimated to be significantly heritable, with a point estimate of 56% (95%; CI, 37–68%) (Lichtenstein et al., 2010). Some estimates for Tourette disorder alone are lower (32%) (Mathews and Grados, 2011).
CHILDHOOD PSYCHIATRIC DISORDERS AS THE EXTREMES OF NORMAL TRAIT VARIATION
A growing body of population-based studies suggests that, like mild intellectual disabilities, ASDs and ADHD may exist as extreme positions of phenotypic continua (Larsson et al., 2011a; Lundstrom et al., 2012; Robinson et al., 2011a). This section reviews some of that evidence, concentrating on studies that link the genetic influences on major behavioral impairment to typical variation in traits of those disorders (e.g., autistic behavior, traits of hyperactivity/inattentiveness).
Because intelligence is measured on a continuum, much like height or weight, it is natural to think of intellectual disability as an extreme position on that distribution. The point below which an individual meets criteria for ID, however, is in many ways arbitrary, a problem common to categorical cutoffs superimposed on continuous distributions. Although an IQ of 70 is typically employed as the clinical threshold (APA, 2000), there is no clear biological or phenotypic break point that indicates where the boundary of “meaningfully” disordered cognition should lie. In other words, identifying the position on the IQ distribution below which a person has ID is no simpler than identifying the position on the height distribution above which a person is tall.
Only recently have researchers begun to consider the degree to which major psychiatric disorders may similarly exist as extreme positions on behavioral distributions. Diseases like autism and ADHD have traditionally been thought of as categorical clinical entities that one either has or does not have. However, traits typical of many behavioral disorders occur commonly at subthreshold levels in the general population as well as “deficits” characterizing many of these clinical phenotypes (Constantino and Todd, 2003; Goodman et al., 2000; Ronald et al., 2008; Skuse et al., 2005).
Autistic traits provide a useful example as they have been studied extensively over the last decade. Autistic trait measures are designed to capture the extent to which an individual manifests characteristics of the clinical ASD phenotype. These measures aim to assess the extent of social impairment, communication impairment, and restricted and repetitive behaviors and interests that a person displays (Constantino et al., 2003; Scott et al., 2002). When autistic trait scores are estimated in large samples of people in the general population, the distribution is typically skewed. The bulk of the population displays few if any autism-like behaviors, with a declining distribution to individuals that endorse a high number of traits, at or above the level that is on average associated with clinical ASD (Robinson et al., 2011a, 2011b). Using these trait measures, however, does not reveal a distinction between the clinical and subclinical levels of autism-like behavior. This pattern mimics the IQ distribution leading to intellectual disability (Constantino and Todd, 2003; Ronald et al., 2006).
Traits of ADHD can be measured in a similar way (Hansson et al., 2005) using a variety of instruments. The Conner’s Rating Scale, for example, aims to assess the presence and severity of symptoms recognized in the clinical phenotype. The Swan Rating Scale, in contrast, attempts to characterize individuals on a more normal scale of activity level, impulsivity, and attention. Consequently, the distribution of the Conner’s ADHD scale tends to be J-shaped, whereas the Swan is considerably more normally distributed. For each of these measures, children with an ADHD diagnosis lie within the most affected tail (Larsson et al., 2011a).
The study of general population traits will only be informative with respect to disease processes if there is an etiological link between the extreme or categorical phenotype (e.g., ASDs) and the behavioral continuum (e.g., autistic traits in the general population). In the case of both ASDs and ADHD, evidence is building that such a link exists. Empirically, there is a high degree of consistency between clinically significant and subclinical levels of impairment. Much like the ASD diagnosis, autistic trait scores in the general population are highly stable over time (Robinson et al., 2011b; St Pourcain et al., 2011). Longitudinal studies of traits of ADHD suggest similar coherence with the clinical diagnosis (Biederman et al., 1996, 2000; Faraone et al., 2006).
On the whole, traits of ADHD are moderately stable across development (Kuntsi et al., 2005; Larsson et al., 2011b).
Many studies suggest a shared etiology between Robinson the clinical phenotypes of ASDs and ADHD and their respective behavioral continua. If autism and ADHD are extreme positions on quantitative distributions, two primary conditions should hold. The estimated heritability of traits of each disorder should be approximately equivalent to the estimated heritability of the clinical phenotype, and a clinical diagnosis should predict increased trait scores in family members of the proband. With respect to overall heritability, there is a substantial body of literature suggesting that this is the case. Like ASDs, autistic traits are moderately to highly heritable (Ronald and Hoekstra, 2011). Similarly, like ADHD, traits of hyperactivity and inattentiveness are consistently highly heritable (Larsson et al., 2011a). Autism spectrum disorders and ADHD also show the predicted pattern of increased trait burden in family members. In the case of ASDs, it has been noted for more than a decade that the family members of people with autism, on average, display more autistic traits than the family members of people without autism (Constantino et al., 2006, 2010; Piven et al., 1997). Twin studies can be used to examine the extent to which that pattern might be secondary to genetic influences that are shared between levels of affectation. Two large, general population twin studies recently reported evidence of strong genetic overlap between severely impairing autistic behavior and subclinical autistic traits (Lundstrom et al., 2012; Robinson et al., 2011a). In a similar analysis, Larsson et al. (2012) evidenced a genetic link between clinically diagnosed ADHD and traits typical of the disorder as they are distributed continuously.
These patterns can be interpreted in a number of ways and, in so doing, it is important to bear in mind the established etiological heterogeneity of neuropsychiatric conditions (Betancur, 2011). On the one hand, etiological overlap between the clinical (extreme) phenotype and the continuum could be interpreted as evidence that relevant common genetic variants act across the behavioral distribution, influencing both normal trait variation in the general population and the probability with which an individual will meet criteria for clinical disorder. On the other, these results are also consistent with the model in which rare (and potentially unique), familial genetic variants that increase risk for autism and ADHD can also predict subclinical behavioral impairment. In other words, pleiotropy may extend to levels of affectation. It is believed that both common and rare genetic variants influence liability toward autism and ADHD, so these possibilities cannot be disentangled in the context of phenotypic family data.
PHENOTYPIC AND ETIOLOGIC RELATIONSHIPS AMONG CHILDHOOD PSYCHIATRIC DISORDERS
Neuropsychiatric disorders of childhood appear together far more often than predicted by chance. Among the phenotypes discussed in this chapter, most individuals with ASDs meet criteria for additional developmental or behavioral disorders such as ADHD, anxiety disorders, or intellectual disabilities (Gillberg and Billstedt, 2000; Lichtenstein et al., 2010; Simonoff et al., 2008; Volkmar et al., 1999). Intellectual disabilities themselves are similarly associated with an increased risk for a wide range of mental health conditions (Einfeld et al., 2011), and Tourette syndrome often co-occurs with ADHD and OCD (Mathews and Grados, 2011).
Twin and molecular genetic studies suggest that shared genetic risk factors are contributing substantially to the disorders’ co-occurrence. Using the twin method, Lichtenstein et al. (2010) estimated that ASDs share approximately three-fourths of their genetic variance with ADHD, one-half of their genetic variance with intellectual disabilities, and one-third of their genetic variance with tic disorder. The authors further noted that the genetic influences on ADHD are highly correlated with those on tic disorder, a finding that has since been replicated in family studies (Mathews and Grados, 2011). Molecular genetic analyses have similarly indicated that the etiological influences on child neuropsychiatric disorders are unlikely to exclusively predict the phenotypes circumscribed by the DSM. For example, virtually every genetic variant that has been associated with ASDs is also associated with intellectual disabilities (Betancur, 2011); several of the notable rare variants are also risk factors for other major psychiatric disorders (McCarthy et al., 2009; Weiss et al., 2008).
Genetic relationships between the disorders raise questions about research approach. More often than not, investigations into ASDs, ADHD, and Tourette are conducted independently. This model is consistent with the classical approach to comorbidity, a term originally designed to indicate distinct, co-occurring medical problems (Feinstein, 1970). The etiological overlap described in the preceding section, however, suggests that conceptualizing multiple psychiatric disorders as comorbid could be “artificially splitting a complex clinical condition into several pieces” (Maj, 2005).
Large multidisorder research initiatives, such as the Psychiatric Genomics Consortium (PGC), are able to investigate etiological factors that may be shared across neuropsychiatric conditions. With the identification of genetic risk factors, broadly phenotyped samples provide the opportunity to potentially shed insight into the disorders’ underlying biological and behavioral processes.
REPRODUCTIVE DISADVANTAGE AND GENETIC ARCHITECTURE
Reproductive disadvantage provides a major influence on the genetic architecture of disorders that are, on average, associated with reduced fecundity. In the simplest case, autosomal-recessive diseases that are lethal in childhood are defined by allelic spectra consisting of rare mutations. This occurs because of the consistently lower than chance probability of the alleles being passed on in each generation. Generally, such genetic variants are seen at less than 0.1% in population frequency, but they can be significantly more frequent in instances in which an advantage is conferred to heterozygote carriers (e.g., Hb mutations that confer resistance to malaria in heterozygotes but sickle cell disease in homozygotes). These alleles may also have been previously favorably selected in alternate environmental conditions or appeared recently in bottlenecked or isolated populations (e.g., Finns, Ashkenazi Jews, Quebecois from Saguenay-Lac St. Jean), where they received a chance boost to high frequency and are still early in the many hundreds of generations selection takes to inexorably push them down. As a consequence, gene mapping efforts in diseases associated with reproductive disadvantage are designed with a focus on rare and spontaneously arising variation.
By contrast, and consistent with classical theory (Kimura and Ota, 1973), humans have limited genetic variation. Considering two copies of the human genome, the heterozygosity rate for single nucleotide polymorphisms (SNPs) is approximately one in 1,000 bases (Sachidanandam et al., 2001) and roughly 90% of those heterozygous sites in each individual are common DNA variants. The variants are typically seen in multiple, diverse populations and show evidence of having existed for thousands of generations (Lewontin, 1972; International Hapmap Consortium, 2005). High frequency of an allele does not by any means rule it out as a contributing factor in diseases with reproductive disadvantage. However, it does indicate that the allele itself is not under strong selective disadvantage and limits the individual impact it could have in a reproductively deleterious phenotype. It is also important to note that mildly deleterious alleles can persist at intermediate allele frequencies, particularly under the conditions described that moderate the relationship between selection and frequency.
With these observations in mind, human genetics has been drawn to the common disease-common variant hypothesis, which suggests a role for common DNA polymorphisms (>1% in population frequency) in the genetic architecture of common and complex human diseases (Collins et al., 1997; Lander and Schork, 1994; Risch and Merikangas, 1996). This hypothesis produced the subsequent proposal that genome-wide association studies (GWAS) of common human diseases might yield, for the first time, specific genetic insights into complex human disease. Genome-wide association studies are powered by massively parallel array–based genotyping technologies that permit the evaluation of hundreds of thousands of common DNA variants simultaneously. Alongside the construction of maps of common variation and linkage disequilibrium patterns across the genome, GWAS has delivered myriad insights into common diseases. As of the end of 2011, more than 1,500 genetic associations have been defined since the first GWAS studies were initiated in 2005–2006 and, in the vast majority of cases, the associations were novel and therefore harbored unique insights into the pathogenesis of disease. It is noteworthy that most of these discoveries emerged in diseases and phenotypes only when very large sample sizes were assessed, often after initial smaller studies had not revealed any replicable, significant associations. Thus, a collective picture that has emerged from GWAS studies is one of extreme polygenicity for the complex disorders studied in which individual contributing alleles are of extremely modest effect and provide little predictive information about individual risk. Collectively, however, they can reveal novel insights into the pathways and processes of disease.
Negative reproductive selection is likely to be acting on severe forms of autism and intellectual disability (Uher, 2009). Although formal studies estimating the specific degree of reproductive disadvantage associated with these conditions have not been conducted, it is widely accepted that individuals with these phenotypes produce offspring far less often than the population average. Bolstered by classical examples of the role of major sporadic chromosomal anomalies in similar phenotypes such as Down syndrome, this observation has led to a substantial and sensible focus on the role of spontaneously arising (so-called de novo) DNA variants in ASD and ID. At the same time, large GWAS studies have also been undertaken, and draw considerable hope from the example of schizophrenia. Schizophrenia has a similar prevalence and estimated selective disadvantage (approximately 50% reduced fecundity) (Svensson et al., 2007). Genome-wide association studies of schizophrenia have proved, at large sample sizes, to be able to demonstrate specific compelling associations and to establish a significant global role for common as well as rare variation. It is not incongruous that common variation can be associated to a common and reproductively deleterious phenotype. In the case of extremely polygenic phenotypes, an obligate relationship between allele frequency and reproductive advantage is precluded by each of: the modest contributions of individual loci; unknown interactions that may exist among genes or between genes and environmental factors; and the other (perhaps positive) phenotypic outcomes that may be influenced by some of the risk variation. Take for example the predominant role of common variation in type I diabetes, and the mix of common, rare, and de novo variation in Hirschsprung disease—both conditions that had extreme selection against them owing to high lethality before modern medical interventions developed in the last century. The genetic architecture of these conditions does not stand in stark violation of fundamental principles but is likely representative of the mix of variation types that will be uncovered in childhood neuropsychiatric disease.
The insights attained to date from the early studies of both common and rare DNA variation are outlined in the next section and more thoroughly delineated in subsequent sections of the book. Of note, the accepted negative selection attributable to ID and ASD does not carry over into ADHD and TD, where no perceived reproductive disadvantage resides. As noted, however, strong etiological overlaps likely exist between each of these disorders. We fully expect that overlap to manifest itself in considerable shared genetic risk factors of both a common and rare variety.
EMPIRICAL INSIGHTS INTO THE GENETIC ARCHITECTURE OF CHILDHOOD PSYCHIATRIC DISORDERS
The most compelling evidence compiled to date with respect to neuropsychiatric disorders of childhood onset surrounds the role of large and predominantly spontaneously arising chromosomal abnormalities and copy number variants. Down syndrome (trisomy 21) is the most common and widely recognized karyotypic abnormality associated with ID but, in recent years, many such findings have emerged as substantial components of both ASD and ID more broadly. Many additional syndromes (e.g., Prader–Willi/Angelman, Williams syndrome, DiGeorge/velocardiofacial syndrome) result from specific recurrent copy number variants. Genome-wide, gross chromosomal abnormalities (Sanders et al., 2011; Vorstman et al., 2006) and even balanced translocations (Talkowski et al., 2012) have been documented as strongly enriched in both ID and ASD. However, as most such events are rare and seen only in individual cases, certainty is generally lacking that individual events are causative. In addition, because these events are most often large and delete, duplicate, or disrupt many genes, biological insights have been slow to emerge in many cases.
Recent surveys (Girirajan et al., 2011) have clearly demonstrated that the burden of CNVs in ID is considerably greater than in ASDs. In particular, large CNVs (>1 Mb) are observed in four to five times as many instances of ID-related phenotypes than in ASDs, although the distinction between syndromic and non-syndromic forms of ID is not always made, and there could be more CNVs associated with syndromic ID (or ASDs). Overall, rare CNVs (seen in roughly 2% of the general population) are observed in 10% of ASD cases and 16% of IDs. A more modest, roughly twofold excess of rare CNVs has also been observed in ADHD (Williams et al., 2010, 2012). Thus, emerging data suggest a convincing role for rare, primarily spontaneous, CNVs but to significantly varying degrees. The highest frequency is in ID, then to lesser degrees respectively in ASD + ID, ASD alone, ADHD, and likely other less deleterious childhood phenotypes. Although it is still early in the use of many genomic analysis techniques (e.g., GWAS, genome sequencing), it seems plausible that these observations represent the relative contribution of extremely rare and de novo variation versus more common, older inherited variation across these phenotypes.
Two other important observations, which are also likely to extend to other variation types, have been made convincingly as sample sizes and studies have grown across many distinct clinical end points. First, the majority of CNVs that have been associated with ASDs have independently been demonstrated to confer risk to other, apparently unrelated phenotypes such as schizophrenia or epilepsy (Sebat et al., 2009). This suggests pointedly that most of these mutations confer risk to a very broad set of phenotypes, rather than a clinically specific one. Second, for most of these CNVs, healthy adult controls are also occasionally seen bearing the mutations. Accordingly, although risk conferred is quite high (approximate odds ratios of 10 to 20), the penetrances for any specific outcome are generally less than 50%. In these two properties, the risk variants discovered in large case-control studies (generally focusing on large, idiopathic population samples of cases) seem distinct from those relevant to the rare mendelian genomic syndromes. Such syndromes usually are caused by high penetrance events and have an extremely specific set of comorbidities and features that make them clinically recognizable often in advance of conclusive genetic testing.
Related to this, a considerable number of specific, single-gene genetic syndromes have been described that feature unusually high rates of ID or ASD (Betancur, 2011). Although some of these are rare and apparently fully penetrant entities such as Rett syndrome, a severe form of autism most often caused by mutations in one copy of MECP2 or CDKL5 on one X chromosome in girls (these mutations are usually lethal prenatally in boys), most have far less than complete penetrance. Noteworthy examples among these in the case of ASDs include Fragile X and tuberous sclerosis complex (TSC) (Folstein and Piven, 1991), specific and long-identified Mendelian syndromes with high rates of ASD. Similar to the CNVs described in the preceding, however, fewer than 50% of cases manifest ASDs. Although many such rare genetic syndromes are now identified, taken in total they likely explain only a small percentage of idiopathic ASDs.
Figure 70.1 shows the relationship between effect size and frequency for CNVs associated to ASD and the first common variants identified in schizophrenia at genome-wide significance. The CNVs clearly confer much greater risk, but are only present in a small fraction of cases. The common variants are selected from schizophrenia, to illustrate the effect sizes of common variants typical of psychiatric phenotypes. As sample sizes continue to increase for GWAS for these traits, further clues are likely to be revealed in the biological basis of these diseases.
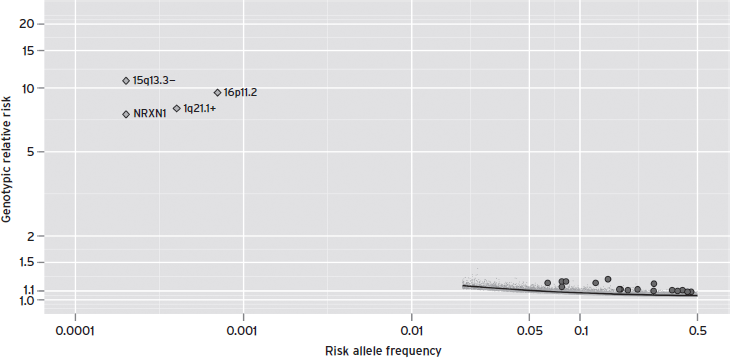
Figure 70.1 The risk allele frequency is plotted against the genotypic relative risk for the copy number variations identified in autism spectrum disorder along with single nucleotide polymorphisms associated at genome-wide significance for schizophrenia. The specific copy number variations identified are 15q33.3 deletion, deletions at NRXN1, 1q21.1 duplications, and both deletions and duplications at 16p11.2 (Malhotra and Sebat, 2012). These events confer strong risk to ASD in contrast to the identified genome-wide significant loci from schizophrenia (Ripke et al., 2011). (Thanks to P.F. Sullivan for concept and underlying data.)
The massively parallel genome sequencing technologies that have become available since 2010 have it possible to sequence the roughly 1% of the human genome that encodes proteins (the exome). Autism spectrum disorders are among the first disorders for which this approach has been utilized in substantial numbers of cases and indeed, by sequencing parents and children, recent studies have documented excesses of de novo loss-of-function mutations in cases when compared with either control individuals (Sanders et al., 2012) or mutational expectations (Neale et al., 2012; O’Roak et al., 2012). Although these early studies suggest that only a minority (<20%) of cases harbor relevant spontaneously arising mutations, they do have the ability to pinpoint specific genes relevant to ASD risk. Overall, however, these studies reinforce estimates from the CNV work that hundreds of genes likely contribute to ASD risk. Although large-scale studies have not yet been performed in the other childhood psychiatric disorders, earlier studies of X-linked mental retardation (Tarpey et al., 2009) similarly concluded that relevant mutations are likely distributed across many genes, resulting in a situation that will inevitably require considerable sample sizes on which to provide clarity.
As noted, GWAS has far and away been the most productive avenue toward insight into the genetics of unquestionably polygenic common diseases. Common variation, as assessed by GWAS, has yet to provide compelling findings in the case of any of the childhood neuropsychiatric disorders. Although the role of rare and spontaneous mutations may well dominate in the case of severe ID, it appears to play a reduced role in idiopathic ASDs and has not been documented to contribute substantially to ADHD or TD. The high heritability of the latter three, particularly ASD and ADHD, suggests some substantial role for inherited and somewhat more common variation. If we contrast the published efforts to date in ASD, ADHD, and TD with those in schizophrenia (as well as particularly polygenic and complex non-psychiatric diseases and phenotypes such as type 2 diabetes, coronary artery disease, and adult stature), a consistent pattern emerges. In all of these cases with similar high heritabilities, early GWAS studies of one to a few thousand cases failed to reject the null hypothesis that any common variants were associated to disease risk. Much larger collaborative studies that followed, however, were able to define many distinct and replicated associated loci. In each case, these studies provided novel insight into the biology of the diseases in question. This is of course not proof that such insights will be forthcoming in childhood neuropsychiatric disorders, but the productivity of schizophrenia GWAS after early “failure” suggests that further investment may be warranted in tandem with the focus on rare variation.
CONCLUSIONS
Research approaches that integrate the levels of evidence introduced in the preceding sections are likely to improve the efficacy of genetic inquiries into child neuropsychiatric conditions. Behaviorally defined disorders are characterized by enormous genetic complexity, and inferences drawn from epidemiological and family data, as well as classic genetic theory, can be used in concert to begin parsing that variation. The ASD literature presented here provides an example of such a collaborative logical trajectory. Autism spectrum disorders as currently defined exist across a broad range of severities, including phenotypes that both do and do not confer substantial reproductive disadvantage. Although the etiological structure of the disorders is unlikely to fall cleanly along these phenotypic lines, family and twin data are consistent with such a pattern in that they suggest that ASDs exist in both familial and non-familial forms (and likely in the space in between). These patterns can be used to justify a tandem approach of genetic investigation, focusing on both common and rare variants, consistent with the relationship between genetic architecture and reproductive disadvantage, as it has been observed more broadly within genomic inquiry.
Large sample, multidisciplinary collaborations designed to uncover the biology underlying neuropsychiatric disorders in children are already in progress. Following lessons from the progress emerging from larger, adult psychiatric consortia, it is very likely that continued efforts along these lines will be necessary to elucidate the complete genetic architecture of each disorder, as well as to progress to a full understanding of the relationship between etiologically linked phenotypes.
DISCLOSURES
Dr. Daly’s research into childhood psychiatric disorders is currently funded through grants from the NIMH and NHGRI and a philanthropic gift from the Gerstner Foundation. In the past year Dr. Daly has consulted with Pfizer, Inc. and Eisai Pharmaceuticals. Grant Support: R01 NS 059727 NIH; 1R01 DK083756-1 NIH/NIEHS; 2P30DK043351-21 NIH/NIDDK; R01AI091649-01AI NIH/NIAID; 1R01MH09443201 NIH/NIMH; 5R01DK064869-09 NIH/NIDDK; 1R01MH089208-01 NIH/NIMH; 1R01MH094469 NIH/NIMH; P40 RR12305 NIH/NCBI; U01HG006569 NIH/NHGRI; I5-A523 Starr Cancer Consortium.
Dr. Neale has no conflicts of interest to disclose. He is funded by the NIMH, the Gerstner Foundation, and the Stanley Center for Psychiatric Research. Grant support: 5R01MH080403-03S1 NIH/NIMH; 1R01MH09443201 NIH/NIMH; 1R01MH089208-01 NIH/NIMH; 1R01MH094469 NIH/NIMH.
Dr. Robinson has no conflicts of interest to disclose. She is funded by Massachusetts General Hospital.
REFERENCES
Anderson, D.K., Lord, C., et al. (2007). Patterns of growth in verbal abilities among children with autism spectrum disorder. J. Consult. Clin. Psychol. 75(4):594–604.
APA. (2000). Diagnostic and Statistic Manual of Mental Disorders, 4th Edition. Washington, DC: American Psychological Association.
Baird, G., Simonoff, E., et al. (2006). Prevalence of disorders of the autism spectrum in a population cohort of children in South Thames: the Special Needs and Autism Project (SNAP). Lancet 368(9531):210–215.
Barbaresi, W.J., and Olsen, R.D. (1998). An ADHD educational intervention for elementary schoolteachers: a pilot study. J. Dev. Behav. Pediatr. 19(2):94–100.
Barkley, R.A., Fischer, M., et al. (2004). Young adult follow-up of hyperactive children: antisocial activities and drug use. J. Child Psychol. Psychiatry 45(2):195–211.
Baron-Cohen, S., Scott, F.J., et al. (2009). Prevalence of autism-spectrum conditions: UK school-based population study. Br. J. Psychiatry 194(6):500–509.
Baumgaertel, A., Wolraich, M.L., et al. (1995). Comparison of diagnostic criteria for attention deficit disorders in a German elementary school sample. J. Am. Acad. Child Adolesc. Psychiatry 34(5):629–638.
Betancur, C. (2011). Etiological heterogeneity in autism spectrum disorders: more than 100 genetic and genomic disorders and still counting. Brain Res. 1380:42–77.
Biederman, J., Faraone, S., et al. (1996). Predictors of persistence and remission of ADHD into adolescence: results from a four-year prospective follow-up study. J. Am. Acad. Child Adolesc. Psychiatry 35(3):343–351.
Biederman, J., Mick, E., et al. (2000). Age-dependent decline of symptoms of attention deficit hyperactivity disorder: impact of remission definition and symptom type. Am. J. Psychiatry 157(5):816–818.
Biederman, J., Petty, C.R., et al. (2010). Adult psychiatric outcomes of girls with attention deficit hyperactivity disorder: 11-year follow-up in a longitudinal case-control study. Am. J. Psychiatry 167(4):409–417.
Brugha, T.S., McManus, S., et al. (2011). Epidemiology of autism spectrum disorders in adults in the community in England. Arch. Gen. Psychiatry 68(5):459–465.
Cavanna, A.E., David, K., et al. (2012). Predictors during childhood of future health-related quality of life in adults with Gilles de la Tourette syndrome. Eur. J. Paediatr. Neurol. 16(6):605–612.
Cavanna, A.E., Servo, S., et al. (2009). The behavioral spectrum of Gilles de la Tourette syndrome. J. Neuropsychiatry Clin. Neurosci. 21(1):13–23.
Centers for Disease Control and Prevention. (2012). Prevalence of autism spectrum disorders: Autism and Developmental Disabilities Monitoring Network, 14 sites, United States, 2008. MMWR Surveill. Summ. 61(3):1–19.
Chakrabarti, S., and Fombonne, E. (2005). Pervasive developmental disorders in preschool children: confirmation of high prevalence. Am. J. Psychiatry 162(6):1133–1141.
Charman, T. (2011a). The highs and lows of counting autism. Am. J. Psychiatry 168(9):873–875.
Charman, T., Pickles, A., et al. (2011b). IQ in children with autism spectrum disorders: data from the Special Needs and Autism Project (SNAP). Psychol. Med. 41(3):619–627.
Charman, T. (2011c). Commentary: glass half full or half empty? Testing social communication interventions for young children with autism: reflections on Landa, Holman, O’Neill, and Stuart. J. Child Psychol. Psychiatry 52(1):22–23.
Coffey, B.J., and Park, K.S. (1997). Behavioral and emotional aspects of Tourette syndrome. Neurol. Clin. 15(2):277–289.
Collins, F.S., Guyer, M.S., et al. (1997). Variations on a theme: cataloging human DNA sequence variation. Science 278(5343):1580–1581.
Constantino, J.N., Davis, S.A., et al. (2003). Validation of a brief quantitative measure of autistic traits: comparison of the social responsiveness scale with the autism diagnostic interview-revised. J. Autism Dev. Disord. 33(4):427–433.
Constantino, J.N., Lajonchere, C., et al. (2006). Autistic social impairment in the siblings of children with pervasive developmental disorders. Am. J. Psychiatry 163(2):294–296.
Constantino, J.N., and Todd, R.D. (2003). Autistic traits in the general population: a twin study. Arch. Gen. Psychiatry 60(5):524–530.
Constantino, J.N., Zhang, Y., et al. (2010). Sibling recurrence and the genetic epidemiology of autism. Am. J. Psychiatry 167(11):1349–1356.
Croen, L.A., Grether, J.K., et al. (2011). Antidepressant use during pregnancy and childhood autism spectrum disorders. Arch. Gen. Psychiatry 68(11):1104–1112.
Dawson, G., Rogers, S., et al. (2010). Randomized, controlled trial of an intervention for toddlers with autism: the Early Start Denver Model. Pediatrics 125(1):e17–23.
Deary, I.J., Yang, J., et al. (2012). Genetic contributions to stability and change in intelligence from childhood to old age. Nature 482(7384):212–215.
Eaves, L.C., and Ho, H.H. (1996). Brief report: stability and change in cognitive and behavioral characteristics of autism through childhood. J. Autism. Dev. Disord. 26(5):557–569.
Einfeld, S.L., Ellis, L.A., et al. (2011). Comorbidity of intellectual disability and mental disorder in children and adolescents: a systematic review. J. Intellect. Dev. Disabil. 36(2):137–143.
Elsabbagh, M., Divan, G., et al. (2012). Global prevalence of autism and other pervasive developmental disorders. Autism Res. 5(3):160–179.
Faraone, S.V., Biederman, J., et al. (2006). The age-dependent decline of attention deficit hyperactivity disorder: a meta-analysis of follow-up studies. Psychol. Med. 36(2):159–165.
Faraone, S.V., Perlis, R.H., et al. (2005). Molecular genetics of attention-deficit/hyperactivity disorder. Biol. Psychiatry 57(11):1313–1323.
Faraone, S.V., Sergeant, J., et al. (2003). The worldwide prevalence of ADHD: is it an American condition? World Psychiatry 2(2):104–113.
Feinstein, A.R. (1970). The pre-therapeutic classification of comorbidity in chronic disease. J. Chron. Dis. (23):455–468.
Folstein, S.E., and Piven, J. (1991). Etiology of autism: genetic influences. Pediatrics 87(5 Pt 2):767–773.
Fombonne, E. (2003). Epidemiological surveys of autism and other pervasive developmental disorders: an update. J. Autism Dev. Disord. 33(4):365–382.
Fountain, C., Winter, A.S., et al. (2012). Six developmental trajectories characterize children with autism. Pediatrics 129(5):e1112–1120.
Ganz, M.L. (2007). The lifetime distribution of the incremental societal costs of autism. Arch. Pediatr. Adolesc. Med. 161(4):343–349.
Gillberg, C., and Billstedt, E. (2000). Autism and Asperger syndrome: coexistence with other clinical disorders. Acta Psychiatr. Scand. 102(5):321–330.
Gilles de la Tourette, G., Goetz, C.G., et al. (1982). Etude sur une affection nerveuse caracterisee par de l’incoordination motrice accompagnee d’echolalie et de coprolalie. Adv. Neurol. 35:1–16.
Gimpel, G.A., and Kuhn, B.R. (2000). Maternal report of attention deficit hyperactivity disorder symptoms in preschool children. Child Care Health Dev. 26(3):163–176; discussion 176–169.
Girirajan, S., Brkanac, Z., et al. (2011). Relative burden of large CNVs on a range of neurodevelopmental phenotypes. PLoS Genet. 7(11):e1002334.
Goodman, R., Ford, T., et al. (2000). Using the Strengths and Difficulties Questionnaire (SDQ) to screen for child psychiatric disorders in a community sample. Br. J. Psychiatry 177:534–539.
Grados, M.A., and Mathews, C.A. (2009). Clinical phenomenology and phenotype variability in Tourette syndrome. J. Psychosom. Res. 67(6):491–496.
Graetz, B.W., Sawyer, M.G., et al. (2001). Validity of DSM-IV ADHD subtypes in a nationally representative sample of Australian children and adolescents. J. Am. Acad. Child Adolesc. Psychiatry 40(12):1410–1417.
Hallmayer, J., Cleveland, S., et al. (2011). Genetic heritability and shared environmental factors among twin pairs with autism. Arch. Gen. Psychiatry 68(11):1095–1102.
Hansson, S.L., Svanstrom Rojvall, A., et al. (2005). Psychiatric telephone interview with parents for screening of childhood autism: tics, attention-deficit hyperactivity disorder and other comorbidities (A-TAC): preliminary reliability and validity. Br. J. Psychiatry 187:262–267.
Haworth, C.M., Wright, M.J., et al. (2010). The heritability of general cognitive ability increases linearly from childhood to young adulthood. Mol. Psychiatry 15(11):1112–1120.
Hudziak, J.J., Heath, A.C., et al. (1998). Latent class and factor analysis of DSM-IV ADHD: a twin study of female adolescents. J. Am. Acad. Child Adolesc. Psychiatry 37(8):848–857.
International HapMap Consortium. (2005). A haplotype map of the human genome. Nature 437(7063):1299–1320.
Irving, C., Basu, A., et al. (2008). Twenty-year trends in prevalence and survival of Down syndrome. Eur. J. Hum. Genet. 16(11):1336–1340.
Kimura, M., and Ota, T. (1973). The age of a neutral mutant persisting in a finite population. Genetics 75(1):199–212.
King, M., and Bearman, P. (2009). Diagnostic change and the increased prevalence of autism. Int. J. Epidemiol. 38(5):1224–1234.
Knapp, M., Romeo, R., et al. (2009). Economic cost of autism in the UK. Autism 13(3):317–336.
Kuntsi, J., Rijsdijk, F., et al. (2005). Genetic influences on the stability of attention-deficit/hyperactivity disorder symptoms from early to middle childhood. Biol. Psychiatry 57(6):647–654.
Kurlan, R., McDermott, M.P., et al. (2001). Prevalence of tics in schoolchildren and association with placement in special education. Neurology 57(8):1383–1388.
Lander, E.S., and Schork, N.J. (1994). Genetic dissection of complex traits. Science 265(5181):2037–2048.
Larsson, H., Anckarsater, H., et al. (2011a). Childhood attention-deficit hyperactivity disorder as an extreme of a continuous trait: a quantitative genetic study of 8,500 twin pairs. J. Child Psychol. Psychiatry 53(1):73–80.
Larsson, H., Anckarsater, H., et al. (2012). Childhood attention-deficit hyperactivity disorder as an extreme of a continuous trait: a quantitative genetic study of 8,500 twin pairs. J. Child Psychol. Psychiatry 53(1):73–80.
Larsson, H., Dilshad, R., et al. (2011b). Developmental trajectories of DSM-IV symptoms of attention-deficit/hyperactivity disorder: genetic effects, family risk and associated psychopathology. J. Child Psychol. Psychiatry 52(9):954–963.
Leibson, C.L., Katusic, S.K., et al. (2001). Use and costs of medical care for children and adolescents with and without attention-deficit/hyperactivity disorder. JAMA 285(1):60–66.
Lejeune, J., Turpin, R., et al. (1959). [Mongolism; a chromosomal disease (trisomy)]. Bull. Acad. Natl. Med. 143(11–12):256–265.
Leonard, H., and Wen, X. (2002). The epidemiology of mental retardation: challenges and opportunities in the new millennium. Ment. Retard Dev. Disabil. Res. Rev. 8(3):117–134.
Lewontin, R. (1972). The apportionment of human diversity. In: Dobzhansky, T., Hecht, M.K., and Steere, W.C., eds. Evolutionary Biology, 6th Edition. New York: Appleton-Century-Crofts, pp. 381–398.
Lichtenstein, P., Carlstrom, E., et al. (2010). The genetics of autism spectrum disorders and related neuropsychiatric disorders in childhood. Am. J. Psychiatry 167(11):1357–1363.
Lord, C., Risi, S., et al. (2006). Autism from 2 to 9 years of age. Arch. Gen. Psychiatry 63(6):694–701.
Lord, C., and Schopler, E. (1985). Differences in sex ratios in autism as a function of measured intelligence. J. Autism Dev. Disord. 15(2):185–193.
Lossie, A.C., and Driscoll, D.J. (1999). Transmission of Angelman syndrome by an affected mother. Genet. Med. 1(6):262–266.
Lundstrom, S., Chang, Z., et al. (2012). Autism spectrum disorders and autisticlike traits: similar etiology in the extreme end and the normal variation. Arch. Gen. Psychiatry 69(1):46–52.
Maj, M. (2005). “Psychiatric comorbidity”: an artefact of current diagnostic systems? Br. J. Psychiatry 186:182–184.
Malhotra, D., and Sebat, J. (2012). CNVs: harbingers of a rare variant revolution in psychiatric genetics. Cell 148(6):1223–1241.
Mathews, C.A., and Grados, M.A. (2011). Familiality of Tourette syndrome, obsessive-compulsive disorder, and attention-deficit/hyperactivity disorder: heritability analysis in a large sib-pair sample. J. Am. Acad. Child Adolesc. Psychiatry 50(1):46–54.
McCarthy, S.E., Makarov, V., et al. (2009). Microduplications of 16p11.2 are associated with schizophrenia. Nat. Genet. 41(11):1223–1227.
McGovern, C.W., and Sigman, M. (2005). Continuity and change from early childhood to adolescence in autism. J. Child Psychol. Psychiatry 46(4):401–408.
Mefford, H.C., Batshaw, M.L., et al. (2012). Genomics, intellectual disability, and autism. N. Engl. J. Med. 366(8):733–743.
Merikangas, K.R., He, J.P., et al. (2010). Lifetime prevalence of mental disorders in U.S. adolescents: results from the National Comorbidity Survey Replication—Adolescent Supplement (NCS-A). J. Am. Acad. Child Adolesc. Psychiatry 49(10):980–989.
Neale, B.M., Kou, Y., et al. (2012). Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature 485(7397):242–245.
Nichols, P.L. (1984). Familial mental retardation. Behav. Genet. 14(3):161–170.
Nolan, E.E., Gadow, K.D., et al. (2001). Teacher reports of DSM-IV ADHD, ODD, and CD symptoms in schoolchildren. J. Am. Acad. Child Adolesc. Psychiatry 40(2):241–249.
O’Roak, B.J., Vives, L., et al. (2012). Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature 485(7397):246–250.
Ozonoff, S., Young, G.S., et al. (2011). Recurrence risk for autism spectrum disorders: a Baby Siblings Research Consortium study. Pediatrics 128(3):e488–495.
Pappert, E.J., Goetz, C.G., et al. (2003). Objective assessments of longitudinal outcome in Gilles de la Tourette’s syndrome. Neurology 61(7):936–940.
Pedersen, A., Pettygrove, S., et al. (2012). Prevalence of autism spectrum disorders in Hispanic and non-Hispanic white children. Pediatrics 129(3):e629–635.
Piven, J., Harper, J., et al. (1996). Course of behavioral change in autism: a retrospective study of high-IQ adolescents and adults. J. Am. Acad. Child Adolesc. Psychiatry 35(4):523–529.
Piven, J., Palmer, P., et al. (1997). Broader autism phenotype: evidence from a family history study of multiple-incidence autism families. Am. J. Psychiatry 154(2):185–190.
Plomin, R., DeFries, J., et al. (2008). Behavioral Genetics, 5th Edition. New York: Worth.
Ripke, S., Sanders, A.R., et al. (2011). Genome-wide association study identifies five new schizophrenia loci. Nat. Genet. 43(10):969–976.
Risch, N., and Merikangas, K. (1996). The future of genetic studies of complex human diseases. Science 273(5281):1516–1517.
Robinson, E.B., Koenen, K.C., et al. (2011a). Evidence that autistic traits show the same etiology in the general population and at the quantitative extremes (5%, 2.5%, and 1%). Arch. Gen. Psychiatry 68(11):1113–1121.
Robinson, E.B., Munir, K., et al. (2011b). Stability of autistic traits in the general population: further evidence for a continuum of impairment. J. Am. Acad. Child Adolesc. Psychiatry 50(4):376–384.
Rohde, L.A., Biederman, J., et al. (1999). ADHD in a school sample of Brazilian adolescents: a study of prevalence, comorbid conditions, and impairments. J. Am. Acad. Child Adolesc. Psychiatry 38(6):716–722.
Ronald, A., Happe, F., et al. (2006). Genetic heterogeneity between the three components of the autism spectrum: a twin study. J. Am. Acad. Child Adolesc. Psychiatry 45(6):691–699.
Ronald, A., and Hoekstra, R.A. (2011). Autism spectrum disorders and autistic traits: a decade of new twin studies. Am. J. Med. Genet. B Neuropsychiatr. Genet. 156B(3):255–274.
Ronald, A., Simonoff, E., et al. (2008). Evidence for overlapping genetic influences on autistic and ADHD behaviours in a community twin sample. J. Child Psychol. Psychiatry 49(5):535–542.
Rosenberg, R.E., Daniels, A.M., et al. (2009). Trends in autism spectrum disorder diagnoses: 1994–2007. J. Autism Dev. Disord. 39(8):1099–1111.
Russell, G., Steer, C., et al. (2010). Social and demographic factors that influence the diagnosis of autistic spectrum disorders. Soc. Psychiatry Psychiatr. Epidemiol. 46(12):1283–1293.
Rutter, M.L. (2011). Progress in understanding autism: 2007–2010. J. Autism Dev. Disord. 41(4):395–404.
Sachidanandam, R., Weissman, D., et al. (2001). A map of human genome sequence variation containing 1.42 million single nucleotide polymorphisms. Nature 409(6822):928–933.
Sanders, S.J., Ercan-Sencicek, A.G., et al. (2011). Multiple recurrent de novo CNVs, including duplications of the 7q11.23 Williams syndrome region, are strongly associated with autism. Neuron 70(5):863–885.
Sanders, S.J., Murtha, M.T., et al. (2012). De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature 485(7397):237–241.
Scott, F.J., Baron-Cohen, S., et al. (2002). The CAST (Childhood Asperger Syndrome Test): preliminary development of a UK screen for mainstream primary-school-age children. Autism 6(1):9–31.
Sebat, J., Levy, D.L., et al. (2009). Rare structural variants in schizophrenia: one disorder, multiple mutations; one mutation, multiple disorders. Trends Genet. 25(12):528–535.
Shattuck, P.T., Roux, A.M., et al. (2012). Services for adults with an autism spectrum disorder. Can. J. Psychiatry 57(5):284–291.
Shattuck, P.T., Seltzer, M.M., et al. (2007). Change in autism symptoms and maladaptive behaviors in adolescents and adults with an autism spectrum disorder. J. Autism Dev. Disord. 37(9):1735–1747.
Simonoff, E., Pickles, A., et al. (2008). Psychiatric disorders in children with autism spectrum disorders: prevalence, comorbidity, and associated factors in a population-derived sample. J. Am. Acad. Child Adolesc. Psychiatry 47(8):921–929.
Singer, H.S. (2005). Tourette’s syndrome: from behaviour to biology. Lancet Neurol. 4(3):149–159.
Skuse, D.H. (2007). Rethinking the nature of genetic vulnerability to autistic spectrum disorders. Trends Genet. 23(8):387–395.
Skuse, D.H. (2009). Is autism really a coherent syndrome in boys, or girls? Br. J. Psychol. 100(Pt 1):33–37.
Skuse, D.H., Mandy, W.P., et al. (2005). Measuring autistic traits: heritability, reliability and validity of the Social and Communication Disorders Checklist. Br. J. Psychiatry 187:568–572.
St Pourcain, B., Mandy, W.P., et al. (2011). Links between co-occurring social-communication and hyperactive-inattentive trait trajectories. J. Am. Acad. Child Adolesc. Psychiatry 50(9):892–902.
Starr, E., Szatmari, P., et al. (2003). Stability and change among high-functioning children with pervasive developmental disorders: a 2-year outcome study. J. Autism Dev. Disord. 33(1):15–22.
Still, G.F. (1902). Some abnormal psychical conditions in children: the Goulstonian lectures. Lancet (1):1008–1012.
Sukhodolsky, D.G., Scahill, L., et al. (2003). Disruptive behavior in children with Tourette’s syndrome: association with ADHD comorbidity, tic severity, and functional impairment. J. Am. Acad. Child Adolesc. Psychiatry 42(1):98–105.
Svensson, A.C., Lichtenstein, P., et al. (2007). Fertility of first-degree relatives of patients with schizophrenia: a three generation perspective. Schizophr. Res. 91(1–3):238–245.
Szatmari, P., Bryson, S., et al. (2009). Similar developmental trajectories in autism and Asperger syndrome: from early childhood to adolescence. J. Child Psychol. Psychiatry 50(12):1459–1467.
Szatmari, P., Bryson, S.E., et al. (2000). Two-year outcome of preschool children with autism or Asperger’s syndrome. Am. J. Psychiatry 157(12):1980–1987.
Szatmari, P. (2011). Is autism, at least in part, a disorder of fetal programming? Arch. Gen. Psychiatry 68(11):1091–1092.
Talkowski, M.E., Rosenfeld, J.A., et al. (2012). Sequencing chromosomal abnormalities reveals neurodevelopmental loci that confer risk across diagnostic boundaries. Cell 149(3):525–537.
Tarpey, P.S., Smith, R., et al. (2009). A systematic, large-scale resequencing screen of X-chromosome coding exons in mental retardation. Nat. Genet. 41(5):535–543.
Uher, R. (2009). The role of genetic variation in the causation of mental illness: an evolution-informed framework. Mol. Psychiatry 14(12):1072–1082.
Volkmar, F., Cook, E., Jr., et al. (1999). Summary of the practice parameters for the assessment and treatment of children, adolescents, and adults with autism and other pervasive developmental disorders. American Academy of Child and Adolescent Psychiatry. J. Am. Acad. Child Adolesc. Psychiatry 38(12):1611–1616.
Vorstman, J.A., Staal, W.G., et al. (2006). Identification of novel autism candidate regions through analysis of reported cytogenetic abnormalities associated with autism. Mol. Psychiatry 11(1):1, 18–28.
Weiss, L.A., Shen, Y., et al. (2008). Association between microdeletion and microduplication at 16p11.2 and autism. N. Engl. J. Med. 358(7):667–675.
Weschler, D., Golombok, J., et al. (1992). Manual for the Weschler Intelligence Scale for Children, 3rd Edition. Sidkup, UK: Psychological Corporation.
Williams, E., Thomas, K., et al. (2008). Prevalence and characteristics of autistic spectrum disorders in the ALSPAC cohort. Dev. Med. Child Neurol. 50(9):672–677.
Williams, N.M., Franke, B., et al. (2012). Genome-wide analysis of copy number variants in attention deficit hyperactivity disorder: the role of rare variants and duplications at 15q13.3. Am. J. Psychiatry 169(2):195–204.
Williams, N.M., Zaharieva, I., et al. (2010). Rare chromosomal deletions and duplications in attention-deficit hyperactivity disorder: a genome-wide analysis. Lancet 376(9750):1401–1408.
Wolraich, M.L., Hannah, J.N., et al. (1998). Examination of DSM-IV criteria for attention deficit/hyperactivity disorder in a county-wide sample. J. Dev. Behav. Pediatr. 19(3):162–168.