CONTENTS
6.1.1 Rationale for Long-Acting Injectables and Implants
6.1.1.2 Commercial Considerations
6.1.2 Selection of the Technology Platform and Type of Delivery
6.1.3.1 Compressed Steroid Pellets
6.1.3.5 Controlled-Release Era
6.2.1 Oil-Based Lipophilic Depots
6.2.2.2 Alzamer® Depot™ Technology
6.2.2.3 SABER® Injectable Depot
6.2.2.4 CLOUD® Injectable Depot
6.2.3 PEGylated Peptides/Proteins
6.2.4.1 Manufacture of PLGA Microspheres
6.2.4.2 PLGA Microsphere Commercial Products
6.2.6 Long-Acting Nanoparticles
6.3.1 Pellet Systems/Animal Health
6.3.2 Implants Based on a Rate-Controlling Membrane
6.3.4.1 ALZET® Implantable Osmotic Minipumps
6.3.4.2 DUROS® Implantable Osmotic System
6.3.5 Surface-Releasing Systems: Drug-Eluting Stents
6.5 Development Considerations for LAIs and Implants
6.5.2 Biology of the Treatment
6.5.3 Host Response/Biocompatibility
6.5.6 Sterilization/Aseptic Processing
Long-acting injectables (LAIs) and implants are designed to release a drug at a controlled rate for a specific duration and thereby achieve therapeutic drug concentrations for a prolonged period. This chapter describes a variety of LAIs and implant platforms that are currently commercially available, or at an advanced stage of development. This chapter is closely related to Chapter 5, which describes parenteral drug delivery from a targeting perspective, as opposed to the controlled-release perspective that is described here.
In a general chapter such as this, it is not possible to include all the LAIs and implants that have been researched and/or developed. The authors chose systems that have been commercialized or seemed highly relevant to the discussion in the chapter, but in the end, the choice was somewhat arbitrary. The authors acknowledge the many investigators in this area whose work was not possible to include due to space limitations.
6.1.1 RATIONALE FOR LONG-ACTING INJECTABLES AND IMPLANTS
There are both therapeutic and commercial benefits associated with the use of LAIs and implants, listed in Box 6.1 and described further here.
LAIs and implant drug delivery technology platforms offer significant benefits for the effective treatment of a broad range of medical conditions. These conditions include treatment for a variety of diseases (e.g., mental health, endocrinology, oncology, and pain management), infections, surgical procedures, local injuries, and prophylactic treatment regimens. The achievement of a safe, effective, and acceptable treatment requires expertise that spans multiple disciplines including formulation scientists, toxicologists, clinicians, and regulatory experts.
Some specific examples are discussed further here. LAIs and implants are beneficial for patients that have trouble maintaining daily oral or injection regimens or have preference for injection over oral dosage forms. For example, LAIs have been useful for patients with mental disease that experience frequent relapses while on oral medications. Schizophrenia can be a severe and chronic illness and can be costly to treat. Relapse and rehospitalization significantly add to the economic burden of this disease. Further emphasizing the rationale for LAIs are studies that provide evidence that LAIs are underutilized, despite strong evidence-based findings that they have a favored impact on treatment adherence, costs, quality, and reduced hospitalizations (Gopalakrisna et al. 2013).
BOX 6.1 RATIONALE FOR SUSTAINED-RELEASE INJECTABLES AND IMPLANTS
Reduction in fluctuation of drug concentrations
Reduction in the number of treatment administrations
Improved patient compliance
Potentially improved efficacy
Improved safety
Product differentiation
Life cycle management
Reduced development time
Ability to leverage safety data of approved active pharmaceutical ingredient
Potential for additional patent claims
Studies by Louza et al. evaluated patients that were on oral antipsychotic medication who had a history of poor treatment adherence. Switching to a long-acting risperidone injection demonstrated significant clinical improvements, reinforcing that the long-acting antipsychotic is a positive alternative in patients in whom compliance is an issue (Louza et al. 2011).
Poor patient compliance is often associated with treatment of opioid addiction. The issue of treatment adherence is complex and limits treatment effectiveness. Naltrexone is a μ-opioid antagonist and has been used for treatment for opioid dependence. An attractive option to overcome poor treatment compliance is the use of sustained-release technology (Kjome and Moeller 2011). A retrospective review of two types of naltrexone implants, oral naltrexone and historical controls, demonstrated differences between the immediate-release naltrexone versus the implants. The opioid-free rates combined for the implants were 82%, compared to 58% in patients in the oral naltrexone and 52% for the historical control groups (Reece 2007).
LAIs have the potential for improved efficacy. A Phase 3 study compared the efficacy of treatment for patients with chronic hepatitis C infection, who were administered interferon alpha-2a (IFN-a2a) subcutaneously (s.c.) 3 times a week, to a once-weekly PEGylated IFN-a2a for 12 weeks. At week 72, patients receiving PEGylated IFN-a2a demonstrated a 69% response rate with undetectable levels of hepatitis C viral RNA (<100 copies/mL). The response rate was 28% in patients receiving the non-PEGylated form. The treatment completion rate was 84% (223 of 267 patients) in the once-weekly treatment group, whereas 60% (161 of 264) of patients completed treatment in the three times per week group (Zeuzem et al. 2000).
LAIs have the potential to reduce drug side effects. For example, exenatide, the first glucagon-like peptide-1 receptor agonist (GLP-1RA), received U.S. market approval in 2005 for the treatment of type 2 diabetes, under the brand name Byetta®. The formulation is administered by s.c. injection, twice daily. A long-acting microsphere formulation of exenatide (Bydureon®) was subsequently developed and approved in 2012 as a once-weekly s.c. injection. A head-to-head clinical trial demonstrated that once-weekly Bydureon® showed significantly greater improvements in hemoglobin A1c reduction than Byetta® given twice daily (Drucker et al. 2008). Furthermore, nausea and vomiting was less frequent with the once-weekly formulation than with twice daily version (DeYoung et al. 2011).
6.1.1.2 Commercial Considerations
LAIs and implant drug delivery technology platforms also offer significant commercial benefits (Box 6.1). A multibillion-dollar industry has developed that is provided by the benefits from extended drug release technologies. The products include both traditional pharmaceutical drugs and biotech drugs. Market reports estimate that sales for LAIs exceeded $8 billion (EvaluatePharma® 2013 worldwide sales; see also Table 6.5, which lists PEGylated drugs, with estimates of their worldwide sales). The therapeutic areas include dental disease, diabetes, drug addiction, growth disorders, oncology, mental health, and pain management (Table 6.1).
Sales for implants are also significant. For example, Zoladex® (goserelin acetate), an implant for prostate cancer treatment, had reached sales of $996 million in 2013 (EvaluatePharma). Other implant treatments include birth control, cardiovascular disease, and cancer (Table 6.2).
The complexities and costs of discovering and bringing new drugs to approval and to the market are ever increasing. The regulatory and drug safety requirements are also becoming more stringent. Life cycle management has become an integral part of the pharmaceutical industry to ensure maximal value for approved drugs. Life cycle management includes providing strategies to improve efficiencies such as getting products to the market faster while reducing development costs. Included in the strategy for life cycle management is “upgrading” the existing product, such as daily oral or injectable, by developing a long-acting dosage form. Patent extensions can also be created through novel formulations as part of product life cycle management.
TABLE 6.1
Examples of Marketed Sustained-Release Injectable Products
Area |
Drug |
Trade Name |
Platform |
Dental disease |
Doxycycline hyclate |
Atridox® |
In situ forming depot |
Diabetes |
Exenatide |
Bydureon® |
Microspheres |
Drug addiction |
Naltrexone |
Vivitrol® |
Microspheres |
Growth disorders |
Octreotide |
Sandostatin® LAR Depot |
Microspheres |
Oncology |
Leuprolide acetate |
Eligard® |
In situ forming depot |
Mental health |
Paliperidone palmitate |
Invega® Sustenna® |
NanoCrystal® |
Pain management |
Bupivacaine |
Exparel® |
Liposome |
TABLE 6.2
Examples of Marketed Implants
Therapeutic Area |
Drug |
Trade Name |
Platform |
Birth control |
Levonorgestrel |
Norplant® system |
Silicone implant |
Cardiovascular |
Everolimus |
Xience Xpedition™ |
Drug-eluting stent |
Ocular |
Dexamethasone |
Ozurdex® |
PLGA implant |
Oncology |
Carmustine |
Gliadel® wafer |
Polyanhydride copolymer |
Oncology |
Goserelin |
Zoladex® |
PLGA implant |
6.1.2 SELECTION OF THE TECHNOLOGY PLATFORM AND TYPE OF DELIVERY
A variety of different LAIs and implant technology platforms are available to effect controlled release of an active pharmaceutical ingredient (API) (Table 6.3).
TABLE 6.3
Examples of Parenteral Sustained-Release Technology Platforms
Platform |
Technology |
Long-acting injectables |
Oil-based lipophilic depots |
In situ forming depots |
|
PEGylation |
|
PLGA microspheres |
|
Liposomes |
|
Polymeric nanoparticles |
|
Implants |
Pellets |
Implants based on an RCM |
|
Polymeric erodible compositions |
|
Osmotic pumps |
|
Drug-eluting stents |
Each platform has unique properties. There are a number of factors that influence the choice of the drug delivery platform. Much of platform selection depends on the product requirement needs. In some cases, more than one type of platform is capable of meeting product requirement needs. However, care and attention to the capability and limits of each technology is critical for selection. For example, drug delivery duration requirements can limit the type(s) of technology that would be suitable:
• Liposomes and micelles have been generally limited to providing circulating drug for up to a few days.
• In situ forming depots can be tailored to provide release rates from a few days to months.
• Microsphere compositions can provide sustained drug release from a week to several months.
• Implants can provide sustained release ranging from months to years.
Other factors affecting the selection of the appropriate platform include the API type (small molecule, peptide, protein), chemical compatibility of the API with the platform, drug solubility, therapeutic indication, patient population, cost-benefits of the platform, understanding the patient and physician needs, manufacturing complexity, and commercial manufacturing costs. An additional decision criterion can be the development stage of the platform, i.e., platforms utilized in marketed products with regulatory approval vs. earlier-stage technologies. The final selection should be based on the knowledge and experience of the development team and other decision makers.
LAIs and implants are delivered via the parenteral route. They can be used for systemic, local, and targeted delivery. The majority of marketed LAIs and implants release drug from the administration site, to be ultimately absorbed into systemic circulation. The administration of the LAIs or implants into the s.c. or intramuscular (i.m.) space avoids the first-pass metabolism effect. This is particularly desirable for drugs that have significant susceptibility to first-pass metabolism that results in poor bioavailability.
In cases where local drug concentrations are required and the anatomical site is accessible, LAIs and implants have the ability to maintain high local concentrations for prolonged periods. High local drug concentrations with reduced systemic drug levels can potentially reduce undesirable systemic toxicity. Examples include treatments for ocular diseases, release of chemotoxic agents to tumor sites, inhibition of cardiovascular restenosis, and management of local pain. Several products for localized delivery have been commercialized, such as a dexamethasone implant for macular edema (Ozurdex®), implants delivering carmustine for malignant gliomas (Gliadel® wafer), and drug-eluting stents (DES, multiple active agents).
In situations where access to the target tissue/organ is not available or not optimal, technologies that provide drug targeting, such as the incorporation of antibodies or receptors for target ligands, offer additional multifunctional advantages. In cases where the therapeutic agents can be highly toxic or potency is limited, targeted delivery offers improved specificity and efficacy. As an example, antibody drug conjugates (ADCs) can widen the therapeutic window by targeting to tumor-specific or overexpressed cell surface antigens. ADCs have been marketed for Hodgkin lymphoma and anaplastic large cell lymphoma (Adcetris™) and HER2-positive metastatic breast cancer (Kadcyla™). Targeting ligands are described further in Chapter 5 (Section 5.3.2).
The history of LAIs and implants constitutes an important part of the general history of the field of controlled release, and the information in this section complements that of Chapter 1.
6.1.3.1 Compressed Steroid Pellets
While sustained parenteral drug delivery began to emerge as a clearly defined subarea of pharmaceutics in the middle of the twentieth century, there was appreciation of the utility of extended delivery via the parenteral route as early as the 1800s. By the 1930s, it was recognized that implanted pellets containing hydrophobic compounds could provide sustained release (Deanesly and Parkes 1937). Examples included pellets containing estradiol for the treatment of prostate cancer and pellets containing testosterone for the treatment of hormone deficiency (Chien 1982). A significant improvement occurred with advances in polymer technology, specifically the development of polymeric materials that possessed significant biocompatibility. Further, the development of the field has been significantly influenced by advances in pharmacokinetics (PK) and pharmacodynamics, which served to highlight the need for controlled, extended drug delivery and sustained drug plasma/tissue levels in achieving desired therapeutic responses.
The Higuchi model (Higuchi 1963; refined by Paul and McSpadden 1976) provided insight into the release kinetics of drug dispersed in an ointment base or dispersed in a polymer matrix. The model predicts that the drug delivery rate will decline with the inverse square root of time. This model was later applied to other matrix-based delivery systems.
In the 1960s, while circulating rabbit blood inside an arteriovenous shunt of silicone rubber, Folkman discovered that if the tubing was exposed to anesthetic gases on the outside, the rabbits would fall asleep (Folkman et al. 1966). He proposed that short, sealed segments of such tubing containing a drug could form the basis of a constant-rate drug delivery system (DDS) and be implanted (Folkman and Long 1964). Further work in the 1960s and 1970s led to the establishment of the concept of the rate-controlling membrane (RCM)/drug reservoir delivery system as yielding a constant delivery rate and producing a zero-order, flat PK profile. A number of systems based on this underlying concept are described in Section 6.3.2.
Biodegradable polymers of poly(hydroxy acids) were investigated for use as sutures in the 1960s (Schmitt and Polistina 1967). This group of polymers encompasses poly(lactides) (PLA), poly(glycolides), and their copolymers, poly(lactide-co-glycolides) (PLGA). The relevant structures are given in Chapter 5 (Figure 5.11). PLGA systems degrade in vivo by hydrolysis of ester linkages into lactic acid and glycolic acid. The in vivo hydrolysis of PLGA polymers depends on the ratio of lactide to glycolide monomer units (Lactel 2014). The rate is fastest at approximately a 50:50 ratio and then decreases as either the proportion of glycolide or lactide unit increases (see, for example, Tables 6.4 and 6.6).
Investigation of PLGAs for drug delivery applications followed and has had a major impact on the field. Biodegradable polymers are attractive for drug delivery applications because of two potential major attributes: (1) if the polymer erodes only at the surface, then it would seem possible to design systems exhibiting a steady drug release rate, and (2) for parenteral applications, the system can be expected to completely erode, thereby eliminating the need for a procedure to remove the system at the end of the delivery lifetime. In the late 1960s, workers at DuPont developed microparticle and pellet depot delivery systems by mixing drugs with PLA (Boswell and Scribner 1973). In parallel, in the 1970s, researchers at Southern Research Institute and the University of Alabama at Birmingham were investigating steroid-containing PLGA microparticles for contraceptive drug delivery (Hoffman 2008). Currently, PLGAs are used for both LAIs (as in situ forming depots and microparticles) and bioerodible implants, as described in Sections 6.2 and 6.3.3, respectively.
TABLE 6.4
Eligard (Leuprolide Acetate) Drug and Polymer Composition
Dosing Interval |
API Dose (mg) |
Polymer DL-Lactide/Glycolide Molar Ratio |
Monthly |
7.5 |
50:50 |
3 months |
22.5 |
75:25 |
4 months |
30 |
75:25 |
6 months |
45 |
85:15 |
6.1.3.5 Controlled-Release Era
The history of drug delivery was dramatically changed when Alza Corporation was founded in 1968 by Alejandro Zaffaroni (Hoffman 2008). At the time, Alza Corporation was unique because its business model was not to create new drugs but to better enable drug therapy by delivering the drug at the right place and at the right time. The ability to control drug release provided multiple benefits including reduced side effects, improved compliance, and better efficacy. The combination of a drug and delivery system had a profound impact not only in medicine but in all the ancillary scientific disciplines that were involved as an integral part of each delivery platform. Alza developed controlled-release systems based on RCM designs, which found applications in a variety of different platform configurations. Ocusert® (pilocarpine) was the first system designed with an RCM and was approved in 1974. A second RCM system, Progestasert® (progesterone) was approved in 1976 as an intrauterine contraceptive (Barnhart 1985). A family of osmotic systems was developed by Dr. F. Theeuwes and colleagues at Alza in the 1970s (Theeuwes 1975, 1978). The ALZET®-implantable osmotic pump was developed for preclinical research (Theeuwes and Yum 1976), and DUROS®, an osmotically driven titanium implant, the size of a matchstick, was later developed for human use. Both technologies are described in detail in Section 6.3.4.
The OROS® (osmotic [controlled] release system) platform was also developed by Theeuwes and coworkers in the 1970s and 1980s. The OROS® is a solid oral tablet with a water-permeable membrane and laser-drilled hole (see also Chapter 1, Figure 1.7). As the tablet passes through the gastrointestinal tract, water is absorbed, creating osmotic pressure that pushes the active drug out. Approved products include Alpress® (prazosin), Acutrim® (phenylpropanolamine), Adalat® CR (nifedipine), Cardura® XL (doxazosin), Concerta® (methylphenidate HCl), Covera HS® (verapamil HCl), Ditropan XL® (oxybutynin chloride), DynaCirc CR® (isradipine), Efidac 24® (pseudoephedrine), Glucotrol XL® (glipizide), Invega® (paliperidone), Jurnista® (hydromorphone), Metoros® (metoprolol), Procardia XL® (nifedipine), Sudafed® 24 Hour (pseudoephedrine), and Volmax® (salbutamol). A number of other innovative controlled drug delivery technologies were also developed by Alza, including passive transdermal delivery, transdermal delivery by iontophoresis (E-Trans) (Phipps et al. 2002), and microneedles (Macroflux®) (Matriano et al. 2002; Cosman et al. 2010).
Several companies spun out from Alza, including Durect, Alexza, and Zosano Pharma. Durect Corporation was founded by Drs. F. Theeuwes and J. Brown and initially focused on applications of the DUROS® system. The ALZET® product line was subsequently acquired from Alza; other Durect Corporation drug delivery platforms include DURIN® (implant) (see Section 6.3.3), the sucrose acetate isobutyrate extended-release (SABER®) and CLOUD® injectable depot DDS (see Sections 6.2.2.3 and 6.2.2.4), and the nonparenteral DDS, ORADUR® (oral-controlled release with abuse resistance) and TRANSDUR® (transdermal drug delivery). Other major contributions to the field of controlled drug delivery with emphasis on LAIs and implants can be found in subsequent sections of this chapter.
6.2.1 OIL-BASED LIPOPHILIC DEPOTS
If a drug can be dissolved (or dispersed) in an oil, then an injectable extended-release dosage form can potentially be designed and developed. Oil-based (lipophilic) depots tend to exhibit maximum utility with hydrophobic small molecule drugs, as there is correlation between drug release and the partitioning (and solubility) of drug between the oil phase and the external aqueous phase (i.e., the in vitro release medium or tissue fluid) (Larsen and Larsen 2009; Larsen et al. 2012).
Oil-based depots have been developed for the extended release of steroids (e.g., testosterone, estradiol) and antipsychotic drugs (e.g., fluphenazine, haloperidol) (Chien 1982; Larsen and Larsen 2009). Oftentimes, a hydrophobic ester of a drug has achieved success, especially for steroids. Oil-based depots are generally administered i.m., although s.c. administration and local administration are also possible.
Oleaginous vehicles utilized for these depots have included castor oil, corn oil, cottonseed oil, medium-chain triglycerides, olive oil, peanut oil, sesame oil, and fractionated coconut oil. Vehicles have also been gelled by the addition of aluminum monostearate and benzoic acid derivatives to the vehicles. Duration of drug release from oil-based depots ranges from several days to 6 weeks or longer.
The partitioning of drug between the oil-based depot and the external aqueous phase is described in terms of oil/water partition coefficients or distribution coefficients (Larsen et al. 2012). The release of drugs from oil-based depots has been modeled by Hirano and coworkers (Hirano et al. 1981). The release was analyzed in terms of the oil/water partition coefficient and mass transfer (boundary layer) coefficients. An exponential relationship with time resulted and was compared to the in vivo release of several compounds.
Several recent oil-based depots include Faslodex® (fulvestrant) injection, indicated for treatment of metastatic breast cancer, and Nebido® (testosterone undecanoate) injection, indicated for treatment of male hypogonadism as indicated by testosterone deficiency. Both formulations use a castor oil–based depot.
In situ forming sustained-release formulations are liquids at ambient conditions that transform into a semisolid when injected into a target tissue site. These in situ forming systems have been used to deliver small molecules, peptides, and proteins. There are multiple formulation platforms that correspond to multiple mechanisms to achieve sustained drug release (Heller 2009; Wright et al. 2012). The compositions generally comprise a carrier and solvent(s) and are formulated as solutions or suspensions. This section provides examples of products that utilize several formulation platforms.
Dunn and coworkers, at the Southern Research Institute in Birmingham, Alabama, developed some of the first marketed in situ forming systems (Dunn 2003; Dadey 2008). The technology was licensed as the Atrigel system (Atrix Laboratories) and comprises PLGA that is dissolved in N-methyl-2-pyrrolidone (NMP); drug is added to form a solution or suspension.
There are a number of marketed products based on the Atrigel technology. Atridox® (doxycycline hyclate) provides site-specific delivery of the antibiotic doxycycline hyclate, for the treatment of chronic adult periodontitis. Upon injection into the periodontal pocket, the liquid product solidifies, allowing for controlled release of doxycycline for a period of 7 days. Atrisorb® GTR barrier does not contain a drug but allows for the provision of a custom-fitted gel barrier, which promotes regeneration of periodontal tissue.
The Atrigel technology platform is also used in Eligard® s.c. injection, which is a suspension of leuprolide acetate for the treatment of prostate cancer. By varying the ratio of DL-lactide to glycolide of the PLGA copolymer, four different controlled-release profiles are possible, allowing a dosing interval that can range from 1 to 6 months (Table 6.4).
6.2.2.2 Alzamer® Depot™ Technology
The Alzamer formulation comprises a drug mixed with a PLGA polymer, dissolved in a low-water-miscible solvent. On administration, the gradual loss of the solvent from the depot and the concurrent ingress of water or interstitial fluid change the physical form in situ, from a viscous gel to a finely porous spongelike structure. Initial drug release occurs through diffusion from the depot. The solvent systems selected have low water miscibility (e.g., benzyl benzoate), which slows down phase inversion and thus significantly reduces porosity of the depot. The reduced porosity slows down drug release, thereby minimizing burst effects. Further drug release is facilitated by PLGA degradation.
PLGA polymers are favored with lactide/glycolide ratios of 50:50. Drug loading ranges from 5% to 30%. By careful selection of the polymer, solvent, and drug characteristics, near zero-order delivery profiles have been demonstrated in vivo with acceptable local and systemic tolerability. Drug delivery durations can vary from days to months. The platform has been evaluated for small molecules, peptides, and proteins (Brodbeck et al. 1999; Chen and Junnarkar 2008).
6.2.2.3 SABER® Injectable Depot
The SABER® depot technology (Durect Corp.) consists of a biodegradable, high-viscosity, nonpolymeric liquid carrier material, formulated with one or more pharmaceutically acceptable solvents and other excipients. Sucrose acetate isobutyrate (SAIB) is a high viscosity agent that has been used as a food additive approved in over 40 countries (Reynolds and Chappel 1998). The use of SAIB as a pharmaceutical excipient is being explored in several investigative human drug products, and SAIB is a component in an FDA-approved veterinary injectable product for the controlled release of the peptide deslorelin (Sucromate™).
Solvents in the SABER® system may include ethanol, N-methyl-2-pyrrolidone (NMP), dimethyl sulfoxide (DMSO), benzyl alcohol, benzyl benzoate, and others. Upon injection, the water-miscible solvents (e.g., ethanol or DMSO) diffuse out of the depot, resulting in an increased in situ viscosity. Water-immiscible solvents (e.g., benzyl benzoate) diffuse at a slower rate: the selection of appropriate solvents thus allows flexibility in the control of the drug release rate. Additives such as biodegradable polymers (e.g., PLA and PLGA) further moderate the levels of drug release. SABER® formulations are compatible with small molecules, peptide, and proteins, for durations ranging from days to months (Okumu et al. 2002; Shah et al. 2014). The formulations can be solutions or suspensions and are used for both systemic and local delivery (Wright et al. 2008).
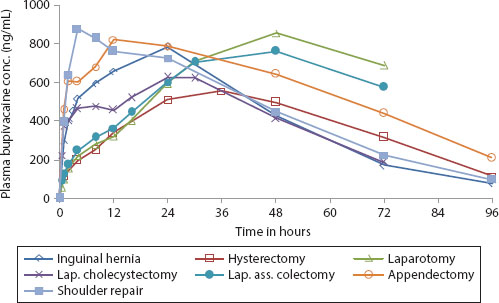
POSIMIRTM (Durect Corp.) is a SABER® formulation for the local administration of the anesthetic agent bupivacaine, for postoperative pain treatment. Administered to the surgical site, it is designed to provide 72 hours of local anesthesia/analgesia. POSIMIR™ comprises bupivacaine, SAIB, and benzyl alcohol. Clinical studies demonstrated 72 hours of sustained bupivacaine delivery, without initial drug burst, in a variety of surgical models (Figure 6.1) (Shah et al. 2014). SABER®-bupivacaine was administered to a variety of injection/instillation sites, in a dose-linear fashion. The absorption of bupivacaine was rapid, measurable within 0.5–1 hour, with a gradual increase in concentration, without initial burst. The differences in the PK profiles were attributable to the different surgeries evaluated and are thought to be associated with differences in local blood perfusion, type of tissue, and patient-to-patient variability. PK was dose linear with administration volumes ranging from 2.5 to 7.5 mL. The bioavailability of bupivacaine was 100%. The product is currently in Phase 3 clinical trials.
Relday™ (Zogenix 2013) is being developed as a once-monthly SABER® depot injection of risperidone, for the management of schizophrenia. Positive single-dose safety and PK profiles from a Phase 1 clinical trial of Relday have been reported. Therapeutic blood concentrations were achieved on the first day of dosing and were maintained throughout the 1-month period. Dose proportionality was demonstrated (Zogenix 2013).
Intra-articular injection of a recombinant human growth hormone SABER® formulation in dogs demonstrated sustained high local concentrations of the protein in the synovial joint for at least 14 days as a potential treatment for osteoarthritis or knee trauma (Sekar et al. 2009). The SABER® system has been evaluated for a variety of other compounds, including small molecules such as steroids and neoplastic agents, peptides, and proteins (Wright et al. 2008).
FIGURE 6.1 Pharmacokinetics of SABER®-bupivacaine across different surgery types. (Courtesy of Durect Corporation.)
6.2.2.4 CLOUD® Injectable Depot
Durect Corporation is also developing the CLOUD® injectable depot platform. This technology involves complexation of the API as an additional rate-controlling element. It is well suited for APIs with good aqueous solubility, confers additional API stability, provides a broader control over biodegradation characteristics, and involves low-formulation viscosities, which potentially allows for injection with finer needles.
6.2.3 PEGYLATED PEPTIDES/PROTEINS
PEGylation is the conjugation of hydrophilic polyethylene glycol (PEG) with the API, e.g., a protein, peptide, or small molecule. The goal of PEGylation is to modify the drug PK, i.e., extending the circulating half-life while maintaining the biological activity. The technology was pioneered in the 1950s and 1960s for protein modifications (Davis 2002). PEG is water soluble, nontoxic, and FDA approved. PEGylation increases the hydrodynamic volume, slows proteolysis, and reduces renal drug clearance. Drug solubility in aqueous and organic environments can also be increased with PEG, and toxicity and immunogenicity to the API is generally reduced (Veronese and Pasut 2005; Zhang et al. 2012).
PEG-adenosine deaminase (Adagen®) was the first approved product, developed by Enzon. PEG is covalently modified to the adenosine deaminase enzyme for the treatment of severe combined immunodeficiency disease. A variety of PEGylated therapeutic agents have now been commercialized, for a variety of treatments including immunodeficiency, cancer, inflammation, and ocular disease; they account for greater than $7 billion in sales (Table 6.5). The current PEGylated products are all large molecules. There are ongoing efforts for PEGylation of small molecules, and although no product has yet been commercialized, several late-stage clinical trials are underway (Riley and Riggs-Sauther 2008; Li et al. 2013; Nektar 2014). PEGylation technology can also be applied to the preparation of long-circulating liposomal and other nanoparticle DDS, as described in Sections 6.2.5 and 6.2.6.
This section provides some of the main considerations on the technology. PEG can be linear or branched and sizes have ranged from 5,000 to greater than 30,000 Da. PEGylation can be heterogeneous, i.e., multiple sites on the molecule can be PEGylated. PEGylation can be reversible or nonreversible. Peptides or proteins that are covalently bound with PEG are considered to be new chemical entities (NCEs).
TABLE 6.5
FDA-Approved PEGylated Drugs
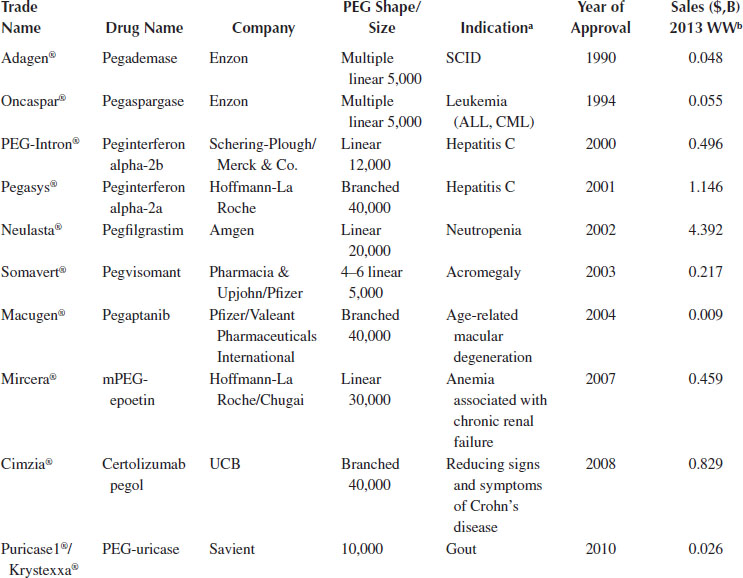
Source: Modified from Li, W. et al., Progr. Polym. Sci., 38, 421, 2013.
a ALL, acute lymphoblastic leukemia; CML, chronic myeloid leukemia; SCID, severe combined immunodeficiency disease.
b Worldwide sales.
PEGylation can be achieved through various platforms such as chemical modification, enzymatic methods, and genetic engineering. A critical factor of PEGylation technology is to maintain biological activity of the API. As a consequence, the type of PEGylation platform to use should be carefully considered in development. Although PEGylation can impact the potency of the molecule, this can be offset by the benefit of the increased circulating half-life. There are a number of good reviews on this topic (Veronese and Pasut 2005, 2012).
Chemical modification of the API with PEG, under mild conditions, is relatively well established and can produce high yields. However, the reactions are not highly specific and can lead to losses in bioactivity. The common reactive groups in polypeptides are nucleophiles (thiols, alpha-amino groups, epsilon-amino groups, carboxylate and hydroxylate groups) (Veronese and Pasut 2005). Nonspecific PEGylation runs the risk of a potential loss of biological activity. Site-specific PEGylation methods/technologies are thus emerging, in order to improve bioactivity. Site-specific conjugations avoid random PEGylation, resulting in a single species. The process can be optimized for both functional activity and half-life. Examples include a transglutaminase method and functionalization of maleimide PEG (Gong et al. 2011).
As outlined in Section 6.1.3.4, research into PLGA microspheres began in the late 1960s and 1970s. The development of biodegradable microspheres based on PLGA has been one of the major successful applications of LAIs and implants. The literature on microspheres is quite extensive and the reader is referred elsewhere for more detailed discussions (Wang and Burgess 2012; Gombotz and Hoffman 2013).
Microspheres can range in size from 1 to 999 μm and may have either a continuous polymeric matrix structure wherein the drug is homogeneously dispersed or a shell-like wall surrounding the drug reservoir/core. PLGA microspheres may have some inherent porosity, and porosity can develop during drug delivery. They are almost always suspended in an aqueous vehicle, prior to administration through a small gauge needle.
As noted earlier, PLGA systems degrade in vivo by hydrolysis into lactic acid and glycolic acid. While initial expectations were that PLGA microspheres would surface erode and therefore release drug at a nearly constant rate, for a number of systems, it was found that surface-available drug was released quickly in an initial burst. The drug would then not diffuse appreciably through the dense PLGA matrix (“lag time”) until the PLGA molecular weight declined to a critical value. At this point, the diffusivity in the microspheres increased substantially, coupled with the loss of physical integrity, resulting in a second, large period of drug release. PLGA degradation can be autocatalytic, as acid moieties accumulate during polymer hydrolysis. Drug release is considered to be a complex phenomena involving diffusion of the drug in a manner dependent on the degradation of the PLGA, polymer erosion, water sorption into the microspheres, diffusion of polymer degradation products, and (aqueous) pore formation in the microspheres. Mathematical modeling of drug delivery from PLGA microspheres has recently been reviewed (Versypt et al. 2013).
6.2.4.1 Manufacture of PLGA Microspheres
There are a number of processes for the manufacture of PLGA microspheres (Wang and Burgess 2012) including the following:
Solvent evaporation/extraction: In this process, the PLGA is dissolved in an organic solvent, and the drug is then dissolved or dispersed in the polymer/solvent solution. This mixture is emulsified in an aqueous solution (or other nonsolvent), and the solvent is removed by evaporation/extraction. The resultant microspheres may be harvested by filtration or centrifugation and further processed. Particle size of the microspheres can be controlled by processing parameters (Freitas et al. 2005).
Coacervation: In this process, drug particles are dispersed in a PLGA/solvent solution. Coacervation can be induced by several methods, usually by addition of a nonsolvent, resulting in deposition of the polymer around the drug particle.
Prolease process: In this process, the drug particles are dispersed into a PLGA solution, which is then atomized into a liquid nitrogen/ethanol bath. The droplets are transformed into microspheres, which can be further processed (Johnson and Herbert 2003).
The processes for microsphere manufacturing do not result in the encapsulation of all the drug, and for commercialization, attention must be paid to the overall process yield of encapsulated drug.
6.2.4.2 PLGA Microsphere Commercial Products
A number of PLGA-based LAI microsphere products have been developed and commercialized for applications ranging from prophylactic treatment of prostate cancer, to growth hormone deficiency, to alcohol and opioid dependency (Table 6.6). By modifying polymer composition, molecular weight, and ratio of lactide to glycolide monomers, a wide range of release profiles can be obtained, from weekly, to as long as 6 months (Table 6.6).
TABLE 6.6
Examples of Marketed Poly(Lactide-co-Glycolide) Microsphere Products
Product |
Company |
Drug |
Indication |
Duration |
Lupron Depot® |
Abbvie |
Leuprolide |
Prostate cancer |
1, 3, 4, and 6 months |
Trelstar® Depot |
Debiopharm/Actavis |
Triptorelin |
Prostate cancer |
4, 12, and 24 weeks |
Sandostatin® LAR |
Novartis |
Octreotide |
Acromegaly |
4 weeks |
Vivitrol® |
Alkermes |
Naltrexone |
Alcohol dependence and opioid dependence relapse |
1 month |
Risperdal® Consta® is a further commercial example. This PLGA microsphere formulation is a prolonged-release suspension for the i.m. delivery of risperidone, for the treatment of schizophrenia and bipolar disorder. This formulation exploits the PLGA degradation pattern of initial release of surface drug (first day), followed by a lag time of approximately 3 weeks, followed by a period of substantial release of drug (approximately 3 weeks). By timing injections at 2-week intervals and administering oral drug for the first several weeks of therapy, sustained plasma levels are achieved in patient populations (Ereshefsky and Mascarenas 2003).
As outlined in Section 6.1.1.1, exanatide was originally marketed as a twice daily s.c. injection. A long-acting microsphere formulation of exenatide was subsequently developed (Bydureon®), based on 50:50 PLGA. This PLGA microsphere formulation only requires a once-weekly s.c. injection. A further PLGA microsphere product, Nutropin® depot, delivered the protein human growth hormone (somatropin) for 2–4 weeks. It was approved by the FDA and marketed by Genentech, but distribution was ceased for commercial reasons.
PLGA microspheres are also used for targeted or local delivery of drugs to specific body sites, if the site is accessible for injection or other administration. One such example is in the dental area for the treatment of periodontitis. Arestin® microspheres (Orapharma) are PLGA microspheres that contain the antibiotic, minocycline HCl. The microspheres are for subgingival administration into periodontal pockets around affected teeth, as an adjunct with scaling and root planning.
As described in detail in Chapter 5 (Section 5.5.1), liposomes are colloidal suspensions consisting of vesicular bilayer structures, which are generally composed of phospholipids and cholesterol. Liposomes offer drug delivery and drug-targeting capabilities. Although discussed here as LAIs, it should be remembered that liposomes are generally limited to providing circulating drug for up to a few days. They are currently unable to provide therapeutic drug levels for a prolonged period (for example, a week or longer).
Their classification can be based on structure, method of preparation, composition, and application. They can be processed to different sizes, composition, charge, and lamellarity (Immordino et al. 2006; Xu and Burgess 2012). Because of their chemical properties, they can incorporate lipid- and water-soluble drugs, including small molecules, peptides, and proteins. In some cases, liposomes can improve drug solubility. Liposomes can also confer (1) protection from degrading enzymes, improving drug stability, (2) improved drug potency for therapy, (3) reduced toxicity, (4) drug-targeting possibilities, and (5) modified PK (e.g., long-acting delivery properties). However, additionally, liposomes can be disrupted by high shear (injection through very fine needles) and temperature excursions during storage. Application of liposomes includes oncology, infectious disease vaccine delivery/adjuvants, pain management, dermatology, and eye disorders. Marketed liposome–based products include treatments for fungal infections, vascular disease, cancer, and pain management. The clearance of liposomes is determined by a number of factors, including their physicochemical properties, membrane fusion events, rate of drug leakage, uptake of liposomes by the reticular endothelial system (RES; also called the mononuclear phagocyte system), mechanical filtration, and interactions with specific serum proteins (e.g., opsonins) (Ishida et al. 2002).
A challenge of liposomes injected intravenously (i.v.) is that they are phagocytosed by the RES and accumulate in the liver and spleen. Circulating liposomes interact with high- and low-density lipoproteins and also result in rapid release of the encapsulated drug into the blood. The addition of saturated phospholipids and cholesterol helps to overcome some of the limitations (Immordino et al. 2006). Further research on the technology led to a significant step in longer-circulating liposomes by the incorporation of PEG into the composition, resulting in sterically stabilized, PEGylated liposomes. PEG has been widely used as a steric stabilizer (Chiu et al. 2001) and, as described in Section 6.2.3 for protein PEGylation, confers other useful properties such as good biocompatibility, improved solubility, low toxicity, and low immunogenicity (Veronese and Pasut 2012; Zhang et al. 2012).
Woodle and Martin developed PEGylated liposomal doxorubicin (Doxil® in the United States, Caelyx® in Europe) (Hoffman 2008). Doxil® is the only approved Stealth (avoids detection by immune system) liposomal product for treatment of ovarian cancer, AIDS-related Kaposi’s sarcoma, and multiple myeloma. The product is administered by slow i.v. infusion. Stealth liposomes can passively target tumor sites by preferential accumulation into the tumor sites that have higher vascular permeability than normal tissue.
A local sustained-release liposome formulation delivering bupivacaine (Exparel®) was approved by the U.S. FDA in 2011 for local postsurgical anesthesia based on clinical studies in bunionectomy or hemorrhoidectomy. This liposomal formulation of bupivacaine is injected into soft tissue to produce local analgesia. The reported half-life of liposomal bupivacaine after a single dose was 23.8 hours in hemorrhoidectomy and 34.1 hours in bunionectomy clinical studies (Exparel 2012), as compared to 2.7 hours for bupivacaine HCl that is formulated in an isotonic aqueous buffer.
6.2.6 LONG-ACTING NANOPARTICLES
Nanoparticles, i.e., particulate drug carriers that have mean particle diameters below 1000 nm, are described in detail in Chapter 5 (Section 5.5.5). Such systems include those made from synthetic polymers such as PLGA (e.g., the Accurins™ technology; see Table 6.8) and poly(alkyl cyanoacrylates), as well as natural polymers such as albumin, gelatin, and chitosan. Similar to liposomal DDS, many are used in conjunction with PEG technology, in order to achieve extended circulation times, avoid RES capture, and facilitate uptake in sites exhibiting enhanced permeability, such as tumor sites. Although polymeric nanoparticles do provide extended circulation times in comparison with a free drug, the times are still limited to hours and days, rather than the longer time frames possible with microsphere-based systems. Thus, polymeric nanoparticles tend to be studied more for their ability to facilitate drug targeting to an active site, rather than to achieve extended release.
As described in Chapter 3 (Section 3.2), nanocrystals are drug crystals that have been size reduced to the nanometer range (Note: NanoCrystals® specifically denote a patented size-reduction technology of Elan Corporation). Dispersions of nanocrystals in liquid media are known as nanosuspensions; the drug crystals are stabilized by polymers and/or surfactants. Production of nanocrystals utilizes the basic principles of milling, precipitation, homogenization, or a combination(s) of these processes. This perceived simple process addresses a major issue for many drugs that are poorly soluble. Particle size reduction increases surface area for greater dissolution, increased drug solubility, and enhanced bioavailability. The particle characteristics are optimized based on target parameters including the desired PK profile and product shelf life. The first marketed products utilizing nanocrystal structures were oral dosage forms (Junghanns and Müller 2008). Parenteral application followed, including Invega® Sustenna® for schizophrenia in 2009. The product composition is a suspension of paliperidone palmitate NanoCrystals® that slowly dissolve after i.m. injection and then hydrolyze into the active moiety, paliperidone, followed by systemic absorption. Initial dosing is required on day 1 and a week later; maintenance doses are monthly (Kim et al. 2012).
Implantable systems can deliver either systemic or local therapy. The systems can be either nonerodible or bioerodible. While nonerodible systems generally require removal or explantation at the end of the delivery lifetime, this attribute is, conversely, a safety feature in that the system can be removed if the need arises to terminate therapy prior to the end of the delivery lifetime.
6.3.1 PELLET SYSTEMS/ANIMAL HEALTH
As outlined in Section 6.1.3, implants of steroid compounds were early sustained-release systems. There are a number of compressed pellet systems that have been utilized in animal health, mainly steroid based, including
• Ralgro® (zeranol)
• Synovex® (estradiol benzoate)
• Revalor® (estradiol-17beta + trenbolone acetate)
• Finaplix® (trenbolone acetate)
These implanted pellet systems have durations of 60–140 days (Ferguson 2001), due to the very slow dissolution and uptake of the compressed steroids.
An additional animal health product is the Compudose® implant that delivers estradiol over periods in excess of 200 days. The Compudose® implant comprises a cylindrical implant that consists of an outer layer of silicone rubber matrix, containing dispersed estradiol, overlaying an inner inert core of silicone rubber (Ferguson et al. 1988; Ferguson 2001). This design, featuring a thin layer of drug-containing matrix and a relatively thick drug-free core, minimizes the tailing in the drug release profile that is typically seen with matrix-release systems.
6.3.2 IMPLANTS BASED ON A RATE-CONTROLLING MEMBRANE
RCM implants consist of an inner core in which drug is dissolved or dispersed and an outer RCM layer, through which drug can diffuse (see also Chapter 1, Figure 1.3). As long as the drug concentrations at the inner and outer membrane surfaces are constant, a constant rate of drug release can be achieved. The first RCM products were insertable DDS including the Ocusert® (delivery of pilocarpine for the treatment of glaucoma from a system placed in the cul-de-sac of the eye) and the Progestasert® (delivery of progesterone to the uterus for contraception), both of which were developed by Alza Corporation (Hoffman 2008).
The Vitrasert® implant releases ganciclovir for the treatment of cytomegalovirus retinitis. The system is implanted intravitreally. The Vitrasert® consists of a compressed tablet of ganciclovir, overcoated with poly(vinyl alcohol) (PVA), further partially overcoated with poly(ethylene vinyl acetate) (PEVA) and then affixed to a PVA suture stub. The system is designed to release the drug over 5–8 months (Lee et al. 2008). An extensive array of other ocular implants is described in Chapter 13.
The Norplant system is a 5-year contraceptive system consisting of the hormone levonorgestrel encapsulated into six thin, flexible silicone capsules (Silastic tubing) for s.c. implantation on the inside of a woman’s upper arm. The silicone rubber copolymer serves as an RCM. The system has been approved in many countries and was approved by the U.S. FDA in 1990. Later in the United States, the 6-rod Norplant system became associated with removal problems and marketing in the United States ceased in 2002 (Kleiner and Wright 2013).
The Implanon® system consists of a single-rod system, 4 cm long and 2 mm wide, which makes for easier insertion and removal than the Norplant system. The system has a PEVA core containing 68 mg of etonogestrel (reservoir). The core is covered with a PEVA RCM and is designed to release drug to prevent ovulation for a 3-year period. The Implanon® system was first approved (ex-US) in 1998 and received U.S. approval in 2006 (Alam et al. 2008).
The advantage of using erodible implants is that a surgical procedure is not required to remove the implant at the end of the product lifetime. The PLGAs are widely used as bioerodible implants. The Zoladex® implant is indicated for the palliative treatment of advanced carcinoma of the prostate and breast cancer. The implant contains the peptide goserelin acetate, a potent synthetic peptide analog of gonadotropin-releasing hormone (GnRH), dispersed in a matrix of PLGA (Hutchinson and Furr 1985, 1990; Dijkman et al. 1990). The Zoladex® implant is in the form of a cylinder. The Zoladex® 3.6 mg implant is implanted s.c. with a 16 G needle and releases drug over a period of 28 days; the Zoladex® 10.8 mg implant is implanted s.c. with a 14 G needle and releases drug over a 3-month period. The encapsulated drug is released by a combination of diffusion- and erosion-controlled mechanisms.
DURIN® (Durect Corp.) is an implant that incorporates either biodegradable (PLGA polymers) or nonbiodegradable polymers. The implant either can be a drug-loaded monolithic design or can incorporate an RCM around a drug reservoir core. The RCM is created through a coextrusion manufacturing process, consisting of different polymer types between the inner core and outer RCM, of a uniform rod-shaped implant. In addition to the RCM, drug diffusion and release is controlled by drug content, polymer type and composition, and other excipients such as permeation enhancers and stabilizers (Gibson et al. 2012).
A biodegradable polyanhydride copolymer was developed for the Gliadel® wafer implant, for the treatment of brain tumors (Brem and Langer 1996). Gliadel wafers are small biodegradable polyanhydride disks, 1.45 cm in diameter and 1.0 mm thick, designed to deliver the chemotherapeutic drug, BCNU (carmustine), directly into the surgical cavity created after the tumor (high-grade malignant glioma) has been surgically excised. Up to eight wafers may be implanted along the walls and floor of the cavity that contained the tumor. This system was approved by the FDA in 1996. The biodegradable polyanhydride copolymer in the Gliadel® wafer is trade named Polifeprosan 20 and consists of poly[bis(p-carboxyphenoxy)propane/sebacic acid] in a 20:80 molar ratio. The chemical structure of Polifeprosan 20 is shown in Figure 6.2.
Upon exposure to the aqueous environment in the surgical cavity, the anhydride bonds hydrolyze to release carmustine and the polymer constituents carboxyphenoxypropane and sebacic acid. More than 70% of the copolymer biodegrades within 3 weeks back to constituent monomers. The Gliadel® wafer was investigated clinically by Brem and coworkers (Brem et al. 1991) and is associated with an approximately 2-month improvement in median survival in the treatment of newly diagnosed malignant glioma (Perry et al. 2007).
FIGURE 6.2 Polyanhydride random copolymer (Polifeprosan 20) chemical structure (ratio n:m = 20:80).
Osmosis is the movement of water through a semipermeable membrane in response to high solute (usually ionic) concentration on the opposing side of the membrane. If the influx of water displaces (pumps) an equal volume of drug solution or suspension from the delivery system, steady, zero-order delivery can be achieved. Osmotic drug delivery systems for oral, animal research, veterinary, and human delivery have been intensively investigated and developed (Eckenhoff et al. 1987; Zingerman et al. 1997; Wong et al. 2003; Wright et al. 2003).
The osmotic transport of water can be described by the following equation:
where
dV/dt is the volume flux of water across the membrane
Δπ and ΔP are, respectively, the osmotic and hydrostatic pressure differences across the semipermeable membrane
Lp is the membrane mechanical permeability coefficient
σ is the reflection coefficient (usually ≈1)
A and l are, respectively, the membrane area and thickness (Theeuwes and Yum 1976)
Equation 6.1 can be utilized to mathematically describe the delivery rate from an osmotic drug delivery system (with assumptions including σ = 1 and negligible ΔP):
where
dM/dt is the delivery rate of drug
c is the drug concentration
From Equation 6.2, it can be appreciated that if the osmotic pressure and the drug concentration are held constant, a constant rate of drug delivery will result. Osmotic pressure can be held constant by utilizing saturated solute solutions with excess solid solute. Some examples of osmotically driven implants are described below.
6.3.4.1 ALZET® Implantable Osmotic Minipumps
The ALZET® osmotic pump was developed for implantable drug delivery to laboratory animals (Theeuwes and Yum 1976; Culwell et al. 2012). It has become a popular research tool for continuous delivery of a wide range of test agents, including small molecules, peptides, proteins, nucleic acids, and lipids, and is cited in over 15,000 research publications. The ALZET® pump has a cylindrical shape and is composed of three concentric layers: the inner drug reservoir, the osmotic layer, and the outer RCM (Figure 6.3). The flexible, inner reservoir is molded from a synthetic elastomer. The osmotic layer contains sodium chloride (NaCl). The outermost, rate-controlling semipermeable membrane is formulated with a blend of cellulose esters. An additional component of the ALZET® system is the flow moderator, a 21-gauge stainless steel tube with a plastic end cap, which prevents random drug diffusion.
Operationally, the pump is filled with drug solution and the flow moderator is inserted through the delivery port. When placed in an aqueous medium (or implanted in an animal), water is transported across the semipermeable membrane into the osmotic layer. The osmotic layer expands, exerting pressure on the flexible reservoir and pumping the drug solution through the flow moderator and out via the delivery portal. Various ALZET® pump sizes have been developed, to provide researchers with a wide range of delivery rates to choose from (ALZET 2014).
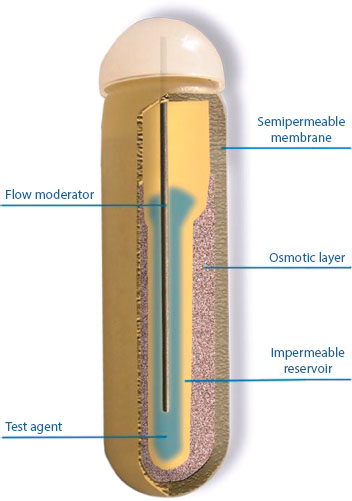
FIGURE 6.3 Cross section of the ALZET® osmotic pump. (Courtesy of Durect Corporation.)
The ALZET® pump can be implanted s.c. or intraperitoneally in unrestrained animals as small as mice and young rats; it has also been used in a variety of other animal species. The pump is particularly useful for continuous delivery of compounds with short half-lives, which are notoriously difficult to reliably deliver by conventional dosing regimens. The pump eliminates the need for repeated animal handling for dosing, thus minimizing unnecessary animal stress and experimental variables, which can interfere with study results. Local or targeted delivery can be achieved either by implantation directly at the desired site of action or via attachment to a catheter delivering to the desired site of action, such as blood vessels, the spinal cord, or into the cerebral ventricles and brain parenchyma. Another important capability of the ALZET® pump is the possibility of delivering solutions in an intermittent fashion, which is achieved by loading alternating series of drug and placebo solutions within the catheter (Culwell et al. 2012).
Some interesting examples of the use of the ALZET® pump in preclinical research include their use in the evaluation of the following:
• The impact of dosing schedules on the anticancer effects and pulmonary toxicity of bleomycin, in an animal model of lung cancer (Sikic et al. 1978).
• The in vivo efficacy of the angiogenesis inhibitor, anginex, to inhibit tumor growth in mouse xenograft models of ovarian carcinoma (Dings et al. 2003). The anti-obesity effects of continuous infusion of fibroblast growth factor 21 in dietinduced obese mice (Coskun et al. 2008).
• Intracerebroventricular (ICV) infusion of the tricyclic pyrone CP2 in a transgenic mouse model of Alzheimer’s disease. The 5X FAD mouse strain is known to manifest the Alzheimer’s phenotype with generous deposition of β-amyloid. A 2-week ICV infusion of CP2 with Alzet pumps reduced β-amyloid 1–42 in brain tissue by 40%–50% in comparison with the control (Hong et al. 2009).
• The role of the cerebellum in a rat model of noise-induced chronic tinnitus. Animals with established tinnitus had the paraflocculus of the cerebellum either surgically removed or inactivated by chronic lidocaine infusion into the subarcuate fossa using ALZET® pumps. Lidocaine treatment via ALZET® pumps gradually eliminated tinnitus over a 14-day infusion period, but these effects were reversed after a 6-week washout period (Bauer et al. 2013).
6.3.4.2 DUROS® Implantable Osmotic System
A schematic of the DUROS® implantable osmotic system is shown in Figure 6.4. The system consists of an outer cylindrical reservoir, an RCM at one end, the “osmotic engine” (tablets containing primarily NaCl, combined with other pharmaceutical excipients), an elastomeric piston, the drug formulation in the drug reservoir, and an exit port at the other end (see diffusion moderator/orifice in Figure 6.4). The outer reservoir can be constructed from titanium alloy or a biocompatible rigid polymer. The RCM is composed of a specially designed semipermeable polyurethane polymer. The drug formulation consists of the drug dissolved or suspended in pharmaceutically acceptable vehicles or solvents. The drug must remain stable in the formulation for the duration of delivery; accordingly, the formulation components are specific to the properties of the drug being delivered.
DUROS® dimensions can range from as small as several millimeters in outside diameter (OD) to 10 mm OD × 60 mm in length. Resulting volumes available for the drug formulation (drug reservoir volume) range from less than 100 μL to slightly more than 1 mL.
When implanted, water is osmotically imbibed from extracellular fluid into the osmotic engine at a rate controlled by the hydraulic permeability of the semipermeable membrane. The osmotic engine expands at a controlled rate, resulting in the displacement of the piston and delivery of the drug formulation at a controlled rate through the diffusion moderator and orifice, as described by Equation 6.2. Sufficient NaCl is usually present in the osmotic engine so that a saturated solution of NaCl is present in the osmotic engine compartment throughout the full delivery stroke of the piston.
The volumetric delivery rate from the DUROS® system depends on the membrane permeability, which, in turn, depends on the choice of the material used for the membrane or, in the case of a copolymer such as polyurethane, the copolymer composition. Additionally, the volumetric delivery rate of the pump can be adjusted by the membrane surface area and the membrane thickness. The resulting delivery rate is then fixed for a given pump design; different doses can be provided by different formulation concentrations.
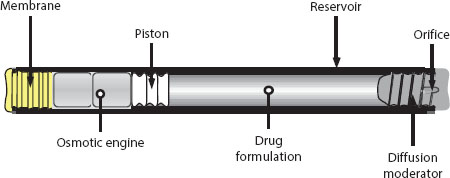
FIGURE 6.4 Schematic of DUROS® implantable osmotic system. (Reprinted from J. Control. Release, 75, Wright, J.C., Leonard, S.T., Stevenson, C.L. et al., An in vivo/in vitro comparison with a leuprolide osmotic implant for the treatment of prostate cancer, 1–10. Copyright 2001, with permission from Elsevier.)
DUROS® systems can be implanted s.c. in body sites where there is sufficient extracellular water and where there are no biomechanical limitations on implant placement or patient comfort restrictions. For systemic delivery, a preferred site is the s.c. space of the inside of the upper arm. Implantation is accomplished under local anesthesia; a small skin incision is made and a trocar, or a custom designed implanter can be used for implant insertion (Wright et al. 2003). For site-specific delivery, the DUROS® system can be implanted directly at the site, or the system can be implanted at an accessible location and a catheter tunneled to the desired site of action. Implant removal (explantation) is accomplished under local anesthesia. A small incision is made at one end of the implant site and in the thin capsule surrounding the implant, and the implant is pushed out.
The Viadur® implant was the first approved product, in March 2000, to incorporate the DUROS® implant technology. The implant contains the peptide drug leuprolide acetate and was developed for the palliative treatment of advanced prostate cancer (Wright et al. 2001). Suppression of circulating testosterone levels is the primary therapeutic approach to the management of advanced prostate cancer (Crawford et al. 1995). Sustained administration of leuprolide, a GnRH agonist, acts on the pituitary–testicular axis to produce an initial, transient increase in circulating testosterone levels, followed by long-term suppression to low concentrations.
The drug reservoir of the Viadur® implant contains a concentrated solution of leuprolide acetate dissolved in DMSO. The system was designed to deliver leuprolide for 1 year; the steady serum leuprolide levels observed during clinical trials are shown in Figure 6.5. In the clinical trials, the expected initial surge in testosterone levels was observed, followed by sustained suppression of testosterone levels (Fowler et al. 2000).
In vivo/in vitro delivery rate correlation was investigated for the Viadur® implant in rats, dogs, and humans. Based on analysis of explanted systems, there was a good correlation between the amount of drug delivered in vitro and the amount delivered in vivo (Wright et al. 2001).
The DUROS® technology is also being investigated for exenatide, a GLP-1RA for the treatment of type 2 diabetes. Twelve-month stability data of an exenatide DUROS® implant (Intarcia) at human body temperature have been reported (Yang et al. 2009). A Phase 2 study with a 3-month s.c. implant that provided steady delivery of exenatide has been completed (Dahms et al. 2011), and a Phase 3 study is in progress. The DUROS® implant delivery system has also been investigated for the s.c. delivery of interferon (Omega DUROS®), sufentanil (Chronogesic® system), and the site-targeted delivery of other opioids (Wright et al. 2003; Yang et al. 2008).
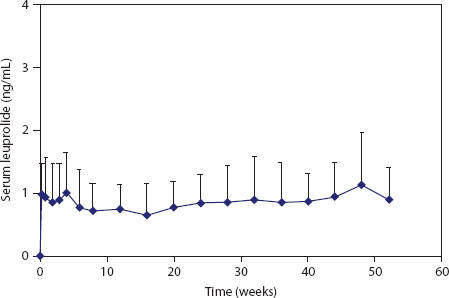
FIGURE 6.5 Serum leuprolide levels for patients treated with the Viadur® implant. (Reprinted from J. Control. Release, 75, Wright, J.C., Leonard, S.T., Stevenson, C.L. et al., An in vivo/in vitro comparison with a leuprolide osmotic implant for the treatment of prostate cancer, 1–10. Copyright 2001, with permission from Elsevier.)
TABLE 6.7
Examples of Drug-Eluting Stents
Trade Name |
Dmg |
Stent |
Polymer |
Cypher® stent |
Sirolimus |
Stainless steel |
PEVA and poly(butyl methacrylate) |
Taxus® stent |
Paclitaxel |
Stainless steel |
Copolymer of poly(styrene-isobutylene-styrene) |
Endeavor® stent |
Zotarolimus |
Cobalt/chromium alloy |
Copolymer of 2-methacryloyloxyethyl phosphorylcholine, lauryl methacrylate, hydroxypropyl methacrylate, and trimethoxysilylpropyl methacrylate |
Xience V® stent |
Everolimus |
Cobalt/chromium alloy |
Copolymer of vinylidene fluoride and hexafluoropropylene |
6.3.5 SURFACE-RELEASING SYSTEMS: DRUG-ELUTING STENTS
Coronary artery disease is a major health problem worldwide. Narrowing of arteries can be treated by balloon angioplasty followed (optionally) by placement of a stent, a bare metal wire device intended to prevent restenosis of the treated artery. However, restenosis is observed in a significant number of patients treated with bare metal stents (Zhao and Alquier 2012).
A major implantable drug delivery system application is the DES, which has revolutionized the treatment of vascular disease by reducing restenosis rates by 60%–75% across all patients when compared to bare-metal stents (Kukreja et al. 2009). DES are classified as combination drug-device products. A DES is normally a three-component system consisting of a scaffold (i.e., the stent that serves to keep the artery patent), a polymer to control the release of drug, and an antiproliferative drug, to inhibit neointimal growth.
A DES is generally designed as a matrix delivery system; drug particles are blended with the polymer and the mode of release from these coatings is diffusion controlled. A successful DES requires careful attention to all three components of the system. Drug release kinetics are governed by the choice of polymer, the specific drug incorporated in the system, and, as matrix systems, a number of other design variables. Drugs are released from DES designs over periods ranging from approximately 10 days to over 30 days (Zhao and Alquier 2012).
The first DES to obtain U.S. approval was the Cypher® stent in 2003 (note that Johnson & Johnson ceased manufacturing the Cypher stent in 2011). The Taxus® stent from Boston Scientific obtained U.S. approval in 2004. Other DES with U.S. approval include the Endeavor® stent (Medtronic) and the Xience V® stent (Abbott Vascular). Their properties are summarized in Table 6.7.
The commercial success of the DES has led to the involvement of other companies in the field and to the investigation of refined design options. Of special note is the concept of a fully bioresorbable DES, which would perform all the needed scaffolding and drug release functions and then be resorbed. Such a design would eliminate any potential for late-stage thrombosis events. A bioresorbable DES design has been tested in clinical trials (Kleiner and Wright 2013). The literature on DES is extensive; more detailed overviews can be found in Zhao and Alquier (2012) and in Kleiner and Wright (2013).
There are a number of emerging platforms that are being developed, summarized in Table 6.8. These technologies include drug encapsulation with targeting capabilities; the addition of a polymer of amino acids to therapeutic peptides and proteins; bioerodible polymer and solvent formulations; incorporation of nonnatural amino acids that allow site-specific chemical modification; and nanoparticle manufacturing based on high-resolution micromolding fabrication. Table 6.8 presents examples of emerging technologies; we have selected several examples of platform technologies and corresponding companies that have advanced their platforms into preclinical proof of concept, clinical testing, or near-term commercialization.
TABLE 6.8
Examples of Emerging Technologies
Platform Name |
Company |
Technology |
Reference |
Accurins™ |
BIND Therapeutics |
Drug-encapsulated targeted nanoparticles with a controlled-release polymer (PLA, PLGA), a stealth layer (PEG), and targeting ligands to bind to cell surface or tissue markers. |
Valencia et al. (2011) Hrkach et al. (2012) |
XTEN® |
Amunix |
Hydrophilic and unstructured polypeptide polymers using natural amino acids designed to mimic properties of PEG (e.g., to increase hydrodynamic volume). XTEN is added to proteins or peptides by genetic fusion or by chemical conjugation. |
Schellenberger et al. (2009) Podust et al. (2013) |
PASylation® |
XL-Protein |
Conformationally disordered polypeptide sequence consisting of proline, alanine, and/or serine (PAS) that are used for attachment to a therapeutic protein. |
Schlapschy et al. (2013) |
Medusa® |
Flamel Technologies |
Amphiphilic polymer comprising a poly-L-glutamate backbone grafted with alpha-tocopherol (vitamin E) molecules. The active agent binds via hydrophobic or electrostatic interactions within the polymer. |
Chan et al. (2007) |
Re-CODE™ |
Ambryx |
Enabling placement of a chemically reactive amino acid into a specific position in coding DNA sequence. The amino acids are nonnative and have selective linkable bioorthagonal chemical groups that allow site-specific modification of therapeutic peptides and proteins. |
Xie and Schultz (2006) |
Fluid Crystal™ |
Camurus |
Injectable depot is based on lipid liquid crystal (LLC) phase formation. Composition includes long-chain lipids such as soy phosphatidyl choline (SPC) and glycerol dioleate (GDO) and solvent (e.g., ethanol, propylene glycol) for viscosity control. |
Tiberg and Joabsson (2010) |
PRINT® |
Liquidia |
A continuous, roll-to-roll, high-resolution molding technique is being developed to fabricate and define micro- and nanoparticles. |
Mack et al. (2012) Perry et al. (2012) |
6.5 DEVELOPMENT CONSIDERATIONS FOR LAIs AND IMPLANTS
A number of factors that influence the development of LAI and implantable DDS are discussed here.
The development and establishment of a target product profile (TPP) is an important element that defines the drug product. The TPP should be based on the unmet medical need, which can be derived from several sources including clinical and scientific analysis, market research, and physician and patient surveys. The TPP should include product description, indication and usage, dosage and administration, dosage forms and strengths, contraindications, warnings and precautions, adverse reactions, drug interactions, use in specific populations, drug abuse and dependence, overdosage, clinical pharmacology, nonclinical toxicology, clinical studies, patient counseling, how supplied, and storage and handling (FDA Guidance TPP 2007).
For controlled-release dosage forms, the TPP should indicate the delivery platform (e.g., LAIs vs. implant, erodible vs. nonerodible implant). The selection of appropriate excipients or materials of construction is a key element to support the TPP requirements. Nonerodible components should exhibit biocompatibility consistent with the intended use. For biodegradable systems, biocompatibility is also very important; careful consideration should be given to the degradation and/or diffusion characteristics of each excipient in the LAI or implant. Disappearance of the depot or biodegradable implant in situ should be consistent with the duration of the drug release kinetics. The depot/implant constituents must be present to provide the functional attributes for sustained release and should disappear in a reasonable time thereafter. With regard to implants, setting specification limits on dimensions can drive specifications on drug loading dose, drug concentration, and formulation excipient composition.
TPPs may evolve as a program develops. Having a TPP helps to set goals and target specifications for formulation attributes for LAIs or implants. Additional benefits from a TPP include assuring that perspectives from all relevant expertise areas (e.g., medical, formulation, regulatory, and marketing) contribute to establish desired product performance characteristics. The TPP provides regulatory agencies the therapeutic indication and the performance attributes for the product (FDA Guidance TPP 2007).
It is important to include additional details on PK and efficacy target duration within the TPP. Drugs that have known therapeutic windows provide an advantage to define the drug release rate targets for controlled-release formulations. However, as with the case of NCEs, the desired therapeutic concentration is often not determined until clinical efficacy studies are performed. The development challenge is whether the delivery system will be able to meet target drug PK parameters.
The injection volume will vary from product to product and is influenced by the drug concentration and total dose in the formulation. The volume administered should be within the maximal allowed volume for s.c., i.m., or other administration routes. Drugs with higher potency generally require lower doses per treatment and allow flexibility in drug-loading dose in the formulation. Drugs with lower potency generally require higher doses to achieve efficacy. It is important to understand the range in loading capacities for the drug delivery platform technology. The TPP should also consider the target needle gauge size such that the formulation can be easily injected. The TPP should also include information on the target product shelf life and indicate storage condition requirements. Additional information on product shelf life is described in Section 6.5.7.
6.5.2 BIOLOGY OF THE TREATMENT
The physicochemical aspects of the formulation may be fairly developed. It is very important that the biology of the disease also be well understood. In addition to the biocompatibility considerations discussed later, the pharmacodynamics and desired temporal pattern of drug delivery should be matched to the underlying medical need. For example, treating a disease with a sustained-release GnRH agonist works well for treating prostate cancer when chronic suppression of testosterone levels is desired. In contrast, sustained release of parathyroid hormone would not be beneficial for osteoporosis treatment because daily pulsatile administration would be warranted (Potter et al. 2005). A perspective of the biology of chronic versus acute treatment and severity of the disease state should also be considered. These attributes can affect product risk/benefit analysis, product design, and the overall development program.
6.5.3 HOST RESPONSE/BIOCOMPATIBILITY
A marketed LAI or implant provides the desired PK and efficacy for a given therapeutic. A successful commercialization outcome may fail if the local tissue response (biological interface) is too severe and other key safety parameters are not met. Biocompatibility is an essential element in the success of LAIs and implants. Thus, a fundamental understanding of what reactions may occur, as well as the severity and the duration of the reaction, is important. Table 6.9 highlights some of the key assessments in addressing biocompatibility. Local site assessments are critically important for biocompatibility testing. The evaluations should include histological as well as clinical findings. Toxicological evaluation of systemic toxicity often focuses on the active agent. It is also important to be able to distinguish drug-related toxicities versus toxicities that may be contributed by the delivery system.
It is vital for scientists and product developers to recognize that host responses (foreign body reactions) to LAI and implants may be elicited and that these reactions can be acceptable transient responses. The perspective of the acceptability of biocompatibility of the LAI and implant varies and should include assessments such as treatment indication, disease severity, acute vs. chronic treatment, and patient population. It should be also noted that go/no-go decisions based on biocompatibility assessment can occur at all stages of development. As each delivery technology is unique, it is not possible to include all the details on this topic in this section, but the discussion is intended to highlight some of the key issues that investigators seek to understand and address for developing LAIs and implants.
The biological response resulting from a LAI or implant results in a series cascade of complex events. The host’s foreign body response includes recognition of the LAI or implant and induction of innate and specific immune inflammatory processes and tissue healing. Work by Anderson and Shive (1997) provides valuable information surrounding the events following microsphere implantation. The temporal change in the local tissue environment after implantation is characterized as a continuum of cellular responses. The formulation composition and residence time can strongly influence the magnitude of the response. The active agents (e.g., cytotoxic anti-inflammatory agents) as well as the concentrations that are incorporated into the delivery system can also influence the foreign body responses (Spilizewski et al. 1985; Ike et al. 1992). Over time, with biocompatible polymers, the fibrosis becomes residual, leaving minimal scarring and fibrotic tissue.
TABLE 6.9
Biocompatibility Assessment
Host Response |
Assessment |
Local tissue response |
Clinical visibility at administration site Redness, induration, swelling, pain Level of severity Duration of response (transient vs. prolonged) Tissue recovery Temporal relationship of the foreign body response vs. the erosion kinetics of depot or implant Potential effect on drug release kinetics Response to therapeutic agent and/or excipient(s) |
Systemic response (hypersensitivity, immunogenicity) |
Clinical safety Level of severity Duration of response Response to therapeutic agent and/or excipient(s) Immunologic mechanism(s) Effect on drug release kinetics Effect on efficacy |
It is important to evaluate biocompatibility preclinically. However, the findings and interpretations from animal studies should be tempered by the understanding that the preclinical models chosen for evaluation have limits on predicting the clinical outcome. A review by Engelhardt (2008) assessed the predictive capability of nonclinical studies that evaluated injection-site reactions from the administration of biopharmaceuticals to the clinical outcome. Based on the available literature and personal observations for some therapeutic agents, the interspecies correlation was poor. In some cases, profound inflammation was observed in animals, whereas relatively mild cutaneous responses were observed in humans. In other cases, minimal inflammation was found in animals, but significant inflammation was observed in humans. In the case of biologic agents, it is important to be aware whether the agent has biologic activity in the host species.
Evaluation of biocompatibility should include experimental designs that allow evaluation of injection site reactions that can discriminate the contribution(s) of the active agent versus delivery system excipients or a combination of multiple components. It is also important to determine the appropriate rodent and nonrodent species used in the conduct of biocompatibility studies. The assessments should include the presence or absence of a response, incidence, onset, duration, severity, and recovery. In some cases, the relationship between observations of a local response versus drug delivery performance (e.g., PK) may be warranted. Biocompatibility studies may include in vitro cytotoxicity studies (ISO 1992).
As part of the development of controlled-release delivery systems, the evaluation of immunogenicity may be relevant. This will depend on a number of factors including the type of therapeutic agent (e.g., small molecule vs. peptide vs. protein), formulation composition, route of administration, duration of treatment, stage of development, therapeutic indication, and patient population.
Immunogenicity reactions are antigen-specific immune responses. The activation of the immune response involves both innate and specific immune systems. The factors that influence the induction and elicitation of unwanted immunogenicity (humoral or cellular) to therapeutic compounds are complex. They include host/patient factors (e.g., genetics, age, sex, and disease state), the drug (e.g., protein structure, sequence homology to host, complexity, purity, and stability), route of administration, dose, dosing frequency, and formulation composition (Hermeling et al. 2004; Schellekens 2005). The FDA and European Medicines Agency have provided guidance on immunogenicity testing for large molecules (EMEA 2007; FDA 2013). The guidance focuses on the testing strategy and the analytical methods to assess anti-drug responses with an emphasis on therapeutic proteins. Because of the regulatory requirements of testing and monitoring for anti-drug responses, appropriate risk assessments should be conducted as part of development activities for sustained-release platforms.
With regard to LAIs and implants, developers should also determine the influence of sustained-release systems on immunogenicity. For example, patients treated with exenatide twice daily in an aqueous formulation (Byetta®) reported 38% incidence of low antibody titers and 6% had high titers. Patients receiving a once-weekly microsphere formulation of exenatide (Bydureon®) reported a 45% incidence of low antibody titers to exenatide and a 12% incidence in patients having high antibody titers. Although a higher incidence was observed with the microsphere-based long-acting formulation, the immunogenicity testing results were acceptable from a drug-approval perspective.
The value of preclinical immunogenicity testing to predict the human response can be questionable but may have merit in some cases. For example, immunogenicity testing in support of good laboratory practice toxicokinetic studies provides value in the interpretation of the PK data that are generated. Studies conducted by Alza Corporation provide a good example of the influence of excipients for a sustained-release dosage form (Alzamer®) on immunogenicity to a therapeutic protein in rats. Rat growth hormone (rGH) was chosen as the model protein so that immunogenicity was not due to sequence homology differences between the animal host and the protein. Antibody responses were not observed in animals receiving injections of rGH formulated in an aqueous bolus or in an Alzamer depot containing benzyl benzoate as the solvent. In contrast, antibodies to growth hormone were observed in rats injected with an rGH Alzamer depot with benzyl alcohol as a cosolvent (Chen and Junnarker 2008). Studies by Schellenberger conducted immunogenicity testing as a screening and selection mechanism to determine amino acid polymer constructs (called XTEN) that demonstrate minimal or no antibody responses in a mouse model. These constructs were then further developed (Schellenberger et al. 2009).
Initially, LAIs and implants are produced on the laboratory bench using research processes where the objective is to produce sufficient amounts of the LAI for preliminary testing and cost is not a primary consideration. It is often possible to use the laboratory bench process for initial development work. However, the laboratory bench process may not scale up easily, may be expensive, and may be difficult to validate.
Ideal processes for LAI’s involved unit operations that are standard for the pharmaceutical industry or the medical device industry, such as lyophilization, fluid mixing, dispensing of solutions into unit-dose containers, and molding and extrusion. Novel delivery systems can involve entirely new process steps that require extensive process development. The investment in process development may be justified based on the therapeutic need or market opportunity.
In the authors’ experience, some manufacturing problems with novel delivery systems do not become evident to the development team until processing at larger scale or until multiple batches have been made at commercial scale. This occurrence can be especially true for raw materials, where variations in the characteristics of the raw material may occur over several years of production by the outside supplier.
6.5.6 STERILIZATION/ASEPTIC PROCESSING
Implantable or injectable drug delivery systems must be sterile and nonpyrogenic and pass compendial tests for sterility and pyrogens/endotoxins (FDA 2012; USP 2013). While nonsterile products can be evaluated in rodents during development, the final commercial product must be able to withstand a sterilization process, or the product must be manufactured via an aseptic process. Common methods of sterilization include steam, dry heat, ethylene oxide, gamma irradiation, and electron beam sterilization. Regulatory agencies prefer terminal sterilization over aseptic processing because of the higher level of sterility assurance (10-6 vs. 10-3), and use of aseptic processing will need to be justified in the regulatory approval process (Yaman 2012).
For commercial products, a 2-year shelf life at the indicated storage temperature is generally desired from distribution and economic considerations. Storage temperatures include refrigerated, frozen, and controlled room temperature. It should be noted that actual storage conditions in other climatic zones (III and IV) may be different. Product stability is usually tested in the final container/closure system at a range of temperatures and humidities (including a temperature greater than the intended storage condition) so that the shelf life can be extrapolated to longer time periods. The impact of the method of sterilization on product degradation must be considered, as often initial stability experiments are performed on nonsterile product, and sterilization can result in accelerated degradation kinetics or new routes of degradation. Regulatory agencies generally request 6–12 months of stability data in the marketing application (FDA 1996, 2003).
Significant strides have been made in developing effective LAIs and implants that utilize a range of underlying delivery technologies. Therapeutic agents married with liposomes, microspheres, in situ forming depots, PEGylation, nanoparticle, and implant platforms have been successfully developed and commercialized. Clinical uses of products leveraging these technologies have improved effectiveness of therapies for the patient, doctor, and healthcare system.
The challenges to develop safe, biocompatible, therapeutic, and cost-effective delivery systems can be similar or unique with each platform. Continued development, refinement, and optimization will be required for each technology. While cost considerations may favor LAIs and implants that are nontar-geted, improvements in efficacy may justify the economics of targeted delivery systems. New product opportunities leveraging LAIs and implant technologies are expected to further improve drug efficacy and overall patient care, as well as to continue to benefit the healthcare system.
We thank Dr. Felix Theeuwes and Dr. WeiQi Lin for their helpful review and insights and Jose Gadea for the assistance with the ALZET® section.
Alam, S., J.B. Baldwin, K.A. Tombros et al. 2008. The single-rod contraceptive implant. In Clinical Proceedings® from Association of Reproductive Health Professionals, pp. 7–9.
ALZET 2014, ALZET® Osmotic Pumps–Implantable pumps for research, Accessed January 19, 2014. http://www.alzet.com.
Anderson, J.M. and M.S. Shive. 1997. Biodegradation and biocompatibility of PLA and PLGA microspheres. Adv. Drug Del. Rev. 28:5–24.
Barnhart, E. ed. 1985. Alza Corporation: Progestasert, pp. 590–602. Oradell, NJ: Medical Economics Company, Inc.
Bauer, C.A., K. Wisner, L.T. Sybert et al. 2013. The cerebellum as a novel tinnitus generator. Hear. Res. 295:130–139.
Boswell, G. and R. Scribner. 1973. Polylactide-drug mixtures. US Patent 3,773,919, Issued November 1973.
Brem, H. and R. Langer. 1996. Polymer-based drug delivery to the brain. Sci. Med. 3:52–56.
Brem, H., M.S. Mahaley Jr., N.A. Vick et al. 1991. Interstitial chemotherapy with drug polymer implants for the treatment of recurrent gliomas. J. Neurosurg. 74:441–446.
Brodbeck, K.J., S. Pushpala, and A.J. McHugh. 1999. Sustained release of human growth hormone from PLGA solution depots. Pharm. Res. 16(12):1825–1829.
Chan, Y.-P., R. Meyrueix, R. Kravtzoff et al. 2007. Review on Medusa®: A polymer-based sustained release technology for protein and peptide drugs. Expert Opin. Drug Deliv. 4:441–451.
Chen, G. and G. Junnarkar. 2008. ALZAMER® Depot™ Bioerodible technology. In Modified Release Drug Delivery Technology, 2nd ed., Vol. 2, M.J. Rathbone, J. Hadgraft, M.S. Roberts, and M.E. Lane (eds.), pp. 215–225. New York: Informa Healthcare Inc.
Chien, Y.W. 1982. Novel Drug Delivery Systems, pp. 247–280. New York: Marcel Dekker, Inc.
Chiu, G.N.C., M.B. Bally, and L.D. Mayer. 2001. Selective interactions with phosphatidylserine containing liposomes alter the steric stabilization properties of poly(ethylene glycol). Biochim. Biophys. Acta 1510:56–59.
Cho, H.S., T. Daniel, Y.J. Buechler et al. 2011. Optimized clinical performance of growth hormone with an expanded genetic code. Proc. Nat. Acad. Sci. 108:9060–9065.
Coskun, T., H.A. Bina, M.A. Schneider et al. 2008. Fibroblast growth factor 21 corrects obesity in mice. Endocrinology 149:6018–6027.
Cosman F., N.E. Lane, M.A. Bolognese et al. 2010. Effect of transdermal teriparatide administration on bone mineral density in postmenopausal women. J. Clin. Endocrinol. Metab. 95:151–158.
Crawford, E.D., E.P. DeAntonio, F. Labrie et al. 1995. Endocrine therapy of prostate cancer: Optimal form and appropriate timing. J. Clin. Endocrinol. Metab. 80:1062–1078.
Culwell, J.A., J.R. Gadea, C.E. Peer et al. 2012. Implantable drug delivery systems based on the principles of osmosis. In Long Acting Injections and Implants, J. Wright and D. Burgess (eds.), pp. 335–357. New York: Springer.
Dadey, E.J. 2008. The Atrigel® drug delivery system. In Modified-Release Drug Delivery Technology, 2nd ed., M.J. Rathbone, J. Hadgraft, M.S. Roberts, and M.J. Lane (eds.), pp. 183–189. New York: Informa Healthcare Inc.
Dahms, J., Y. Chandrasekher, R. Fielding et al. 2011. Pharmacokinetic and pharmacodynamic assessments with ITCA 650, continuous subcutaneous delivery of exenatide, in Type 2 diabetes. Presented at the European Association for the Study of Diabetes, September 14, 2011, Lisbon, Portugal.
Davis, F.F. 2002. The origin of pegnology. Adv. Drug Deliv. Rev. 54:457–458.
Deanesly, R. and A.S. Parkes. 1937. Biological properties of some new derivatives of testosterone. Biochem. J. 31:1161–1164.
DeYoung, M.B., L. MacConell, V. Sarin et al. 2011. Encapsulation of exenatide in poly-(D,L-lactide-co-glycolide) microspheres produced and investigational long-acting once-weekly formulation for type-2 diabetes. Diab. Technol. Therap. 13:1–10.
Di Cesare, S., U. Binder, T. Maier et al. 2013. High-yield production of PASylated human growth hormone using secretory E. coli technology. BioProcess Int. 11:30–38.
Dijkman, G.A, P.F. del Moral, J.W. Plasman et al. 1990. A new extra long acting depot preparation of the LHRH analogue Zoladex. First endocrinological and pharmacokinetic data in patients with advanced prostate cancer. J. Steroid Biochem. Mol. Biol. 37:933–936.
Dings, R.P.M., D.W.J. van der Schaft, B. Hargittai et al. 2003. Anti-tumor activity of the novel angiogenesis inhibitor anginex. Cancer Lett. 194:55–66.
Drucker, D.J., J.B. Buse, K. Taylor et al. 2008. Exenatide once weekly versus twice daily for the treatment of type 2 diabetes: A randomised, open-label, non-inferiority study. Lancet 372:1240–1250.
Dunn, R.L. 2003. The Atrigel drug delivery system. In Modified-Release Drug Delivery Technology, M.J. Rathbone, J. Hadgraft, and M.S. Roberts (eds.), pp. 647–655. New York: Marcel Dekker, Inc.
Eckenhoff, B., F. Theeuwes, and J. Urquhart. 1987. Osmotically actuated dosage forms for rate-controlled delivery. Pharm. Tech. 11:96–105.
EMEA Guidance. 2007. Guideline on immunogenicity assessment of biotechnology-derived therapeutic proteins. Accessed May 19, 2014. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003946.pdf.
Engelhardt, J.A. 2008. Predictivity of animal studies for human injection site reactions with parenteral drug products. Exp. Toxicol. Pathol. 60:323–327.
Ereshefsky, L. and C.A. Mascarenas. 2003. Comparison of the effects of different routes of antipsychotic administration on pharmacokinetics and pharmacodynamics. J. Clin. Psychiatry 64:18–23.
EvaluatePharma®. 2013. Evaluate Ltd. www.evaluate.com.
Exparel 2014. Package Insert. Pacira Pharmaceuticals Inc. San Diego, CA. 2012. Accessed June 2, 2014. http://www.exparel.com/pdf/EXPAREL_Prescribing_Information.pdf.
FDA. 1996. Guidance for industry—Q1C stability testing for new dosage forms. Accessed June 2, 2014. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm073374.pdf.
FDA. 2003. Guidance for industry Q1A(R2) stability testing of new drug substances. Accessed June 2, 2014. http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm073369.pdf.
FDA. 2007. Guidance for industry and review staff, target product profile—A strategic development process tool. Accessed June 2, 2014. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm080593.pdf.
FDA. 2012. Guidance for industry—Pyrogen and endotoxins testing: Questions and answers. Accessed January 26, 2014. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM310098.pdf.
FDA. 2013. Draft guidance for industry—Immunogenicity assessment for therapeutic protein products. Accessed May 19, 2014. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM338856.pdf.
Ferguson, T.H. 2001. Implants: Overview of technology. In AAPS Workshop, Assuring Quality and Performance of Sustained and Controlled Release Parenterals, April 19, 2001, Washington, DC.
Ferguson, T.H., G.F. Needham, and J.F. Wagner. 1988. Compudose®: An implant system for growth promotion and feed efficiency in cattle. J. Control. Release 8:45–54.
Folkman, J. and D.M. Long. 1964. The use of silicone rubber as a carrier for prolonged drug therapy. J. Surg. Res. 4:139–142.
Folkman, J., D.M. Long, and R. Rosenbaum. 1966. Silicone rubber: A new diffusion property useful for general anesthesia. Science 154:148–149.
Fowler, J.E., M. Flanagan, D.M. Gleason et al. 2000. Evaluation of an implant that delivers leuprolide for 1 year for the palliative treatment of prostate cancer. Urology 55:639–642.
Freitas, S., H.P. Merkle, and B. Gander. 2005. Microencapsulation by solvent extraction/evaporation: Reviewing the state of the art of microsphere preparation process technology. J. Control. Release 102:313–332.
Gibson, J.W., A.J. Tipton, R.J. Holl et al. 2012. Zero-order prolonged release coaxial implants. US 8,263,108 B2, Issued September 2012.
Gombotz, W.R. and A.S. Hoffman. 2013. Injected depot DDS. In Biomaterials Science, 3rd ed., B.B. Ratner, A.S. Hoffman, F.J. Schoen, and J.E. Leomons (eds.). Waltham, MA: Elsevier/Academic Press.
Gong, N., A.I. Ma, L.J. Zhang et al. 2011. Site-specific PEGylation of exenatide analogues markedly improved their glucoregulatory activity. Brit. J. Pharmacol. 163:399–412.
Gopalakrishna, G., A. Aggarwal, and J. Lauriello. 2013. Long-acting injectable Aripiprazole: How might it fit in our tool box? Clin. Schizophr. Relat. Psychoses 7:87–92.
Heller, J. 2009. Patient-friendly bioerodible drug delivery systems. J. Control. Release 133:88–89.
Hermeling, S., D.J.A. Crommelin, H. Schellekens et al. 2004. Structure–immunogenicity relationships of therapeutic proteins. Pharm. Res. 21:897–903.
Higuchi, T. 1963. Mechanism of sustained-action medication. J. Pharm. Sci. 52:1145–1149.
Hirano, K., T. Ichihashi, and H. Yamada. 1981. Studies on the absorption of practically water-insoluble drugs following injection. I. Intramuscular absorption from water-immiscible oil solutions in rats. Chem. Pharm. Bull. 29:519–531.
Hoffman, A.S. 2008. The origins and evolutions of controlled drug delivery systems. J. Control. Release 132:153–163.
Hong, H.-S., S. Rana, L. Barrigan et al. 2009. Inhibition of Alzheimer’s amyloid toxicity with a tricyclic pyrone molecule in vitro and in vivo. J. Neurochem. 108:1097–1108.
Hrkach, J., D. Von Hoff, M. M. Ali et al. 2012. Preclinical development and clinical translation of a PSMA-targeted Docetaxel nanoparticle with a differentiated pharmacological profile. Sci. Transl. Med. 4:1–11.
Hutchinson, F.G. and B.J. Furr. 1985. Biodegradable polymers for the sustained release of peptides. Biochem. Soc. Trans. 13:520–523.
Hutchinson, F.G. and B.J. Furr. 1990. Biodegradable polymer systems for the sustained release of polypeptides. J. Control. Release 13:279–294.
Ike, O., Y. Shimizu, R. Wasa et al. 1992. Controlled cisplatin delivery system using poly(DL-lactic acid). Biomaterials 13:230–234.
Immordino, M.L., F. Dosio, and L. Cattel. 2006. Stealth liposomes: Review of the basic science, rationale, and clinical application, existing and potential. Int. Natl. J. Nanomed. I:297–315.
Intarcia Therapeutics, Inc. 2014. ICTA 650. Accessed June 3, 2014. http://www.intarcia.com/products-technology/type2-diabetes.html.
Ishida, T., H. Harashima, and H. Kiwada. 2002. Liposome clearance. Biosci. Rep. 22:197–224.
ISO Document 10993. 1992. Biological compatibility of medical devices. Part 5. Tests for cytotoxicity: In vitro methods.
Johnson, O.L. and P. Herbert. 2003. Long-acting protein formulations—ProLease technology. In Modified-Release Drug Delivery Technology, M.J. Rathbone, J. Hadgraft, and M.S. Roberts (eds.), New York: Marcel Dekker, Inc., pp. 671–677.
Junghanns, J.-U.A.H. and R.H. Müller. 2008. Nanocrystal technology, drug delivery and clinical applications. Int. J. Nanomed. 3:295–309.
Kim, S., H. Solari, P.J. Weiden, and J.R. Bishop. 2012. Paliperidone palmitate injection for the acute and maintenance treatment of schizophrenia in adults. Patient Prefer. Adherence 6:533–545.
Kjome, K.L. and F.G. Moeller. 2011. Long-acting injectable naltrexone for the management of patients with opioid dependence. Subst. Abuse: Res. Treat. 5:1–9.
Kleiner, L.W. and J.C. Wright. 2013. Implants and Inserts. In Biomaterials Science: An Introduction to Materials in Medicine, 3rd ed., B.D. Ratner, A.S. Hoffman, F.J. Schoen, and J.E. Lemons (eds.), pp. 1062–1071. New York: Academic Press/Elsevier.
Kukreja, N., Y. Onuma, and P.W. Serruys. 2009. Future directions of drug-eluting stents. J. Interven. Cardiol. 22:96–105.
Lactel 2014. Lactel® Absorbable Polymers. Accessed January 20, 2014. http://www.absorbables.com/technical/biodegradation.html.
Larsen, S.W. and C. Larsen. 2009. Critical factors influencing the in vivo performance of long-acting lipophilic solutions—Impact on in vitro release method design. AAPS J. 11(4):762–770.
Larsen, S.W., A. Mette, M.A. Thing et al. 2012. Oily (lipophilic) solutions and suspensions. In Long Acting Injections and Implants, J. Wright and D. Burgess (eds.). New York: Springer.
Lee, S.S., P. Yuan, and M.R. Robinson. 2008. Ocular implants for drug delivery. In Encyclopedia of Biomaterials and Biomedical Engineering, 2nd ed., G.E. Wnek and G.L. Bowlin (eds.), pp. 1981–1995. New York: Informa Healthcare.
Li, W., P. Zhan, E. De Clerq et al. 2013. Current drug research on PEGylation with small molecular agents. Progr. Polym. Sci. 38:421–444.
Louza, M.R., H. Elkis, S. Ruschel et al. 2011. Long-acting injectable risperidone in partially adherent and nonadherent patients with schizophrenia. Neuropsychiatric Dis. Treat. 7:391–398.
Mack, P., K. Horvath, A. Garcia et al. 2012. Particle engineering for inhalation formulation and delivery of biotherapeutics. Accessed June 3, 2014. http://www.liquidia.com/Publications.html.
Matriano, J., M. Cormier, J. Johnson et al. 2002. Macroflux® microprojection array patch technology: A new and efficient approach for intracutaneous immunization. Pharm. Res. 19:63–70.
Nektar 2014. Accessed June 3, 2014. http://www.nektar.com/product_pipeline/all_phases.html.
Okumu, F.W., L.N. Dao, P.J. Fielder et al. 2002. Sustained delivery of human growth hormone from a novel gel system: SABER™. Biomaterials 23:4353–4358.
Paul, D.R. and S.K. McSpadden. 1976. Diffusional release of a solute from a Polymer Matrix. J. Membr. Sci. 1:33–48.
Perry, J., A. Chambers, K. Spithoff et al. 2007. Gliadel wafers in the treatment of malignant glioma: A systematic review. Curr. Oncol. 14:189–194.
Perry, J.L., K. Reuter, M.P. Kai et al. 2012. PEGylated PRINT Nanoparticles: The impact of PEG density on protein binding, macrophage association, biodistribution, and pharmacokinetics. Nano Letters 12:5304–5310.
Phipps, J.B., R. Padmanabhan, W. Young et al. 2002. E-TRANS technology. In Modified-Release Drug Delivery Technology, M.J. Rathbone, J. Hadgraft, and M.S. Roberts (eds.). New York: Marcel Dekker.
Podust, V., B.C. Sim, D. Kothari et al. 2013. Extension of in vivo half-life of biologically active peptides via chemical conjugation to XTEN protein polymer. Protein Eng. Des. Sel. 26:743–753.
Potter, L.K., L.D. Greller, C.R. Cho et al. 2005. Response to continuous and pulsatile PTH dosing: A mathematical model for parathyroid hormone receptor kinetics. Bone 37:159–169.
Reece, A.S. 2007. Psychosocial and treatment correlates of opiate free success in a clinical review of a naltrexone implant program. Subst. Abuse Treat. Prev. Policy 2:35–49.
Reynolds, R.C. and C.I. Chappel. 1998. Sucrose acetate isobutyrate (SAIB): Historic aspects of its use in beverages and a review of toxicity studies prior to 1988. Food Chem. Toxicol. 36:81–93.
Riley, T. and J. Riggs-Sauther. 2008. The benefits and challenges of PEGylating small molecules. Pharm. Technol. 32. Accessed June 3, 2014. https://www.nektar.com/pdf/PharmTechnologySmallMoleculePEG.July2008.pdf.
Schlapschy, M., U. Binder, C. Börger et al. 2013. PASylation: A biological alternative to PEGylation for extending the plasma half-life of pharmaceutically active proteins. Protein Eng. Des. Sel. 26:489–501.
Schmitt, E. and R. Polistina. 1967. Surgical sutures. US Patent 3,297,033, Issued 1967.
Schellekens, H. 2005. Factors influencing the immunogenicity of therapeutic proteins. Nephrol. Dial. Transplant. 20:vi3–vi9.
Schellenberger, V., C.W. Wang, N.C. Geething et al. 2009. A recombinant polypeptide extends the in vivo half-life of peptides and proteins in a tunable manner. Nat. Biotechnol. 27:1186–1190.
Sekar, M., F. Okumu, W. van Osdol et al. 2009. SABER™ formulation for intra-articular delivery of recombinant human growth hormone. In AAPS National Biotechnology Conference. Poster #NBC-09-00476.
Shah, J., D. Ellis, and N. Verity. 2014. Pharmacokinetic characteristics of SABER®-Bupivacaine in humans demonstrate sustained drug delivery for up to 72 hours in a variety of surgical models. In 39th Annual American Society of Regional Anesthetic and Pain Medicine Meeting, April 3–6. Accessed June 5, 2014. http://www.durect.com/pdf/101100947-03_PK_Poster_L1d.pdf.
Sikic, B.I., J.M. Collins, and E.G. Mimnaugh. 1978. Improved therapeutic index of bleomycin when administered by continuous infusion in mice. Cancer Treat. Rep. 62:2011–2017.
Spilizewski, L.K., R.E. Marchant, C.R. Hamlin et al. 1985. The effect of hydrocortisone acetate loaded poly(DL-lactide) films on the inflammatory response. J. Control. Release 2:197–203.
Theeuwes, F. 1975. The elementary osmotic pump. J. Pharm. Sci. 64:1987–1991.
Theeuwes, F. 1978. Osmotic system for the controlled and delivery of agent over time. US Patent 4,111,202, Issued September 5.
Theeuwes, F. and S.I. Yum. 1976. Principles of the design and operation of generic osmotic pumps for the delivery of semisolid or liquid drug formulations. Ann. Biomed. Eng. 4:343–353.
Tiberg, F. and Joabsson, F. 2010. Lipid liquid crystals for parenteral sustained-release applications: Combining ease of use and manufacturing with consistent drug release control. On Drug Delivery. Accessed June 5, 2014. http://www.ondrugdelivery.com/publications/Injectable%20Formulations%202010/Camurus.pdf.
United States Pharmacopeia 37/National Formulary 32. 2013. General requirements for tests and assays, <1> Injections. 1:33–38. Rockville, MD: United States Pharmacopeial Convention.
Valencia, P.M., M.H. Hanewich-Hollatz, W. Gao et al. 2011. Effects of ligands with different water solubilities on self-assembly and properties of targeted nanoparticles. Biomaterials 32: 6226–6233.
Veronese, F.M. and G. Pasut. 2005. PEGylation, successful approach to drug delivery. Drug Discov. Today 10:1451–1458.
Veronese, F.M. and G. Pasut. 2012. Protein PEGylation. In Long Acting Injections and Implants, J. Wright and D. Burgess (eds.). New York: Springer.
Versypt, A.N.F., D.W. Pack, and R.D. Braatz. 2013. Mathematical modeling of drug delivery from autocatalytically degradable PLGA microspheres—A review. J. Control. Release 165:29–37.
Wang, Y. and Burgess D.J. 2012. Microsphere technologies. In Long Acting Injections and Implants, J. Wright and D. Burgess (eds.). New York: Springer.
Wong, P.S.L., S.K. Gupta, and B.E. Stewart. 2003. Osmotically controlled tablets. In Modified-Release Drug Delivery Technology, 1st ed., M.J. Rathbone, J. Hadgraft, and M.S. Roberts (eds.). New York: Marcel Dekker Inc.
Wright, J.C., R.M. Johnson, and S.I. Yum. 2003. DUROS® osmotic pharmaceutical systems for parenteral and site-directed therapy. Drug Del. Tech. 3:3–11.
Wright, J.C., S.T. Leonard, C.L. Stevenson et al. 2001. An in vivo/in vitro comparison with a leuprolide osmotic implant for the treatment of prostate cancer. J. Control. Release 75:1–10.
Wright, J.C., M. Sekar, W. van Osdol et al. 2012. In Situ forming systems (Depots). In Long Acting Injections and Implants, J. Wright and D. Burgess (eds.). New York: Springer.
Wright, J.C., A.N. Verity, and F.W. Okumu. 2008. The SABER™ delivery system for parenteral administration. In Modified-Release Drug Delivery Technology, 2nd ed., M.J. Rathbone, J. Hadgraft, M.S. Roberts, and M.E. Lane (eds.), pp. 151–158. New York: Informa Healthcare.
Xie, J. and P.G. Schultz. 2006. A chemical toolkit for proteins—An expanded genetic code. Nat. Rev. Mol. Cell Biol. 7:775–782.
Xu, X. and D.J. Burgess. 2012. Liposomes as carriers for controlled drug delivery. In Long Acting Injections and Implants, J. Wright and D.J. Burgess (eds.), pp. 195–220. New York: Springer.
Yaman, A. 2012. Methods of sterilization for controlled release injectable and implantable preparations. In Long Acting Injections and Implants, J. Wright and D. Burgess (eds.), pp. 459–473. New York: Springer.
Yang, B., C. Negulescu, R. D’vaz et al. 2009. Stability of ITCA 650 for continuous subcutaneous delivery of exenatide at body temperature for 12 months. In Presentation at the Ninth Annual Diabetes Technology Meeting, San Francisco, CA. Accessed January 2013. http://www.intarcia.com/documents/11609DUROSPosterDiabetesTechMtg.pdf.
Yang, B., C. Rohloff, R. Mercer et al. 2008. Presentation of continuous delivery of stabilized proteins and peptides at consistent rates for at least 3 months from the DUROS® device. In American Association of Pharmaceutical Scientists Annual Meeting.
Zeuzem, S., S.V. Feinman, J. Rasenack et al. 2000. Peginterferon alfa-2a in patients with chronic hepatitis C. N. Engl. J. Med. 343:1666–1672.
Zhang, C., X.L. Yang, Y.H. Yuan et al. 2012. Site-specific PEGylation of therapeutic proteins via optimization of both accessible reactive amino acid residues and PEG derivatives. Biodrugs 26:209–215.
Zhao, J. and L. Alquier. 2012. Drug-eluting stents. In Long Acting Injections and Implants, J. Wright and D. Burgess (eds.). New York: Springer.
Zingerman, J.R., J.R. Cardinal, R.T. Chern et al. 1997. The in vitro and in vivo performance of an osmotically controlled delivery system-IVOMEC SR® bolus. J. Control. Release 47:1–11.
Zogenix 2013. Zogenix Reports Positive Top-Line Results From Extended ReldayTM Phase 1 Clinical Trial. Accessed May 19, 2014. http://ir.zogenix.com/phoenix.zhtml?c=220862&p=irol-newsArticle&ID=1814238&highlight.