The Phosphorus Cycle
Phosphorus is one of the most important elements on Earth. It participates in, influences, or controls many of the biogeochemical processes occurring in the biosphere. Feedbacks between the P and other global chemical cycles have been suggested to control many basic characteristics of the biosphere such as the oxygen content of the atmosphere. To understand the interaction between P and other biogeochemical processes and elemental distributions, it is necessary to understand the distribution of P on the Earth’s surface and the processes that control its distribution. The strategy of this chapter, therefore, is to (1) discuss the chemical forms in which P is present in the environment; (2) describe the processes that control its distribution in terrestrial, aquatic, and oceanic systems; and (3) define the major P reservoirs on the Earth’s surface and the rate at which P is exchanged between these reservoirs. Because all of these subjects must be addressed within this single chapter, the discussion is somewhat superficial and intended to expose the reader to the individual topics rather than to provide a thorough discussion of each. References in each section provide more detailed presentations of the individual topics.
14.1 Occurrence of Phosphorus
The global occurrence of P differs from that of the other major biogeochemical elements, C, N, S, O, and H in several very important aspects. First, while gaseous forms of P can be produced in the laboratory, none have ever been found in significant quantities in the natural environment. Thus, although some P is transported within the atmosphere on dust particles and dissolved in rain and cloud droplets, the atmosphere generally plays a minor role in the global P cycle. It should be noted, however, that at certain locations this small atmospheric source of P could be important. An example is the surface waters in the central gyres of the oceans where extremely low standing stocks of P are observed and the transport of P from other potential sources is very slow.
The second significant difference between P and the other major biogeochemical elements is that oxidation-reduction reactions play a very minor role in controlling the reactivity and distribution of P in the natural environment. While several oxidation states for P are chemically possible, these forms are generally restricted to controlled laboratory settings. In natural systems, therefore, P is almost exclusively present in the +V oxidation state where it is found as phosphate (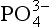 ), a tetrahedral oxy-anion. Nearly all dissolved and particulate forms of P are combined, complexed or slightly modified forms of this ion. In general, the biogeochemical cycle of P is synonymous with that of phosphate.
), a tetrahedral oxy-anion. Nearly all dissolved and particulate forms of P are combined, complexed or slightly modified forms of this ion. In general, the biogeochemical cycle of P is synonymous with that of phosphate.
Finally, P also differs from other elements in that it is overwhelmingly dominated by a single, stable isotopic form containing 15 protons and 16 neutrons. There are only two naturally occurring radioactive forms of P: 32P and 33P, which are produced in the atmosphere by nuclear reactions with argon. A small amount of 32P is also contributed by 32Si decay. Because these isotopes have extremely short half-lives (32P half-life, 14.3 days; 33P half-life, 25.3 days), their activities in the environment are always very low and account for a minute portion of the total P in the Earth. Nevertheless, modern analytical capabilities have made the study of these isotopes possible, providing new insights in aquatic biogenic processes (Waser et al., 1996; Lai and Lee, 1988; Benitez-Nelson and Buessler, 1999).
14.1.1 Dissolved Inorganic Forms of Phosphorus
Phosphate,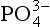 , is the fully dissociated anion of triprotic phosphoric acid,
, is the fully dissociated anion of triprotic phosphoric acid,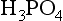 :
:
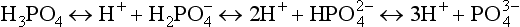
The dissociation constants for these equilibria for freshwater and seawater are listed in Table 14-1. The proportion of the individual protonated species in distilled water and seawater over the pH range of 2 to 10 is shown inFig. 14-1. At the pH of freshwater systems (very roughly 6–7),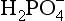 is the dominant phosphate species. The high ionic strength of seawater and the presence of cations such as Ca2+, Mg2+, and Na+, which form ion pairs with the
is the dominant phosphate species. The high ionic strength of seawater and the presence of cations such as Ca2+, Mg2+, and Na+, which form ion pairs with the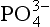 species, significantly alter the dissociation of phosphoric acid. In seawater at a pH of 8,
species, significantly alter the dissociation of phosphoric acid. In seawater at a pH of 8,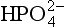 dominates. The importance of ion pairs on the
dominates. The importance of ion pairs on the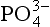 activity in seawater is further demonstrated inFig. 14-2. It is clear that ion pairs with dissolved cations play a central role in controlling the aqueous
activity in seawater is further demonstrated inFig. 14-2. It is clear that ion pairs with dissolved cations play a central role in controlling the aqueous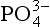 speciation and that free
speciation and that free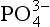 ions constitute a very small proportion of the total present (0.01% at standard seawater conditions). The chemical reactivity of
ions constitute a very small proportion of the total present (0.01% at standard seawater conditions). The chemical reactivity of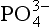 in aqueous systems is therefore highly dependent on the composition and pH of the solution. Acid-base and complexation reactions are not only important in seawater systems, but also influence the reactivity of
in aqueous systems is therefore highly dependent on the composition and pH of the solution. Acid-base and complexation reactions are not only important in seawater systems, but also influence the reactivity of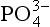 in groundwater and freshwater systems.
in groundwater and freshwater systems.
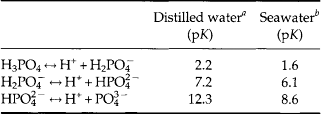
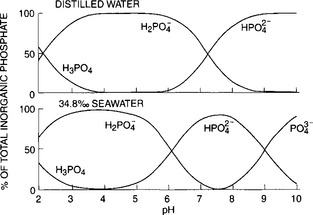
Fig. 14-1 Extent of dissolution of phosphoric acid species as a function of pH in distilled and sea waters (Atlas, 1975).
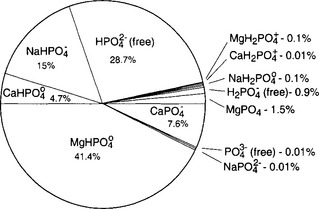
Fig. 14-2 Calculated speciation of in seawater of 34.8 parts per thousand salinity at 20¯C and a pH of 8.0 (Atlas, 1975).
in seawater of 34.8 parts per thousand salinity at 20¯C and a pH of 8.0 (Atlas, 1975).
Another important class of inorganic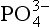 compounds are the condensed or polyphosphates. In these compounds, two or more phosphate groups bond together via P–O–P bonds to form chains or in some cases cyclic compounds. Although polyphosphates generally account for only a small portion of the total P present in natural waters, they are extremely reactive compounds and are routinely used in industrial and commercial applications. Condensed phosphates form soluble complexes with many metal cations and are used, therefore, as water softeners.
compounds are the condensed or polyphosphates. In these compounds, two or more phosphate groups bond together via P–O–P bonds to form chains or in some cases cyclic compounds. Although polyphosphates generally account for only a small portion of the total P present in natural waters, they are extremely reactive compounds and are routinely used in industrial and commercial applications. Condensed phosphates form soluble complexes with many metal cations and are used, therefore, as water softeners.
14.1.2 Particulate Inorganic Forms of Phosphorus
Phosphorus is the tenth most abundant element on Earth with an average crustal abundance of 0.1% and may be found in a wide variety of mineral phases. There are approximately 300 naturally occurring minerals in which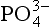 is a required structural component. Phosphate may also be present as a trace component in many minerals either by the substitution of small quantities of
is a required structural component. Phosphate may also be present as a trace component in many minerals either by the substitution of small quantities of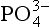 into the crystal structure or by the adsorption of
into the crystal structure or by the adsorption of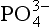 onto the mineral surface (Nriagu and Moore, 1984; Slansky, 1986).
onto the mineral surface (Nriagu and Moore, 1984; Slansky, 1986).
By far the most abundant phosphate mineral is apatite, which accounts for more than 95% of all P in the Earth’s crust. The basic composition of apatite is listed in Table 14-2. Apatite exhibits a hexagonal crystal structure with long open channels parallel to the c-axis. In its pure form, F−, OH−, or Cl− occupies sites along this axis to form fluorapatite, hydroxyapatite, or chlorapatite, respectively. However, because of the “open” nature of the apatite crystal lattice, many minor substitutions are possible and “pure” forms of apatite as depicted by the general formula in Table 14-2 are rarely found. Of the possible substituting ions,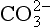 ion is by far the most important followed by Na+,
ion is by far the most important followed by Na+,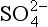 and Mg2+. The most common form of natural apatite in sedimentary rocks is francolite, a substituted form of carbonate fluorapatite deposited in marine systems. The substitution of
and Mg2+. The most common form of natural apatite in sedimentary rocks is francolite, a substituted form of carbonate fluorapatite deposited in marine systems. The substitution of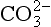 ions into the mineral lattice has a substantial effect on apatite solubility (Jahnke, 1984). More studies are required, however, before the effects of all substituting ions are understood and an accurate assessment of the solubility of complex, natural apatites can be made.
ions into the mineral lattice has a substantial effect on apatite solubility (Jahnke, 1984). More studies are required, however, before the effects of all substituting ions are understood and an accurate assessment of the solubility of complex, natural apatites can be made.
Table 14-2
The chemical formula of apatites
General formula: Ca10(PO4)6X2
X = F− Fluorapatite
OH− Hydroxyapatite
Cl− Chlorapatite
Possible substitutes for Ca2+:
Na+, K+, Ag+, Sr2+, Mn2+, Mg2+, Zn2+, Cd2+, Ba2+, Sc3+, Y3+, rare earth elements, Bi3+, U4+
Possible substitutes for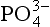 :
:
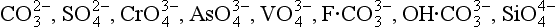
The importance of this mineral is perhaps best demonstrated by the diversity of its sources. Assuming that nearly all of the P present is in the form of apatite, igneous rocks contain between 0.02% and 1.2% apatite. Apatite is also produced by organisms (including man) as structural body parts such as teeth, bones, and scales. After an organism dies, these components tend to accumulate in sediments and soils. In some locations, these constituents are reworked and concentrated by physical processes to form economically important deposits. By far the largest accumulations of P on the Earth’s surface are massive sedimentary apatite deposits (phosphorites). The mining of these deposits provides 82% of the total world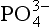 production and 95% of the total remaining reserves (Howard, 1979). Phosphate rock may also form by the accumulation of bird or bat droppings (guano) and subsequent diagenetic alteration and crystallization. The mining of guano can be important locally, although this constitutes a negligible fraction of the world’s phosphate rock production.
production and 95% of the total remaining reserves (Howard, 1979). Phosphate rock may also form by the accumulation of bird or bat droppings (guano) and subsequent diagenetic alteration and crystallization. The mining of guano can be important locally, although this constitutes a negligible fraction of the world’s phosphate rock production.
In general, the major phosphorite deposits are of marine origin and occur as sedimentary beds ranging from a few centimeters to tens of meters in thickness. There is still debate concerning the exact mechanism or mechanisms by which phosphorites form (Sheldon, 1981). The relative roles of direct inorganic precipitation, solid-phase replacement of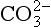 by
by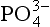 in biogenic calcium carbonates, biologically mediated precipitation and concentration of dispersed apatite grains through physical reworking processes may vary considerably among different deposits (Froelich et al., 1988). Ultimately, however, the
in biogenic calcium carbonates, biologically mediated precipitation and concentration of dispersed apatite grains through physical reworking processes may vary considerably among different deposits (Froelich et al., 1988). Ultimately, however, the 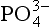 that is incorporated into the authigenic apatite is thought to be supplied primarily via the decomposition of organic materials at the sea floor. A variety of additional processes, such as cycling at redox boundaries or incorporation by microbial communities, may act to elevate pore water
that is incorporated into the authigenic apatite is thought to be supplied primarily via the decomposition of organic materials at the sea floor. A variety of additional processes, such as cycling at redox boundaries or incorporation by microbial communities, may act to elevate pore water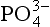 concentrations, promoting precipitation (Schuffert et al., 1994; 1998; Follmi, 1996). Because of the ultimate connection to organic matter input, modern phosphorite formation tends to be located along continental margins which exhibit strong coastal upwelling and high biological primary production (Follmi, 1996). While these deposits are most obvious, finely dispersed apatite appears to be forming at many continental margin locations (Ruttenberg and Berner, 1993).
concentrations, promoting precipitation (Schuffert et al., 1994; 1998; Follmi, 1996). Because of the ultimate connection to organic matter input, modern phosphorite formation tends to be located along continental margins which exhibit strong coastal upwelling and high biological primary production (Follmi, 1996). While these deposits are most obvious, finely dispersed apatite appears to be forming at many continental margin locations (Ruttenberg and Berner, 1993).
The occurrence of other phosphate minerals is also important in certain locations. In sediments, the formation of vivianite, Fe3(PO4)2·H2O, and struvite, NH4MgPO4·6H2O, have been reported (Emerson and Widmer, 1978; Murray et al., 1978). Minerals that may form as secondary weathering products in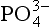 deposits include crandallite, CaAl3(PO4)2(OH)5·8H2O, brushite, CaHPO4·2H2O, whitlockite, Ca3(PO4)2, vivianite, and others. Nriagu (1976) has described with thermodynamic arguments the weathering of
deposits include crandallite, CaAl3(PO4)2(OH)5·8H2O, brushite, CaHPO4·2H2O, whitlockite, Ca3(PO4)2, vivianite, and others. Nriagu (1976) has described with thermodynamic arguments the weathering of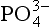 minerals in terrestrial systems. His calculations suggest that in neutral to acidic soils, calcareous
minerals in terrestrial systems. His calculations suggest that in neutral to acidic soils, calcareous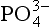 minerals may be progressively weathered to forms richer in aluminum, passing first through crandallite, to eventually wavellite, Al3(OH)3(PO4)2·5H2O.
minerals may be progressively weathered to forms richer in aluminum, passing first through crandallite, to eventually wavellite, Al3(OH)3(PO4)2·5H2O.
Phosphate is also ubiquitous as a minor component within the crystal lattices of other minerals or adsorbed onto the surface of particles such as clays, calcium carbonate, or ferric oxyhydroxides (Ruttenberg, 1992). Therefore, in general, transport of these other particulate phases represents an important transport pathway of P as well.
14.1.3 Organic Forms of Phosphorus
Many of the most fundamental biochemicals required for life contain P generally linked to long, complicated organic molecules by phosphate ester bonds. Among the impressive list of compounds in which phosphate is a necessary constituent are the nucleic acids, DNA and RNA, discussed in Chapter 3. In these compounds (Fig. 14-3a), phosphates covalently link the mononucleotide units forming long polymers which, depending on the composition of the attached base, encode all necessary genetic information.
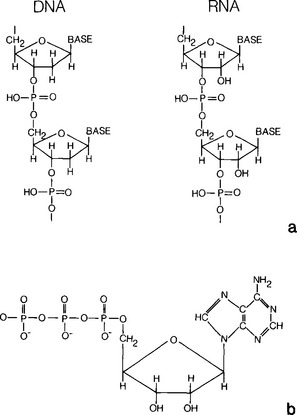
Phosphate also plays a central role in the transmission and control of chemical energy within the cells primarily via the hydrolysis of the terminal phosphate ester bond of the adenosine triphosphate (ATP) molecule(Fig. 14-3b). In addition, phosphate is a necessary constituent of phospholipids, which are important components in cell membranes, and as mentioned before, of apatite, which forms structural body parts such as teeth and bones. It is not surprising, therefore, that the cycling of P is closely linked with biological processes. This connection is, in fact, inseparable as organisms cannot exist without P, and their existence controls, to a large extent, the natural distribution of P.
Because these P-containing compounds are abundant in all organisms, P is one of the major components of all organisms. In general, marine microorganisms contain 105 to 125 carbon atoms for every P atom (Redfield et al., 1963; Peng and Broecker, 1987; Anderson and Sarmiento, 1994). Because of the increased abundance of structural parts not involving phosphorus, the average C:P ratio of benthic macroalgae and sea grasses and terrestrial plants is much higher, approximately 550:1 and 800:1, respectively (Atkinson and Smith, 1983; Deevey, 1970).
Dissolved organic compounds that contain P also constitute an important pool of reactive P, particularly in the euphotic zone where it may exceed dissolved inorganic phosphate concentrations (Karl and Yanagi, 1997; Smith et al., 1986). The chemical form of the dissolved organic P is not well known but is reported to be dominated by phosphate esters and phosphonates and differs significantly from fresh organic materials, suggesting considerable remineralization and transformation (Clark et al., 1998; Nanny and Minear, 1997). The dissolved organic P pool appears to be readily available to organisms (Bjorkman and Karl, 1994) and may provide a “buffer,” providing utilizable phosphate to phytoplankton between episodic inputs from other sources (Jackson and Williams, 1985).
14.2 Sub-Global Phosphorus Transfers
A global representation of the P cycle, by necessity, will be general. It will combine a wide variety of P-containing components into relatively few reservoirs and will parameterize intricate processes and feedback mechanisms into simple first-order transfers. To appreciate the rationale behind the construction of such a model and to understand its limitations, the transfers of P within a hypothetical terrestrial ecosystem and in a generalized ocean system will be discussed first.
14.2.1 Freshwater Terrestrial Ecosystems
The dominant processes controlling the movements of P through terrestrial ecosystems are schematically presented inFig. 14-4. In a general way, the overall movement of P on the continents may be envisioned as the constant erosion of P from continental rocks and transport in both dissolved and particulate form by rivers to the ocean, stopping occasionally along this pathway to interact with biological and mineralogical systems.
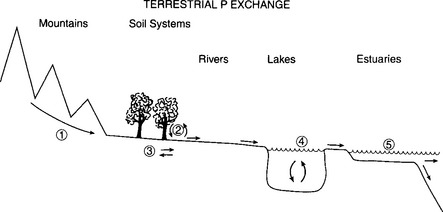
Fig. 14-4 Schematic representation of the transport of P through the terrestrial system. The dominant processes indicated are: (1) mechanical and chemical weathering of rocks, (2) incorporation of P into terrestrial biomass and its return to the soil system through decomposition, (3) exchange reactions between soil interstitial waters and soil particles, (4) cycling in freshwater lakes, and (5) transport through the estuaries to the oceans of both particulate and dissolved P.
Physical and chemical erosion of continental rocks (1) introduces particulate and dissolved P to the soil system. Approximately 5% of the mobilized P is present in dissolved form and is readily available to enter biological systems (2) and to react with soil particles (3). The majority (approximately 95%) of the P in rivers remains in particulate form, with approximately 40% in organic phases and the remainder either trapped in the mineral lattices of the particulate matter or sorbed onto particle surfaces (such as FeOOH coating mineral grains or as colloids). Much of this P will be transported with the suspended material or bedload downstream until it eventually reaches the estuaries and the oceans, never having entered the biological cycles. However, even a small release from the particulate phase would significantly add to the reactive dissolved pool. Release from particles appears to occur in estuaries in response to increasing salinity. Developing a quantitative understanding of this release remains an important research topic (Kaul and Froelich, 1984; Conley et al., 1995; Chambers et al., 1995).
As reactive P is transported through the terrestrial system, it is assimilated into plants and subsequently into the rest of the biosphere (2). Although many elements are required for plant life, in many ecosystems P is the least available and, therefore, limits overall primary production (Schindler, 1977; Smith et al., 1986). Thus, in many instances the availability of P influences or controls the cycling of other bioactive elements. When organisms die, the organic P compounds decompose and the P is released back into the soil-water system. This cycle of uptake and release may be repeated numerous times as P makes its way to the oceans.
Inorganic reactions in the soil interstitial waters also influence dissolved P concentrations. These reactions include the dissolution or precipitation of P-containing minerals or the adsorption and desorption of P onto and from mineral surfaces. As discussed above, the inorganic reactivity of phosphate is strongly dependent on pH. In alkaline systems, apatite solubility should limit groundwater phosphate whereas in acidic soils, aluminum phosphates should dominate. Adsorption of phosphate onto mineral surfaces, such as iron or aluminum oxyhydroxides and clays, is favored by low solution pH and may influence soil interstitial water concentrations. Phosphorus will be exchanged between organic materials, soil interstitial waters, and mineral phases many times on its way toward the ocean.
Lakes (4) also constitute an important component of the terrestrial P system. Because much of mankind’s activities occur on or adjacent to lakes and because P availability has such a major influence on the biological community, P cycling in lakes has been extensively studied. The hypothetical P and temperature profiles for a temperate lake in summer are displayed inFig. 14-5. Briefly, in summer, warming of the surface layers produces strong stratification which restricts exchange between the lighter, warm surface water and the colder, denser deep water. During photosynthesis, the dissolved P in the photic zone is incorporated into plants and is eventually transported below the thermocline on sinking particles. Most of this P is released back to the lake’s deep water during the degradation of the organic matter but due to the stratification, this P is transported back to the surface photic zone very slowly. The constant stripping of P from the surface layers by production and subsequent sinking of particles results in extremely low levels of P in the surface waters which often limit overall biological productivity.
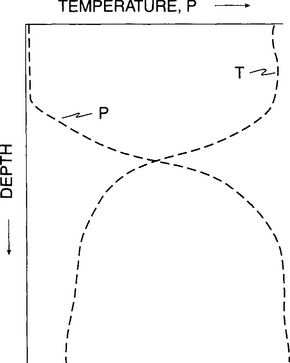
Fig. 14-5 Typical distribution of P and temperature in a temperate lake in summer. Thermal stratification restricts exchange between surface and deep waters. Phosphorus is depleted in the surface waters by the sinking of biologically produced particles.
In this context, it is easy to envision the potential influence of increased P input to the surface layers via anthropogenic sources. If P is continually added to the photic zone, productivity will not be limited but will simply continue unchecked. This results in large amounts of organic particles sinking below the thermocline. As this material decomposes, it consumes oxygen. Since exchange with the surface layer is restricted, the deep waters will become depleted in oxygen (cf. Lehman, 1988). If enough organic materials sink into the deep waters, the oxygen will be thoroughly consumed via decomposition. In extreme conditions, this can result in the formation of anoxic deep waters and fish kills.
As cooling occurs in the late fall and early winter, the thermal stratification breaks down, permitting mixing of the deep and surface layers. This allows the surface layers to be replenished with P. During the winter months, biological productivity in a temperate lake is limited by the availability of light rather than nutrients.
In the hypothetical terrestrial system depicted inFig. 14-4, the P eroded from the land is eventually transported to the estuaries. As in lakes, soils, and rivers, many chemical and biological processes act to control the transport of P within and from the estuary (Lucotte and d’Anglejan, 1988; Jonge and Villerius, 1989). Dissolved P may be removed from solution onto the particulate phase and deposited in the sediments. On the other hand, the change in the solution composition may cause P to be released from the particulate load. The P that is transported from the estuaries to the ocean bonded to particles will rapidly settle to the sea floor and be incorporated into the sediments. The reactive forms of P, primarily dissolved but including particulate forms that can be easily solubilized, will enter the surface ocean and participate in the biological cycles. Determining what proportion of the transported P is reactive is a critical step in the elucidation of the marine P budget (Froelich et al., 1982).
14.2.2 The Oceanic System
Over much of the ocean (exclusive of upwelling regions, high-latitude areas and specific high-nutrient, low-chlorophyll regions) the vertical distribution of dissolved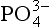 is represented by the shape of the profile displayed inFig. 14-6, which is similar to the shape observed for the temperate lake in summer. Also included in the figure are the major processes responsible for controlling this shape. In general, dissolved P is nearly undetectable in the euphotic zone (generally the upper 20–100 m) and increases to maximum concentrations of 1–3 µM at approximately 1000 m. The distribution can best be envisioned as the balance between the incorporation of P into organisms with the eventual sinking of some fraction of this P from the surface waters and the constant slow rate of return of P to the surface layer by physical processes. The majority of ocean deep water is formed in the North Atlantic and slowly spreads sequentially to the South Atlantic, Indian, and finally to the Pacific Oceans. Because of the continuous rain of particulate P into the deep waters from the surface layers, the deepwater
is represented by the shape of the profile displayed inFig. 14-6, which is similar to the shape observed for the temperate lake in summer. Also included in the figure are the major processes responsible for controlling this shape. In general, dissolved P is nearly undetectable in the euphotic zone (generally the upper 20–100 m) and increases to maximum concentrations of 1–3 µM at approximately 1000 m. The distribution can best be envisioned as the balance between the incorporation of P into organisms with the eventual sinking of some fraction of this P from the surface waters and the constant slow rate of return of P to the surface layer by physical processes. The majority of ocean deep water is formed in the North Atlantic and slowly spreads sequentially to the South Atlantic, Indian, and finally to the Pacific Oceans. Because of the continuous rain of particulate P into the deep waters from the surface layers, the deepwater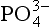 concentration increases progressively from the North Atlantic to the Pacific.
concentration increases progressively from the North Atlantic to the Pacific.
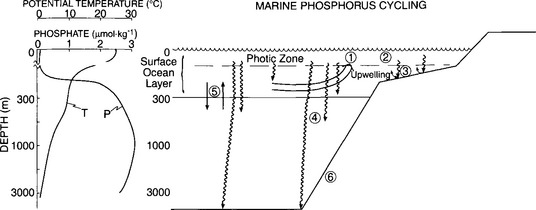
Fig. 14-6 Profiles of potential temperature and phosphate at 21¯ 29’N, 122¯ 15’W in the Pacific Ocean and a schematic representation of the oceanic processes controlling the P distribution. The dominant processes shown are: (1) upwelling of nutrient-rich waters, (2) biological productivity and the sinking of biogenic particles, (3) regeneration of P by the decomposition of organic matter within the water column and surface sediments, (4) decomposition of particles below the main thermocline, (5) slow exchange between surface and deep waters, and (6) incorporation of P into the bottom sediments.
Unlike the temperate lake, stratification in the ocean does not completely break down in the winter. Except for specific high-latitude regions of the ocean, processes other than deep convective overturn are responsible for returning the P stored in the deep waters to the surface. A relatively slow exchange occurs between water layers everywhere in the ocean and this supplies some P to the surface ocean(process 5 in Fig. 14-6) from the intermediate layers below. More important sources of P to the photic zone are the major upwelling regions generally located adjacent to the western, sub-tropical continental margins (1) and in equatorial divergence zones. In the western margins, the prevailing winds tend to transport surface water offshore. This water is replaced by nutrient-rich water from below. At these locations P input (along with other required nutrients) is not limited by slow diffusive transport processes but is enhanced many-fold by the upward advection of water. For this reason, upwelling areas are capable of supporting extremely high rates of biological primary production and abundant populations of higher organisms. Thus, the major fisheries of the world are concentrated in upwelling regions such as off Peru. A significant amount of P is also returned to the surface ocean in cold, high-latitude regions where decreased stratification results in greater vertical mixing than in the temperate and equatorial regions.
Once in the photic zone, P is readily incorporated into biogenic particles (2) via the photosynthetic activities of plants and some fraction of the biogenic materials subsequently sinks. Increasingly, improved technologies permit the dynamics of this system to be followed by studying 32P and 33P distributions. The majority of the particles decompose in the surface layer or in shallow sediments and the P is recycled directly back into the photic zone (3) to be reincorporated into biological particles. A small portion of the particles produced in the surface layers, however, does escape the surface layers and sinks into the deep ocean. Most of these particles eventually decompose (4) and the cycle is repeated. A very small fraction of these particles, however, escapes decomposition and is incorporated into the sediment (6). P appears to be buried in sediments primarily as organic P, apatitic P (including both authigenic apatite and fish debris), P associated with other mineral phases (primarily CaCO3 and FeOOH) and P loosely sorbed on to other solid phases (Ruttenberg, 1993; Follmi, 1996; Anschutz et al., 1998).
14.3 The Global Phosphorus Cycle
The main reservoirs and exchange pathways of the global P cycle are schematically presented inFig. 14-7. This representation is primarily taken from Lerman et al. (1975) and modified to include atmospheric transfers. The mass of P in each reservoir and rates of exchange are taken from Mackenzie et al. (1993) and Follmi (1996).
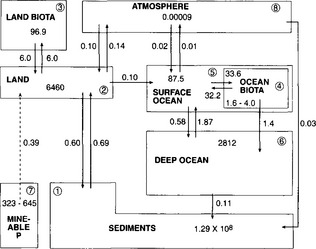
Fig. 14-7 The global phosphorus cycle. Values shown are Tmol and Tmol/yr for reservoirs and fluxes, respectively. (T = 1012).
In choosing these reservoirs to describe the P cycle, compromises were made to maintain a general focus and global scale and yet avoid being too general and hence lose information about important transfers and reservoirs. The following is a brief discussion of the rationale behind the choice of the reservoir definitions and their estimates. For the purpose of discussion, the reservoirs have been numbered as presented in Lerman et al. (1975) with the addition of the atmosphere (reservoir 8). The total P content of each reservoir and comments concerning the estimate are provided in Table 14-3.
Table 14-3
The mass of P in the major reservoirs (mol P × 10−12)
| Reservoir | P content | Comments |
| 1. Sediments | 1.29 × 108 | Van Wazer (1961) |
| 2. Land | 6460 | Computed from land area of 133 × 106 km2, soil thickness of 60 cm, density of 2.5 g/cm3 and mean P content of 0.1% (Taylor, 1964) |
| 3. Land biota | 96.9 | Computed from an estimate of the N in land biota (12 × 104 tons N; Delwiche, 1970); and a mean P:N atomic ratio in land plants (1.8:16; Deevey, 1970) |
| 4. Oceanic biota | 1.6–4.0 | Mackenzie et al. (1993) |
| 5. Surface Ocean | 87.5 | Computed from assumed mean concentration of 25 mg/m3 of dissolved P, 300 m thick surface layer and area of 3.61 × 108 km2 |
| 6. Deep ocean | 2812 | Computed from assumed mean concentration of 80 mg/m3 of dissolved P, a deep water thickness of 3000 m, and same surface area as above. |
| 7. Mineable P | 323–645 | Mean values reported by Stumm (1973), Ronov and Korzina (1960) and Van Wazer (1961) and the value reported by Mackenzie et al. (1993) |
| 8. Atmospheric | 0.00009 | Graham (1977) |
The reservoir representing the land (2) is defined as the amount of P contained in the upper 60 cm of the soil. This rather narrow definition of the land reservoir is made because it is through the upper portions of the soil system that the major interactions with the other P reservoirs occur. Specifically, most plants receive their nutritive P needs from the upper soil horizons and the return of P to the soil system by the decomposition of plant matter is also concentrated in this upper soil zone. Similarly, the major interactions with the atmosphere, ground waters, and rivers occur near the soil surface. Finally, phosphate in the form of fertilizer is applied directly to the soil surface. Thus, in attempting to represent the land and its interaction with other reservoirs, the surface soil horizon most directly interacts with all components and best represents the dynamic nature of this reservoir. Phosphorus in soils deeper than 60 cm and in crustal rocks is included in the sediment reservoir (1). This reservoir accounts for all of the particulate P that exchanges with the other reservoirs only on longer time scales.
The land biota reservoir (3) represents the phosphorus contained within all living terrestrial organisms. The dominant contributors are forest ecosystems with aquatic systems contributing only a minor amount. Phosphorus contained in dead and decaying organic materials is not included in this reservoir. It is important to note that although society most directly influences and interacts with the P in lakes and rivers, these reservoirs contain little P relative to soil and land biota and are not included in this representation of the global cycle.
The ocean system is separated into three major reservoirs that best represent the dominant pools and pathways of P transport within the ocean. The surface ocean reservoir (5) is defined as the upper 300 m of the oceanic water column. As discussed in an earlier section and displayed inFig. 14-6, the surface layer roughly corresponds to the surface mixed layer where all the photosynthetic uptake of P and the majority of the decomposition and release of P from sinking organic matter occur. Therefore, the most active exchange of P between solution and ocean biota occurs in this zone. Also, the 300 m depth roughly corresponds to the top of the main thermocline, which restricts exchange between surface and deep waters, thus representing a natural boundary. Additionally, dissolved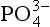 brought to the ocean via rivers is introduced directly to the surface layer.
brought to the ocean via rivers is introduced directly to the surface layer.
The oceanic biota reservoir (4) is also within the surface layers. Although organisms reside at all depths within the ocean, the overwhelming majority reside within the photic zone where phytoplankton dominate. The oceanic biota reservoir only contains roughly 1/30 as much P as the land biota reservoir. This is primarily because oceanic biomass is composed of relatively short-lived organisms, while land biomass is dominated by massive long-lived forests.
The deep ocean (6) is the portion of the water column from 300 m to 3300 m and is the largest ocean reservoir of dissolved P. However, since the deep ocean is devoid of light, this P is not significantly incorporated into ocean biota. Mostly, it is stored in the deep waters until it is eventually transported back into the photic zone via upwelling or eddy diffusive mixing.
The sediment reservoir (1) represents all phosphorus in particulate form on the Earth’s crust that is (1) not in the upper 60 cm of the soil and (2) not mineable. This includes unconsolidated marine and fresh water sediments and all sedimentary, metamorphic and volcanic rocks. The reason for this choice of compartmentalization has already been discussed. In particulate form, P is not readily available for utilization by plants. The upper 60 cm of the soil system represents the portion of the particulate P that can be transported relatively quickly to other reservoirs or solubilized by biological uptake. The sediment reservoir, on the other hand, represents the particulate P that is transported primarily on geologic time scales.
The atmospheric reservoir (8) represents P contained on dust particles. Because the mean residence time of dust in the air is very short, the standing stock of P in the atmosphere is relatively small. Mineable P (7) is simply an estimate of the total amount of P stored in economic deposits.
14.3.1 Fluxes between Reservoirs
A summary of the estimated fluxes between reservoirs and the methods of calculation is presented in Table 14-4. The fluxes between reservoirs are chosen to represent the principal pathways by which P is transported between reservoirs. The notation used here is the same as that presented by Lerman et al. (1975) with the first number representing the reservoir from which the P originates and the second number representing the receiving reservoir. It is important for the reader to understand that the evaluation of the fluxes is extremely difficult. This is, in part, caused by the introduction of P from anthropogenic sources obscuring natural levels. Also, as stated earlier, most P is present in particulate form and is not biologically active. Thus, the evaluation of the P that is actively transferred between the inorganic reservoirs and the biota or as the dissolved component in rivers must always be made in the presence of a large background. Small exchanges between the relatively non-reactive particulate P and reactive (primarily dissolved) P could significantly alter the flux estimate.
Table 14-4
Summary of the flux of P between reservoirs (mol P × 1012)
| Transfer | Flux | Comments |
| F12 | 0.69 | Computed from combined rates of mechanical and chemical denudation of continents (2 × 109 tons/yr; Garrels and Mackenzie, 1971) and a mean P content of crustal material of 0.1% (Taylor, 1964) |
| F23, F32 | 6.00 | Computed from total C fixed annually (560 Pg C/yr; Sundquist, 1993) and a mean P:C atomic ratio of land biota of 1:510 (Delwiche and Likens, 1977) |
| F25 | 0.10 | Garrels et al. (1973) and adjusting upward 33% to account for release from particles (after Kaul and Froelich, 1984) |
| F54 | 33.6 | Computed from rate of N fixation of oceanic biota of 7.5 × 109 tons N/yr (Vaccaro, 1965) and a mean P:N atomic ratio in ocean biota of 1:16 (Redfield et al., 1963) |
| F45 | 32.2 | Computed assuming that 96% of oceanic biota recycled within upper 300 m |
| F46 | 1.40 | Difference between fluxes F54 and F45 |
| F56 | 0.58 | Computed from the P content of surface layer given in Table 14-3 and a water exchange rate between surface and deep ocean of 2 m/yr (Broecker, 1971) |
| F65 | 1.87 | Computed as F56 using the P content of the deep ocean given in Table 14-3 |
| F61 | 0.11 | Calculated that the ocean is in steady state |
| F72 | 0.39 | Stumm (1973); Mackenzie et al. (1993) |
| F21 | 0.60 | Mackenzie et al. (1993) |
| F28 | 0.14 | Graham (1977) |
| F82 | 0.10 | Graham (1977) |
| F58 | 0.01 | Graham (1977) |
| F85 | 0.02 | Graham (1977) |
| F81 | 0.03 | Graham (1977) |
The transfer of P from land to terrestrial biota (F23) represents the sum of terrestrial biological productivity. There is no significant gaseous form of P, nor is there a major transfer of living organisms between the freshwater-terrestrial system and the oceans. The terrestrial biota system is, therefore, essentially a closed system where the flux of P to the biota (F23) is balanced by the return of P to the land from the biota (F32) due to the decay of dead organic materials.
The transfer of P from the continents to the ocean is separated into two distinct pathways. The flux of reactive P (F25) is estimated via measurements of dissolved organic and inorganic P in rivers. A small correction (33%; after Kaul and Froelich, 1984) is added to the measured values to account for P released from particles within the estuaries. This P is transported directly to the surface ocean and is available for biological uptake. The other pathway by which P is transported to the oceans is that associated with particulate materials, either suspended in river waters or simply transported to the ocean as bedload. The P in these materials is considered locked in the solid structure and not available for biological uptake. In general, these particles rapidly settle to the ocean bottom and are incorporated into the sediments. This removal is rapid enough that this flux is represented as the direct transport of P from the land reservoirs to the sediments (F21).
Separating “reactive”and “non-reactive” P in the estuarine system is extremely difficult. Since the majority of P is associated with the particles, a small exchange between the particulate and the dissolved fraction would alter the “reactive” flux (F25) significantly. Kaul and Froelich (1984) have investigated the fate of dissolved and particulate P in a small estuary. They determined that although P is readily incorporated into plants in the estuary, all of the dissolved P brought to the estuary by the river is eventually transported to the ocean. In addition, they found that one-third of the “reactive” P that leaves the estuary is derived from fluvial particulate matter. Thus, estimates of the flux of “reactive” P from the land to the surface ocean based on dissolved P concentration data may be 50% too low. While uncertainty exists, the value of F25 has been increased by 33% as a rough attempt to account for this release.
A small flux is shown between the land and atmosphere. This represents the transport of dust particles to the atmosphere (F28) and the deposition of these particles back on land either as dry deposition or associated with atmospheric precipitation (F82). Similarly, fluxes that represent the transport of seasalt from the surface ocean to the atmosphere (F58) and the deposition of soluble (F85) and insoluble (F81) atmospheric forms are also shown. As already discussed for the river fluxes, the insoluble particulate flux is represented as a direct transport of P to the sediment reservoir.
The natural circulation of the oceans also exchanges waters between the deep and surface ocean reservoirs. Because biological uptake constantly strips P from the surface layers, the P concentration is much less in the surface reservoir than the deep reservoir. Thus, although the continuity of water demands equal volumes of water to be exchanged between the reservoirs, far more P is carried by the upwelling water (F65) than by the downwelling waters (F56).
By far, the largest exchange of P between reservoirs occurs between the surface ocean and ocean biota. These large numbers attest to the tremendous role of primary production in controlling P dynamics in the ocean. However, the relatively small standing stock also attests to the short life-span and rapid turnover of these organisms. Most of the recycling occurs in the surface waters and, hence, the flux of P into the biota (F54) is nearly equaled by the release of P from the organisms (F45). However, a small fraction (4%) of the P incorporated into the biota sinks out of the surface layers before being solubilized by decomposition. Although this particulate flux to the deep ocean reservoir (F46) is a small fraction of the total amount incorporated into oceanic biota, ocean productivity is so great that this flux is quantitatively quite large.
Nearly all of the detrital particles sinking into the deep ocean decompose and release the associated P. A small percentage (approximately 8%), however, do survive and accumulate on the sea floor. This P is then buried in the sediments (F61) and represents the ultimate removal of P from the ocean.
The basic cycle of P transport from continents to oceans to sediments is then completed by the slow geologic processes that eventually return marine sediments to the continents (F12). One additional flux is considered which represents the P mined and placed on agricultural fields in the form of fertilizers (F72). Notice that there are no fluxes into the reservoir marked mineable P. This is undoubtedly incorrect. However, at time scales important to human beings, the formation of mineable P deposits is assumed to be too slow to be included. Notice also that because of this, the representation of the P cycle is not in a steady state.
14.3.2 The Steady-State Cycle
The total burden, sum of inputs or exports, and average residence times for the reservoirs are listed in Table 14-5. As discussed in Chapter 4, the residence time of an element within a reservoir reflects the reactivity and exchange of that element with other reservoirs. A short residence time suggests that removal processes are rapid and significant over short time scales compared to the amount in the reservoir.
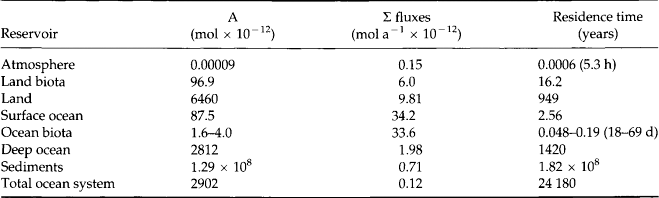
From Table 14-5 it is obvious that the residence time of P in the atmosphere is extremely short. This does not represent chemical reaction and removal of P from the atmosphere but rather the rapid removal of most phosphorus-containing particles that enter the atmosphere.
More informative is the comparison between the residence times of P in the land and ocean biota. Although there is 5.6 times more biological incorporation of P in the oceans, the standing stock is only 1.7–4.1% of that on land. The residence time of a P atom incorporated into oceanic biota is relatively short (2–7 days) compared to the 16-year-residence time of P in land biota. This disparity represents a fundamental difference in the types of organisms in the two reservoirs. Whereas oceanic biota are dominated by single celled, short-lived planktonic plants and microbes, terrestrial biomass is dominated by forests.
The residence time of P in the deep ocean is 1400 years and is dependent primarily on the rate at which P is transported to the surface waters via upwelling. The ocean system also demonstrates an important characteristic of global reservoir models. The residence times of P in the oceanic biota, surface ocean, and deep ocean are 2–7 days, 2.6 years, and 1400 years, respectively. These relatively short times suggest that P is recycled rapidly throughout the ocean. However, since this cycling is almost exclusively among oceanic reservoirs and not to outside reservoirs, the residence time of P in the entire ocean system is relatively long, 24 180 years. This value is well within the range 16 000–38 000 years estimated by Ruttenberg (1993) but much shorter than the 80 000 years estimated earlier by Froelich et al. (1982). Increased estimates on input and burial rates account for this decrease in the estimated residence time. Based on these values, the average P atom is cycled approximately 17 times between the deep water and surface waters before being removed to the sediments. Also, each time a P atom reaches the surface waters, it is cycled between the oceanic biota and the dissolved inorganic pool 14–52 times before being transported to the deep water. Thus, the average P atom is incorporated into the ocean biota a total of 238–884 times during its stay in the ocean.
14.3.3 Perturbations
One of the main goals in establishing a global understanding of P cycling is to evaluate the influence of changing conditions on the P distribution. Perhaps the most obvious and visible mechanism by which the P cycle may be altered is through anthropogenic input. This includes P applied to the soil in the form of fertilizers as well as P used in detergents and various industrial applications. The amount of P added to the environment in this manner may be estimated from the amount of P mined each year. We have all witnessed or have read about the detrimental effects of large P inputs on some freshwater systems. The influence of this added P on the global cycle is less obvious.
Lerman et al. (1975) considered several cases in which mankind’s activities perturbed the natural cycle. If we assume that all mined P is supplied to the land as fertilizer and that all of this P is incorporated into land biota, the mass of the land biota will increase by 20%. This amount is small relative to the P stored in the land reservoir. Since P incorporated into land biota must first decompose and be returned to the land reservoir before being transported further, there is essentially no change in the other reservoirs. Thus, although such inputs would significantly alter the freshwater-terrestrial ecosystem locally where the P release is concentrated, the global cycle would be essentially unaffected.
A greater perturbation results if one assumes that the rate at which P is mined doubles every 10 years and that the dissolved riverborne flux to the ocean increases in proportion to the mining rate. At the time of Lerman et al.’s calculation, known reserves implied that mineable P would be exhausted in 60 years under these conditions. Since then, additional deposits have been identified. Nevertheless, this calculation still effectively demonstrates the sensitivity of the P reservoirs to changes of this magnitude. At the end of the 60-year period, the P contained in the surface ocean has increased by 38% and the P present in ocean biota is 30% greater than present (assuming no other limiting factors for biological production). The other reservoirs will not change significantly. After 60 years, increased P input will then cease and after approximately 150 years, the system will then return to present-day levels. Additional discussion of the impacts of perturbations to the P cycle due to increased land weathering rates and decreased terrestrial productivity are discussed in Mackenzie et al. (1993).
There may also be natural fluctuations within this cycle that occur over time scales ranging from thousands of years (glacial–interglacial) to millions of years (Follmi, 1996). Overall, the global P burial rate is estimated to have varied by one order of magnitude or less over the last 160 million years. Maxima in P burial are estimated for the Callovian–Oxfordian boundary (approx. 157 Myr ago), Valanginian–Hauterivian boundary (approx. 135 Myr ago), late Albian (approx. 98 Myr ago), Campanian (approx. 82 Myr ago), and Paleocene (approx. 60 Myr ago). The changes in total P burial rates appear to be correlated with changes in the burial of biogenic P. These variations suggest that there have been relative variations in continental weathering rates, proportion of mobilized P that is dissolved, and overall sediment accumulation.
Because P is such an important element in biological systems, the P cycle may influence other biogeochemical cycles and processes. One example is the potential link between the P content of deep ocean water and atmospheric CO2. Atmospheric CO2 levels are increasing rapidly presently due to the burning of fossil fuels and deforestation. Because CO2 influences the efficiency with which solar radiation is absorbed in the atmosphere and, hence, the temperature at the Earth’s surface, there is great concern as to how this increase will influence the global climate. However, analyses of gas pockets trapped in ice cores taken from Greenland and Antarctica suggest that there have been natural fluctuations in the concentration of atmospheric CO2 and that during the last glacial period, atmospheric CO2 was 80 ppmv less than the modern pre-industrial value (Neftel et al., 1982). Thus, to predict the consequences of the build-up of CO2 in the atmosphere, one must understand the natural variations. Broecker (1982) has suggested that one mechanism by which atmospheric CO2 may be altered is that of changing the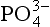 concentration in the deep waters of the ocean. His argument is as follows.
concentration in the deep waters of the ocean. His argument is as follows.
The present average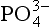 concentration of deep ocean water is 2.2 µmol/kg. When a parcel of deep water is transported to the photic zone, this
concentration of deep ocean water is 2.2 µmol/kg. When a parcel of deep water is transported to the photic zone, this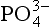 is completely incorporated into plants. Note that this assumes that net primary productivity is not limited by the availability of other micronutrients. In short-term laboratory studies, this assumption is clearly not true in that it has been demonstrated that the availability of fixed N or iron can also limit production. Because certain organisms are capable of fixing N from the large N2 gas pool whereas there is no alternative source or substitute for
is completely incorporated into plants. Note that this assumes that net primary productivity is not limited by the availability of other micronutrients. In short-term laboratory studies, this assumption is clearly not true in that it has been demonstrated that the availability of fixed N or iron can also limit production. Because certain organisms are capable of fixing N from the large N2 gas pool whereas there is no alternative source or substitute for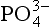 , it is likely that on longer time scales,
, it is likely that on longer time scales,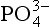 limits productivity. Because the overall chemical composition of marine organisms is relatively constant, the complete utilization of the up welled
limits productivity. Because the overall chemical composition of marine organisms is relatively constant, the complete utilization of the up welled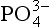 also determines the amount of dissolved inorganic carbon and alkalinity that is removed from the surface waters and transported downward on sinking particles. This, in turn, alters the solution chemistry and partial pressure of CO2 in the surface waters and, in the long term, the partial pressure of CO2 in the atmosphere as well. If the
also determines the amount of dissolved inorganic carbon and alkalinity that is removed from the surface waters and transported downward on sinking particles. This, in turn, alters the solution chemistry and partial pressure of CO2 in the surface waters and, in the long term, the partial pressure of CO2 in the atmosphere as well. If the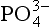 concentration of the water entering the photic zone were to change, the resulting partial pressure of CO2 would also be affected. In fact, Broecker (1982) speculates that if the
concentration of the water entering the photic zone were to change, the resulting partial pressure of CO2 would also be affected. In fact, Broecker (1982) speculates that if the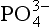 concentration in the deep water during the last glacial period averaged 3.2 µmol/kg, the resulting atmospheric CO2 would be 80 ppmv less than the modern pre-industrial value.
concentration in the deep water during the last glacial period averaged 3.2 µmol/kg, the resulting atmospheric CO2 would be 80 ppmv less than the modern pre-industrial value.
Recently, it has also been suggested that the burial rates of organic carbon and phosphorus in marine sediments are inversely correlated (Ingall and Jahnke, 1994, 1997; Van Cappellen and Ingall, 1994). This provides a mechanism for stabilizing atmospheric oxygen through geological time by providing a controlling feedback mechanism between organic carbon burial and photosynthetic oxygen production (Van Cappellen and Ingall, 1996; Coleman and Holland, in press). Understanding such potential linkages are fundamental to understanding Earth systems and to predicting how they may change in the future. Thus, natural variations in the transfer rates of P between reservoirs may have profound effects on other geochemical cycles and climate. The elucidation of such interactions between cycles is the focus of many exciting research programs currently underway.
Questions
14-1. Due to anthropogenic inputs, the mean pH of a lake has decreased from 6.5 to 6.0. Assuming no significant changes in the chemical composition and ionic strength of the lake waters and a total dissolved inorganic P concentration of 1 µM, by what factor will the free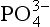 change?
change?
14-2. A farmer in the Imperial Valley has been irrigating and fertilizing fields for many years. Because of the generally hot conditions, much of this irrigation water is lost to evaporation, increasing the salt content of the soil and groundwater. Assuming that the composition of the salt is roughly similar to that of seawater, how might this salt buildup influence the availability of P to the plants? Will the farmer need to increase the amount of fertilizer used?
14-3. Because of a primitive sewage system and the use of fertilizers, the citizens of a small community have added a significant amount of P to a nearby lake. Recognizing that this will stimulate biological production and may cause anoxia in the deep waters when the lake is stratified, the community decides to install a pumping system to continually exchange the deep water with the surface water. What will this do to the productivity of the lake? Will this prevent the deep water from becoming anoxic?
14-4. In recent years, many of the world’s forests have been cut down and replaced with short-lived crops. What effect, if any, might this have on the: (1) P stored in the land biota reservoir; (2) exchange rate of P between the land biota and the land reservoirs; (3) exchange rate between the land reservoir and the surface ocean?
14-5. It has been suggested by Broecker (1982) that glacial-interglacial pCO2 differences could be explained if the glacial ocean contained 1.5 times more dissolved P than at present. If the exchange fluxes with the non-oceanic reservoirs remained the same, what would be the residence time of P in the ocean during the last glacial period? How does this compare to glacial-interglacial time scales and what does it suggest about the exchange fluxes?
Anderson, L. A., Sarmiento, J. L. Redfield ratios of remineralization determined by nutrient data analysis. Global Biogeochem. Cycles. 1994; 8:65–80.
Anschutz, P., Zhong, S., Sundby, B., Mucci, A., Gobeil, C. Burial efficiency of phosphorus and the geochemistry of iron in continental margin sediments. Limnol. Oceanogr. 1998; 43:53–64.
Atkinson, M. J., Smith, S. V. C:N:P ratios of benthic marine plants. Limnol. Oceanogr. 1983; 28:568–574.
Atlas, E. L. Phosphate equilibria in seawater and interstitial waters. Ph. D. Thesis, Oregon State University. 1975.
Benitez-Nelson, C. R., Buesseler, K. O. Variability of inorganic and organic phosphorus turnover rates in the coastal ocean. Nature. 1999; 398:502–505.
Bjorkman, K., Karl, D. M. Bioavailability of inorganic and organic phosphorus compounds to natural assemblages of microorganisms in Hawaiian coastal waters. Mar. Ecol. Prog. Ser. 1994; 111:265–273.
Broecker, W. S. A kinetic model for the chemical composition of seawater. Quatern. Res. 1971; 1:188–207.
Broecker, W. S. Ocean chemistry during glacial time. Geochim. Cosmochim. Acta. 1982; 46:1689–1706.
Chambers, R. M., Fourquean, J. W., Hollibaugh, J. T., Vink, S. M. Importance of terrestrially-derived particulate phosphorus to phosphorus dynamics in a west coast estuary. Estuaries. 1995; 18:518–526.
Clark, L. L., Ingall, E. D., Benner, R. Marine phosphorus is selectively remineralized. Nature. 1998; 393:426–428.
in press, January. Coleman, A. S., Holland, H. D., The global diagenetic flux of phosphorus from marine sediments to the oceans: redox sensitivity and the control of atmospheric oxygen levelsGlenn C. R., Prevot-Lucas L., Lucas J., eds. Marine Authigenesis: from Microbial to Global. SEPM Publication No. 66. 2000.
Conley, D. J., Smith, W. M., Cornwell, J. C., Fisher, T. R. Transformation of particle-bound phosphorus at the land-sea interface. Estuar. Coast. Shelf Sci. 1995; 40:161–176.
Deevey, E. S., Jr. Mineral cycles. Scient. Am. 1970; 223:149–158.
Delwiche, C. C. The nitrogen cycle. Scient. Am. 1970; 223:137–146.
Delwiche, C. C., Likens, G. E. Biological response to fossil fuel combustion products. In: Stumm W., ed. Global Chemical Cycles and Their Alterations by Man. New York: Dahlem Konferenzen; 1977:73–88.
Emerson, S., Widmer, G. Early diagenesis in anaerobic lake sediments II. Equilibrium and kinetic factors controlling the formation of iron phosphate. Geochim. Cosmochim. Acta. 1978; 42:1307–1316.
Follmi, K. B. The phosphorus cycle, phosphogenesis and marine phosphate-rich deposits. Earth-Sci. Rev. 1996; 40:55–124.
Froelich, P. N., Bender, M. L., Luedtke, N. A., Heath, G. R., DeVries, T. The marine phosphorus cycle. Am. J. Sci. 1982; 282:474–511.
Froelich, P. N., Arthur, M. A., Burnett, W. C., Deakin, M., Hensley, V., Jahnke, R., Kaul, L., Kim, K. -H., Roe, K., Soutar, A., Vathakanon, C. Early diagenesis of organic matter in Peru continental margin sediments: phosphate precipitation. Mar. Geol. 1988; 80:309–343.
Garrels, R. M., MacKenzie, F. T. Evolution of Sedimentary Rocks. Berlin: W. W. Norton & Co., Inc. ; 1971.
Garrels, R. M., MacKenzie, F. T., Hunt, C. A. Chemical Cycles and the Global Environment. New York: W. Kaufmann; 1973.
Graham, W. F., Atmospheric pathways of the phosphorus cycle. Ph. D. Thesis, University of Rhode Island. 1977.
Howard, P. F. Phosphate. Econ. Geol. 1979; 74:192–194.
Ingall, E. D., Jahnke, R. A. Evidence for enhanced phosphorus regeneration from marine sediments overlain by oxygen depleted waters. Geochim. Cosmochim. Acta. 1994; 58:2571–2575.
Ingall, E., Jahnke, R. Influence of water-column anoxia on the elemental fractionation of carbon and phosphorus during sediment diagenesis. Mar. Geol. 1997; 139:219–229.
Jackson, G. A., Williams, P. M. Importance of dissolved organic nitrogen and phosphorus in biological nutrient cycling. Deep Sea Res. 1985; 32:223–235.
Jahnke, R. A. The synthesis and solubility of carbonate fluorapatite. Am. J. Sci. 1984; 284:58–78.
Jonge de, V. N., Villerius, L. A. Possible role of carbonate dissolution in estuarine phosphate dynamics. Limnol. Oceanogr. 1989; 34:332–340.
Karl, D. M., Yanagi, K. Partial characterization of the dissolved organic phosphorus pool in the oligotrophic North Pacific Ocean. Limnol. Oceanogr. 1997; 42:1398–1405.
Kaul, L. W., Froelich, P. N., Jr. Modeling estuarine nutrient geochemistry in a simple system. Geochim. Cosmochim. Acta. 1984; 48:1417–1434.
Lal, D., Lee, T. Cosmogenic 32P and 33P used as tracers to study phosphorus recycling in the upper ocean. Nature. 1988; 333:752–754.
Lehman, J. T. Hypolimnetic metabolism in Lake Washington: Relative effects of nutrient load and food web structure on lake productivity. Limnol. Oceanogr. 1988; 33:1334–1347.
Lerman, A., MacKenzie, F. T., Garrels, R. M. Modeling of geochemical cycles: Phosphorus as an example. Geo. Soc. Am. Mem. 1975; 142:205–218.
Lucotte, M., d’Anglejan, B. Seasonal changes in the phosphorus-iron geochemistry of the St. Lawrence estuary. J. Coastal Res. 1988; 4:339–349.
Mackenzie, F. T., Ver, L. M., Sabine, C., Lane, M., Lerman, A. C, N, P, S global biogeochemical cycles and modeling of global change. In: Wollast R., Mackenzie F. T., Chou L., eds. Interactions of C, N, P, and S Biogeochemical Cycles and Global Change. Los Altos, CA: Springer-Verlag; 1993:1–61.
Murray, J. W., Grundmanis, V., Smethie, W. M., Jr. Interstitial water in the sediments of Saanich Inlet. Geochim. Cosmochim. Acta. 1978; 42:1011–1026.
Nanny, M. A., Minear, R. A. Characterization of soluble unreactive phosphorus using 31P nuclear magnetic resonance spectroscopy. Mar. Geol. 1997; 139:77–94.
Neftel, A., Oeschger, H., Swander, J., Stauffer, B., Zumbrunn, R. New measurements on ice core samples to determine the CO2 content of the atmosphere during the last 40 000 years. Nature. 1982; 295:220–223.
Nriagu, J. O. Phosphate-clay mineral relations in soils and sediments. Can. J. Earth Sci. 1976; 13:717–736.
Nriagu, J. O., Moore, P. B. Phosphate Minerals. New York: Springer-Verlag; 1984.
Peng, T. H., Broecker, W. S. C:P ratios in marine detritus. Global Biogeochem. Cycles. 1987; 1:155–162.
Redfield, A. C., Ketchum, B. H., Richards, F. A. The influence of organisms on the composition of seawater. In: Hill M. N., ed. The Sea. New York: Interscience Publishers; 1963:26–77.
Ronov, A. B., Korzina, G. A. Phosphorus in sedimentary rocks. Geochemistry. 1960; 8:805–829.
Ruttenberg, K. C. Development of a sequential extraction method for different forms of phosphorus in marine sediments. Limnol. Oceanogr. 1992; 37:1460–1482.
Ruttenberg, K. C. Reassessment of the oceanic residence time of phosphorus. Chem. Geol. 1993; 107:405–409.
Ruttenberg, K. C., Berner, R. A. Authigenic apatite formation and burial in sediments from non-upwelling, continental margin environments. Geochim. Cosmochim. Acta. 1993; 57:991–1007.
Schindler, D. W. Evolution of phosphorus limitation in lakes. Science. 1977; 195:260–262.
Schuffert, J. D., Jahnke, R. A., Kastner, M., Leather, J., Sturtz, A., Wing, M. R. Rates of formation of modern phosphorites off western Mexico. Geochim. Cosmochim. Acta. 1994; 58:5001–5010.
Schuffert, J. D., Kastner, M., Jahnke, R. A. Carbon and phosphorus burial associated with modern phosphorite formation. Mar. Geol. 1998; 146:21–31.
Sheldon, R. P. Ancient marine phosphorites. Ann. Rev. Earth Planet. Sci. 1981; 9:251–284.
Slansky, M. Geology of Sedimentary Phosphates. New York: Elsevier; 1986.
Smith, S. V., Kimmerer, W. J., Walsh, T. W. Vertical flux and biogeochemical turnover regulate nutrient limitation of net organic production in the North Pacific Gyre. Limnol. Oceanogr. 1986; 31:161–167.
Stumm, W. The acceleration of the hydrogeochemical cycling of phosphorus. Water Res. 1973; 7:131–144.
Stumm, W., Morgan, J. J. Aquatic Chemistry. New York: Wiley-Interscience; 1981.
Sundquist, E. T. The global carbon dioxide budget. Science. 1993; 259:934–941.
Taylor, S. R. Abundance of chemical elements in the continental crust: A new table. Geochim. Cosmochim. Acta. 1964; 28:1273–1285.
Vaccaro, R. F. Inorganic nitrogen in seawater. In: Riley J. P., Skirrow G., eds. Chemical Oceanography. New York: Academic Press; 1965:365–408.
Van Cappellen, P., Ingall, E. D. Benthic phosphorus regeneration, net primary production, and ocean anoxia: A model of the coupled marine biogeochemical cycles of carbon and phosphorus. Paleoceanography. 1994; 9:677–692.
Van Cappellen, P., Ingall, E. D. Redox stabilization of the atmosphere and oceans by phosphorus-limited marine productivity. Science. 1996; 271:493–496.
Van Wazer F., ed. Phosphorus and Its Compounds. New York: Interscience Publishers, 1961.
Waser, N. A. D., Bacon, M. P., Michaels, A. F. Natural activities of 32P and 33P and the 32P/33P ratio in suspended particulate matter and plankton in the Sargasso Sea. Deep-Sea Res. 1996; 43:421–436.