Genome Evolution
Sara J. Hanson and John M. Logsdon Jr.
OUTLINE
1. Evolution of genome architecture
2. Genome expansion and restructuring
3. Drivers of genome evolution
The entirety of an organism’s DNA content—its genome—is a heritable storage system containing all information a cell needs to dictate the organism’s growth, development, and phenotypic characteristics. Throughout all forms of life, huge variation exists in the size and content of genomes, demonstrating the highly flexible, dynamic, and complex nature of their evolution. The frequently striking amounts of noncoding DNA present in eukaryotic genomes—largely absent in prokaryotic genomes—is particularly striking. This includes intragenic (introns and untranslated regions) and extragenic (regulatory sequences) as well as transposable elements: features that dominate eukaryotic genomes and usually make up the vast majority of nuclear DNA. Processes including recombination and transposition of mobile genetic elements have been hypothesized as mechanisms for the expansion of eukaryotic nuclear genomes. Both adaptive and neutral processes have been implicated in the origin and evolution of these genomic elements, and understanding the nature of such mechanisms for genome evolution can provide important insights into the evolution of prokaryotic and eukaryotic diversity.
GLOSSARY
Alternative Splicing. The generation of mRNA isoforms through differential use of splice donor and acceptor sites, retention of introns, and/or exon skipping.
Constructive Neutral Evolution. Conditions that decrease the efficacy of selection make it more likely that novel elements such as introns, untranslated regions, and modularity in gene expression will become fixed in a population. As a result, the increased genome size and content in eukaryotes derives from the fact that they have smaller population sizes that stem from increased cell size relative to prokaryotes.
C-Value Paradox. The mass of DNA in a haploid cell—or C-value—corresponds to an organism’s genome size in base pairs but displays no clear correlation with organismal complexity.
Modularity. In eukaryotic organisms, a gene is expressed under the control of its own promoter and a combination of trans-acting factors that interact with other regulatory sequences. This is in contrast to prokaryotes, where a single promoter and set of regulatory sequences and few trans-acting factors dictate the coordinated expression of groups of linked genes.
Mutation Bias. Processes that generate unequal outcomes for seemingly reciprocal mutational events. For example, small deletions in genomic DNA occur at higher frequency than small insertions, resulting in smaller genome size over time.
Noncoding DNA. Genomic region that does not encode a protein or functional RNA product. These include introns (intragenic sequences removed following transcription), untranslated regions (transcribed sequences upstream of the translation start codon and downstream of the translation stop codon), and all other intergenic DNA.
Nucleoskeletal Hypothesis. The size of an organism’s genome shapes the size of the nucleus required to contain it (the genome serves as a “nucleoskeleton”). Cell size and nucleus size coevolve such that increased cell size corresponds with increased genome size.
Recombination Hot Spot. Genomic regions where crossovers occur at much higher rates than in other regions of the genome.
Selfish DNA Hypothesis. Increased genome size is attributed to proliferation of transposable elements. Transposable elements multiply until they begin to affect (reduce) host fitness, thereby natural selection prevents their further proliferation.
Transposable Element. Mobile DNA segments that are capable of self-proliferation—within and between genomes—through either “cut-and-paste” or “copy-and-paste” mechanisms.
1. EVOLUTION OF GENOME ARCHITECTURE
Before the advent of high-throughput sequencing technologies and the resulting plethora of available genome sequence data, studies of genome evolution concentrated on comparing genome sizes across the tree of life. Such early studies focused on estimates of the mass of DNA within haploid cells, termed the C-value, which can be extrapolated to an estimate of the number of base pairs composing an organism’s genome. The initial—and seemingly reasonable—hypothesis was that the number of genes contained in an organism’s genome would increase with the increasing complexity of organisms. As such, prokaryotes and single-celled eukaryotes would possess fewer genes than multicellular eukaryotes; however, this relationship was not observed, and it was instead found that genome sizes ranged greatly, even within relatively closely related groups of taxa. In 1971, C. A. Thomas Jr. described these perplexing observations as the “C-value paradox,” as genome size apparently did not account for increasing organismal complexity.
Although a clear correlation between genome size and organismal complexity was not realized, two generalizable genome configurations are evident. First, in prokaryotes, genomes are small and compact, comprising circular pieces of DNA. Intergenic space in these organisms is limited, and blocks of genes—called operons—which largely encode for genes with functions in the same pathway or process, are cotranscribed using the same promoter and regulatory sequence(s). Second, in eukaryotes, genomes are dramatically larger and contained on one or more linear chromosomes. This difference (presumably due to expansion) results from modular gene regulation (in which each gene is transcribed separately, with limited overlapping use of regulatory elements) as well as sometimes-massive amounts of noncoding DNA, including introns, untranslated regions (UTRs), and repetitive elements. Furthermore, the linear nature of eukaryotic chromosomes requires additional elements for proper maintenance and segregation of chromosomes; these include centromeres and telomeres, generally comprising repetitive DNA sequences, needed for segregation during mitosis and meiosis and the maintenance of chromosome ends, respectively.
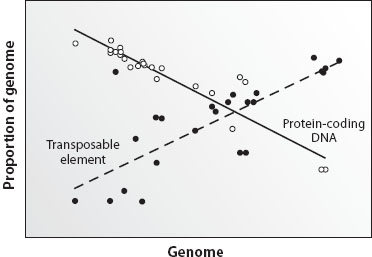
Genome sequence data have revealed clear patterns relating to variation in genome size, most notably in the characterization of noncoding DNA elements in a wide range of prokaryotes and eukaryotes. The general trend appears to be that genome size increases with genome complexity, which, in turn, is correlated with increasing organismal complexity (although there are notable outliers). This pattern is well illustrated by comparisons among individual genome components, including the observed correlation between genome size and intron and intergenic DNA content. In fact, noncoding DNA is nearly exclusively responsible for differences in eukaryotic genome size: a 10,000-fold difference in the range of genome size exists between prokaryotes and eukaryotes, but only a 100-fold range in the amount of their protein-coding DNA. As shown in figure 1, the relative amount of protein-coding DNA decreases with increasing genome size while other genomic elements, such as transposons, increase.
Figure 1. Relative contributions of two components of eukaryotic genomes. As genome size increases, the relative amount of protein-coding DNA decreases (white circles). In contrast, transposable element content increases in larger genomes. Thus, larger genomes contain proportionately fewer genes and more transposable elements than smaller genomes. (Adapted from Gregory 2005b.)
2. GENOME EXPANSION AND RESTRUCTURING
How does genome restructuring occur? What processes result in changes in genome size? Several mechanisms are thought to play a role in large-scale changes in genome architecture, including those that shuffle genotypes and, thus, alter the structure of chromosomes, as well as processes that result in addition or relocation of new DNA sequences in the genome.
Recombination
Recombination, the repair of double-stranded breaks (DSBs) in DNA, has important influences on organismal evolution, including both generating and reducing genetic variation (see chapter IV.4). DSBs may be incurred exogenously through exposure to environmental agents at any point during an organism’s life cycle, or endogenously during meiosis in eukaryotes. Repair of these breaks frequently involves using a homologous piece of DNA as template—typically a sister chromatid or homologous chromosome. When a reciprocal exchange of DNA between homologous chromosomes occurs, it is referred to as a crossover event. Efficient repair of these breaks is critical, because their presence will disrupt replication and transcription. Errors in recombination can be devastating to an organism, but when they occur in the germ line, they also provide heritable restructuring events in genomes that contribute to genomic evolution in eukaryotes.
Repetitive elements and self-replicating mobile elements are present throughout eukaryotic genomes. Because these elements have multiple homologous templates in the genome, recombination can potentially occur between any two, even if located on different chromosomes. When ectopic recombination occurs between these elements as a result of their sequence similarity, large-scale chromosomal rearrangements can occur, including sequence duplications, deletions, or inversions of large sections of chromosomes, and translocation of a chromosomal section from one chromosome to another. These changes can disrupt protein-coding sequences directly, as well as remove or add regulatory sequences that can result in aberrant expression of genes.
During meiosis, DSBs are induced and repaired in a process mediated by a cell’s machinery. Because of the inherent risk associated with the formation of these breaks, it is unsurprising that meiotic recombination appears a tightly regulated and evolutionarily constrained process. More unexpected are the constraints limiting the number of these breaks that result in a crossover event. Furthermore, these crossovers do not occur equally across the genome; rather, they are concentrated at hot spots, where rates of recombination are higher by several orders of magnitude than their flanking genome regions, or cold spots, in which crossover rates are extremely low. These hot spots are rapidly evolving and dynamic. Organisms as closely related as humans and chimpanzees share no overlap in the genomic locations of hot spots, and intraspecific variation has even been observed within humans. Despite this fast rate of evolution of hot spots, there is mounting evidence that their location is sometimes dictated by specific sequence motifs. In the fission yeast Schizosaccharomyces pombe, several discrete sequences seven base pairs in length have been identified at active hot spots. In humans, one degenerate thirteen base-pair motif has been characterized at 41 percent of identified hot spots. Furthermore, in humans, the transcription factor Prdm9 is required for activation of these hot spots, and the amino acids that interact with the thirteen base-pair motif are under strong positive selection. Prdm9 may therefore act as a driver in hot spot evolution, or may be evolving rapidly in response to changes in hot-spot sequence motifs.
This observed specificity in DNA sequence at active hot spots is puzzling. If a specific sequence is required for increased recombination, that sequence should also be lost as a result of the very recombination that it induces. A model was recently proposed to explain this apparent paradox. If a specific sequence is required for hot-spot activation, then a single base-pair change will inactivate it. Conversely, there are many sites in the genome that require a single base-pair change in order to become an activated hot spot; therefore, an evolutionary equilibrium may exist in which hot spots are degraded and introduced through these single base-pair changes. This explanation, coupled with the rapid evolution of hot-spot activators like Prdm9, may explain the dynamic nature of genomic hot-spot locations even in very closely related organisms.
Transposable Elements
An astonishingly large fraction of many eukaryotic genomes is composed of mobile DNA elements. These self-replicating pieces of DNA, which frequently contain their own protein-coding and regulatory sequences, make up about 50 percent of the human genome. Generally, there are two classes of mobile elements characterized primarily by their mode of replication. First there are DNA transposons, which replicate through a “cut-and-paste” mechanism, in which an enzyme (transposase)—which may be encoded by the transposon itself or by a separate transposable element—excises the DNA sequence prior to its insertion into a new genomic location. Proliferation of these elements relies on the horizontal transfer of new elements from one organism to another, such as the transmission of small circular chromosomes containing the elements between prokaryotes. The second class of mobile elements is collectively referred to as retrotransposons. These elements replicate by “copy-and-paste” mechanisms, in which an RNA intermediate is produced and reverse transcribed (by a retrotransposon-encoded reverse transcriptase) before insertion. Such elements can proliferate horizontally, as described for DNA transposons, as well as vertically, when they proliferate within cells in the germ line and can then be transmitted to the next generation.
It is easy to see how the replication and insertion of mobile elements in a host genome could be slightly or strongly deleterious. For example, transposon insertion into a protein-coding region would most likely result in a frameshift, premature stop codon, or otherwise-aberrant protein sequence. Potentially, for this reason, a host has mechanisms to defend its genome from such elements. These include transcriptional silencing of the elements by chromatin modifications and transcription of small interfering RNA molecules that target mRNA produced by the element for destruction, thus depriving the transposable element of the machinery it needs for proliferation. Because successful proliferation of a mobile element depends on the success of a host genome, it is also possible that transposable elements have built-in self-regulatory mechanisms preventing them from uncontrolled proliferation that would drive a host to extinction; however, such mechanisms have not been characterized.
Importantly, as with all forms of mutation, mobile element insertion can on rare occasion give rise to evolutionary novelty. Because mobile elements encode their own machinery, multiple consequences can arise following their insertion into a new location. First, the elements contain protein-coding sequences and thus can introduce new coding regions into the genome (see chapter V.6). Second, these protein-coding sequences in mobile elements frequently have their own regulatory elements that can modify gene expression patterns of sequences, especially when adjacent to the insertion site. For example, the promoter region of a gene in the transposable element may recruit transcriptional machinery to a location near a host gene that has tight temporal or spatial regulation, causing it to be transcribed when it is normally silent. Indeed, it is hypothesized that centromeres and telomeres are often derived from mobile elements, and in some cases (e.g., Drosophila), mobile elements provide a mechanism for telomere maintenance. Finally, there is also evidence that mobile elements play a role in DNA double-strand break repair by using double-strand breaks as sites of insertion.
Noncoding Elements
Noncoding DNA sequences are those that do not determine a functional product. This chapter will consider the evolution of two types of noncoding elements, untranslated regions (UTRs) and introns. UTRs are parts of genes that are transcribed but not translated into an amino acid sequence, and are found both preceding the translation initiation site (5′ UTRs) and following the termination of translation (3′ UTRs). The addition of 5′ UTRs to eukaryotic genes is a risky prospect when the potential inclusion of an alternative translation initiation site is considered. Mutation of the 5′ UTR to contain such a site could have dramatic effects on the resulting amino acid sequence, resulting in a nonfunctional product. Because of this increased mutation risk, it is not clear what, if any, advantage eukaryotes gain through the addition of 5′ UTRs, but their presence and length are consistent across eukaryotic diversity. Although the addition of 3′ UTRs to eukaryotic genes does not appear to carry the same risks as 5′ UTRs, these elements are important in several aspects of mRNA regulation. The 3′ UTRs are critical for mRNA stability and nuclear export, and they have important regulatory functions in several aspects of translation. It is likely these features arose subsequent to the evolution of the 3′ UTR itself; therefore they cannot provide an explanation for the addition of this element.
The mechanisms for evolution and origins of introns are much better understood than 5′ UTRs. Despite the similarity in the length and number of protein-coding genes across eukaryotic diversity, there is substantial variation in the amount of intronic DNA. In eukaryotes, introns in nuclear genes (spliceosomal introns) are processed by a nucleoprotein complex—the spliceosome—which is present in all eukaryotes and thus likely present in the most recent eukaryotic ancestor. In humans, an average gene contains 7.7 introns, with an average intron length of 4.66 kilobases (kb). Compared to the average length of a human exon sequence (0.15 kb), it is clear that the total length of a human (and in general any eukaryotic) gene is dominated by introns. This density of introns allows for a large number of potential transcripts per locus through alternative splicing, which in humans is responsible for the average 2.6 transcripts produced per gene. Although the current importance of introns is at least partly understood (alternative splicing, regulatory element content, etc.), the origin and evolutionary mechanisms responsible for the proliferation of introns in eukaryotes remains unclear.
Debate over spliceosomal intron origin has been divided into two camps: those that propose the early evolution of introns prior to the divergence of eukaryotes and prokaryotes, and those that posit a later origin exclusively in eukaryotes. The resolution of this debate rests primarily on the hypothesized relationship of eukaryotic spliceosomal introns with the self-splicing group II introns found in some prokaryotes, which some argue are homologous. Whether spliceosomal introns arose early or late, there has been massive divergence in intron content in eukaryotes, making our understanding of the mechanisms underlying intron gain and loss of great importance.
Both intron loss and gain can be mediated by recombination, with intron loss hypothesized to result from replacement of a genomic gene copy with a reverse transcribed mRNA transcript of that gene (see chapter V.6), while hypotheses for mechanisms of intron gain include ectopic insertion of DNA fragments during an alternative DNA repair mechanism known as nonhomologous end joining (NHEJ). During NHEJ, fragments of DNA with very little sequence identity (microhomology) may be joined to repair DSBs, and aberrant insertion of a DNA fragment within a coding region may explain the origin of novel introns. NHEJ may be an intron loss mechanism as well, if microhomology between an intron’s splice junctions is used for repair. Consistent with this, species that are intron poor have high conservation of their splice sites, whereas intron-rich species have more degeneracy in their intron splice sites. The hypothesized role of NHEJ in intron gain is supported by the observation that intron-rich species use NHEJ more frequently during DNA repair.
3. DRIVERS OF GENOME EVOLUTION
A challenge that remains for our understanding of genome evolution is explaining how the addition of DNA to the genome and the existence of more complex genomic elements are possible. The presence of these elements is inherently risky, as they provide additional locations at which deleterious mutations can occur. For example, the addition of an intron to a protein-coding region now adds splice junctions, a branch point, and other regulatory elements that are evolutionarily constrained. One could argue that such genomic complexity is necessary for the evolution of organismal complexity; however, the diversity in content of these complex elements suggests otherwise. Adaptive and neutral arguments for the evolution of genomic complexity are described below.
Adaptive Evolution
What evolutionary pressures might be acting on genome size? Some data suggest that the forces may be mutation bias, such that small (<400 kb) deletions occur more frequently than insertions, resulting in reduction in genome size over time. For example, work performed in Caenorhabditis elegans demonstrated that at genomic sites not under selective constraints (i.e., pseudogenes), the rate of deletion was 2.8-fold higher than the rate of insertion. These data offer an explanation for the relatively compact size of the C. elegans genome, and suggest a more generalizable trend of deletions outnumbering insertions: selective pressures may favor a smaller genome.
Some other, less generally supported hypotheses suggest selective pressures might underlie the evolution of genome size as a result of the phenotypic consequences of these differences, primarily the effect of genome size on cell size. For example, the nucleoskeletal hypothesis proposes that increasing genome size requires an increase in the size of the nucleus, which coevolves with cell size. According to this hypothesis, a larger cell has greater requirements for transcription and translation, and thus requires a larger nucleus and genome to meet its needs; however, the nucleoskeletal hypothesis does not account for accommodation of a larger cell’s needs through increased rates of transcript production as opposed to increased DNA content.
Because beneficial outcomes are extremely unlikely for the majority of transposable element insertions, adaptive hypotheses for the existence of these elements can be excluded for the most part. These elements are more frequently thought of as parasitic or selfish because of their lack of dependence on host machinery for replication, and their likely detrimental effects on host fitness. The role of mobile elements in genome evolution is therefore referred to as the selfish DNA hypothesis, which posits that genome expansion is mediated by proliferation of mobile elements, and that such elements will spread until the point at which their impact on host fitness is so great that natural selection prohibits their further proliferation. This hypothesis does not account for the role of other elements present in eukaryotic genomes, such as introns, and therefore cannot fully explain the increased genome size in eukaryotes.
There are also several hypotheses for adaptive mechanisms underlying intron evolution in eukaryotes. First, large introns within genes increase the likelihood that incorrect splicing will result in the introduction of a premature stop codon that will be recognized early and will result in the degradation of the mRNA—a process known as nonsense-mediated decay. Second, the presence of one or more introns allows for alternative splicing to occur, in which introns can be excised or retained, exons can be skipped, or exon length can vary depending on the usage of specific splice junctions. This diversity in mRNA products from a single locus greatly increases the number of potential protein products resulting from that locus and allows for increased variation and complexity in molecular pathways (see chapter V.3). Further, the modular nature of genes that result from the inclusion of introns may have allowed for exon shuffling, in which mixing of domains from several different genes gives rise to genes with novel functions (see chapter V.6).
Neutral Evolution
Because eukaryotic genome expansion likely gave rise to sources of vast phenotypic novelty, it is tempting to develop adaptive hypotheses for their origination, such as those described above. However, the main explanation for the existence of these novel features may lie in neutral evolutionary processes—those that result from changes that have little or no effect on host fitness, but arise and become fixed in a population through genetic drift. A general framework for understanding the origin and evolution of such genomic novelties was proposed by Arlin Stoltzfus in 1999, which he called “constructive neutral evolution.” Expanding on this, Michael Lynch proposed a synthetic hypothesis that posits neutral processes as being largely responsible for the origin of genomic elements that, in turn, gave rise to the expanded genome size observed in eukaryotes. Since eukaryotic cells are typically much larger than prokaryotic cells, which generally result in much smaller population sizes for eukaryotes, the effects of genetic drift are amplified, making it much more likely that neutral or even slightly deleterious mutations—including unusual genetic features—will become fixed in a population (see chapter IV.1).
As described above, incorporation of the features most responsible for increased genome size would have been a very risky prospect for early eukaryotes. In particular, noncoding elements like introns and UTRs dramatically increase the number of sites at which deleterious mutation may occur. Further, the origin of these elements would have been extremely dangerous, as their addition would interrupt protein-coding regions, potentially causing frameshifts, premature stop codons, or alternative translation start sites. The inclusion of these elements therefore would likely have immediate deleterious effects on an organism, or at best would not confer an immediate benefit to be acted on by natural selection.
Instead, neutral processes may account for the initial fixation of these features in early eukaryotic populations. Small eukaryotic populations increased the impact of genetic drift and reduced the efficacy of selection such that these genomic elements could become fixed despite not conferring an advantage on a cell. Any beneficial effects these elements currently have were therefore subsequently acquired and may contribute to their maintenance in a population, but adaptive arguments are unlikely to explain their original fixation in eukaryotes.
Our understanding of the evolution of genome structure and content has substantially improved in the past decade. Fast-moving advances in DNA sequencing technologies have provided unfettered access to complete genomes from across the entire tree of life. Decoding the content of these genomes has been only a first step in understanding their biology. A deeper and more satisfying view of genome biology is emerging in which genomes are not only repositories of genes but also evolving entities with emergent and sometimes-unusual properties that are increasingly explicable within a solid theoretical framework.
FURTHER READING
Denver, D. R., K. Morris, M. Lynch, and W. K. Thomas. 2004. High mutation rate and predominance of insertions in the Caenorhabditis elegans nuclear genome. Nature 430: 679–682. An exemplar study using an experimental approach to examine the evolution of genome size.
Farlow, A., E. Meduri, and C. Schlotterer. 2011. DNA double-strand break repair and the evolution of intron density. Trends in Genetics 27: 1–6. Proposed model for role of introns in recombination and double-strand break repair.
Gregory, T. R, ed. 2005a. The Evolution of the Genome. London: Elsevier Academic Press. A comprehensive overview of genome diversity and evolution, including the evolution of specific genomic features.
Gregory, T. R., 2005b. Synergy between sequence and size in large-scale genomics. Nature Reviews Genetics 6: 699–708. Discussion of the impact of genome sequencing technology on the analysis of genome content at a large scale.
Kazazian, H. H., Jr. 2004. Mobile elements: Drivers of genome evolution. Science 303: 1626–1632. This review is a concise introduction to the impact of transposable elements on genomes.
Lynch, M. 2007. The Origins of Genome Architecture. Sunderland, MA: Sinauer. Detailed description of the mechanisms of genome evolution in the context of the theory of constructive neutral evolution.
Roy, S. W., and W. Gilbert. 2006. The evolution of spliceosomal introns: Patterns, puzzles, and progress. Nature Reviews Genetics 7: 211–221. Overview of the origin and maintenance of introns in eukaryotes.
Stolzfus, A. 1999. On the possibility of constructive neutral evolution. Journal of Molecular Evolution 49: 169–181. Initial presentation of constructive neutral evolution theory for the evolution of eukaryotic genomes.
Thomas, C. A., Jr. 1971. The genetic organization of chromosomes. Annual Review of Genetics 5: 237–256. Original description of the C-value paradox for chromosome size and organismal complexity.
Wahls, W. P., and M. K. Davidson. 2011. DNA sequence-mediated, evolutionarily rapid redistribution of meiotic recombination hotspots. Genetics 189: 685–694. Presentation of a model encompassing the rapid evolution of recombination hot spots and the protein Prdm9.
Webster, M. T., and L. D. Hurst. 2012. Direct and indirect consequences of meiotic recombination: Implications for genome evolution. Trends in Genetics 28: 101–109. Summary of the effects of recombination on genome structure and content from the perspective of population genetics.